
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dip-coated PbS/PVP nanocomposite films with tunable band gap
Mitesh H. Patela,
Tapas K. Chaudhuri *a,
Vaibhav K. Patelb,
T. Shripathic,
U. Deshpandec and
N. P. Lallac
*a,
Vaibhav K. Patelb,
T. Shripathic,
U. Deshpandec and
N. P. Lallac
aDr. K. C. Patel Research and Development Centre, Charotar University of Science and Technology Changa, Anand District, Gujarat 388421, India. E-mail: tkchaudhri.rnd@gmail.com
bP. D. Patel Institute of Applied Sciences, Charotar University of Science and Technology, Changa, Anand District, Gujarat 388421, India
cUGC-DAE Consortium for Scientific Research, Khandwa Road, Indore, Madhya Pradesh 452001, India
First published on 16th January 2017
Abstract
PbS/polyvinylpyrrolidone (PVP) nanocomposite films with tunable band gap are synthesized by solid state in situ thermolysis. Precursor films are dip-coated on glass substrates from a methanolic solution of Pb2+–thiourea complex and PVP and then heated in air at 110 °C for 10 minutes to obtain shiny brown clear films of PbS/PVP. The formation of PbS nanoparticles in PVP matrix is confirmed by X-ray diffraction studies. The size of PbS nanoparticles varied from 2 to 8 nm depending of the weight fraction of the Pb2+–thiourea complex in the PVP. Transmission electron microscopy shows that the nanoparticles are spherical. The transmission spectra of the PbS/PVP films in the wavelength range of 300 to 2600 nm showed absorption edges near 900 nm and below due to the presence of PbS nanoparticles. The band gaps of PbS/PVP films, as determined from Tauc plots, varied from 0.8 to 1.92 eV as the weight fraction of the complex decreased from 82 to 36%. Fourier transformed infrared (FTIR) spectroscopy, X-ray photoelectron spectroscopy (XPS) and 13C Nuclear Magnetic Resonance (NMR) studies reveal that there is strong interaction between PbS and PVP which limits the growth of the nanoparticles.
1. Introduction
Organic–inorganic nanocomposites (NCs) consisting of PbS nanoparticles (NPs) or quantum dots (QDs) implanted in polymers have received considerable attention due to the significant quantum size effect of PbS itself.1 Semiconducting conjugated or insulating polymers have been used as the host in PbS/polymer nanocomposites. Conjugated polymer hosts include poly[2-methoxy-5-(2′-ethylhexyloxy-p-phenylenevinylene)] (MEH-PPV),2–8 poly(2-(6-cyano-6′-methyl heptyloxy)-1,4-phenylene) (CN-PPP),2 poly(3-hexylthiophene-2,5-diyl) (P3HT),4,9–12 polypyrrole (PPy),13 and poly[2-methoxy-5-(3′,7′-dimethyloctyloxy)-1,4-phenylenevinylene] (MDMO-PPV).14 Insulating polymers, such as, polyacryl amide (PAM),15,16 poly-p-xylylene (PPX),17 polyacrylonitrile (PAN),18 poly(methyl methacrylate) (PMMA),19 polystyrene (PSt),7,20,21 poly thiourethane (PTU),22 polyvinyl alcohol (PVA),23–28 poly(vinyl acetate) (PVAc),29 poly(vinyl pyrrolidone) (PVP),30,31 polyvinylidene fluoride (PVDF),32 and poly(ethylene oxide) (PEO)33 have been employed as hosts. Films of PbS/conjugated polymer NCs have been basically investigated as a photoactive component of bulk heterojunction solar cells,6,10,12,14,32,34 and photo detectors. Application of PbS/insulated polymer NC films as photo-detectors,3,20,27 high refractive index material,22 non-linear optical device,19,23,29 have been also explored. However, studies on interaction between PbS NPs and host polymer or control of band gaps of PbS NPs by polymer matrix are very few. Further, deposition of PbS/polymer NC thin films by simple dip-coating technique from a single precursor solution have not been studied. Also there is no detailed study on PbS/PVP nanocomposite.PbS is an earth-abundant low band gap (0.41 eV at 300 K) semiconductor with extraordinary size dependent properties at nanoscale. PbS has a large excitionic Bohr radius of 18 nm because of high dielectric constant 17.2 and low effective mass of electrons (≈0.1me). Hence, it exhibits a strong quantum size effect for larger NPs/QDs (<18 nm) as compared to CdS or CdSe (<4 nm). Thus, the band gap of PbS NPs can be easily tuned from 0.41 (bulk) to 4 eV by selecting appropriate size.
To synthesize nanocomposites (NCs), two different schemes are generally employed depending on where the nanoparticles (NPs) are processed ex situ or in situ. In the ex situ method, NPs are made separately and then blended either with a monomer before the polymerization or directly with the polymer. The in situ method of NCs consists of the synthesis of inorganic NPs either in monomers or in polymers. Both the routes are generally liquid based and solid NCs are obtained by co-precipitation or solvent evaporation. Although both ex situ and in situ solution-based routes have been utilized for obtaining PbS–polymer nanocomposites, the latter has been favoured the most. Nanocomposites were synthesized from a solution of polymer and lead salt by introducing sulphur ions from different sources. Sulphur ions was added in form of H2S gas,15,22,24,25,33 elemental sulphur,4–7,9,11 Na2S,23,26,27,29 thioacetamide14,19,21 and dithiooxamide.16 In a different approach, Qiao et al.18 produced PbS/PAN nanocomposite by irradiating the solution by γ-ray. Pateland Chaudhuri30 reported the synthesis of PbS/PVP nanocomposite by in situ decomposition of Pb2+–TU complex into PbS in PVP solution at room temperature (300 K). All the in situ methods reported earlier are in liquid state. That is, PbS nanoparticles are first synthesized in host polymer solution and then solid nanocomposite films are deposited by solution casting. Alternatively, nanocomposites can be also prepared by synthesizing nanoparticles in situ solid host polymer.34 In this solid state in situ (SSIS) process, sulphide precursor and a host polymer is first mixed in a solvent and then solid polymer–precursor film is cast by solvent evaporation or dip-coating or spin-coating. The polymer–precursor film is subsequently heated in air at a specific temperature to synthesize sulphide NPs in situ by thermolysis and sulphide–polymer nanocomposite (NC) is formed. During thermolysis step, gaseous side products are formed which readily escape from the polymer matrix. The solid polymer matrix restricts the growth of sulphide NPs and prevents agglomeration. Homogeneous distribution of NPs is most likely in this process because the sulphide–precursor and the host polymer are mixed at the molecular level. Further, the proportion of NPs in polymer matrix can be easily varied by changing the concentration of sulphide precursor without any concern about non uniformity of NPs. Review of earlier work on PbS/polymer NCs reveals that only Chaudhuri et al.20 have utilized SSIS process to prepare PbS/polystyrene NC.
