
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of a Fe3O4@P4VP@metal–organic framework core–shell structure and studies of its aerobic oxidation reactivity†
Zongcheng Miaoa,
Xin Shu *b and
Daniele Ramellac
*b and
Daniele Ramellac
aXijing University, Xi'an, Shaanxi Province 710123, P. R. China
bCollege of Science, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: shuxin@mail.buct.edu.cn
cTemple University-Beury Hall, 1901, N. 13th Street, Philadelphia, PA 19122, USA
First published on 12th January 2017
Abstract
A novel core–shell magnetic Fe3O4@P4VP(poly(4-vinylpyridine))@MIL-100(Fe) composite material has been successfully synthesized. The Fe3O4@P4VP core was initially prepared via polymerization of P4VP on the surface of pre-made Fe3O4 microspheres. The MIL-100(Fe) (MIL, Matérial Institut Lavoisier) metal–organic framework shell was then introduced through a layer by layer strategy. The obtained Fe3O4@P4VP@MIL-100(Fe) catalyst was applied in the selective oxidation of alcohols using molecular oxygen as the oxidant, and a variety of substrates were tolerated. The initial activity of the Fe3O4@P4VP@MIL-100(Fe) core–shell catalyst was maintained after ten reuses.
1. Introduction
Catalytic oxidations, such as conversions of alcohols to their corresponding carbonyl compounds, are of considerable importance in fine and industrial organic chemistry.1 Recently, aerobic transition metal-promoted selective oxidations have received increasing attention because of the inexpensiveness and environmental friendliness of molecular oxygen.2 Studies about homogenous catalysts are abundant in the literature.3 However, homogeneous catalysts are inherently difficult to recover from the reaction mixture, causing industrial wastes and additional costs.4 For this reason, heterogeneous catalysts are preferable for organic transformations, as they display several advantages during the purification and recovery steps of products and catalysts.5As a result, numerous heterogeneous catalysts, such as molecular sieve,6 polymer supported7 and inorganic microspheres supported ones, have been developed in recent years.8 Among these solid catalysts, core–shell structured ones have been considered the most ideal ones, thanks to their unique physicochemical and multi-functional properties.9 Core–shell structures bearing magnetic functionality are of particular interest as an effective separation method because of their easy and quick response in an external magnetic field.10 A variety of magnetic core–shell composites have been developed utilizing different coatings, such as silica, carbon, polymer and porous materials on a Fe3O4 core.11 Several hybrid magnetic core–shell nanocatalysts have been utilized in a variety of catalytic reactions including reductions, oxidations, epoxidations, coupling reactions and photocatalysis.12 Many studies have reported immobilization of transition metal salts into core–shell structures.13 On the other hand, significant metal leaching and poor catalyst recyclability have been recurring issues in these metal immobilized systems. It is difficult to address such limitations because of the low molecular interactions present in the coordination bonds between the metal and the core–shell structure (Scheme 1).14
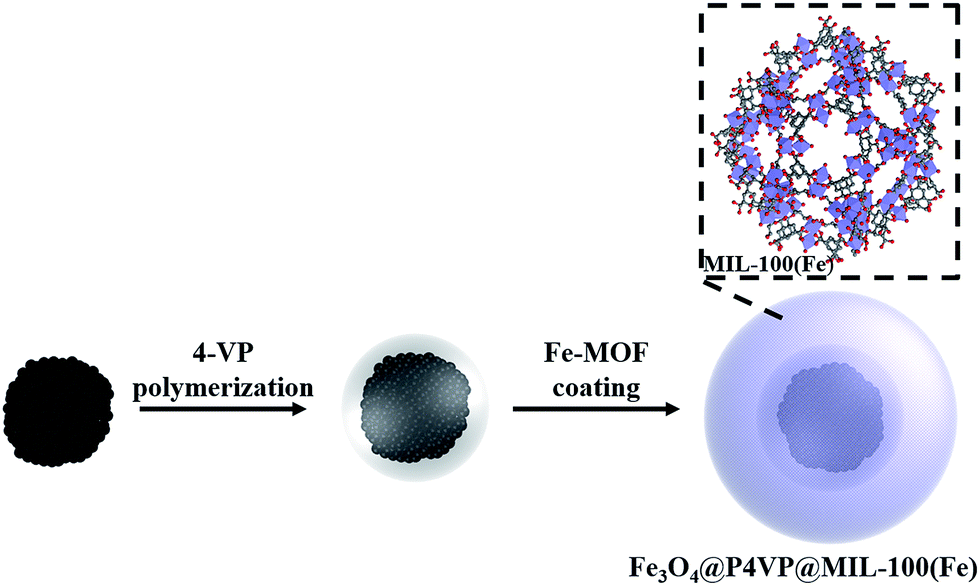 | ||
| Scheme 1 Schematic illustration of the synthesis of Fe3O4@P4VP@MIL-100(Fe). | ||
Metal–organic frameworks (MOFs)15 have gained increasing focus in catalysis, as a kind of porous materials with extremely large surface area, a high number of active sites and good stability.16 Therefore, fabrication of MOFs coated magnetic core–shell catalysts could address the problem of catalyst leaching and the need for recyclability.17 In this work, we wish to report a core–shell–shell structured Fe3O4@P4VP@MIL-100(Fe) catalyst, which was assembled by coating MIL-100(Fe) on the surface of Fe3O4@P4VP microspheres.18 The inner shell of the P4VP polymer layer played an important role in the absorption of Fe3+ ions for the formation of the MIL-100(Fe) shell, as well as in protecting the magnetic core from destruction and aggregation during the oxidation process. The synthesized Fe3O4@P4VP@MIL-100(Fe) was characterized by TEM, field-emission SEM (FESEM), powder X-ray diffraction (PXRD), nitrogen adsorption/desorption analysis, FTIR spectroscopy, thermogravimetric analysis (TGA) and vibrating sample magnetometry (VSM). The aerobic oxidation of alcohols was investigated using our synthesized Fe3O4@P4VP@MIL-100(Fe) catalyst and great yields were achieved. A variety of alcohols was compatible with our oxidation system and the core–shell catalyst could be rapidly separated by an externally applied magnetic field.
2. Experimental section
2.1 Materials
Ferric chloride hexahydrate (FeCl3·6H2O), polyvinyl pyrrolidone (PVP; Mw = 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), 4-vinyl pyridine (4-VP, 96%), divinylbenzene (DVB, 80%), 1,3,5-benzenetricarboxylic acid (H3BTC), potassium persulfate (KPS, 99%), 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), trimethylacetaldehyde, benzyl alcohol, substituted benzyl alcohols, 2-pyridinemethanol, cinnamic alcohol, 3-methyl-2-buten-1-ol, 3-methyl-2-buten-1-ol, 1-octyl alcohol, 1-phenylethanol, 2-cyclohexen-1-ol, cyclopentanol, cyclooctene, cyclohexene, cyclododecene, norbornene and α-pinene were purchased from Alfa Aesar. Poly(acrylic acid) (PAA; Mw = 1800) was obtained from Sigma-Aldrich. Anhydrous ethanol, ethylene glycol (EG), sodium acetate and potassium nitrite were purchased from Beijing Chemical Reagents Company (China). 4-Vinyl pyridine was purified by distillation under reduced pressure to remove inhibitor hydroquinone before use.
