
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simultaneous determination of ultra-low traces of lead and cadmium in food and environmental samples using dispersive solid-phase extraction (DSPE) combined with ultrasound-assisted emulsification microextraction based on the solidification of floating organic drop (UAEME-SFO) followed by GFAAS
Marzieh Sadeghi *a,
Ehsan Rostamia,
Davood Kordestanib,
Hojat Veisic and
Mojtaba Shamsipura
*a,
Ehsan Rostamia,
Davood Kordestanib,
Hojat Veisic and
Mojtaba Shamsipura
aDepartment Chemistry, Razi University, Kermanshah, Iran. E-mail: negarsade@gmail.com; m.sadeghi@razi.ac.ir; Fax: +98 8334274559; Tel: +98 8334274559
bDepartment Chemistry, Amir Kabir University of Technology, Tehran, Iran
cDepartment of Chemistry, Payame Noor University, 19395-4697 Tehran, Iran
First published on 24th May 2017
Abstract
Dispersive solid-phase extraction (DSPE) combined with ultrasound-assisted emulsification microextraction (UAEME) has been developed as a novel approach for the determination of lead and cadmium in food and environmental samples prior to graphite furnace atomic absorption spectrometry. SBA-15/CCMet was synthesized and used as a new sorbent for the extraction of metal ions in DSPE. It was characterized by TEM, and FT-IR techniques. After the DSPE step, stripped metal elements were complexed with diethyldithiophosphate (DDTP), and then the complexes were extracted into 1-undecanol using UAEME. Variables affecting the performance of both steps were thoroughly investigated and their effects on the selectivity and efficiency of the whole sample preparation process are discussed. Some parameters such as the type and volume of the extraction solvent, sonication time, pH, ionic strength and centrifugation time were evaluated and optimized. Under the optimized conditions, the limits of detection were 0.2 ng L−1 for Pb2+ and 0.5 ng L−1 for Cd2+, with a preconcentration factor of 1800. The optimized method exhibited a good precision level with relative standard deviation (RSD%) values of 4.8% and 5.1% for 0.1 μg L−1 Pb2+ and Cd2+ respectively (n = 7). The proposed method was successfully applied to the extraction of Pb and Cd in different food and environmental samples. Application of the proposed method to the analysis of fish certified reference material produced results that were in good agreement with the certified values.
1. Introduction
Heavy metal contamination presents a significant threat to the ecosystem due to severe toxicological effects on living organisms. The determination of hazardous metal ions (HMIs) is of interest in different areas including environmental analysis, pharmaceutical and food processes, biology and medicine. Since heavy metals in the environment can enter food chains, and finally accumulate in living organisms to produce biological toxicity even at a very trace level, the strict monitoring of heavy metals in environmental substances is of great importance to evaluate the risk of heavy metals for human health; in this case, accurate analytical methods are required.Lead and cadmium are two of the most hazardous elements to human health, because both cause adverse effects on metabolic processes. Cadmium is the sixth most poisonous substance jeopardizing human health. Cd can be harmful to plants and, through accumulation into the food chain, to human health.1 Lead is a serious cumulative body poison and enters our body system through food, water, and air. The main target for lead toxicity is the nervous system. The World Health Organization has reported tolerable weekly intakes of Cd and Pb as 0.007 and 0.025 mg kg−1 body weight, respectively.2
Several advance analytical techniques are available for the determination of trace metals with sufficient sensitivity such as electrothermal atomic absorption spectrometry (ETAAS) and inductively coupled plasma optical emission spectrometry (ICP OES) as well as other methods.3 Because of electrothermal atomic absorption spectrometry (ETAAS) only requires a few microliters of sample to carry out the determination, and DLLME involves preconcentration in a small volume of solvent, the DLLME-ETAAS combination is of a particular interest. In this way a procedure able to compete with ICP-MS as regards sensitivity results, as demonstrated in a number of recent articles.4–6
The analysis of elements in trace levels is a difficult analytical task, mostly due to the complexity of the matrix and the low concentration of these elements, which requires sensitive instrumental techniques and often a preconcentration step.4,7–9 For this reason, a preliminary separation and preconcentration step is often required to enhance the sensitivity of the method. Several methods involving preconcentration techniques, such as liquid–liquid extraction (LLE) or solid phase extraction (SPE) have been developed for the quantification of trace elements. However, conventional LLE consumes large amounts of the high costing and potentially hazardous organic solvents. In addition, in trace analysis, a large volume of sample is often required and its handling can be extremely time-consuming besides being tedious. The liquid phase microextraction (LPME) technique efficiently overcomes these problems by reducing the amount of organic solvent and combining extraction, preconcentration and sample introduction in one step.
Modern trends in analytical chemistry are towards the simplification and miniaturization of sample preparation procedures. LPME has emerged in these last years as a powerful tool for preconcentration and matrix separation prior to detection. Dispersive liquid–liquid microextraction (DLLME) is based on ternary component solvent systems, in which a mixture of extracting and dispersive solvent is rapidly injected into an aqueous sample containing the analytes of interest, which causes the formation of a cloudy solution. In DLLME, disperser solvent is miscible in both aqueous and organic phases.10 On the other hand, adding disperser solvent such as methanol in water, the interfacial tension of mixture decreases which it seems play an important role in dispersion of organic solvent in water by increasing the surface area between the organic and aqueous sample.11 Hereon, the surface area between the extraction solvent and aqueous phase (sample) is infinitely large and phase transition of analyte into the organic phase is fast. Hence, equilibrium state achieves fast, leading to a very short extraction time.
Despite of undeniable benefits of DLLME, the use of disperser solvent usually decreases the partition coefficient of analytes into the extractant solvent and also it can complicate the phase separation process. To overcome these problems, a new mode of liquid-phase microextraction, namely ultrasound-assisted emulsification microextraction (USAEME) has been developed.12 In this method, an extraction solvent is dispersed into aqueous solution by ultrasound radiation without addition of any water-miscible organic dispersive solvent. Ultrasound radiation can lead to a process named cavitation. In cavitation process, bubble in a liquid rapidly collapses, producing a shock wave. Sufficient energy of this shock can break down the droplet of dispersed phase and generate a smaller droplet size immediately after disruption, thus enhancing the emulsification.13 As a consequence, the ultrasound radiation is an efficient tool to accelerate the mass-transfer process between two immiscible phases, leading to an increment in the extraction efficiency of the technique in a minimum amount of time.14 The method is also modified by the advantages of solidification of a floating organic (SFO) droplet (USAEME-SFO). In this technique, low density solvents having near room temperature melting points was used as extraction solvent, solidified in ice bath after its centrifugation and can be collected easily without using any specific holders.
Dispersive solid phase extraction (DSPE)15 is another novel sample preparation technique which offers unique advantages such as simplicity, low solvent use, high cost effectiveness, safety, and automation. The sorbent in DSPE is directly added into the sample solution containing the target analytes without forming a column or conditioning. The choice of appropriate sorbent is a critical factor to obtain good recovery and high enrichment factor in SPE procedure.
Ordered mesoporous materials have gained considerable interest due to their potential applications in adsorption, catalysis and preparation of advanced nanostructured materials with remarkable physical and chemical properties.16,17 Among mesoporous silica materials, SBA-15 has relatively good hydrothermal stability, and possesses hexagonal arrays of uniform pores with high special surface area and large pore volume.18
Newly synthesized amine-functionalized SBA-15 was used as DSPE sorbent. The structural evaluation studies of sorbent were performed with various experimental techniques such as transmission electron microscopy (TEM), Fourier transform infrared spectroscopy (FT-IR).
In this study, functionalized SBA-15/CCmet was synthesized and used as a new sorbent for the extraction of metal ions. The aim of this study was the combination of dispersive solid phase extraction (DSPE) with ultrasound-assisted emulsification microextraction (USAEME) for the highly efficient extraction and determination of ultra trace amounts of Pb2+ and Cd2+ ions using graphite furnace atomic absorption spectrometry (GFAAS). The influence of different experimental parameters on the performance of both the steps were thoroughly investigated and discussed. Finally, the applicability of the proposed method was tested by the determination of lead and cadmium in food and environmental samples. In addition, the accuracy of the proposed methodology was evaluated by analyzing a standard reference material.