In this paper we report the synthesis of PbS/PVP NC films by solid state in situ method. Precursor films are first deposited on glass by dip coating from a precursor solution of Pb2+–TU complex and PVP in methanol. Shiny brown PbS/PVP nanocomposite films are formed by heating them at 110 °C in air. The band gap of these films could be tuned from 0.8 to 1.92 eV by varying the concentration of Pb2+–TU complex in PVP. Fourier transform infrared spectroscopy (FTIR), X-ray photoelectron spectroscopy (XPS) and 13C nuclear magnetic resonance (NMR) have been used to examine in detail interaction between PbS NPs and PVP. It has been found there exists strong interaction between PbS and PVP.
2. Materials and method
PbS/PVP NC films were deposited on glass substrates by dip coating from precursor solution of lead acetate trihydrate (Pb(OAc)2), thiourea (TU) and poly(vinyl pyrrolidone) (PVP) in methanol. The precursor solution was prepared as follows: equimolar of Pb(OAc)2 and TU was first dissolved in methanol with stirring to form Pb2+–TU complex and then PVP powder (1% wt/vol) was added to form the precursor solution.The final volume of precursor solution was 100 mL. Chaudhuri et al.35 and Vankhade et al.36 have shown that Pb(OAc)2 combines with equimolar TU in methanol to form Pb2+–TU complex which on thermolysis at around 100 °C converts to equimolar PbS. Precursor solutions with different concentrations of Pb(OAc)2 and TU (hence Pb2+–TU complex) with PVP were prepared as shown in Table 1. The table shows the weight fractions of Pb2+–TU complex in solid precursor films along with expected weight and volumetric ratio of PbS nanoparticles to PVP after formation of NC.
| Methanolic precursor solution (100 mL) | Pb2+–TU complex in precursor film (wt%) | PbS in PbS/PVP film | |||
|---|---|---|---|---|---|
| Pb(OAc)2 (M) | TU (M) | PVP (g) | Wt% | Vol% | |
| 0.0125 | 0.0125 | 1 | 36 | 23 | 4.5 |
| 0.025 | 0.025 | 1 | 52 | 37 | 8.5 |
| 0.05 | 0.05 | 1 | 70 | 54 | 16 |
| 0.1 | 0.1 | 1 | 82 | 70 | 27 |
Weight of PbS was deduced from the fact that Pb2+–TU complex produced equimolar PbS on heating. The weight ratio was then converted to volumetric ratio using the relation:
| VNP(PbS) = WNP/ρNP(WNP/ρNP + WP/ρP) | (1) |
The volumetric ratio of PbS and PVP in NC for different concentrations of Pb2+–TU complex is presented in Table 1. In the present investigation the NC samples are assumed to contain PbS NPs in volumetric ratios as indicated in Table 1. The PbS/PVP samples are designated as PbS(36)/PVP, PbS(52)/PVP, PbS(70)/PVP and PbS(82)/PVP where the value in parenthesis denotes the weight fraction (WF) of Pb2+–TU complex.
PbS/PVP NC films were deposited on glass substrates by dip coating as shown in Fig. 1. At first, glass substrates were dip-coated with precursor films from above mentioned precursor solutions (Pb2+–TU and PVP) and dried at ∼70 °C in an oven. The precursor films were then heated at 110 °C in the air for 10 min. The transparent precursor films (containing Pb2+–TU complex) on heating converted to clear shiny brown films. Scrupulously cleaned glass slides of size 75 × 25 × 3 mm3 were used as substrates. All the chemicals used were of analytical grade supplied by Merck Limited, India.
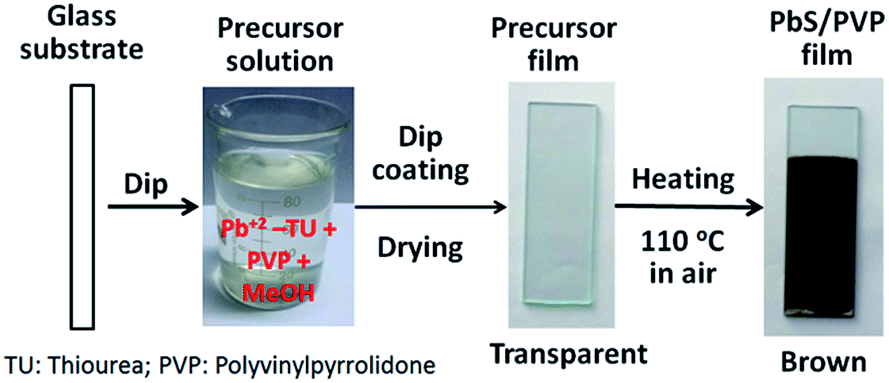 | ||
| Fig. 1 Deposition of PbS/PVP nanocomposite films by dip-coating from precursor solution of Pb2+–TU complex. | ||
The composition of the films was determined by X-Ray Diffractometer (XRD, Bruker, D2 PHASER) having Ni-filtered CuKα radiation (λ = 0.15418 nm) and operated in the 2θ range from 10 to 60°. For optical studies PbS/PVP films were deposited on fused quartz substrates and transmission spectra of the films were measured in the wavelength range of 200–2600 nm by using a UV-VIS-NIR Spectrophotometer (Shimadzu, UV-3600). The influence of PbS NPs on PVP matrix was determined by recording the IR transmittance spectra (400 to 4000 cm−1) of PbS/PVP samples with Fourier Transformed Infrared (FTIR) Spectrophotometer (Thermo Nicolet 6700). NC samples were examined by X-ray Photoelectron Spectrometer (XPS) (ESCA System, SPECS GmbH, Germany) with Al Kα radiation (1486.6 eV). Carbon-13 Nuclear Magnetic Resonance (NMR) Spectrometer (Avance III, 400 MHz, BrukerBiospin, Switzerland) was used to probe the NC films. The effect of PbS NPs on melting point of PVP was studied by Thermo gravimetric Analyser (TGA, Mettler-Toledo, TGA/DSC 1) operated from 50 to 650 °C in nitrogen with a heating rate of 5 °C min−1. The thicknesses of films were ascertained by performing cross sectional Scanning Electron Microscopy (SEM, LEO S-440i). The surface morphology of the films was studied by Atomic Force Microscopy (AFM, Nanosurf, Easyscan2). The size and shape of PbS NPs in NCs were determined with a Transmission Electron Microscope (TEM, Philips, Tecnai 20).