000), 4-vinyl pyridine (4-VP, 96%), divinylbenzene (DVB, 80%), 1,3,5-benzenetricarboxylic acid (H3BTC), potassium persulfate (KPS, 99%), 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), trimethylacetaldehyde, benzyl alcohol, substituted benzyl alcohols, 2-pyridinemethanol, cinnamic alcohol, 3-methyl-2-buten-1-ol, 3-methyl-2-buten-1-ol, 1-octyl alcohol, 1-phenylethanol, 2-cyclohexen-1-ol, cyclopentanol, cyclooctene, cyclohexene, cyclododecene, norbornene and α-pinene were purchased from Alfa Aesar. Poly(acrylic acid) (PAA; Mw = 1800) was obtained from Sigma-Aldrich. Anhydrous ethanol, ethylene glycol (EG), sodium acetate and potassium nitrite were purchased from Beijing Chemical Reagents Company (China). 4-Vinyl pyridine was purified by distillation under reduced pressure to remove inhibitor hydroquinone before use.
2.2 Synthesis of Fe3O4 nanoparticles with PAA
The Fe3O4 nanoparticles modified on the surface with poly(acrylic acid) (PAA) were synthesized by solvothermal method according to reported literature. Firstly, FeCl3·6H2O (1.08 g) and PAA (0.108 g) were dissolved in ethylene glycol (40 mL) under magnetic stirring at 60 °C, followed by adding sodium acetate (9 g) under vigorous stirring to form a uniform solution. Then, the solution was decanted to a stainless-steel autoclave with Teflon-lining (50 mL) and maintained at 200 °C for 12 h. After the autoclave was cooled to room temperature, the black product was collected using a magnet. The Fe3O4 nanoparticles modified with PAA were washed with deionized water and ethanol several times, collected and dried under vacuum.2.3 Preparation of core–shell Fe3O4@P4VP microspheres
The core–shell Fe3O4@P4VP microspheres were prepared by coating P4VP shell on the surface of Fe3O4 nanoparticles through radical initiated polymerization. Fe3O4 nanoparticles modified with PAA (0.1 g) were dispersed in 100 mL of a 0.15 wt% PVP aqueous solution under ultrasonication. The uniform solution was mixed with emulsion of 4-VP (0.125 g) and DVB (0.125 g) in aqueous solution (20 mL) that contains PVP (0.05 g). Then it was emulsionized by ultrasonic after placed in a 250 mL four-necked flask. The liquid was mechanical stirred for 4 hours under a flow of nitrogen and then polymerization of 4-VP was initiated by KPS (10 mg); at this point, the solution was heated to 70 °C. Core–shell Fe3O4@P4VP microspheres were obtained after 4 h, washed with deionized water and ethanol three times and dried under vacuum.2.4 Synthesis of core–shell–shell Fe3O4@P4VP@MIL-100(Fe) microspheres
The core–shell–shell Fe3O4@P4VP@MIL-100(Fe) microspheres were synthesized by a step-by-step strategy. Typically, 0.05 g of Fe3O4@P4VP microspheres were dispersed in 10 mL of a 4.0 mM FeCl3·6H2O ethanol solution for 15 minutes and collected with a magnet. Then the microspheres were dispersed in a H3BTC ethanol solution (4 mL, 10 mM) and stirred at 70 °C for 30 minutes. The product was washed with ethanol and vacuum dried after twenty cycles.2.5 Typical catalytic procedure for aerobic oxidation of alcohol
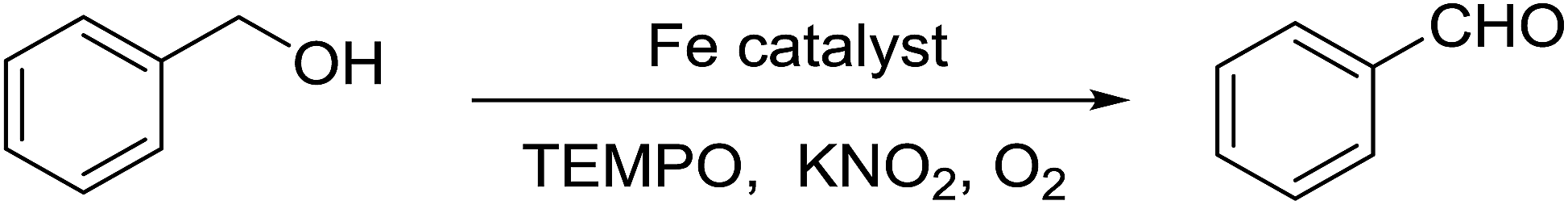
Alcohol (1.0 mmol), Fe3O4@P4VP@MIL-100(Fe) catalyst (5 mol%) were mixed in acetonitrile (5 mL); TEMPO (0.5 equiv.) and KNO2 (0.2 equiv.) were added in a 25 mL flask equipped with a condenser. The catalytic reaction was stirred at 75 °C under 1 atm of O2. After the reaction, the Fe3O4@P4VP@MIL-100(Fe) catalyst was recovered by applying an external magnetic field and the yield of product was determined by GC-MS.2.6 Leaching test and catalyst recycling
The hot filtration test for the aerobic oxidation of benzyl alcohol was performed on a sample taken from the mixture after letting the reaction run for 4 hours. The catalyst was separated by a magnet and the supernatant solution was transferred into a new flask letting the reaction continue in absence of the catalyst. After certain reaction time, the reaction solution was analyzed by GC-MS. The recyclability of the heterogeneous catalyst was tested for the oxidation of alcohols and epoxidation of olefins. The reaction run with a batch of recycled catalyst was performed under the conditions described above.3. Results and discussion
The Fe3O4 microspheres modified with PAA were synthesized by a solvothermal method reported in literature.19 These microspheres were composed of tiny Fe3O4 nanocrystals within 10 nm (Fig. 1a). The P4VP shell was successfully grafted on Fe3O4 to form core–shell composite microspheres held together by hydrogen bonds between carboxyl groups of PAA chains and pyridine. The diameters of the magnetic core and polymer shell were about 200 and 38 nm, respectively (Fig. 1b). Fe3O4@P4VP@MIL-100(Fe) core–shell–shell catalysts were prepared by a layer-by-layer method with 5 to 20 assembling cycles (Fig. 1c–f). The Fe3+ ions were adsorbed on the surface of P4VP through pyridine coordination, followed by reacting with 1,3,5-benzenetricarboxylic acid to complete each assembling cycle. The thickness of the MIL-100(Fe) shell was controlled by adjusting the assembling cycle. The MOF shells could be increased significantly to achieve a thickness ranging between 15 and 90 nm. | ||
| Fig. 1 HRTEM images of (a) Fe3O4, (b) Fe3O4@P4VP and Fe3O4@P4VP@MIL-100(Fe) core–shell after (c) 5, (d) 10, (e) 15, (f) 20 assembling cycles. | ||
To further confirm the crystalline structures of the Fe3O4, Fe3O4@P4VP and Fe3O4@P4VP@MIL-100(Fe) microspheres, powder XRD were performed and the patterns are shown in Fig. 2. The diffraction peaks of Fe3O4 (Fig. 2a) agree with standard JCPDS 75-1609.39 indicating a face-centered cubic lattice. After coating with P4VP, the peaks of the core–shell Fe3O4@P4VP microspheres are almost the same as those for Fe3O4, because of the low-crystalline nature of the polymeric shell (Fig. 2b). However, after the introduction of the MIL-100(Fe) layer, several new peaks appeared on the XRD pattern (Fig. 2c). These new peak match the peaks present in the simulated MIL-100(Fe) pattern (Fig. S1†), which indicates that the formation of MIL-100(Fe) was successful.