2. Experimental
2.1. Instrumentation
Transmission electron microscopy (TEM) measurements were performed using a Philips CM10 operated at 100 kV electron beam accelerating voltage. Scanning electron microscopy (SEM) measurements were obtained using a Philips XL30. Thermo gravimetric analyses (TGA) were performed on a NETZSCH TG 209 F1 Iris analyzer with a dynamic temperature program of 25–800 °C. All measurements were carried out using a Model nov AA400 atomic absorption spectrometer (Analytik Jena AG, Jena, Germany), equipped with deuterium background correction, a transversely heated graphite tube atomizer and MPE 60 auto-sampler. Pyrolytic graphite coated graphite tubes with integrated PIN platform (Analytik Jena Part No. 407-A81.026) were used for all measurements. The inert gas was argon flowing at 250 mL min−1. Lead and cadmium hollow cathode lamps (Analytik Jena, Jena, Germany) were used as the radiation sources. The instrumental parameters were as follows: wavelength 283.3 nm (Pb2+) and 228.8 nm (Cd2+) slit width: 0.8 nm and lamp current: 3.0 mA. The optimum operating parameters for GFAAS determination of Pb2+ and Cd2+ are given in Table 1. The pH values were measured with a Metrohm pH-meter (Model: 692, Herisau, Switzerland) supplied with a glass-combined electrode. A Hettich Zentrifugen (EBA20, Tuttlingen, Germany) was used for centrifugations.| Steps | Temperature (°C) | Time (s) | Argon flow rate (mL min−1) | ||
|---|---|---|---|---|---|
| Pb | Cd | Ramp time (s) | Hold time (s) | ||
| Drying | 250 | 250 | 2 | 10 | 500 |
| Pyrolysis | 600 | 650 | 250 | 10 | 500 |
| Atomization | 1200 | 1200 | 1500 | 5 | 0 |
| Cleanout | 2300 | 2300 | 500 | 4 | 500 |
2.2. Reagents and samples
Analytical-grade tetraethylorthosilicate (TEOS), 3-aminopropyltrimethoxysilane (APTES), THF, acetonitrile, hydrogen peroxide, nitric acid, hydrochloric acid and ammonia were supplied from Merck (Darmstadt, Germany). 1-Undecanol, 2-undecanol, 1-octanol, n-hexane as extraction solvents were acquired from Merck. The chelating agent, diethyl dithiophosphate (DDTP) with the density of 1.17 g mL−1 was supplied from Merck. Pluronic P123 triblock copolymer, metformin hydrochloride, 3-aminoropropyl trimethoxysilane (APTMS), diisopropylethylamine and cyanoric chloride were purchased from Sigma-Aldrich.Analytical grade nitrate salts of Pb2+ and Cd2+ (from Merck) were of the highest purity available and used without any further purification. All solutions were prepared in double-distilled deionized water. The stock metal ion solutions (1000 mg L−1) of Pb2+ and Cd2+ were prepared by dissolving the appropriate amounts of nitrate salts. Working standard solution was obtained by appropriate dilution of the stock standard solution.
2.3. Synthesis of amine denderon groups in SBA-15 (SBA-15/CCMet)
The synthesis of SBA-15 was performed following a well-known procedure.19,20 In short explanation for the SBA-15 synthesis, 4.0 g of pluronic P123 was dissolved in 50 mL of water and stirred for 5 h at room temperature. The mixture was added to 120 mL of 2 mol L−1 hydrochloric acid solution and remained for 2 h. Then, 8.5 g of TEOS was added to that solution and stirred for 24 h at 35 °C. The mixture was then aged at 80 °C for 24 h without stirring. After completion of the reaction, the solid products were filtered, washed with deionized water, and air-dried overnight. The P123 was removed thoroughly with hot ethanol/water (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) using a Soxhlet apparatus for 24 h. It was dried in air at 100 °C overnight.
:2) using a Soxhlet apparatus for 24 h. It was dried in air at 100 °C overnight.
The route employed for the synthesis of the SBA/CCMet are shown in Scheme 1. To a 100 mL of round-bottom flask were introduced 30 mL of anhydrous toluene and 3.0 g of SBA-15 and 0.18 g (1.5 mmol) of 3-aminoropropyl trimethoxysilane (APTMS) were added. The solution was refluxed for 24 h under an inert atmosphere, filtered and washed subsequently with toluene, dichloromethane, and methanol, and dried under reduced pressure at 80 °C for 10 h. In another 100 mL round-bottom flask, to a solution of 3 g of aminopropyl functionalized SBA-15 in 35 mL of THF, 0.5 mL of diisopropylethylamine was added. Then, 0.46 g (3 mmol) of cyanoric chloride was added in 0 °C. After 2 h, the solution was decanted and washed with 2 × 25 of fresh THF and decanted. Then, 25 mL of acetonitrile and 1 mL of diisopropylethylamine was added to residue. 0.91 g of free metformin (7 mmol) was added to the mixture and stirred. After completion of the reaction, the solid products were filtered, washed with deionized water and then acetone and dried at 50 °C for 12 h. The clung metformin on triazine functionalized SBA-15 (SBA-15/CCMet) was obtained through this simple procedure.21
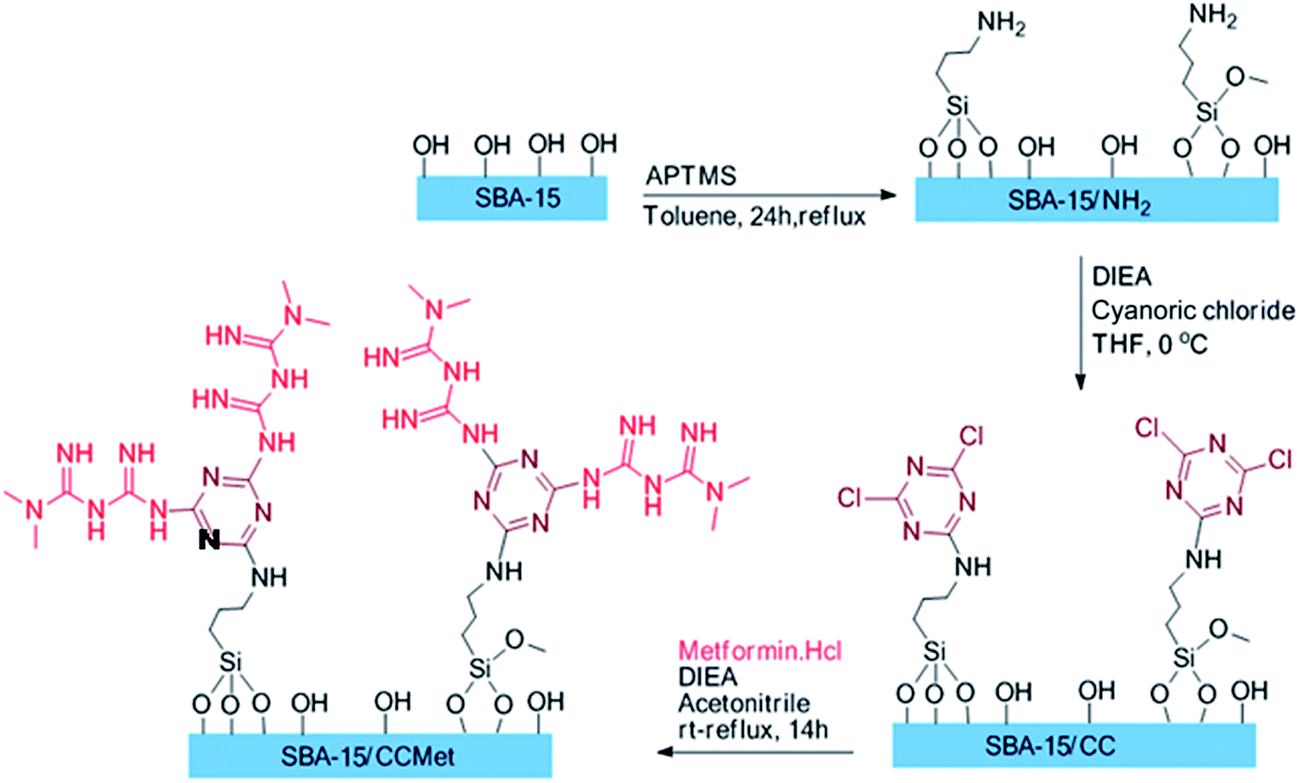 | ||
| Scheme 1 Route employed for the synthesis of the SBA/CCMet. | ||
2.4. Extraction procedure