3. Results and discussion
3.1 X-ray diffraction
Dip-coated transparent precursor films on glass converted to shiny brown films of PbS/PVP NC on heating at 110 °C for 10 min in air. The Pb2+–TU complex in PVP thermolysis to PbS at 110 °C (ref. 31) and thus imparts brown colour to the NC films. The thicknesses of once-coated PbS/PVP films are around 120, 150, 180 and 200 nm for 36, 52, 70 and 82% weight fraction of complex, respectively. Fig. 2 shows the XRD plots of typical PbS/PVP NC films obtained with different weight fraction of Pb2+–TU complex. The figure also includes XRD of PVP film which shows a hump at around 23° due to the amorphous nature of polymer.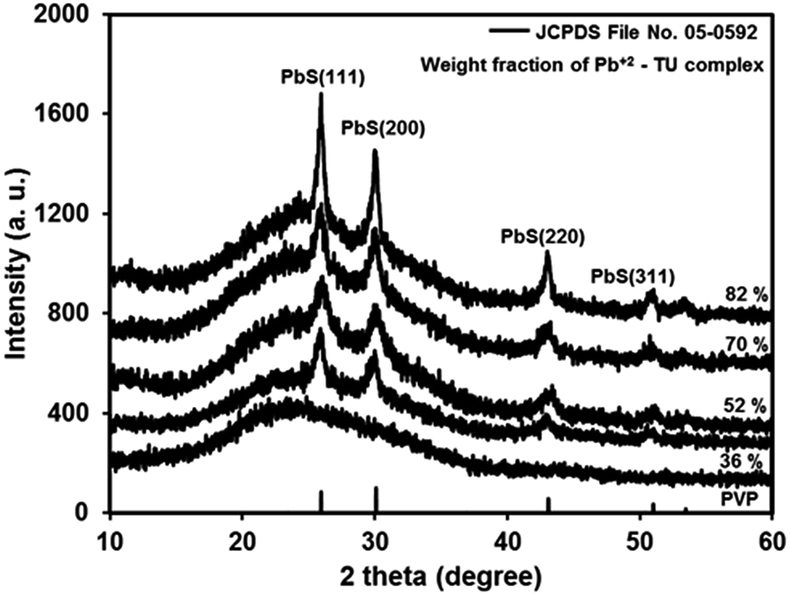 | ||
| Fig. 2 X-ray diffractogram of PVP film and PbS/PVP nanocomposite films on glass with different weight fraction of Pb2+–TU complex. | ||
The PbS/PVP films yield distinct but broad XRD lines at 2θ values of 25.3°, 29.9° 41.9°, and 51.2° superimposed on PVP hump. The inter-planar distances (d) deduced from 2θ of the XRD lines are 0.3411, 0.2966, 0.2099 and 0.1788 nm. These are in good agreement with standard (JCPDS: 05-592) inter-planar distances of 0.3412, 0.2967, 0.2099, and 0.1790 nm corresponding to (111), (200), (220) and (311) planes of cubic PbS. Further, broadening of XRD lines imply that PbS nanoparticles have formed in the PVP matrix. The intensity of PbS XRD lines increases with increase in weight fraction of Pb2+–TU complex in PVP matrix. This is because as the WF of complex is increased in the NC, the density of PbS NPs is also increased. Consequently, the number of PbS NPs encountered by the X-ray beam also increases giving rise to increment in intensity of XRD lines.
The average crystallite size (D) of PbS NC in PVP was deduced from the broadening of PbS (200) line using the Scherrer equation:
D = 0.9λ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ cosθ
| (2) |
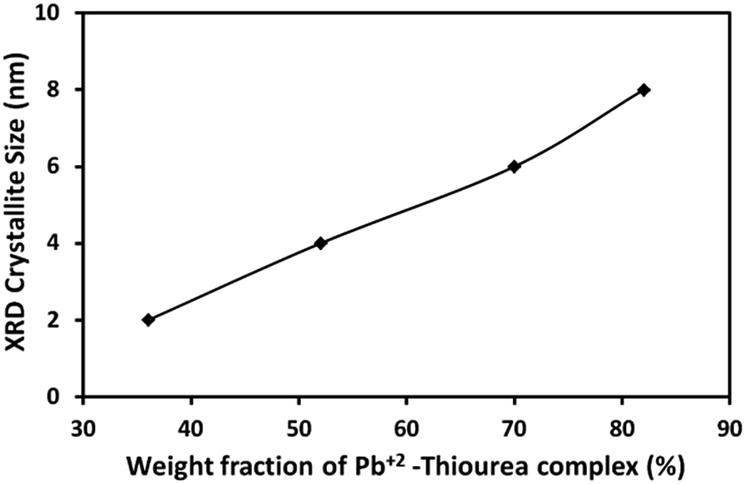 | ||
| Fig. 3 Average crystallite size (Scherrer) of PbS nanoparticles in PbS/PVP nanocomposite films as a function of weight fraction of Pb2+–TU complex. | ||
3.2 Microscopy
TEM was used to physically determine the size and shape of PbS nanoparticles in PbS/PVP NC films. For this NC film on glass was dissolved in methanol and sonicated for an hour. A drop of the resulting solution was placed on carbon-coated TEM grid and dried for observation under TEM. Typical TEM images of PbS nanoparticles in PbS(82)/PVP nanocomposite are shown in Fig. 4. | ||
| Fig. 4 Transmission electron micrograph of PbS nanoparticles in PbS/PVP film produced from PVP containing 82% by weight of Pb2+–TU complex. Inset shows electron diffraction pattern of PbS nanoparticles. | ||
Fig. 4(a) presents low magnification view of PbS NPs along with the electron diffraction (ED) of PbS NPs (inset). The ED lines are in good agreement with the standard lines (JCPDS: 05-592) of cubic PbS. Viewing at higher magnification [Fig. 4(b)] reveals that PbS NPs are spherical in shape having diameters of 7–8 nm. This is in good agreement with the crystallite size determined by Scherrer relation from X-ray diffractograms.
The SEM micrograph of top and cross-section of one-time dip-coated PbS(82)/PVP NC film is depicted in Fig. 5(a) and (b), respectively. Fig. 5(a) shows rice-like growths on the surface of the film. Cross-sectional view [Fig. 5(b)] reveals that film is continuous without any grains or pores and thickness is about 200 nm.
 | ||
| Fig. 5 Scanning electron micrograph of dip-coated PbS/PVP film on glass: (a) top view and (b) cross-sectional view. | ||
The surface morphology of PbS(82)/PVP film was examined by AFM as shown in Fig. 6. AFM also showed that surface had worm-like overgrowths [Fig. 6(a)]. Further magnification [Fig. 6(a)] disclosed that film consists of spherical grains of different sizes along with larger spherical overgrowths connected to each other.