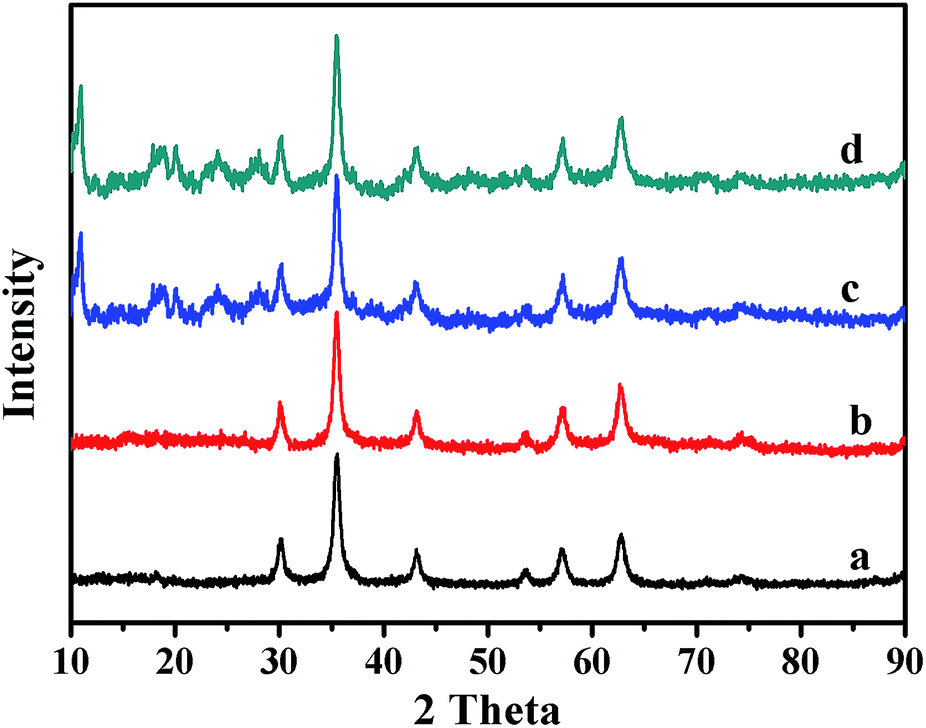 | ||
| Fig. 2 XRD patterns of (a) Fe3O4, (b) Fe3O4@P4VP, (c) Fe3O4@P4VP@MIL-100(Fe) and (d) recycled Fe3O4@P4VP@MIL-100(Fe). | ||
The EDX elemental maps further support the successfulness of the synthesis of Fe3O4@P4VP@MIL-100(Fe), and that the iron MOF was well grafted on the surface of Fe3O4@P4VP (Fig. 3). The iron elemental maps of the Fe3O4@P4VP@MIL-100(Fe) also showed an excellent distribution of iron content on the surface. The existence of iron in the Fe3O4 core structure was also observed, as shown in Fig. 3.
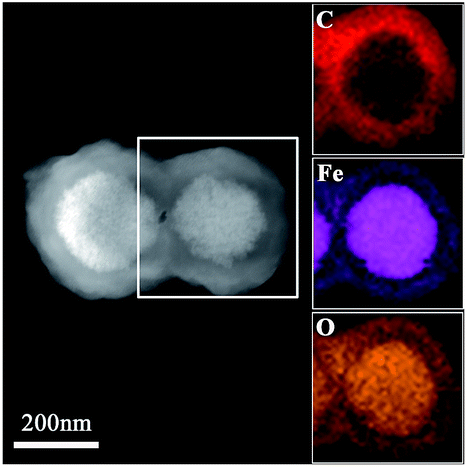 | ||
| Fig. 3 The TEM images of the Fe3O4@P4VP@MIL-100(Fe) and EDX elemental maps of Fe, O and C, respectively. | ||
The FTIR spectrum of the product is shown in Fig. 4. The relatively high intensity of the band at 592 cm−1 is characteristic of Fe–O vibrations. The characteristic absorption bands at 1707, 1250, and 1165 cm−1 correspond to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the carboxylic group, to the in-plane bending of C–O–H, and to the –(C–O)H stretching, proving the existence of rich –COOH functional groups on the surface of the Fe3O4 particles. As for the Fe3O4@P(4-VP-DVB) microspheres, the characteristic absorptions at 1603, 1562, and 1417 cm−1 are attributed to the vibrational modes of the pyridine ring. The band at 1603 cm−1 corresponds to the stretching vibration absorption of the C–N bond, and the bands at around 1562 and 1417 cm−1 are attributed to the stretching vibration absorption of the CC bond; this further proves the encapsulation of P4VP on the surface of the Fe3O4 particles. The peaks at 1435 and 1575 cm−1 are due to the stretching vibrations of the C–C bonds of the benzene ring, while the peaks at 1376 and 1618 cm−1 are attributed to the C–O stretching vibrations of the carboxylic moiety, suggesting that MIL-100 (Fe) shell has been successfully grafted on the surface of Fe3O4@P4VP.
O stretching of the carboxylic group, to the in-plane bending of C–O–H, and to the –(C–O)H stretching, proving the existence of rich –COOH functional groups on the surface of the Fe3O4 particles. As for the Fe3O4@P(4-VP-DVB) microspheres, the characteristic absorptions at 1603, 1562, and 1417 cm−1 are attributed to the vibrational modes of the pyridine ring. The band at 1603 cm−1 corresponds to the stretching vibration absorption of the C–N bond, and the bands at around 1562 and 1417 cm−1 are attributed to the stretching vibration absorption of the CC bond; this further proves the encapsulation of P4VP on the surface of the Fe3O4 particles. The peaks at 1435 and 1575 cm−1 are due to the stretching vibrations of the C–C bonds of the benzene ring, while the peaks at 1376 and 1618 cm−1 are attributed to the C–O stretching vibrations of the carboxylic moiety, suggesting that MIL-100 (Fe) shell has been successfully grafted on the surface of Fe3O4@P4VP.