The general procedure for extraction of ions by SBA-15/CCMet was as follows: SBA-15/CCMet as solid-phase extraction sorbent was used for the extraction of Pb2+ and Cd2+ from the aqueous samples. To a suitable volume (100 mL) solution of Pb2+, Cd2+ and (at pH = 7.5), 10 mg of SBA-15/CCMet was added and the mixture was stirred for at least 10 min. In the case of food and environmental sample, the final volume after digestion reached to 100 mL in a volumetric flask. Then 10 mg of SBA-15/CCMet was added to final solution and the mixture was stirred for at least 10 min. Then the resulting mixture was centrifuged for 5 min at 5000 rpm. After the extraction, the extracted ions by the SBA-15/CCMet were stripped using 5 mL of 0.2 mol L−1 solution of nitric acid. The eluent pH was adjusted to pH 3 with sodium acetate solution (0.1 mol L−1). DDTP was utilized as the chelating agent for extraction of Pb2+ and Cd2+. The optimum concentration of chelating agent (0.03% (v/v) of DDTP) was added into the metal solution. Pb2+ and Cd2+ reacted with DDTP in the form of hydrophobic complex which could be extracted into the dispersed fine droplets. Then 40 μL of extractant (1-undecanol) was injected quickly into the solution by using a syringe. The mixture was homogenized by shaking with hand for a few seconds, then, the solution was sonicated in ultrasonic bath for 3 min. A cloudy solution, resulting from the dispersion of the fine 1-undecanol droplets in the aqueous solution, was formed in the test tube. In this step, the metal ion complexes were extracted into the fine droplets of 1-undecanol. The mixture was then centrifuged for 5 min at 5000 rpm. Accordingly, the dispersed fine droplets of the extraction phase were collected on the top of the conical test tube. The test tube was transferred into a beaker containing ice pieces and the organic solvent was solidified, then the solidified solvent was transferred into a vial, where it melted immediately. Finally, 10 μL of collected extraction solvent using an auto-sampler was injected into the GFAAS. The extraction set-up for DSPE-UAEME-SFO is illustrated in Scheme 2.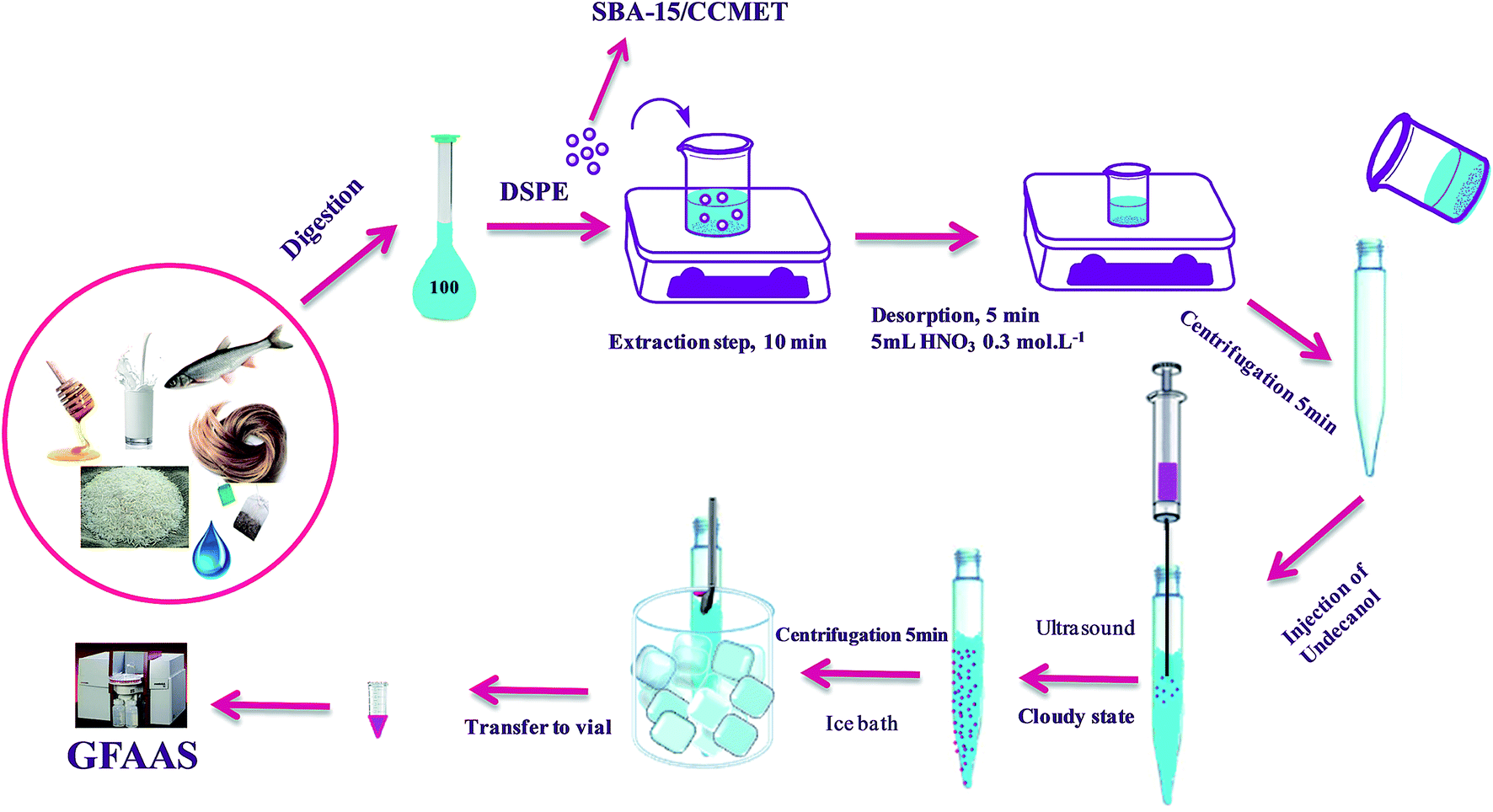 | ||
| Scheme 2 Schematic diagram of DSPE-UAEME-SFO extraction method. | ||
2.5. Sample collection and preparation
2.5.3.1. Live subject statement. Method of digestion of human hair was done according to pervious method in our department. Human hair sample was collected voluntarily and it is provided from waste haircut of hair dresser.
Two certified reference material (DORM-2-dogfish muscle, NRC, Canada; Muscle tissue-NIST SRM 2976) were prepared according above procedure.
3. Results and discussion
3.1. Characterization of functionalized SBA-15
Fig. 1 shows the FT-IR spectra obtained for SBA-15/CCMet. As it is seen, the typical silica bands associated with the main inorganic backbone can be clearly observed in spectrum. The sharp band at 1052 cm−1 is corresponding to Si–O–Si anti-symmetric stretching vibration, being indicative of the existence of a silica material.24 Fig. 1 shows the spectrum of SBA-15/CCMet and signal appeared at 3350 cm−1 is attributed to the presence of N–H stretching. The bending vibration of amine group is also observed at 1650 cm−1. This assignment is supported by the analysis of the IR spectrum of melamine.25 The band in the spectral region of 1251 cm−1 can be assigned to the side chain in plane C–N–C and N–C–N bending of triazine ring.26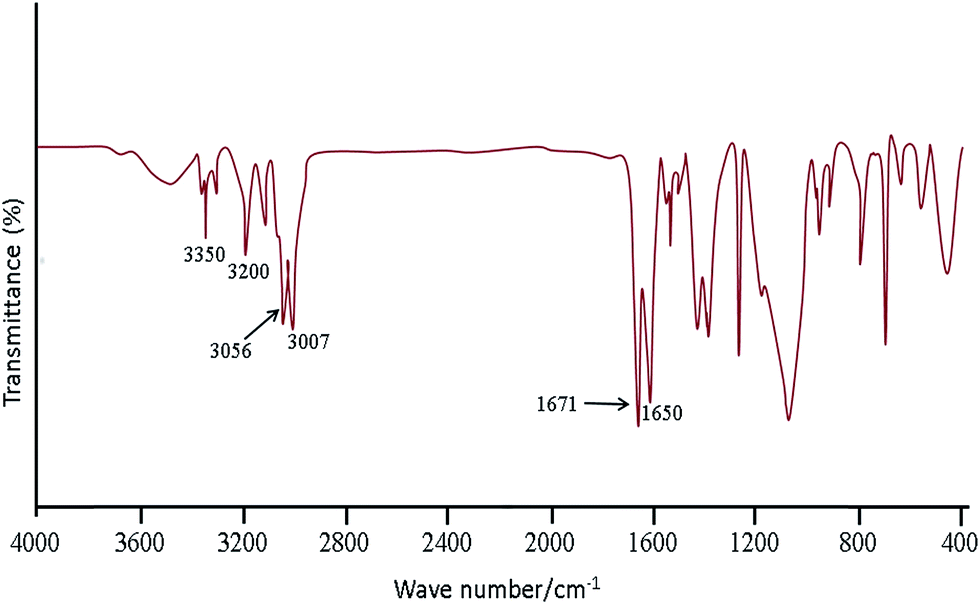 | ||
| Fig. 1 FT-IR spectra obtained for SBA-15/CCMet. | ||
The morphology and size of the SBA-15/CCMet was determined by TEM images. Typical TEM images obtained for SBA-15/CCMet is shown in Fig. 2.
 | ||
| Fig. 2 TEM image of SBA-15/CCMet. | ||
3.2. Optimization of DSPE-SA-DLLME parameters
In the present work, DSPE-USAEME combined with GFAAS was developed for the determination of lead and cadmium in food and environmental samples. This combination lead to very high enrichment factor and could be used in complex matrices. In order to obtain a high extraction recovery and enrichment factor with the employment of DSPE-USAEME, the DSPE and USAEME conditions were optimized. In order to reach optimum experimental conditions for quantitative extraction of lead and cadmium ions via DSPE-UASEME, the influence of different parameters affecting the extraction efficiency such as, amount of SBA-15/CCMet, pH, type and volume of extraction and elution solvent, concentration of chelating agent, ultrasonic and centrifugation time, and concentration of salt were investigated.3.2.1.1. Effect of the breakthrough volume. In the analysis of heavy metals, the sample volume is one of the important parameters influencing the extraction recovery. In order to achieve a high extraction recovery for sample with very low analyte concentration, a large volume of samples solution is required. The effect of breakthrough volume on the extraction of 0.1 μg of Pb2+ and Cd2+ from different sample volumes (50.0 to 2000.0 mL) was investigated. The results showed that an acceptable extraction recovery was observed when sample volumes were increased to 1500 mL, which seemed to be the tolerated volume for breakthrough (Fig. 3). Considering the analytical time and trace level of Pb2+ and Cd2+ in water samples, 100 mL was used as the optimized breakthrough volume.