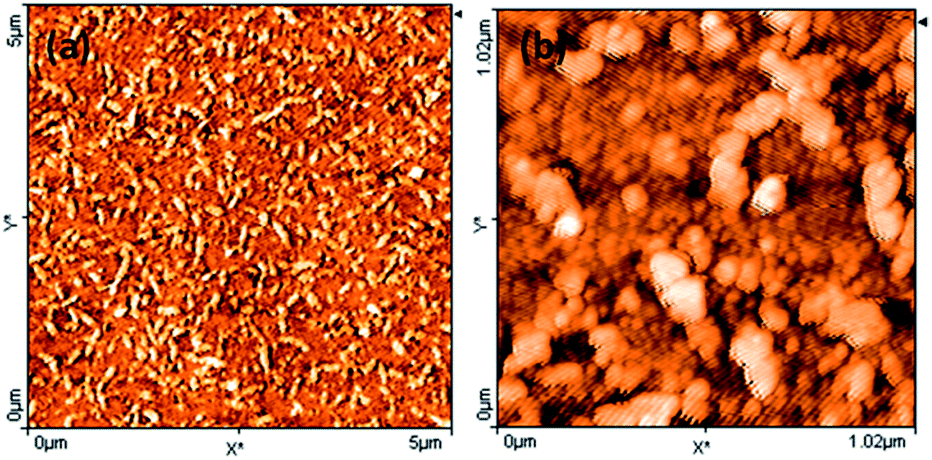 | ||
| Fig. 6 Atomic force microscopic image of the surface of PbS(82)/PVP film on glass: (a) 5 μm × 5 μm area (b) 1 μm × 1 μm area (Z-scale denotes height). | ||
3.3 Band gap tunability
The transmittance spectra of PVP and different PbS/PVP films deposited on quartz are shown in Fig. 7. Only PVP film is highly transparent in the wavelength range of 230 nm to 2600 nm. However, the transmission spectrum of PbS/PVP films shows drastic decrease in transmission below about 600 nm [PbS(36)/PVP] to about 1000 nm [PbS(82)/PVP]. Since, PVP is transparent in this region the reduction in transmission is due to absorption by the PbS nanoparticles which also impart brown colour to the films (depending on weight fraction of Pb2+–TU complex). The absorption edge of PbS/PVP NC films shifts towards longer wavelengths as the gravimetric ratio of Pb2+–TU complex increases. The band gaps of PbS nanoparticles were determined by using Tauc relation for direct band gap semiconductor:| αhν = A(hν − Eg)1/2 | (3) |
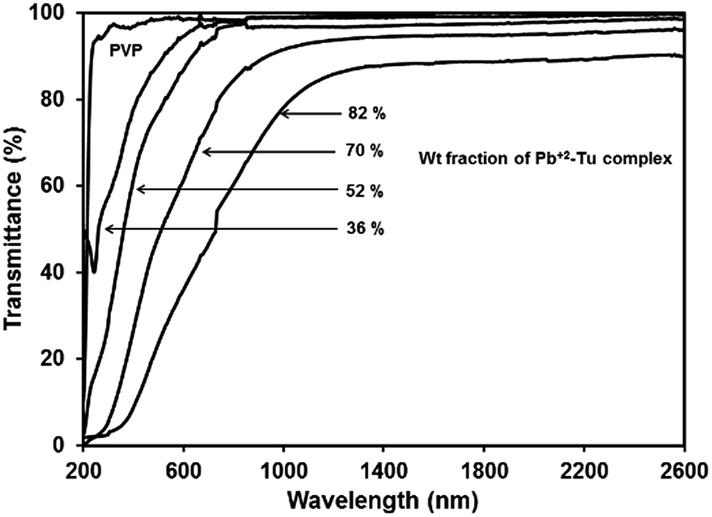 | ||
| Fig. 7 Transmission spectra of PbS/PVP nanocomposite and PVP films. | ||
The absorption coefficient (α) at each wavelength is calculated from the transmittance spectra (Fig. 5). Fig. 8 presents the Tauc plots [(αhν)2 vs. hν] for PbS/PVP films made from precursor containing different weight fractions of Pb2+–TU complex. The band gaps of PbS/PVP films were determined by extrapolating the linear parts of the plots to zero. The band gaps deduced are 0.8, 1.18, 1.58 and 1.92 eV for PbS(82)/PVP, PbS(70)/PVP, PbS(52)/PVP and PbS(32)/PVP films, respectively. This implies that the band gap of the PbS/PVP films decreases as the weight fraction of Pb2+–TU complex increases as shown Fig. 9. It is well known that PbS nanoparticles exhibit quantum size effect below Bohr radius of 18 nm.37 In the size quantization regime, band gaps of PbS NPs increases as the size decreases. Further, bulk PbS has a band gap of 0.41 eV at 300 K. The band gaps observed in PbS/PVP films (Fig. 8) are basically due to the PbS nanoparticles incorporated in PVP matrix. The decrease in band gaps of PbS/PVP films (Fig. 9) implies that the sizes of the PbS NPs are increasing as weight fractions of Pb2+–TU complex are increasing. Thus, optical studies of PbS/PVP nanocomposite films reveal that sizes of PbS NPs increases as the weight fractions of Pb2+–TU complex in precursor film increases. This is also supported by XRD analysis of the films (Fig. 3). Further, Fig. 9 also indicates that the band gap of PbS/PVP nanocomposite films can be tuned to a desired value between 0.8 to 1.92 eV by mixing appropriate weight fraction of Pb2+–TU complex in precursor film. Hence, the PbS/PVP nanocomposite films in the present study have tunable band gaps.