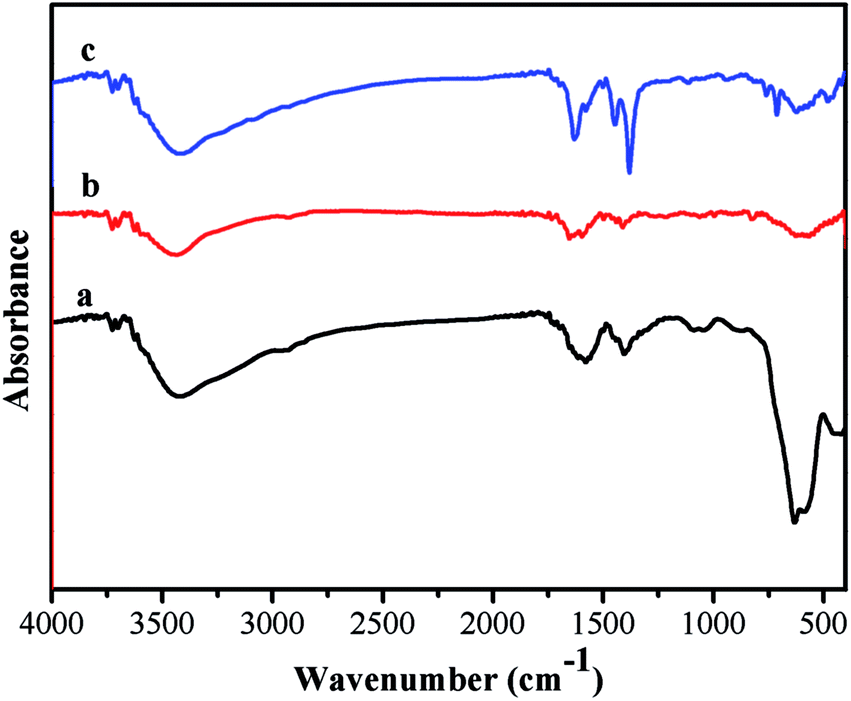 | ||
| Fig. 4 FTIR spectra of (a) Fe3O4, (b) Fe3O4@P4VP and (c) Fe3O4@P4VP@MIL-100(Fe). | ||
Surface area and pore size distribution were characterized by nitrogen adsorption/desorption experiment. The N2 adsorption/desorption isotherms are shown in Fig. 5 and the curves are formed by the type I isotherms (Fig. 5). In addition, the isotherms are of type II at the range of high P/P0. The surface area and pore volume of Fe3O4@P4VP@MIL-100(Fe) were calculated by the BET model and the values were determined to be 346.2 and 2.31 m3 g−1, respectively. The surface area is significant improved, when compared to its Fe3O4@P4VP precursor, which has a surface area of 31.1 m2 g−1.
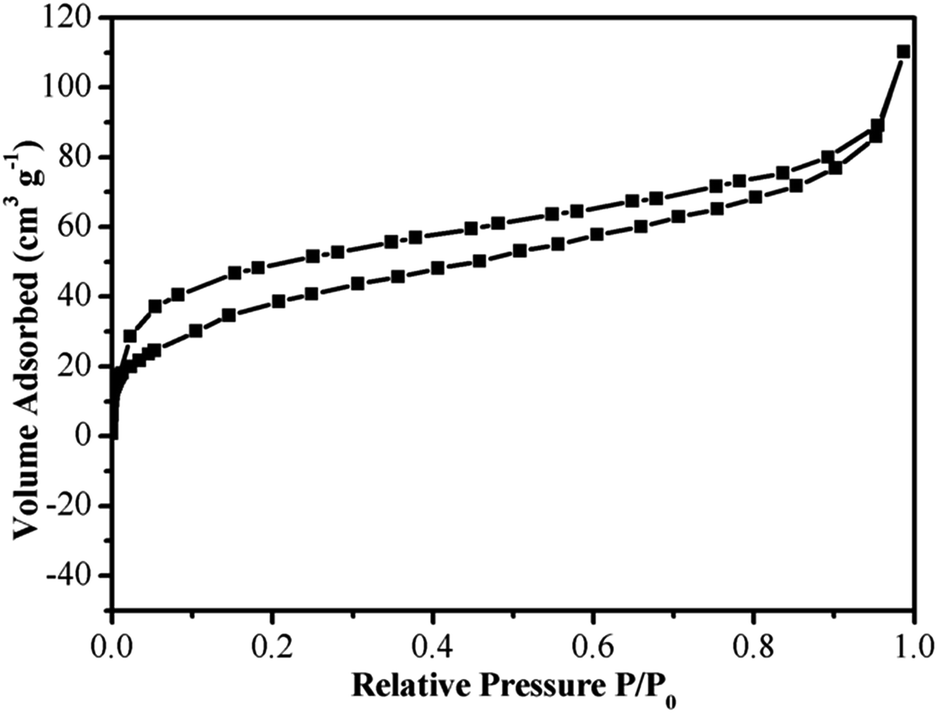 | ||
| Fig. 5 N2 adsorption–desorption isotherms of the Fe3O4@P4VP@MIL-100(Fe) catalyst. | ||
TGA curves of Fe3O4, Fe3O4@P4VP and Fe3O4@P4VP@MIL-100(Fe) are shown in Fig. 6. For PAA modified Fe3O4 microspheres, the weight loss was about 17 wt% over two steps, attributed to the desorption of adsorbed water and PAA chains in Fe3O4 microspheres (Fig. 6a). The TGA curves of the Fe3O4@P4VP microspheres exhibited three steps of weight loss with 52 wt% of the initial weight remaining. Elimination of water, decomposition of the oligomeric structure and the desorption of P4VP shell correspond to the three weight losses (Fig. 6b). As expected, the mass loss of Fe3O4@P4VP@MIL-100(Fe) was lower than that of Fe3O4@P4VP, likely due to the formation of iron oxide through calcination (Fig. 6c).