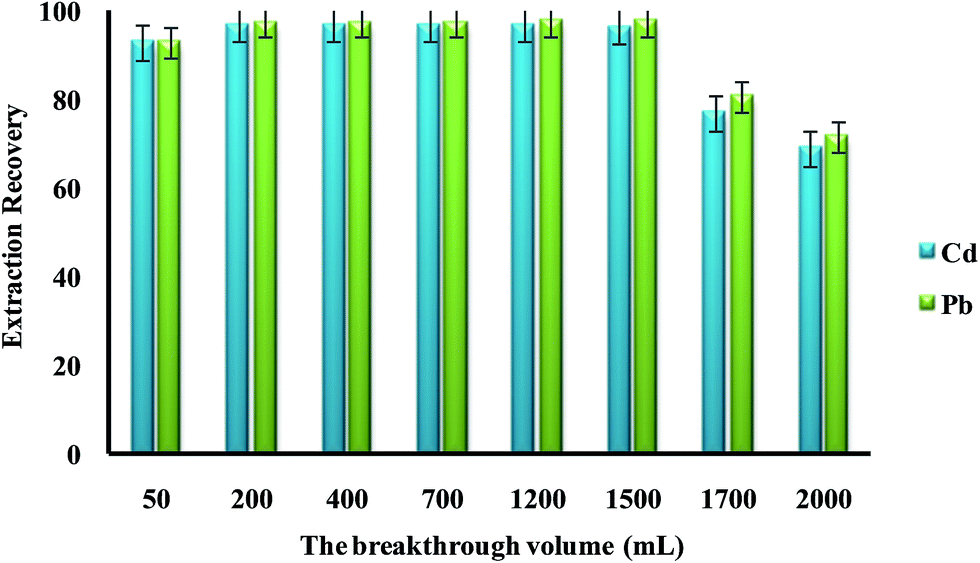 | ||
| Fig. 3 The effect of the breakthrough volume on the extraction recovery of Pb2+ and Cd2+ by dispersive solid phase extraction. Extraction conditions: amount of SBA-15/CCMet, 10 mg; elution solvent (nitric acid) volume, 5.0 mL; concentration of DDTP, 0.04% (v/v); extraction solvent (1-undecanol) volume, 40.0 μL; room temperature; concentration of Pb2+ and Cd2+, 0.1 μg L−1. | ||
3.2.1.2. The influence of the pH on the Pb2+ and Cd2+ ions extraction recovery. The existence of electron donating nitrogen atoms in biguanide and triazide structures was expected to increase the stability of its transition and heavy metal ions complexes over other metal ions, especially alkali and alkaline earth cations. The effect of pH on the extraction of Pb2+ and Cd2+ from water samples by SBA-15/CCMet was investigated and the results are illustrated in Fig. 4. The pH of solution was adjusted by either HNO3 or concentrated ammonia. As can be seen from Fig. 4, Pb2+ and Cd2+ can be extracted quantitatively by the SBA-15/CCMet in the pH ≥ 7.5. It could be due to the charge–dipole interaction between metal ions and nitrogen atoms of SBA-15/CCMet. But at lower pH (pH ≤ 4), the nitrogen atoms could be protonated and reduces the stability of the complexes, leading to decrease in extraction efficiency. Thus, in subsequent experiments, a solution of pH 7.5 was used for further studies.
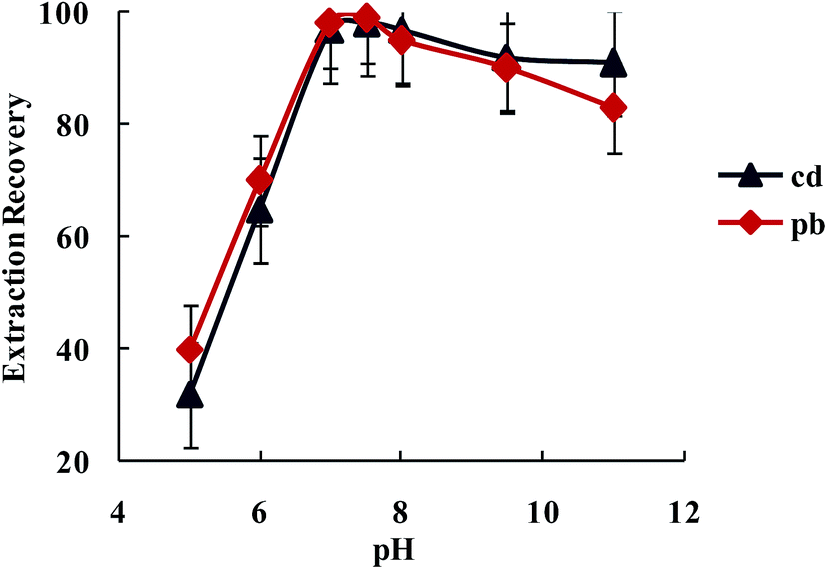 | ||
| Fig. 4 The effect of the solution pH on the extraction recovery of Pb2+ and Cd2+ by dispersive solid phase extraction. Extraction conditions: water sample volume, 100 mL, amount of SBA-15/CCMet, 10 mg; elution solvent (nitric acid) volume, 5.0 mL; concentration of DDTP, 0.04% (v/v); extraction solvent (1-undecanol) volume, 40.0 μL; room temperature; concentration of Pb2+ and Cd2+, 0.1 μg L−1. | ||
3.2.1.3. Optimization amount of SBA-15/CCMet. In order to study the effect of the amount of SBA-15/CCMet on the quantitative extraction of metal ions, the extraction was conducted by varying the amounts of the SBA-15/CCMet from 1 to 20 mg. As shown in Fig. 5, the results indicated that Pb2+ and Cd2+ could be extracted quantitatively using 10 mg of SBA-15/CCMet. Therefore 10 mg of adsorbent selected for subsequent studies.
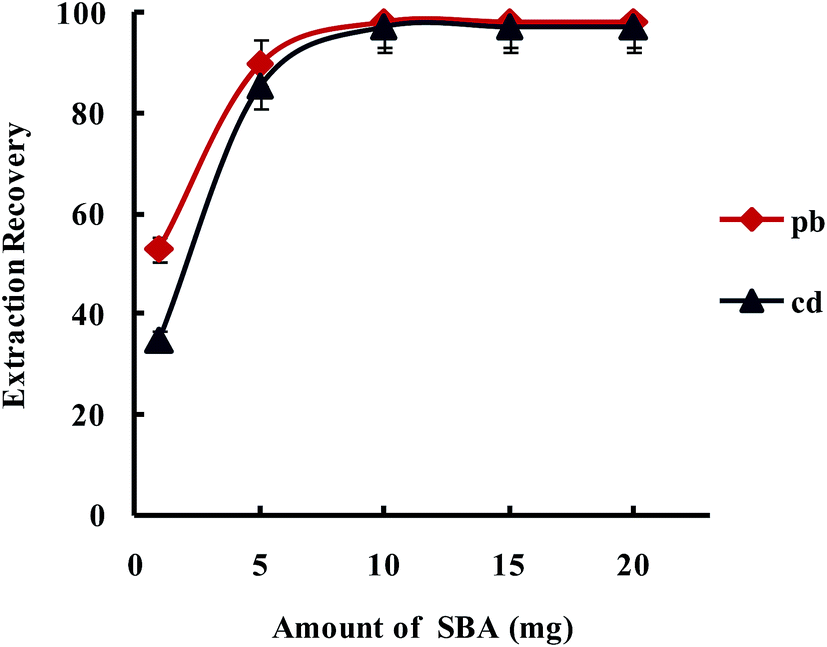 | ||
| Fig. 5 Effect of amount of SBA-15/Met on the extraction recovery of Pb2+ and Cd2+ with a concentration of 0.1 μg L−1 obtained from DSPE-USAEME-SFO. Extraction conditions: similar to those in Fig. 4, except for a sample solution pH of 7.5. | ||
3.2.1.4. Effect of concentration and volume of eluent. Nitric acid was selected as the eluent and the effect of its concentration in the range of 0.1–2.0 mol L−1 on desorption of heavy metal ions was studied. Results showed that 0.2 mol L−1 nitric acid solution can accomplish the quantitative elution of Pb2+ and Cd2+ ions from the SBA-15/CCMet. Therefore, an optimum concentration of 0.2 mol L−1 of nitric acid was used in the future studies. Some experiments were carried out in order to choose a proper volume of nitric acid for the recovery of Pb2+ and Cd2+ ions after extraction by the SBA-15/CCMet. The effect of the volume of eluent on the desorption of metals ions from the SBA-15/CCMet was studied in the range of 1.0–10.0 mL and 5.0 mL of nitric acid (0.2 mol L−1) was found to be sufficient for the quantitative recovery of the analyte.
3.2.1.5. Effect of the extraction time. The effect of the contact time on the efficiency of the extraction for a series of solutions containing 0.1 μg L−1 of Pb2+ and Cd2+ ions was studied. The quantitative recoveries of the Pb2+ and Cd2+ were obtained using 7 min of stirring time. Thus, the mixtures were stirred for 7 min for all experiments.