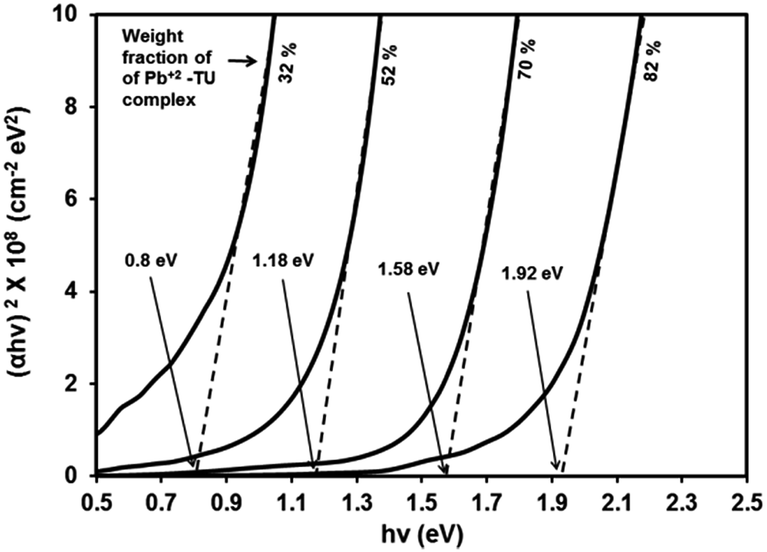 | ||
| Fig. 8 Tauc plots of PbS/PVP nanocomposite films. | ||
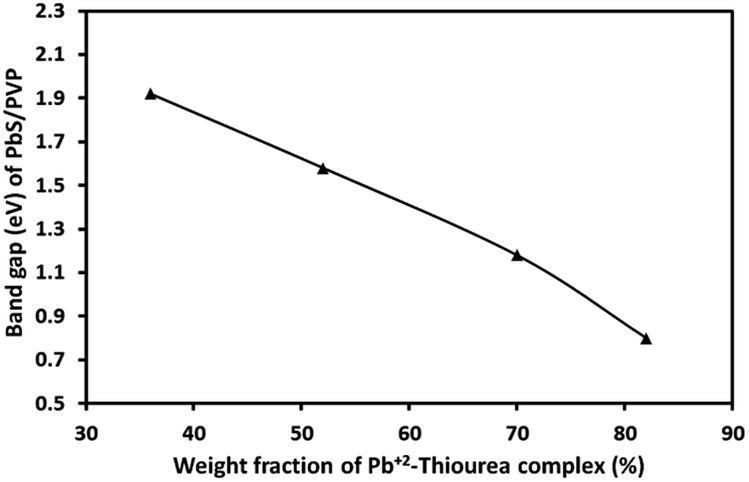 | ||
| Fig. 9 Band gaps of PbS/PVP nanocomposite films as a function of weight fractions of Pb2+–TU complex in precursor. | ||
The XRD studies (Fig. 2) illustrate that PbS/PVP nanocomposite films are readily formed by heating solid precursor films at 110 °C. The precursor films are basically molecular mixture of Pb2+–TU complex and PVP. On heating, Pb2+–TU complex thermally decomposes into PbS and NPs are formed in PVP matrix. The size of the NPs depends on the weight fraction of Pb2+–TU complex in precursor film with respect to PVP (Fig. 3). The higher the weight fraction larger is the size of PbS NPs. UV-VIS-NIR spectroscopy (Fig. 7) confirms the formation of PbS NPs in PbS/PVP nanocomposite films by observing blue shift (Fig. 8) in band gaps of PbS NPs. The band gap of PbS NPs depends on the weight fraction of Pb2+–TU complex in precursor film with respect to PVP (Fig. 9). The higher the weight fraction smaller is the band gap of PbS NPs which in turn means larger is the size of NPs.
Further investigations involve relationship between PbS NPs and PVP. Hence, the PbS/PVP nanocomposite samples are now labelled with corresponding volumetric fractions of PbS with respect to PVP as mentioned in Table 1.
3.4 Thermal analysis
The TGA of PbS/PVP nanocomposites and pristine PVP are shown in Fig. 10(a). There are two temperature regions where weight loss is observed. The first one is from 100 to 150 °C which corresponds to removal of adsorbed H2O. The second region is from 350 to 440 °C where considerable weight loss occurs due to decomposition of PVP.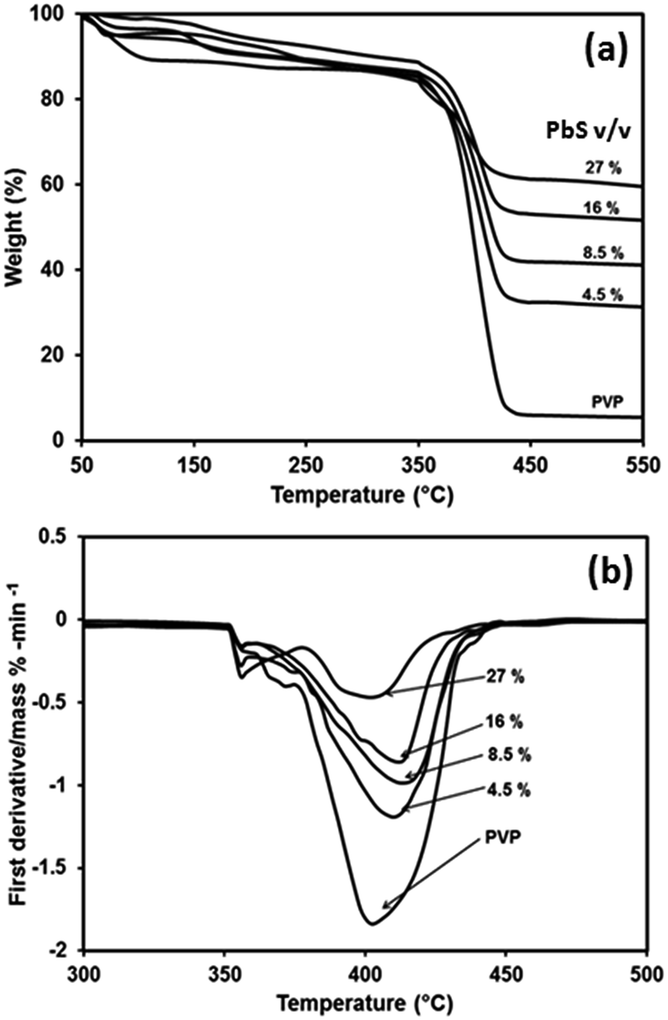 | ||
| Fig. 10 Thermal analysis of PVP and PbS/PVP nanocomposites with different volume fractions of PbS (a) thermogravimetric analysis (b) differential thermogravimetric analysis. | ||
After 440 °C there is no more weight loss in case of PbS/PVP NCs because of residual PbS NPs which do not decompose.
As expected the residual weight of PbS/PVP NCs increases as the PbS content increases. However, there is a residue of 5% for PVP even after total thermal decomposition. The degradation of polymer starts with free radical formations at weak bonds. The improved in thermal stability of PbS/PVP nanocomposite (Fig. 10(a)) can be explained through the reduced mobility of the free radicals. Thus chain transfer reaction will be blocked38 and degradation process will be slowed down resulting in higher decomposition of temperature. The residual weight is due to the weight of PbS nanoparticle. Both TGA and DTG graphs clearly indicated that as the PbS concentration increases in PVP, the decomposition temperature increases compared to virgin polymer. In general, for all composite samples, the decomposition temperature is higher by 5 to 15 °C compared to virgin PVP.
3.5 PbS–PVP interaction by spectroscopy
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching. The shifting of this peak to lower wave number in all the PbS nanocomposites indicates interaction of PbS with –CO carbonyl stretching in pyrrolidone ring. The band at 1295 cm−1 attributed to stretching of –C–N adjacent to –CO group becomes less intense and shows blue shift in the nanocomposites. This is because of interaction of –CO with PbS, resulting in more lone pair electron density of N – atom to be shifted to –C–N. This will lead to increase in –C–N stretching frequency and decrease in –CO stretching. FTIR studies thus confirm that there is strong interaction between PbS nanoparticles and PVP. The nanoparticle–polymer interaction in nanocomposites through FTIR has been investigated earlier in case of PbS/PAM,15,16 PbS/PVAc,29 PbS/PVP30 and PbS/PSt20 nanocomposites. No interaction was observed in PbS/PAM and PbS/PVAc systems while there was strong interaction in PbS/PVP and PbS/PSt nanocomposites. Such strong interaction between nanoparticles and polymer has been also observed for CdS/PVP39 and ZnS/PVP40 nanocomposites.