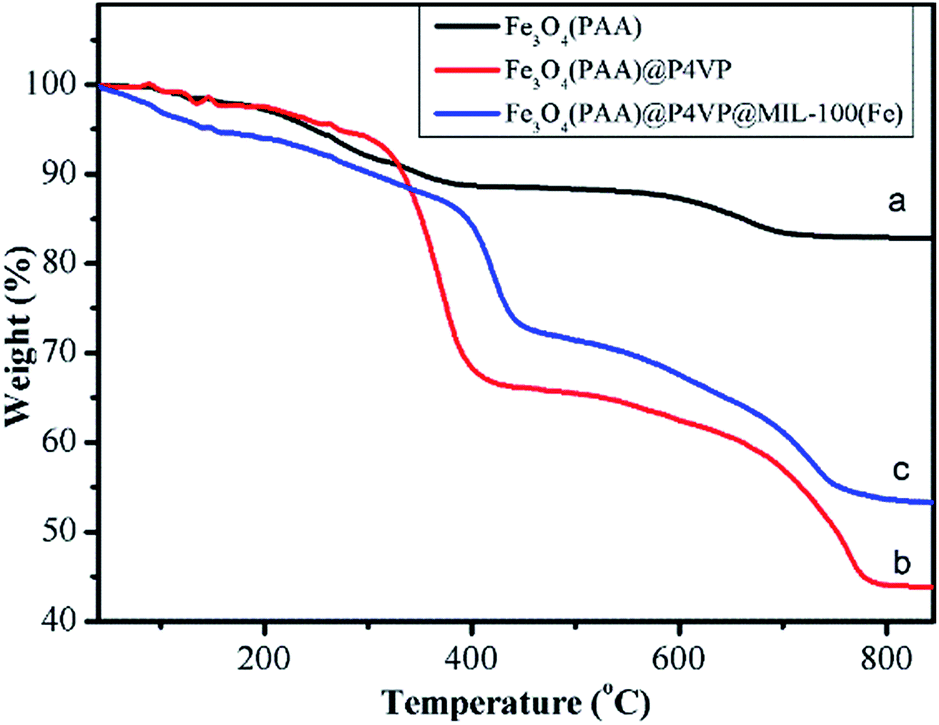 | ||
| Fig. 6 TGA curves of (a) Fe3O4, (b) Fe3O4@P4VP and (c) Fe3O4@P4VP@MIL-100(Fe). | ||
The magnetic properties of Fe3O4 (PAA), Fe3O4@P4VP and Fe3O4@P4VP@MIL-100(Fe) were measured by vibrating sample magnetometry (VSM). All three samples exhibited superparamagnetism, presenting no remanence or coercitive forces (Fig. 7). The resultant magnetization saturation values of Fe3O4 (PAA), Fe3O4@P4VP and Fe3O4@P4VP@MIL-100(Fe) were 73.90, 47.35 and 28.21 emu g−1, respectively, indicating strong magnetic response by the materials. The saturation magnetic moment of Fe3O4@P4VP and Fe3O4@P4VP@MIL-100(Fe) are lower than that of Fe3O4 because the P4VP and MOF shells are non-magnetic materials. Moreover, the saturation magnetic moment shows linear dependence on the content of magnetite. Therefore, the amount of P4VP and MIL-100(Fe) calculated according to the VSM results were 21.41 wt% and 40.42 wt%, respectively.
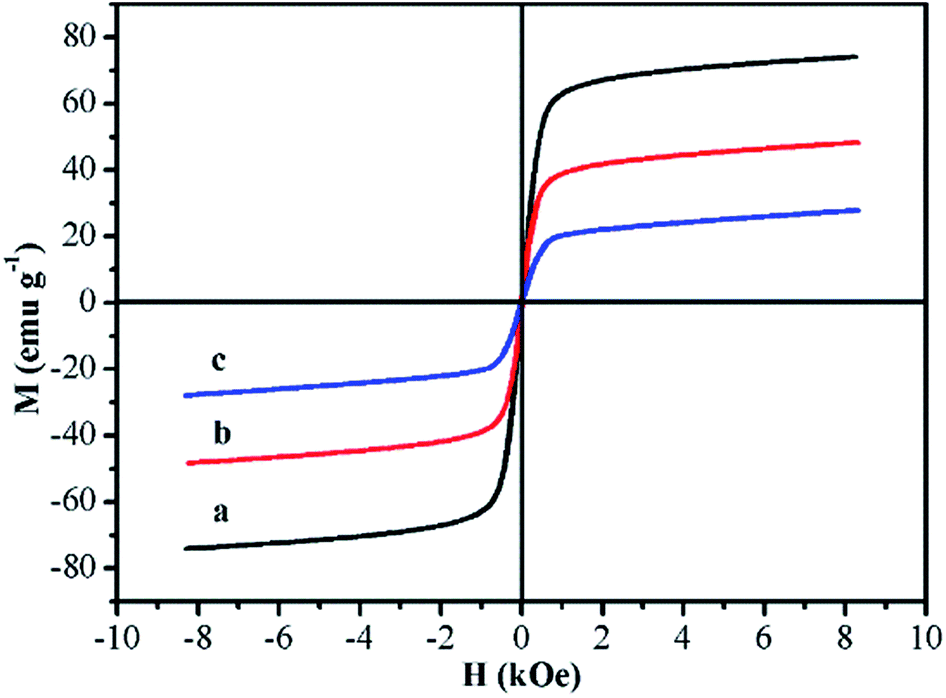 | ||
| Fig. 7 Room-temperature magnetic hysteresis loops of (a) Fe3O4, (b) Fe3O4@P4VP and (c) Fe3O4@P4VP@MIL-100(Fe). | ||
The catalytic activity of the synthesized Fe3O4@P4VP@MIL-100(Fe) toward the aerobic oxidation reaction was evaluated employing alcohol substrates, and using TEMPO as the radical initiator. No benzaldehyde was detected in the absence of the catalyst under our reaction conditions (Table 1, entry 1). Also, the employment of Fe3O4@P4VP as the catalyst failed to oxidize any benzyl alcohol to its corresponding aldehyde (Table 1, entry 2). Homogeneous iron catalysts were evaluated and almost quantitative conversion was observed. However, slightly lowered selectivity was observed due to the formation of benzoic acid (Table 1, entries 3 and 4). 82% conversion and 99% selectivity were achieved using MIL-100(Fe) MOF catalyst at 5 mol% catalyst loading (Table 1, entry 5). The amounts of MIL-100(Fe) and Fe3O4@P4VP@MIL-100(Fe) added in this reaction were 5 mol%, based on the same iron loading (Table 1, entries 5 and 6). The Fe3O4@P4VP@MIL-100(Fe) catalyst gave an excellent conversion and yield due to the efficient utilization of the porous MOF structure, which was coated as a nanolayer on the magnetic core. Hydrogen peroxide has been utilized as the oxidant, only low yield was obtained (Table 1, entry 7). Our reaction condition is not compatible with aqueous hydrogen peroxide oxidant. Furthermore, air was also tested as the low concentration form of oxygen and moderate yield was obtained (Table 1, entry 8). Further solvent screenings showed that CH3CN is the most suitable solvent for the benzyl alcohol oxidation (Table 1, entries 6, 9–11). Aromatic solvents such as toluene gave slightly compromised yields, presumable due to their lower polarity (Table 1, entry 9). Moreover, oxygen containing solvents, such as THF and ethanol, were not suitable oxidation reaction solvent due to possible metal coordination (Table 1, entries 10 and 11).