3.2.2.1. Effect of concentration of chelating reagent. Diethyldithiophosphate (DDTP) is a well-known chelating agent for lead and cadmium extraction with the formation of a stable 2
:1 complex with Pb2+ and Cd2+ ions. The effect of concentration of DDTP on extraction recovery of Pb2+ and Cd2+ ions was investigated in the range of 0.002–0.040% (v/v) and the results are shown in Fig. 6. As shown in Fig. 6, upon increase of DDTP up to 0.04% (v/v), the extraction efficiency increased and kept constant upon further increase in concentration. Therefore, a concentration of 0.04% (v/v) DDTP was selected as the best choice to prevent any interference.
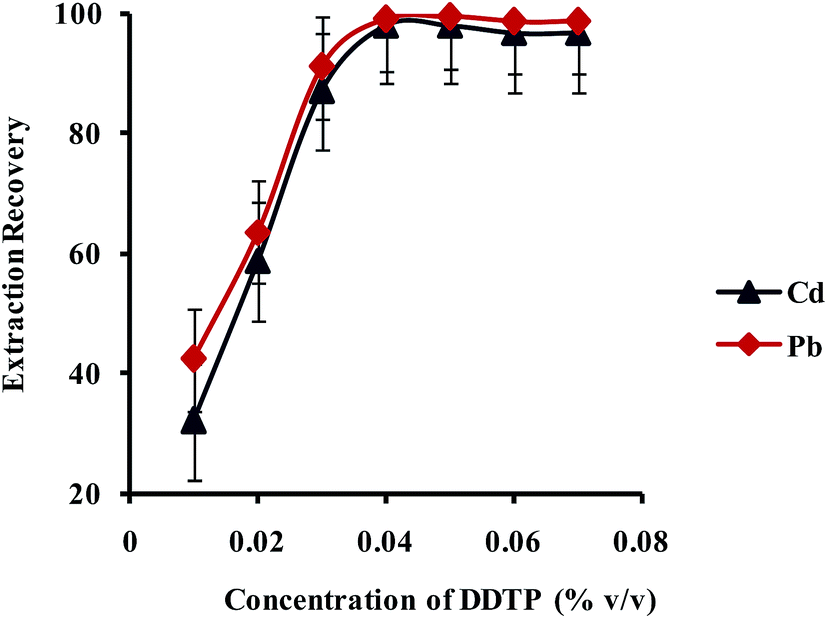 | ||
| Fig. 6 Effect of concentration of DDTP on the extraction recovery of Pb2+ and Cd2+ obtained from DSPE-UAEME-SFO. Extraction conditions are similar to those of Fig. 4. | ||
3.2.2.2. Effect of type and volume of extraction solvent. Select a proper extraction solvent in UAEME is crucial. Extraction solvent should possess some properties as extraction capability of analytes, low solubility in water, low volatility, proper melting point around room temperature, no interference with the analytical techniques used for the determination of analyte, and a density lower than water. In this study, three different organic solvents were tested; including 1-decanol, 1-dodecanol and 1-undecanol. Average extraction recovery (n = 5) and standard deviation (SD) for different extraction solvents are shown in Fig. 7. The results revealed that 1-undecanol has the highest extraction recovery in comparison with the other tested solvents. Therefore, 1-undecanol was selected for further experiments.
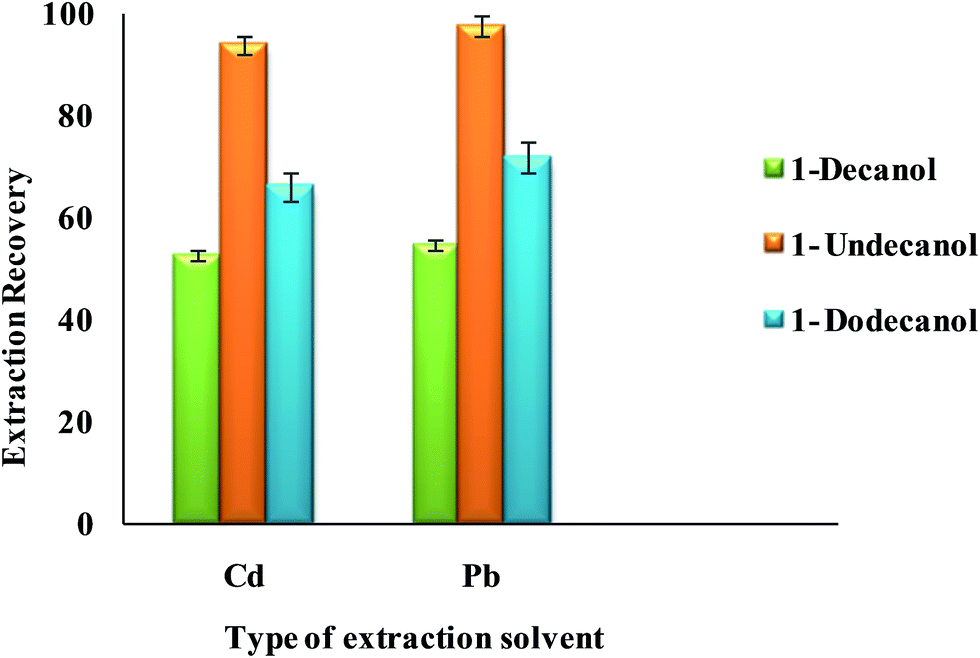 | ||
| Fig. 7 Effect of type of extraction solvent on the extraction recovery of Pb2+ and Cd2+ obtained from DSPE-UAEME-SFO. Extraction conditions are similar to those of Fig. 4. | ||
In order to examine the effect of the volume of the extraction solvent on the performance of the presented UAEME procedure, different volumes of 1-undecanol (20, 30, 40, 50, 60, 80 μL) were used. By increasing the volume of 1-undecanol from 20 to 80 μL, the volume of the floating organic drop increases from 16 to 74 μL. It is clear that, by increasing the volume of 1-undecanol, the extraction recovery increases and by further increasing of the extraction solvent volume, the extraction recovery slightly decreases because of dilution effect (Fig. 8) at higher volumes than 40 μL, extraction recovery slightly decreases whereas the enrichment factor found to decrease significantly. The volume of extractant solvent has to be selected to obtain high enrichment factor and extraction recovery. In the subsequent studies, 40 μL of 1-undecanol was used as the optimal volume of the extraction solvent.
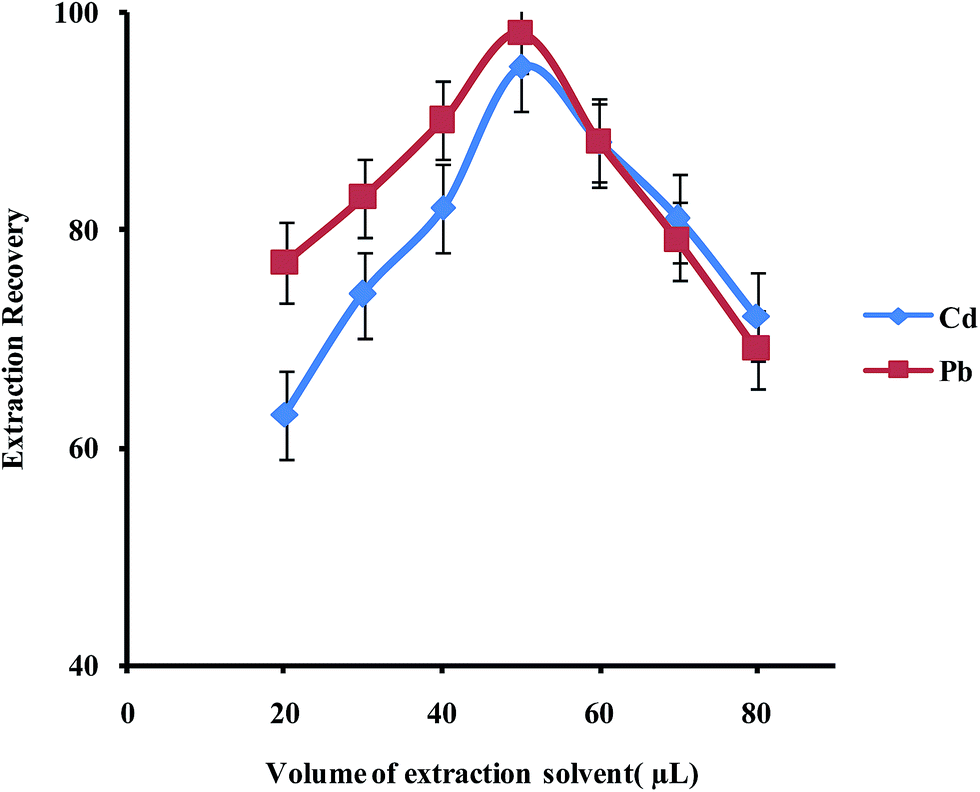 | ||
| Fig. 8 Effect of volume of extraction solvent on the extraction recovery of Pb2+ and Cd2+ obtained from DSPE-UAEME-SFO. Extraction conditions are similar to those of Fig. 4. | ||
3.2.2.3. Effect of pH. In the extraction of metal ions, the complexing and extraction efficiency are closely related to the pH of the solution system.