O stretching. The shifting of this peak to lower wave number in all the PbS nanocomposites indicates interaction of PbS with –CO carbonyl stretching in pyrrolidone ring. The band at 1295 cm−1 attributed to stretching of –C–N adjacent to –CO group becomes less intense and shows blue shift in the nanocomposites. This is because of interaction of –CO with PbS, resulting in more lone pair electron density of N – atom to be shifted to –C–N. This will lead to increase in –C–N stretching frequency and decrease in –CO stretching. FTIR studies thus confirm that there is strong interaction between PbS nanoparticles and PVP. The nanoparticle–polymer interaction in nanocomposites through FTIR has been investigated earlier in case of PbS/PAM,15,16 PbS/PVAc,29 PbS/PVP30 and PbS/PSt20 nanocomposites. No interaction was observed in PbS/PAM and PbS/PVAc systems while there was strong interaction in PbS/PVP and PbS/PSt nanocomposites. Such strong interaction between nanoparticles and polymer has been also observed for CdS/PVP39 and ZnS/PVP40 nanocomposites.
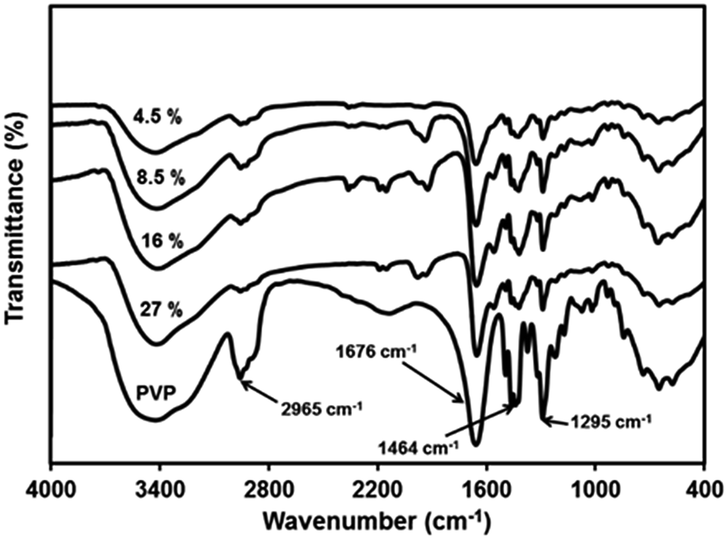 | ||
| Fig. 11 Infrared spectra of PVP and PbS/PVP nanocomposites with different volume fractions of PbS. | ||
O and nano PbS, the effective nuclear charge on O atom increases, there by needing more energy for the core electrons in oxygen.
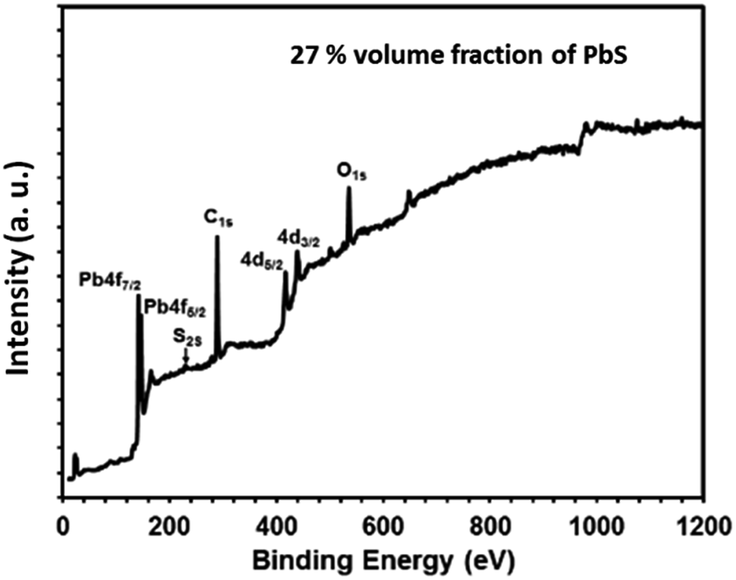 | ||
| Fig. 12 X-ray photoelectron survey scan spectrum of PbS/PVP nanocomposite film with 27% volume fraction of PbS. | ||
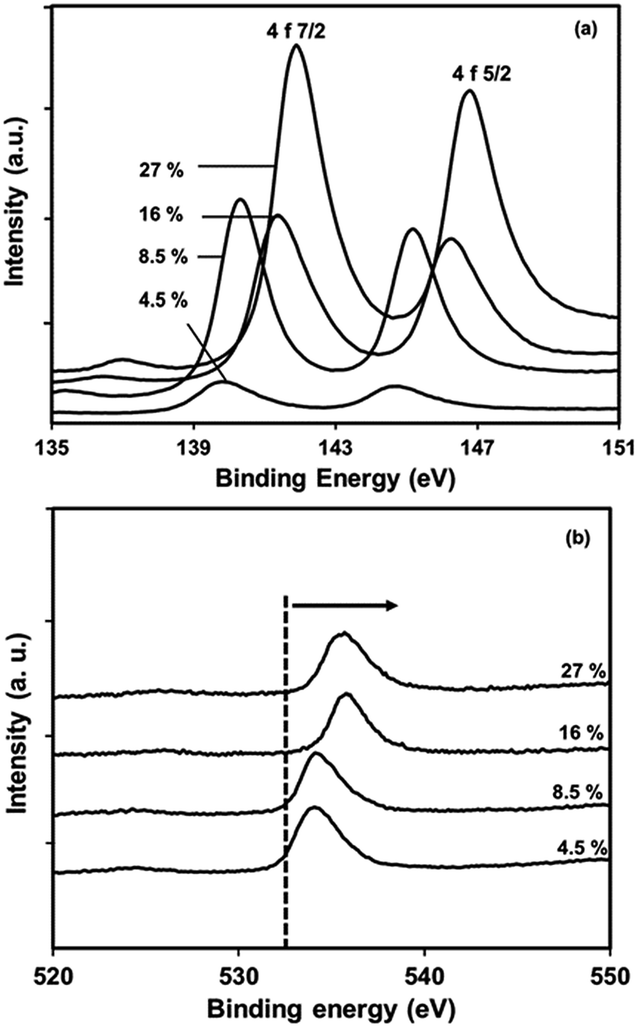 | ||
| Fig. 13 X-ray photoelectron spectra of PbS/PVP nanocomposite films with different volume fractions of PbS in PVP (a) Pb core 4f, (b) O1s core. | ||
The fact indicates that a strong interaction occurs between the Pb atoms and O1s to the weakness in electron density around O atoms in the carbonyl groups in PVP. As the amount of PbS increases the effective nuclear charge on oxygen atom increases that's why the energy requires to knock off core electron is more. It is interesting to know that effective nuclear charge on oxygen atoms in 27% and 16% PbS will not be very much different. This is evident from the ESCA peak of O1s. It needs to be pointed out that –CO stretching frequency is also affected in FTIR spectra due to such interaction. The observed chemical shift for Pb 4f is close to the value of Pb–O bonding.41,42 Thus, the results indicate that, the chemical interaction between PbS and PVP play a key role in the growth of NP and also prevent them from the environmental oxidation.