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Conv.b | Sel.b |
| a Reaction conditions: 1.0 mmol of benzyl alcohol, 5 mol% of Fe3O4@P4VP@MIL-100(Fe) catalyst, 5.0 mL of solvent, 0.2 mmol of TEMPO, and 0.2 mmol of KNO2; 60 °C, 12 h under 1 atm O2.b Conversions and selectivities were calculated by GC-MS using nitrobenzene as the internal standard.c H2O2 was used as the oxidant.d Air was used as the oxidant. | ||||
| 1 | — | CH3CN | <5% | — |
| 2 | Fe3O4@P4VP | CH3CN | <5% | — |
| 3 | FeCl3 | CH3CN | 99% | 92% |
| 4 | Fe(NO3)3 | CH3CN | 99% | 93% |
| 5 | MIL-100(Fe) | CH3CN | 86% | 99% |
| 6 | Fe3O4@P4VP@MIL-100(Fe) | CH3CN | 99% | 99% |
| 7c | Fe3O4@P4VP@MIL-100(Fe) | CH3CN | 36% | 99% |
| 8d | Fe3O4@P4VP@MIL-100(Fe) | CH3CN | 62% | 99% |
| 9 | Fe3O4@P4VP@MIL-100(Fe) | PhCH3 | 82% | 99% |
| 10 | Fe3O4@P4VP@MIL-100(Fe) | THF | 67% | 99% |
| 11 | Fe3O4@P4VP@MIL-100(Fe) | EtOH | 25% | 99% |
Conversion was monitored over time employing various catalyst loadings of Fe3O4@P4VP@MIL-100(Fe); the results are summarized in Fig. 8. Relatively low conversion was observed at 1–3 mol% of catalyst loading, indicating the crucial role of the Fe-derived catalyst. However, conversion and yield were not satisfying until the catalyst loading was increased to 5 mol%. As a result, 5 mol% loading of Fe3O4@P4VP@MIL-100(Fe) was chosen to be the optimal reaction condition for future studies.
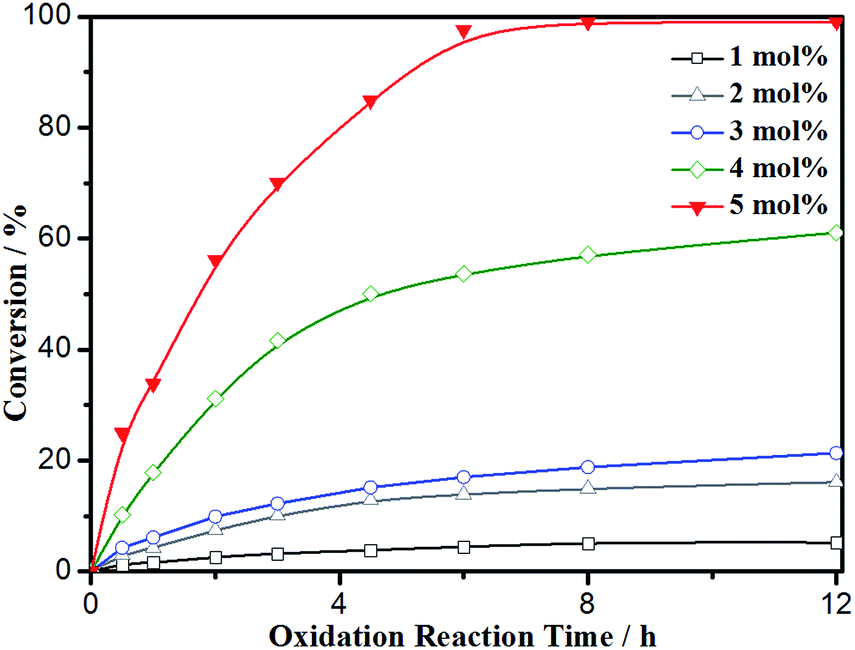 | ||
| Fig. 8 Conversion vs. time at various core–shell catalyst loading. | ||
With the optimal reaction conditions in hand, several alcohol substrates were chosen to study the aerobic oxidation catalytic performance of Fe3O4@P4VP@MIL-100(Fe). Benzyl alcohol was transformed to the corresponding benzaldehyde in 99% yield after 12 h (Table 2, entry 1). For the aerobic oxidation of benzyl alcohol in presence of Fe3O4@P4VP@MIL-100(Fe) as the catalyst, the turnover number (TON) was calculated to be 20. Electron-rich benzyl alcohols, such as p-methyl benzyl alcohol and p-methoxy benzyl alcohol were reactive under the optimal reaction conditions, affording 99% and 92% yield, respectively (Table 2, entries 2 and 3). Alcohol substrates bearing electron-withdrawing functional groups were also tolerated; although a slightly lowered yield was observed for p-fluoro benzyl alcohols (Table 2, entry 4). The heterocyclic alcohol pyridin-2-ylmethanol was converted smoothly to its corresponding aldehyde in the presence of the Fe derived core–shell catalyst (Table 2, entry 5). Cinnamyl alcohol was also evaluated as an example of allylic alcohol; cinnamaldehyde was formed as the only product in 92% yield (Table 2, entry 6). In order to test the transformation of secondary alcohols to ketones, 1-phenylethanol and cyclohexanol were both tested; unfortunately, only low conversions were observed (Table 2, entries 7 and 8).
| a Reaction conditions: 1.0 mmol of alcohol, 5 mol% of Fe3O4@P4VP@MIL-100(Fe) catalyst, 5.0 mL of solvent, 0.5 mmol of TEMPO, and 0.2 mmol of KNO2; stirred at 60 °C for 12 h under 1 atm O2. |
|---|
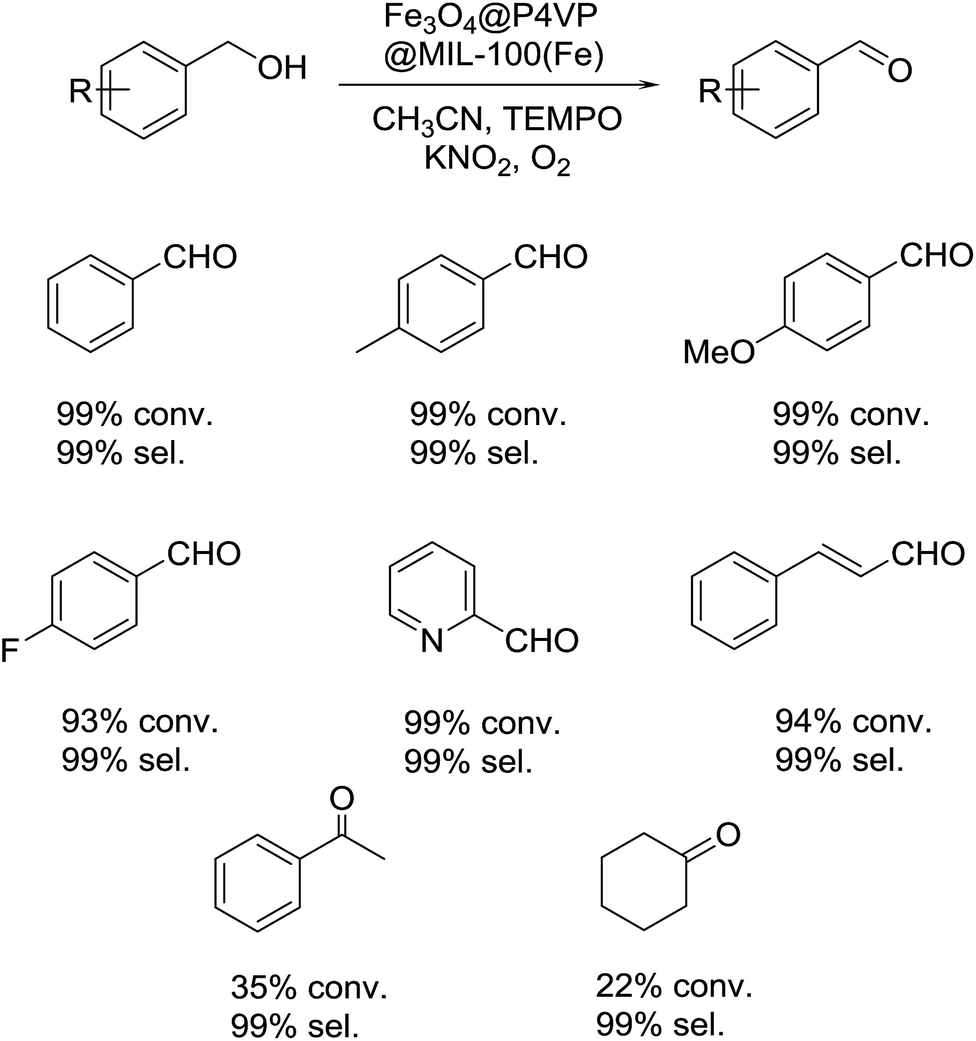 |
The recyclability of the catalyst was studied applying the optimal reaction conditions and utilizing 5 mol% of Fe3O4@P4VP@MIL-100(Fe) catalyst in acetonitrile (Fig. 9). The same batch of the Fe3O4@P4VP@MIL-100(Fe) catalyst was reused over ten reaction cycles; conversion and selectivity were retained at 99%, and the yield of benzaldehyde was compromised only slightly after ten cycles (Fig. 6). The retaining of high conversions and selectivities after ten cycles indicates that the core–shell MOF heterogeneous catalyst is highly stable. FT-IR and XRD spectra showed no significant difference between fresh Fe3O4@P4VP@MIL-100(Fe) and the batch analyzed after being used ten times (Fig. S2 and S3†).