Extraction of metal ions by UAEME involves complex formation with adequate hydrophobicity to be extracted into small volume of organic phase. It is clearly indicated that pH of the aqueous solution is an important factor for the quantitative recoveries of analytes and play a unique role on metal-chelate formation and subsequent extraction. In order to obtain the favorable complexing and preconcentration efficiency, the pH values were studied in the range of 1.0–7.0. The obtained results indicated that the extraction recovery of Pb2+ and Cd2+ remained more or less constant when the sample pH increased from pH 1 to 3.0, and decreased significantly at higher pH values (Fig. 9). On the other hand, since an aqueous solution of DDTP is nearly acidic, within the optimized pH range (i.e., pH 3.1 in 100 mL aqueous solution), in this work, the use of an acidic solution for the pH adjustment was not needed.
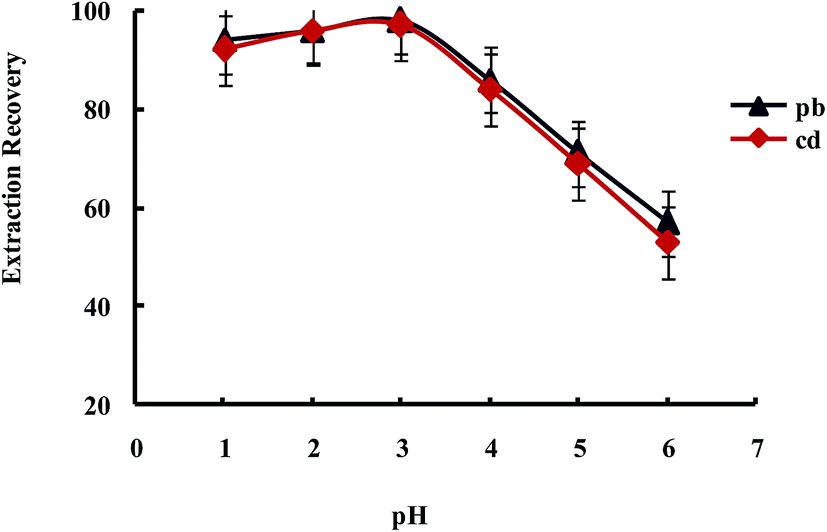 | ||
| Fig. 9 Effect of pH on the extraction recovery of Pb2+ and Cd2+ obtained from DSPE-UAEME-SFO. Extraction conditions are similar to those of Fig. 4. | ||
3.2.2.4. Effect of ionic strength. Generally in liquid phase microextraction methods, addition of salt to aqueous solution causes a decrease in solubility of the analytes and enhances their partitioning into the organic phase (salting-out effect). To investigate the effect of ionic strength on UASEME (extraction recovery of Pb2+ and Cd2+) performance, different amounts of sodium chloride in a range of 0–10% (w/v) were added to the sample solution. The obtained result indicated that the salt addition has no significant effect on the extraction recovery of Pb2+ and Cd2+. Therefore, subsequent experiments were done without any salt addition.
3.2.2.5. Effect of sonication time. In traditional dispersive liquid–liquid extraction, an organics solvent is applied to disperse the extraction solvent, but, as a result of the toxicity of these solvents, ultrasonic bath was used for the dispersion process. The ultrasound radiation is an effective tool to assist the emulsification phenomenon and accelerates the mass-transfer process between two immiscible phases, leading to an increase in the extraction efficiency of the technique in a minimum amount of time.27 Sonication time is one of the important factors in UAEME. The effect of the sonication time was studied in the range of 0–7 min. The results showed that the extraction recoveries are increased by increasing the sonication time before 4 min, and after that, remained nearly constant. Therefore, 4 min of sonication time was chosen in subsequent experiments.
3.2.2.6. Effect of centrifugation time. A centrifugation step was necessary for the phase separation. The effect of centrifugation times on extraction recovery of Pb2+ and Cd2+ ions was studied in the range of 1 to 10 min at a rate of 5500 rpm. The extraction recovery for Pb2+ and Cd2+ ions was lower when the centrifugation time was shorter than 3 min. But, longer centrifugation has no remarkable effect on the extraction recovery of Pb2+ and Cd2+ ions. Hence 3 min (5500 rpm) was chosen in the subsequent experiments.
3.3. Interference study
The selectivity of the DSPE-UASEME for determination of Pb2+ and Cd2+ was evaluated. For this purpose, a series of experiments were designed using 100 mL of solution that contains 0.1 μg L−1 of Pb2+ and Cd2+ and various amounts from interfering ions, were preconcentrated and determined. A given spices was considered to interfere if it resulted in a ±5% variation of the GFAAS signal. The results obtained are given in Table 2. The results showed that interferences possess no obvious influence on the signal intensity of the analytes (Table 2).| Interferents | Foreign ions to the analytes ratio | Recovery (%) ± SD | |
|---|---|---|---|
| Pb2+ | Cd2+ | ||
| Na+ | 1500 | 98.9 ± 1.5 | 98.7 ± 2.8 |
| K+ | 1000 | 97.6 ± 3.0 | 97.5 ± 2.2 |
| Mg2+, Ca2+, Fe2+ | 500 | 98.3 ± 1.3 | 96.8 ± 2.0 |
| Co2+, Ni2+, Zn2+, Mn2+ | 400 | 98.7 ± 1.2 | 96.6 ± 2.1 |
| Hg2+ | 300 | 97.8 ± 2.1 | 97.1 ± 3.9 |
| Cu2+, Fe3+ | 200 | 95.4 ± 2.0 | 96.2 ± 2.5 |
| Cr3+, Cr6+ | 100 | 96.7 ± 1.2 | 97.8 ± 2.4 |
| Parameter | Analytical feature | |
|---|---|---|
| Pb2+ | Cd2+ | |
| Linear range (μg L−1) | 0.001–15 | 0.002–10 |
| R2 | 0.988 | 0.991 |
| Limit of detection (ng L−1) (3σ, n = 7) | 0.2 | 0.5 |
| RSD% (inter-day, n = 7) | 4.8 | 5.1 |
| RSD% (inter-day, n = 7) | 5.3 | 5.5 |
| Enrichment factor | 1800 | 1800 |
| Sample volume (mL) | 100 | 100 |
3.4. Analytical figures of merit
The analytical performance was investigated in terms of linearity, correlation coefficient, limit of detection (LOD), enrichment factor (EF) and reproducibility under optimized experimental conditions (Table 3). The calibration graphs were linear in the range 0.001–15 μg L−1 Pb2+, 0.002–10 μg L−1 Cd2+ under the optimum conditions of the proposed method. The limits of detection were 0.2 ng L−1 for Pb2+ and 0.5 ng L−1 for Cd2+, with a preconcentration factor of 1800. It is noteworthy these limits of detections for the proposed extraction method are in the same order of magnitude as ICP-MS. While capital cost is somewhat lower for the method. Thus, the proposed extraction method seems to be preferable to other existing technology for determination of these elements (Table 3).The repeatability (intra-day) and reproducibility (inter-day) of method were evaluated by carrying out seven replicate extraction and determination of Pb2+ and Cd2+ at a concentration level of 0.1 μg L−1 during a day (intra-day) and seven replicates at seven subsequent days (inter-day). The values of intraday RSD were 4.9 and 5.1 for Pb2+ and Cd2+ respectively. Regarding the inter-day RSD, 5.3 and 5.5 was obtained for Pb2+ and Cd2+ respectively.