O moiety. However, the spectrum of PbS/PVP in Fig. 14(b) shows splitting of 175 ppm resonance peak indicating strong interaction between PbS nanoparticles and PVP. Thus NMR investigations corroborates with the findings of FTIR and XPS studies on PbS/PVP nanocomposites.
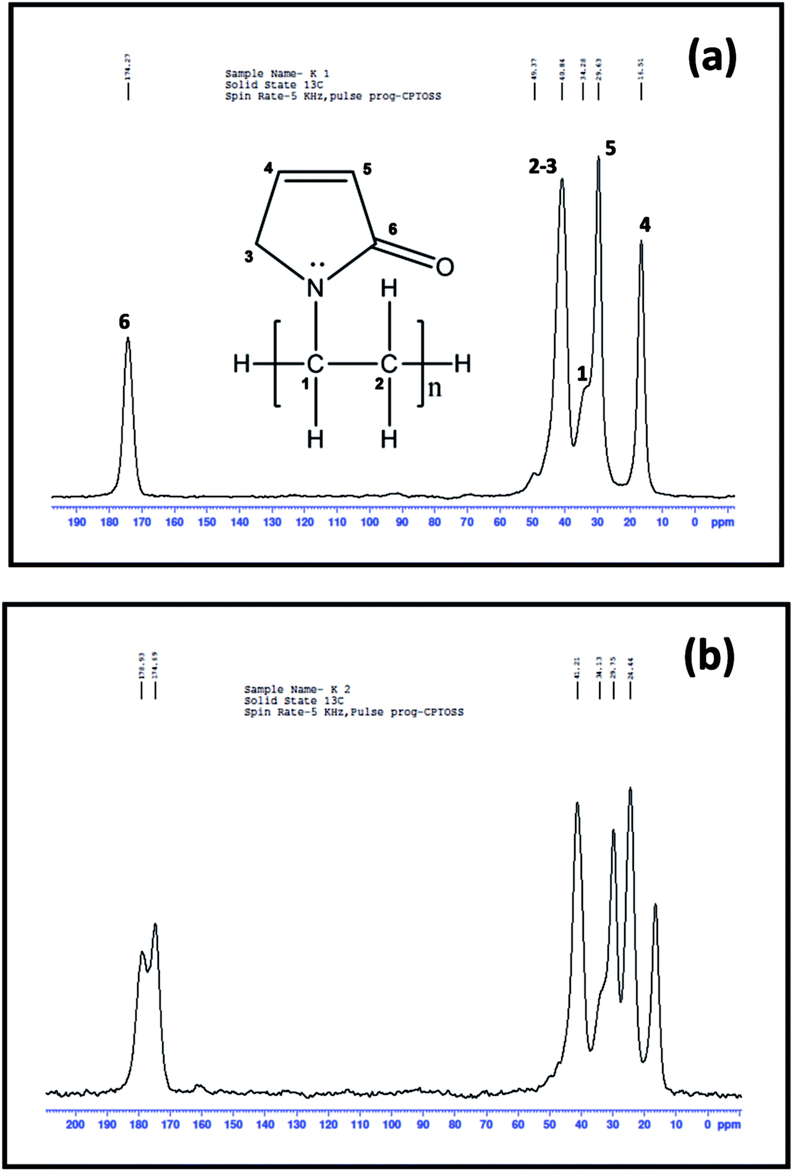 | ||
| Fig. 14 13C nuclear magnetic resonance spectrum of (a) pure PVP and (b) PbS/PVP nanocomposite with 27% volume fraction of PbS. | ||
The above results reveal that PbS/PVP nanocomposite films can be synthesized by adding Pb2+–TU in precursor methanolic solution along with PVP. Precursor films were directly deposited on substrate by dip-coating. Heating of precursor films at 110 °C in air results in the in situ thermolysis of Pb2+–TU complex to PbS in the PVP matrix as given by the reaction:
| Pb(H2NCSNH2)(CH3COO)2 → PbS + CH3COOH↑ + NCNH2↑ |
PbS molecules thus generated merge to form PbS nanoparticles. Solid PVP restricts the growth (size) of these nanoparticles. As the concentration of complex in PVP increases the size of the nanoparticles increases as there are more number of PbS molecules. The PbS nanoparticles are distributed uniformly because the complex (precursor to PbS) is mixed in molecular level with PVP. The PbS nanoparticles impart band gaps to PbS/PVP nanocomposite films. The band gaps of PbS/PVP films depend on the sizes of PbS nanoparticles embedded in PVP matrix which lead to tunability of band gaps from 0.8 to 1.92 eV. Hence, tunable band gap PbS/PVP nanocomposite films can be easily synthesized without taking the trouble of making PbS nanoparticles first.
4. Conclusion
PbS/PVP nanocomposite films with tunable band gaps have been prepared by simple solid state in situ method. Dip-coated precursor films containing Pb2+–thiourea complex and PVP are heated at 110 °C in air to obtain PbS nanoparticles in PVP matrix. The band gaps of PbS/PVP films can be tuned from 0.8 to 1.92 eV by varying the weight fraction of Pb2+–thiourea complex in precursor film. PbS nanoparticles of sizes 2 to 8 nm is formed in PVP as confirmed by XRD and TEM. XPS, 13C NMR and FTIR studies show that there is strong interaction between PbS with –CO of pyrrolidone ring of PVP which restricts the growth of nanoparticles.
Acknowledgements
The authors are thankful to the UGC-DAE Consortium for Scientific Research, Indore for funding under Collaborative Research Scheme (CSR-IC/CRS-92/2014-15/598). Thanks also to the Facilitation Centre for Industrial Plasma Technologies (FCIPT), Gandhinagar for helping in SEM characterization.References
- Y. Wang, W. Suna, R. Mahler and R. J. Kasowski, J. Chem. Phys., 1987, 87, 7315–7322 CrossRef CAS
.