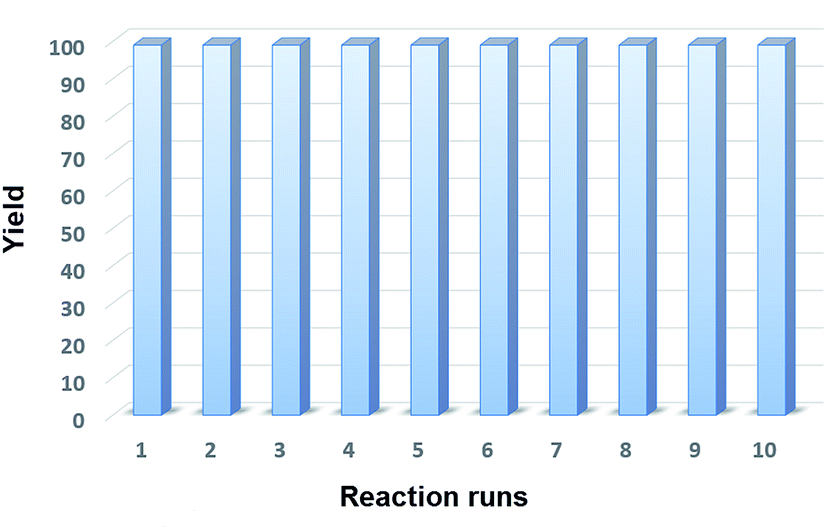 | ||
| Fig. 9 Core–shell Fe-MOF catalyst recyclability test. | ||
The hot filtration test was conducted to confirm the heterogeneous nature of the catalytic aerobic oxidation (Fig. 10). The Fe3O4@P4VP@MIL-100(Fe) catalyst was isolated through magnet after 2 h of reaction and the mixture was stirred for another 10 h. Conversion to product stopped after the catalyst was removed. This result indicates that the strong covalent bond ensures the stability of Fe3O4@P4VP@MIL-100(Fe) material during the oxidation process. And that there is almost no Fe3+ leaching into the reaction solution, as also confirmed by ICP-AAS analysis.
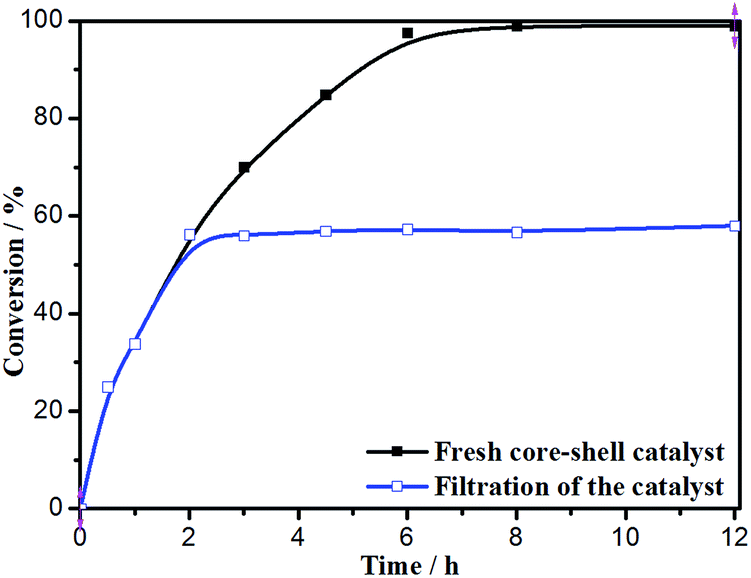 | ||
| Fig. 10 Hot filtration test of Fe3O4@P4VP@MIL-100(Fe). | ||
4. Conclusions
During this study a novel magnetic core–shell Fe3O4@P4VP@MIL-100(Fe) catalyst was designed, prepared and fully characterized. The composite microspheres were composed by a magnetic Fe3O4(PAA) core, a P4VP middle layer, and a MIL-100(Fe) MOF shell. Taking advantage of their porous Fe-derived layer, the magnetic composite microspheres were utilized as an efficient catalyst for the selective aerobic oxidation of alcohols. A variety of alcohol substrates were tolerated under our optimized conditions. The initial catalytic activity of the Fe3O4@P4VP@MIL-100(Fe) catalyst was retained after at least ten consecutive reaction cycles. A hot filtration test suggested extremely low leaching of iron into the solution. Further applications of the prepared core–shell MOF catalyst are currently under investigation.Acknowledgements
We thank the Science Research Foundation of Xijing University (Grant No. XJ16T02), the National Natural Science Foundation of China (No. 51673157) and the BUCT Fund for Disciplines Construction and Development (No. XK1529) for financial support.Notes and references
- (a) Q. Cao, L. M. Dornan, L. Rogan, N. L. Hughes and M. J. Muldoon, Chem. Commun., 2014, 50, 4524–4543 RSC; (b) J. Hou, Y. Luan, J. Tang, A. M. Wensley, M. Yang and Y. Lu, J. Mol. Catal. A: Chem., 2015, 407, 53–59 CrossRef CAS.
- B. Chen, L. Wang and S. Gao, ACS Catal., 2015, 5, 5851–5876 CrossRef CAS.