3.5. Analysis of fish certified reference material
The accuracy of the proposed method for the extraction and determination of Pb2+ and Cd2+ ions was evaluated by determining the concentration of these ions in two fish certified reference material (DORM-2-dogfish muscle, NRC, Canada; Muscle tissue-NIST SRM 2976). The CRM samples were subjected to the DSPE-UAEME procedure. The obtained results are in good agreement with the certified values (Table 4).| Certified reference material | Element | Certified values ± SD (μg kg−1) | Measured values ± SD (n = 3, μg kg−1) | Recovery (%) |
|---|---|---|---|---|
| DORM-2-dogfish muscle, NRC, Canada | Pb | 65 ± 7 | 62.3 ± 4.8 | 95.8 |
| Cd | 43 ± 8 | 44.1 ± 5.2 | 102.5 | |
| Muscle tissue-NIST SRM 2976 | Pb | 1190 ± 180 | 1127 ± 101 | 94.7 |
| Cd | 820 ± 160 | 794 ± 110 | 96.8 |
3.6. Analysis of real samples
The proposed DSPE-UAEME combined with GFAAS methodology was applied for determination of Pb and Cd in different food and environmental samples. The results for fish, honey, Iranian rice, black tea, cow milk, human hair and water samples are given in Table 5. These samples was then spiked with the standards of Pb2+ and Cd2+ at different concentration levels and all experiments were performed in five replicate (n = 5). According to the results, the spiked Pb and Cd can be quantitatively recovered from all of the samples studied by the proposed procedure. These results demonstrate the applicability of the procedure for Pb and Cd determination in real samples.| Sample | Ion | Added (μg g−1) | Found mean ± SD (μg g−1) | Recovery% |
|---|---|---|---|---|
| Human hair | Pb | — | 26.47 ± 0.12 | — |
| 23 | 27.16 ± 0.15 | 98.9 | ||
| Cd | — | 11.32 ± 0.09 | — | |
| 10 | 12.42 ± 0.10 | 100.8 | ||
| Cow milk | Pb | — | 13.65 ± 0.11 | — |
| 10 | 14.67 ± 0.11 | 100.1 | ||
| Cd | — | 23.41 ± 0.16 | — | |
| 23 | 24.32 ± 0.20 | 99.6 | ||
| Honey | Pb | — | 0.13 ± 0.04 | — |
| 1.0 | 1.11 ± 0.06 | 98.2 | ||
| Cd | — | 0.05 ± 0.01 | — | |
| 1.0 | 1.04 ± 0.06 | 99.0 | ||
|
||||
| Tea | ||||
| Ahmad | Pb | — | 2.61 ± 0.05 | — |
| 1.0 | 3.64 ± 0.06 | 100.8 | ||
| Cd | — | 0.21 ± 0.03 | — | |
| 1.0 | 1.19 ± 0.04 | 98.3 | ||
| Lipton | Pb | — | 1.54 ± 0.05 | — |
| 1.0 | 2.48 ± 0.05 | 97.6 | ||
| Cd | — | 0.12 ± 0.02 | — | |
| 1.0 | 1.10 ± 0.05 | 98.2 | ||
| Ckakoshsabz | Pb | — | 0.53 ± 0.03 | — |
| 1.0 | 1.49 ± 0.06 | 97.4 | ||
| Cd | — | 0.06 ± 0.01 | — | |
| 1.0 | 1.08 ± 0.04 | 101.8 | ||
|
||||
| Water | ||||
| Well | Pb | — | n.d. | — |
| 1.0 | 0.97 ± 0.06 | 97.0 | ||
| Cd | — | n.d. | — | |
| 1.0 | 0.98 ± 0.07 | 0.98 | ||
| Garasoo river | Pb | — | 1.25 ± 0.08 | — |
| 1.0 | 2.24 ± 0.09 | 99.5 | ||
| Cd | — | 0.67 ± 0.05 | — | |
| 1.0 | 1.65 ± 0.06 | 98.8 | ||
| Saymareh | Pb | — | 0.53 ± 0.04 | — |
| 1.0 | 1.56 ± 0.06 | 101.9 | ||
| Cd | — | 0.18 ± 0.02 | — | |
| 1.0 | 1.15 ± 0.04 | 97.4 | ||
|
||||
| Rice | ||||
| Feridonkenar | Pb | — | 0.78 ± 0.03 | — |
| 1.0 | 1.75 ± 0.04 | 98.3 | ||
| Cd | — | 0.03 ± 0.01 | — | |
| 1.0 | 1.04 ± 0.04 | 100.9 | ||
| Domsiah | Pb | — | 4.17 ± 0.08 | — |
| 1.0 | 5.15 ± 0.09 | 99.6 | ||
| Cd | — | 0.16 ± 0.01 | — | |
| 1.0 | 1.12 ± 0.05 | 97.4 | ||
| Tarom | Pb | — | 1.52 ± 0.05 | — |
| 1.0 | 2.43 ± 0.06 | 96.4 | ||
| Cd | — | 0.07 ± 0.01 | — | |
| 1.0 | 1.09 ± 0.03 | 101.9 | ||
|
||||
| Fish river | ||||
| 1 | Pb | — | 0.83 ± 0.04 | — |
| 1.0 | 1.78 ± 0.05 | 97.2 | ||
| Cd | — | 0.35 ± 0.02 | — | |
| 1.0 | 1.32 ± 0.06 | 97.8 | ||
| 2 | Pb | — | 0.68 ± 0.04 | — |
| 1.0 | 1.66 ± 0.05 | 98.8 | ||
| Cd | — | 0.19 ± 0.01 | — | |
| 1.0 | 1.21 ± 0.05 | 101.7 | ||
3.7. Comparison of DSPE-UAEME-SFO with previously reported methods
In Table 6 are compared the performance characteristics of the proposed DSPE-UAEME-SFO method for determination of Pb2+ and Cd2+ in real samples with those of some other reported methods. It is clearly seen that the proposed method possesses a good sensitivity together with a suitable dynamic linear range and an improved limit of detection. In fact, the DSPE-UAEME-SFO has higher enrichment factor (except ref. 35) and lower detection limit, than all the other methods (except ref. 37). The RSD values in the DSPE-UAEME-SFO are relatively low, the extraction time is relatively short and its enrichment factor is much higher than that of the previous methods. These characteristics are of great interest for the routine laboratories in the trace analysis of metal ions.| Method | LODa (ng L−1) | R.S.D.b (%) | E.F.c or R% | Sample volume (mL) | Metal ion (s) | L.R.d (μg L−1) | Extraction time (min) | Reference |
|---|---|---|---|---|---|---|---|---|
| a LOD, limit of detection.b RSD, relative standard deviation.c E.F., enrichment factor.d LR, linear range.e UASEME-FAAS, ultrasound-assisted surfactant-enhanced-microextraction-flame atomic absorption spectrometry.f NNGT-MSA-DLLME-FAAS, narrow neck glass tube-magnetic stirring-assisted dispersive liquid–liquid microextraction-flame atomic absorption spectrometry.g TIL-UDLLμE-ETAAS, temperature-controlled ionic liquid-based ultrasound-assisted microextraction-electro thermal atomic absorption spectrometry.h DLLME-ICP, dispersive liquid–liquid microextraction-inductively coupled plasma.i US-TC-IL-ME-FAAS, ultrasound-assisted temperature-controlled ionic liquid microextraction-flame atomic absorption spectrometry.j UA-IL-DLLME-FAAS, ultrasonic-assisted and ionic liquid-based dispersive liquid–liquid microextraction-flame atomic absorption spectrometry.k USAEME-GF-AAS, ultrasound-assisted emulsification microextraction-graphite furnace-atomic absorption spectrometry.l USAEME-GFAAS, ultrasound-assisted emulsification-microextraction-graphite furnace atomic absorption spectrometry.m HF-SPME-DPASV, hollow-fiber solid/liquid phase microextraction-differential pulse anodic stripping voltammetry.n NDILME-GFAAS, non-dispersive ionic liquid microextraction-graphite furnace atomic absorption spectrometry.o EB-DLLME-FAAS, emulsification based-dispersive liquid–liquid microextraction-flame atomic absorption spectrometry.p DLLME-FAAS, dispersive liquid–liquid microextraction-flame atomic absorption spectrometry.q In situ DIL-EME-μSPE-FAAS, in situ dicationic ionic liquid-emulsification microextraction-micro solid phase extraction flame atomic absorption spectrometry.r Solid-phase extraction-dispersive liquid-liquid microextraction-graphite furnace atomic absorption spectrometry.s Dispersive liquid–liquid microextraction-atomic absorption spectrometry.t Dispersive liquid–liquid microextraction-electro thermal atomic absorption spectrometry.u IL-UAEME-FAAS, ionic liquid-ultrasound assisted emulsification microextraction-flame atomic absorption spectrometry.v SPE-SD-DLLME-FAAS, solid phase extraction-solvent-based de-emulsification-dispersive liquid–liquid microextraction-flame atomic absorption spectrophotometry.w Dispersive solid phase extraction-ultrasound assisted emulsification microextraction-solidification of organic droplet-electrothermal atomic absorption. | ||||||||
| UASEME-FAASe | 110 | 1.98 | 64 | 500 | Cd, Ni | 0.3–100 | 2 | 28 |
| NNGT-MSA-DLLME-FAASf | 1400 | 1.15 | 280 | 50 | Cd | 10–1000 | 9 | 29 |
| TIL-UDLLμE-ETAASg | 50 | 3.97 | 87 | 10 | Cd, Ni | 5–20 | 30 | 30 |
| DLLME-ICPh | 200 | 0.9, 1.3 | 13, 11 | 40 | Cd, Co, Pb, Ni | — | 3 | 31 |
| US-TC-IL-ME-FAASi | 100 | 1.5 | 40 | 5 | Cd | 330–2000 | 1 | 32 |
| UA-IL-DLLME-FAASj | 950 | 2.6 | 100 | 10 | Cu, Ni, Pb | 500–1000 | 10 | 33 |
| USAEME-GF-AASk | 20–30 | ≤8 | 70–500 | 10 | Pb, Cr, Cd | 0.05–20 | — | 27 |
| USAEME-GFAASl | 20 | 11 | 70 | 5 | Pb | 0.1–10 | 8 | 34 |
| HF-SPME-DPASVm | 12–15 | 2.10, 4.82 | 2440, 3710 | 5 | Pb, Cd | 0.05–500 | 60 | 35 |
| NDILME-GFAASn | 0.5 | <5 | 150 | 25 | Cd | 0.02–15 | — | 36 |
| EB-DLLME-FAASo | 0.03 | 4.6 | 151 | 10 | Cd | 0.1–25 | — | 37 |
| DLLME-FAASp | 20, 140 | <20 | 100 (% R) | 10 | Cd, Pb | 0.3–10, 22–100 | — | 38 |
| In situ DIL-EME-μSPE-FAASq | 700 | 4.1 | 93 (% R) | 10 | Pb | 2.5–150 | 2 | 39 |
| SPE-DLLME-GFAASr | 1 | 5.2 | 1800 | 5 | Pb | 0.003–0.06 | <30 | 8 |
| DLLME-AASs | 2.600 | 5.2 | Up to 100 (% R) | 15 | Pb | — | — | 40 |
| DLLME-ETAASt | 0.6–10 | 3.5–4.0 | 146 | 10 (g) | Pb, Cd | 2–40 | — | 41 |
| IL-UAEME-FAASu | 400 | 4.29 | 100 | 10 | Cd | 10–500 | 1.5 | 42 |
| SPE-SD-DLLME-FAASv | 30 | 5.1 | 165 | 5 | Cd | 0.1–50 | — | 43 |
| DSPE-UAEME-ETAASw | 0.2, 0.5 | 4.8, 5.1 | 1800 | 5 | Pb, Cd | 0.001–15, 0.002–10 | 11 | Our work |
4. Conclusions
SBA-15/CCMet was synthesized and used as a new sorbent for simultaneous preconcentration and extraction of Pb2+ and Cd2+ in DSPE. It was characterized by TEM, and FT-IR techniques. In the present study, DSPE method was combined with UAEME-SFO technique followed by GFAAS determination as a highly sensitive, accurate and reliable method for the preconcentration and determination of Pb2+ and Cd2+ in food and environmental samples. The limits of detection were 0.2 ng L−1 for Pb2+ and 0.5 ng L−1 for Cd2+, with a preconcentration factor of 1800. It is noteworthy these limits of detections for the proposed extraction method are in the same order of magnitude as ICP-MS. While capital cost is somewhat lower for the method. Thus, the proposed extraction method seems to be preferable to other existing technology for determination of these elements. This technique provides good precision, simplicity, multi-element enrichment capability, ease of operation, environmentally friendly, good recovery and low detection limits within a relatively short time. The other benefits of the developed method were none-use of the organic disperser solvent, low cost and good accuracy and selectivity. As a conclusion, the proposed method possesses great potential in analysis of ultra trace Pb and Cd in real samples.Acknowledgements
The authors thank the research council of Razi University of Kermanshah (Iran) for the financial support.References
- L. Abramsson-Zetterberg, A. C. Vikstrom, M. Tornqvist and K. E. Hellenas, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2008, 653, 50 CrossRef CAS PubMed.