- L. Bakueva, S. Muskin, M. A. Hines, T. Chang, M. Tzolov, G. D. Scholes and E. H. Sargent, Appl. Phys. Lett., 2003, 82, 17 CrossRef
- S. A. Mcdonald, P. W. Cyr, L. levina and E. H. Sargent, Appl. Phys. Lett., 2004, 85, 2089–2091 CrossRef CAS
- A. A. R. Watt, H. Rubinsztein-Dunlop and P. Meredith, Chem. Commun., 2005, 154, 1–3 Search PubMed
- A. Watt, H. Rubinsztein-Dunlop and P. Meredith, Mater. Lett., 2005, 59, 3033–3036 CrossRef CAS
- A. A. R. Watt, D. Blake, J. H. Warner, E. A. Thomesen, E. L. Tanvenner, H. Rubinsztein-Dunlop and P. Meredith, J. Phys. D: Appl. Phys., 2005, 38, 2006–2012 CrossRef CAS
- D. J. Asunskis and L. Hanley, Surf. Sci., 2007, 601, 4648–4656 CrossRef CAS
- P. Piatkowski, W. Gadomski, P. Przybylski and B. Ratajska, J. Photochem. Photobiol., A, 2010, 215, 69–75 CrossRef CAS
- J. H. Warner and A. A. R. Watt, Mater. Lett., 2006, 60, 2375–2378 CrossRef CAS
- J. Seo, S. J. Kim, W. J. Kim, R. Singh, M. Samoc, A. N. Cartwright and P. N. Prasad, Nanotechnology, 2009, 20, 095202–095208 CrossRef PubMed
- M. Zhou, B. Wang, X. Jiang, A. A. Zakhidov, J. P. Ferraris, D. Azunskis and L. Hanley, Int. J. Nanosci., 2011, 10, 521–532 CrossRef CAS
- A. Guchhait, A. K. Rath and J. Pal, Sol. Energy Mater. Sol. Cells, 2011, 95, 651–656 CrossRef CAS
- S. Jing, S. Xing and C. Zhou, Mater. Lett., 2008, 62, 41–43 CrossRef CAS
- Z. Wang, S. Qu, X. Zeng, J. Liu, C. Zhang, M. Shi, F. Tan and Z. Wang, Curr. Appl. Phys., 2009, 9, 1175–1179 CrossRef
- M. Mukherjee, A. Datta and D. Chakravorty, Appl. Phys. Lett., 1994, 64, 1159–1161 CrossRef CAS
- P. S. Nair, T. Radhakrishan, N. Revaprasadu, G. A. Kolawole, A. S. Luyt and V. Djokovic, Appl. Phys. A, 2005, 81, 835–838 CrossRef
- E. V. Nikolaeva, S. A. Ozerin, A. E. Grigoriev, E. I. Grigoriev, S. N. Chvalun, G. N. Gerasimov and L. I. Trakhtenberg, Mater. Sci. Eng., C, 1999, 8–9, 217–223 CrossRef
- Z. Qiao, Y. I. Xie, Y. Zhu and Y. J. Qian, J. Mater. Chem., 1999, 9, 1001–1002 RSC
- B. Liu, H. Li, C. H. Chew, W. Que, Y. L. Lam, C. H. Kam, L. M. Gan and G. Q. Xu, Mater. Lett., 2001, 51, 461–469 CrossRef CAS
- T. K. Chaudhuri, A. J. Kothari, D. Tiwari and A. Ray, Phys. Status Solidi B, 2013, 2, 356–360 CrossRef
- W. Lim, P. Low and H. Y. W. S. Chin, J. Phys. Chem. B, 2004, 108, 13093–13099 CrossRef CAS
- C. Lu, C. Guan, Y. Liu, Y. Cheng and B. Yang, Chem. Mater., 2005, 17, 2448–2454 CrossRef
- V. R. Lyakhovetsky, V. L. Volkov, A. A. Borshch, M. S. Brodyn, M. I. Strashnikova and V. Reznichenko, Mol. Cryst. Liq. Cryst., 2005, 426, 205–217 CrossRef CAS
- J. Kuljanin, M. I. Comor, V. Djokovic and J. M. Nedeljkovic, Mater. Chem. Phys., 2006, 95, 67–71 CrossRef CAS
- R. Kostic, M. Romcevic, D. Markovic, J. Kuljanin and M. I. Comor, Sci. Sintering, 2006, 38, 191–195 CrossRef CAS
- W. E. Mahmoud and S. H. Heniti, Mater. Res. Bull., 2011, 46, 1366–1371 CrossRef CAS
- S. Sarma and P. Datta, Nanosci. Nanotechnol. Lett., 2012, 4, 86–89 CrossRef CAS
- M. H. Patel, T. K. Chaudhuri, T. Shripathi, U. Deshpande and V. K. Patel, J. Mater. Sci.: Mater. Electron., 2016, 27, 12627–12632 CrossRef CAS
- P. A. Kurian, C. Vijayan and S. Sandeep, Nanotechnology, 2007, 18, 075708–075715 CrossRef PubMed
- J. D. Patel and T. K. Chaudhuri, Mater. Res. Bull., 2009, 44, 1647–1651 CrossRef CAS
- M. H. Patel, T. K. Chaudhuri, V. K. Patel, T. Shripathi and U. Deshpande, AIP Conf. Proc., 2016, 1728, 020106 CrossRef
- P. I. Devi and K. Ramachandran, AIP Conf. Proc., 2010, 1276, 183–188 CrossRef CAS
- P. K. Singh, S. K. Tomar and B. J. Bhattacharya, Nanosci. Nanotechnol., 2011, 1, 36–39 CrossRef
- T. Rath and G. Trimmel, Hybrid Mater., 2013, 15–36 Search PubMed
- T. K. Chaudhuri, H. N. Acharya and B. B. Nayak, Thin Solid Films, 1981, 83, L169 CrossRef
- D. Vankhade, A. Kothari and T. K. Chaudhuri, J. Electron. Mater., 2016, 45, 2789 CrossRef CAS
- S. Gallardo, M. Gutierrez, A. Henglein and E. Janata, Ber. Bunsen-Ges., 1989, 93, 1080 CrossRef CAS
- J. Kuljanin, M. Vuckovic, M. I. Comor, N. Bibic, V. Djokovic and J. M. Nedeljkovc, Eur. Polym. J., 2002, 38, 1659–1662 CrossRef CAS
- T. K. Chaudhuri and M. Patel, AIP Conf. Proc., 2010, 1313, 275–277 CrossRef CAS
- T. K. Chaudhuri and M. Patel, J. Exp. Nanosci., 2013, 10, 135–147 CrossRef
- K. Laajalehto, R. Smart, J. Ralston and E. Suoninen, Appl. Surf. Sci., 1993, 64, 29–39 CrossRef CAS
- A. Lobo, T. Möller, M. Nagel, H. Borchert, S. G. Hickey and H. Weller, J. Phys. Chem. B, 2005, 109, 17422–17428 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2017 |