- (a) C. Parmeggiani and F. Cardona, Green Chem., 2012, 14, 547–564 RSC; (b) C. Qi, Y. Xiong, V. Eschenbrenner-Lux, C. Cong and J. A. Porco Jr, J. Am. Chem. Soc., 2016, 138, 798–801 CrossRef CAS PubMed; (c) C. Qi, T. Qin, D. Suzuki and J. A. Porco Jr, J. Am. Chem. Soc., 2014, 136, 3374–3377 CrossRef CAS PubMed; (d) Y. Luan, K. S. Barbato, P. N. Moquist, T. Kodama and S. E. Schaus, J. Am. Chem. Soc., 2015, 137, 3233–3236 CrossRef CAS PubMed.
- (a) M. Stratakis and H. Garcia, Chem. Rev., 2012, 112, 4469–4506 CrossRef CAS PubMed; (b) Y. Luan, Y. Qi, Z. Jin, X. Peng, H. Gao and G. Wang, RSC Adv., 2015, 5, 19273–19278 RSC.
- Z. Guo, B. Liu, Q. Zhang, W. Deng, Y. Wang and Y. Yang, Chem. Soc. Rev., 2014, 43, 3480–3524 RSC.
- (a) M. A. Chari and K. Syamasundar, Synthesis, 2005, 5, 0708–0710 CrossRef; (b) M. R. Farsani and B. Yadollahi, J. Mol. Catal. A: Chem., 2014, 392, 8–15 CrossRef CAS; (c) Y. Kuang, Y. Nabae, T. Hayakawa and M. Kakimoto, Appl. Catal., A, 2012, 423–424, 52–58 CrossRef CAS; (d) P. Das, N. Aggarwal and N. R. Guha, Tetrahedron Lett., 2013, 54, 2924–2928 CrossRef CAS.
- P. R. Likhar, R. Arundhathi, S. Ghosh and M. L. Kantam, J. Mol. Catal. A: Chem., 2009, 302, 142–149 CrossRef CAS.
- J. Mao, X. Hu, H. Li, Y. Sun, C. Wang and Z. Chen, Green Chem., 2008, 10, 827–831 RSC.
- R. G. Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef PubMed.
- M. B. Gawande, A. Goswami, T. Asefa, H. Guo, A. V. Biradar, D. L. Peng, R. Zboril and R. S. Varma, Chem. Soc. Rev., 2015, 44, 7540–7590 RSC.
- (a) C. Hui, C. Shen, J. Tian, L. Bao, H. Ding, C. Li, Y. Tian, X. Shi and H. J. Gao, Nanoscale, 2011, 3, 701–705 RSC; (b) Y. D. Chiang, S. Dutta, C. T. Chen, Y. T. Huang, K. S. Lin, J. C. S. Wu, N. Suzuki, Y. Yamauchi and K. C. W. Wu, ChemSusChem, 2015, 8, 789–794 CrossRef CAS PubMed; (c) Z. Dong, X. Le, C. Dong, W. Zhang, X. Li and J. Ma, Appl. Catal., B, 2015, 162, 372–380 CrossRef CAS; (d) X. Cui, W. Zuo, M. Tian, Z. Dong and J. Ma, J. Mol. Catal. A: Chem., 2016, 423, 386–392 CrossRef CAS.
- (a) Q. Zhang, I. Lee, J. B. Joo, F. Zaera and Y. Yin, Acc. Chem. Res., 2013, 46, 1816–1824 CrossRef CAS PubMed; (b) P. Hu, J. V. Morabito and C. K. Tsung, ACS Catal., 2014, 4, 4409–4419 CrossRef CAS; (c) Y. Long, M. Xie, J. Niu, P. Wang and J. Ma, Appl. Surf. Sci., 2013, 277, 288–292 CrossRef CAS; (d) X. Zhang, J. Wu, G. Meng, X. Guo, C. Liu and Z. Liu, Appl. Surf. Sci., 2016, 366, 486–493 CrossRef CAS; (e) Z. Sun, H. Li, G. Cui, Y. Tian and S. Yan, Appl. Surf. Sci., 2016, 360, 252–262 CrossRef CAS.
- W. Guo, G. Wang, Q. Wang, W. Dong, M. Yang, X. Huang, J. Yu and Z. Shi, J. Mol. Catal. A: Chem., 2013, 378, 344–349 CrossRef CAS.
- (a) S. Hübner, J. G. de Vries and V. Farina, Adv. Synth. Catal., 2016, 358, 3–25 CrossRef; (b) X. S. Zhao, X. Y. Bao, W. Guo and F. Y. Lee, Mater. Today, 2006, 9, 32–39 CrossRef CAS.
- (a) H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed; (b) S. M. Cohen, Chem. Sci., 2010, 1, 32–36 RSC; (c) Y. Luan, M. Yang, Q. Ma, Y. Qi, H. Gao, Z. Wu and G. Wang, J. Mater. Chem. A, 2016, 4, 7641–7649 RSC.
- (a) J. Gascon, A. Corma, F. Kapteijn and F. X. L. I. Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS; (b) Y. Luan, Y. Qi, H. Gao, R. S. Andriamitantsoa, N. Zheng and G. Wang, J. Mater. Chem. A, 2015, 3, 17320–17331 RSC; (c) C. Qi, D. Ramella, A. M. Wensley and Y. Luan, Adv. Synth. Catal., 2016, 358, 2604–2611 CrossRef CAS.
- (a) F. Ke, L. G. Qiu, Y. P. Yuan, X. Jiang and J. F. Zhu, J. Mater. Chem., 2012, 22, 9497–9500 RSC; (b) C. F. Zhang, L. G. Qiu, F. Ke, Y. J. Zhu, Y. P. Yuan, G. S. Xua and X. Jiang, J. Mater. Chem. A, 2013, 1, 14329–14334 RSC; (c) F. Ke, L. Wang and J. Zhu, Nanoscale, 2015, 7, 1201–1208 RSC; (d) W. W. Zhan, Q. Kuang, J. Z. Zhou, X. J. Kong, Z. X. Xie and L. S. Zheng, J. Am. Chem. Soc., 2013, 135, 1926–1933 CrossRef CAS PubMed.
- (a) D. W. Kim, H. G. Kim and D. H. Cho, Catal. Commun., 2016, 73, 69–73 CrossRef CAS; (b) P. Horcajada, S. Surblé, C. Serre, D. Y. Hong, Y. K. Seo, J. S. Chang, J. M. Grenèche, I. Margiolakid and G. Férey, Chem. Commun., 2007, 2820–2822 RSC; (c) A. Dhakshinamoorthy, M. Alvaro, P. Horcajada, E. Gibson, M. Vishnuvarthan, A. Vimont, J. M. Grenèche, C. Serre, M. Daturi and H. Garcia, ACS Catal., 2012, 2, 2060–2065 CrossRef CAS.
- H. Deng, X. Li, Q. Peng, X. Wang, J. Chen and Y. Li, Angew. Chem., Int. Ed., 2005, 44, 2782–2785 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25820d |
| This journal is © The Royal Society of Chemistry 2017 |