- R. E. Rivas, I. López-García and M. Hernández-Córdoba, Microchim. Acta, 2009, 166, 355 CrossRef CAS.
- M. Brkljača, J. Giljanović and A. Prkić, Anal. Lett., 2013, 46, 2912 CrossRef.
- M. Sadeghi, Z. Nematifar, M. Irandoust, N. Fattahi, P. Hamzei, A. Barati, M. Ramezani and M. Shamsipur, RSC Adv., 2015, 5, 100511 RSC.
- A. Bidari, E. Z. Jahromi, Y. Assadi and M. R. M. Hosseini, Microchem. J., 2007, 87, 6 CrossRef CAS.
- M. T. Naseri, M. R. M. Hosseini, Y. Assadi and A. Kiani, Talanta, 2008, 75, 56 CrossRef CAS PubMed.
- M. Shamsipur, N. Fattahi, Y. Assadi, M. Sadeghi and K. Sharafi, Talanta, 2014, 130, 26 CrossRef CAS PubMed.
- M. Shamsipur, N. Fattahi, M. Sadeghi, M. Pirsaheb and J. Iran, Chem. Soc. Rev., 2014, 11, 249 CAS.
- M. Shamsipur, M. Ramezani and M. Sadeghi, Microchim. Acta, 2009, 166, 235 CrossRef CAS.
- M. Rezaee, Y. Assadi, M. R. M. Hosseini, E. Aghaee, F. Ahmadi and S. Berijani, J. Chromatogr. A, 2006, 1116, 1 CrossRef CAS PubMed.
- M. Moradi, Y. Yamini, A. Esrafili and S. Seidi, Talanta, 2010, 82, 1864 CrossRef CAS PubMed.
- J. Regueiro, M. Liompart, C. Garcia-Jares, J. C. Garcia Monteagudo and R. Cela, J. Chromatogr. A, 2008, 1190, 27 CrossRef CAS PubMed.
- S. Nojavan, T. Gorji, S. Hosseiny Davarani and A. Morteza-Najarian, Anal. Chim. Acta, 2014, 838, 51 CrossRef CAS PubMed.
- M. Dehghani, M. Abadi, N. Ashraf, M. Chamsaz and F. Shemirani, Talanta, 2012, 99, 1 CrossRef PubMed.
- M. Anastassiades, S. J. Lehotay, D. Stajnbaher and F. J. Schenck, J. AOAC Int., 2003, 86, 412 CAS.
- F. Hoffmann, M. Cornelius, J. Morell and M. Froba, Angew. Chem., Int. Ed., 2006, 45, 3216 CrossRef CAS PubMed.
- M. E. Davis, Nature, 2002, 417, 813 CrossRef CAS PubMed.
- Y. Shao, J. Guan, S. Wu, H. Liu, B. Liu and Q. Kan, Microporous Mesoporous Mater., 2010, 128, 120 CrossRef CAS.
- D. Y. Zhao, Q. S. Huo, J. L. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024 CrossRef CAS.
- A. Alizadeh, M. M. Khodaei, D. Kordestani, A. H. Fallah and M. Beygzadeh, Microporous Mesoporous Mater., 2012, 159, 9 CrossRef CAS.
- H. Veisi, D. Kordestani and A. R. Faraji, J. Porous Mater., 2014, 21, 141 CrossRef CAS.
- M. Shamsipur, M. Sadeghi, M. H. Beyzavi and H. Sharghi, Mater. Sci. Eng., C, 2015, 48, 424 CrossRef CAS PubMed.
- M. Tuzen and M. Soylak, J. Food Drug Anal., 2005, 13, 343 CAS.
- F. Schuth and W. Schmidt, Adv. Mater., 2002, 14, 629 CrossRef CAS.
- W. J. Jones and W. J. Orville-Thomas, Trans. Faraday Soc., 1959, 55, 203 RSC.
- P. J. Larkin, M. P. Makowski, N. B. Colthup and L. A. Flood, Vib. Spectrosc., 1998, 17, 53 CrossRef CAS.
- M. Aghamohammadi, M. Faraji, P. Shahdousti, H. Kalhora and A. Saleh, Phytochem. Anal., 2015, 26, 209 CrossRef CAS PubMed.
- M. Ezoddin, T. Taghizadeh and B. Majidi, Environ. Technol., 2014, 35, 2401 CrossRef CAS PubMed.
- A. Mohammadzadeh, M. Ramezani and A. Niazi, Desalin. Water Treat., 2016, 57, 9745 CrossRef CAS.
- S. S. Arain, T. Gul Kazi, A. J. Arain, H. I. Afridi, J. A. Baig, K. D. Brahman, N. Khan and S. A. Arain, Spectrochim. Acta, Part A, 2014, 138, 387 CrossRef PubMed.
- E. Silva, L. Correia, L. Santos, E. Vieira and V. Lemos, Microchim. Acta, 2012, 178, 269 CrossRef.
- V. Lemos and L. Oliveira, Food Control, 2015, 50, 901 CrossRef CAS.
- N. Jalbani and M. Soylak, Food Chem., 2015, 167, 433 CrossRef CAS PubMed.
- H. Jiang, Y. Zhang, B. Qiu and L. Wenhu, Clean: Soil, Air, Water, 2012, 40, 438 CrossRef CAS.
- Z. Es'haghi, H. Hoseini, S. Mohammadi-Nokhandani and J. Ebrahimi, J. Adv. Res., 2014, 5, 685 CrossRef PubMed.
- N. Khan, T. Gul Kazi, M. Tuzen, F. Shah, H. I. Afridi and D. Citak, Anal. Chim. Acta, 2014, 812, 59 CrossRef PubMed.
- M. Rahimi-Nasrabadi, M. Zahedi, S. Pourmortazavi, Z. Nazari, A. Banan and A. Asghari, Microchim. Acta, 2013, 180, 973 CrossRef CAS.
- F. Rosa, F. Duarte, J. Paniz, G. Heidrich, M. Nunes, E. Flores and V. Dressler, Microchem. J., 2015, 123, 211 CrossRef CAS.
- M. Shokri, A. Beiraghi and S. Seidi, Anal. Chim. Acta, 2015, 889, 123 CrossRef CAS PubMed.
- Z. A. Alothman, M. A. Habila, E. Yilmaz, I. Warad and M. Soylak, J. Anal. Chem., 2015, 70, 691 CrossRef CAS.
- I. López-García, Y. Vicente-Martínez and M. Hernández-Córdoba, Talanta, 2014, 124, 106 CrossRef PubMed.
- S. Khan, T. Gul Kazi and M. Soylak, Spectrochim. Acta, Part A, 2014, 123, 194 CrossRef CAS PubMed.
- M. Behbahani, A. Esrafili, S. Bagheri, S. Radfar, M. Kalate Bojdi and A. Bagheri, Measurement, 2014, 51, 174 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |