DOI:
10.1039/C6RA26038A
(Paper)
RSC Adv., 2017,
7, 1299-1304
A competitive excited state dynamical process for the 2,2′-((1E,1′E)-((3,3′-dimethyl-[1,1′-biphenyl]-4,4′-diyl)-bis(azanylylidene))bis(methanylylidene))-diphenol system
Received
30th October 2016
, Accepted 9th December 2016
First published on 9th January 2017
Abstract
By applying density functional theory (DFT) and time-dependent DFT (TDDFT) methods, we theoretically investigate the excited state dynamical process for the 2,2′-((1E,1′E)-((3,3′-dimethyl-[1,1′-biphenyl]-4,4′-diyl)-bis(azanylylidene))bis(methanylylidene))-diphenol (YT) system. Our results show that two intramolecular hydrogen bonds in YT strengthen in the S1 state, which may trigger an excited state proton transfer (ESPT) process. By exploring the frontier molecular orbitals (MOs), we confirm that charge redistribution indeed has effects on excited state dynamical behavior. Furthermore, this implies the tendency of the ESPT reaction. Analyzing the constructed S1-state potential energy surface (PES), we find three excited state potential barriers which are low enough for a complete excited state double proton transfer (ESDPT) process. Comparing barriers, we confirm a competitive process for stepwise and simultaneous ESDPT pathways.
1. Introduction
Excited state intramolecular proton transfer (ESIPT), due to its relevance in various chemical and biological processes,1–5 has become a topic of much attention in recent years.6–10 Currently, a significant photochemical property of molecules that undergo ESIPT is that their emission spectra exhibit large Stokes shifts because of the formation of new proton-transferred species.11–13 By exploiting these properties, several molecules have been strategically synthesized in order to explore their application as fluorescence probes,14–17 luminescent materials,18,19 light emitting diodes,20–22 molecular switches,23,24 and so forth. Most excited-state proton-transferred molecules involve a six membered ring type with strong intramolecular hydrogen bonds, where the barrier for proton transfer is much less.25–31
Nowadays, more and more attention has been directed to excited state double proton transfer processes by researchers because of their intrinsic property of mimicking proton relay in vital biosystems.32–35 Thus, molecules containing two or more intramolecular hydrogen bonds are of paramount interest in the fields of chemical or biological materials. Recently, Yan et al. reported a new fluorescent chemosensor 2,2′-((1E,1′E)-((3,3′-dimethyl-[1,1′-biphenyl]-4,4′-diyl)-bis(azanylylidene))bis(methanylylidene))-diphenol (YT) containing two intramolecular hydrogen bonds.36 Based on the combination of phenolic hydroxy and imine groups, they found that the new sensor YT has the ability to probe fluoride anions. In fact, the YT molecule could be a good choice for studying excited state dynamics, especially the excited state proton transfer aspect. We believe that investigation of this kind of molecules having two intramolecular hydrogen bonds could deepen our understanding of the ESPT process and fluorescence probe mechanism. Therefore, a YT sensor, wherein two N atoms are acceptors and two O atoms of the hydroxyl groups are donors (shown in Fig. 1), has been selected by us to further study excited state processes. It is well known that spectroscopic techniques, such as steady-state absorption spectroscopy or time resolved fluorescence spectroscopy, can only provide indirect information about photochemical properties and reflect few excited-state dynamic processes.37–50 In addition, the ESIPT process commonly occurs on an ultrafast time scale, which means that achieving an experimental understanding of the correlation between hydrogen bond strength and ESIPT dynamics is difficult. Therefore, in the present work, density functional theory (DFT) and time-dependent DFT (TDDFT) have been selected to help comprehend the detailed mechanism and clarify the fundamental aspects concerning the different electronic states and structures occurring in the PT and ESPT reactions. The minima in both the S0 and S1 states were optimized; vertical excitation energies, infrared (IR) vibrational spectra, frontier molecular orbitals (MOs), and corresponding potential energy surfaces (PESs) were also calculated and analyzed to provide a clear and definite excited-state process.
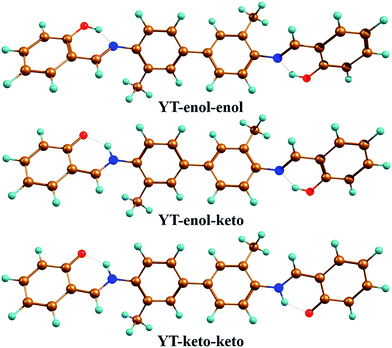 |
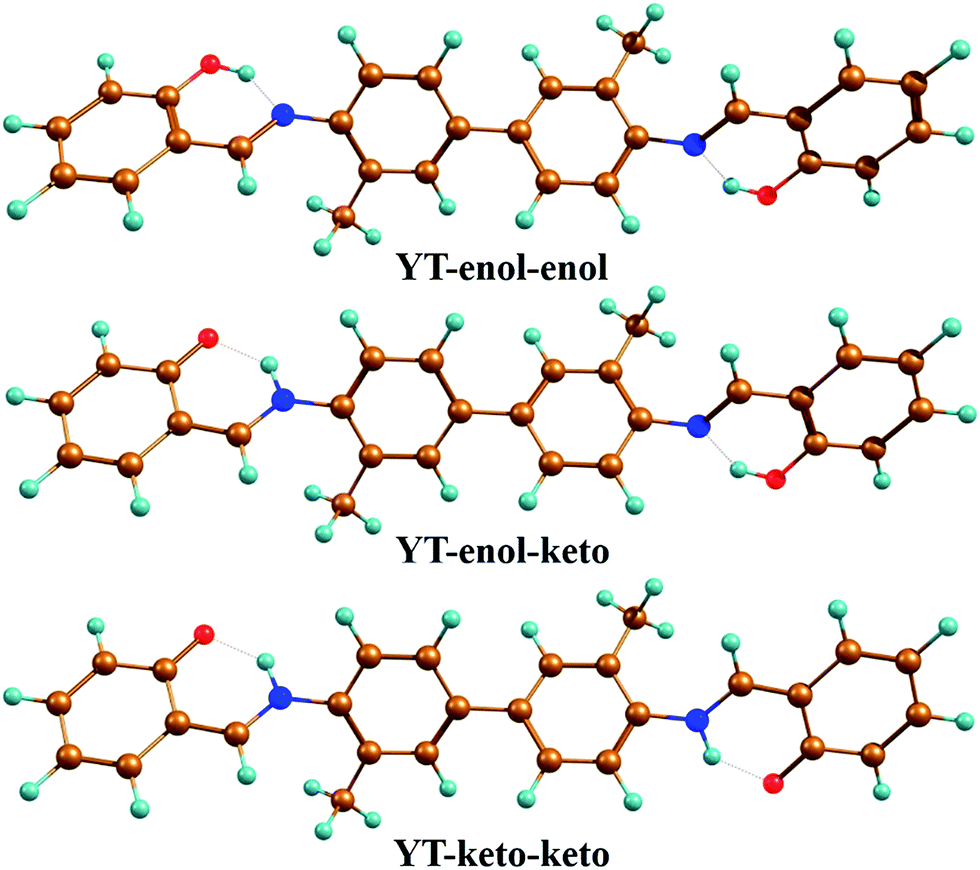
| | Fig. 1 Optimized geometrical configurations of the YT–enol–enol (the normal form); YT–enol–keto (the single proton-transferred tautomeric form); YT–keto–keto (the double proton-transferred tautomeric form). Brown: C atoms; blue: N atoms; red: O atoms; turquoise: H atoms. | |
The remainder of our paper is organized such that the next section describes the computational details. Section 3 presents the discussion about the results of the YT calculations including geometry analyses, electronic spectra, conferences about charge distribution and potential energy surfaces. The final section summarizes the conclusions of this work.
2. Computational details
All of the stationary points in the ground state have been calculated using Becke’s three-parameter hybrid exchange function with the Lee–Yang–Parr gradient-corrected correlation functional (B3LYP)51–54 as well as the triple-ζ valence quality with one of polarisation functions (TZVP)55 basis set based on the density functional theoretical (DFT) method in the Gaussian 09 program.56 The corresponding stable structures of the excited states were accomplished using the time-dependent DFT (TDDFT)57,58 method at the B3LYP/TZVP theoretical level. It is worth mentioning that all the optimized processes were without constraint. In addition, solvent effect was also considered in our work, and herein, all calculations were based on the Polarizable Continuum Model (PCM) using the integral equation formalism variant (IEFPCM) model with dimethylsulfoxide (DMSO) solvent.59–62 In the optimized structures, vibrational frequencies were analyzed to obtain thermodynamic corrections and confirm whether the structures were minima (no imaginary frequency) or transition states (only one imaginary frequency). Zero-point energy corrections and thermal corrections to the Gibbs free energy were also preformed according to the harmonic vibrational frequencies. Vertical excitation energy calculations were obtained from the S0 state optimized configuration using TDDFT with six low-lying absorbing transitions. To further study the excited state dynamic process of ESIPT, the potential energy surface of both the S0 and S1 states were also carried out along the reaction coordinate by constraining optimizations and frequency analyses with a fixed series of configurable values.
3. Results and discussion
3.1 Structures
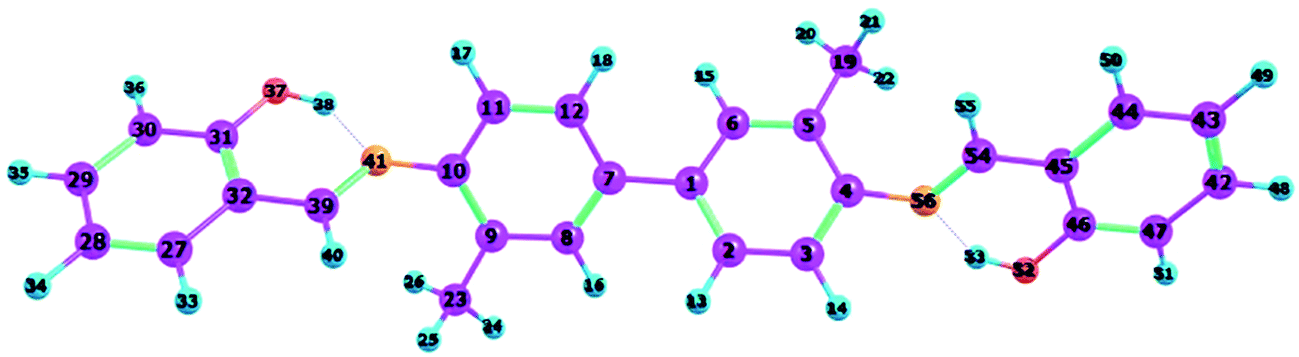
To confirm that all of the stable configurations of the YT tautomers had been obtained, the optimized structures of YT–enol–enol, YT–enol–keto and YT–keto–keto forms were optimized using the B3LYP functional with TZVP base set in DMSO solvent according to the IEFPCM solvation model (displayed in Fig. 1). In order to display factually the numbering scheme of the atoms in YT tautomer, Scheme 1 shows the labelling of the atomic numbers). The important geometric parameters involved in the two intramolecular hydrogen bonds are listed in Table 1. Lengths of O37–H38, H38–N41, O52–H53, H53–N56 bonds in YT–enol–enol are 1.000, 1.709, 1.000 and 1.709 Å in accordance with our theoretical results, respectively. Upon photo-excitation, these bond lengths changed to 1.003, 1.693, 1.003, 1.693 Å in the S1 state. Meanwhile, the bond angle of δ(O37–H38–N41) and δ(O52–H53–N56) are also increased from 148.1° to 150.2° in the S1 state. Even though these changes are not very large, these variations of bond length and bond angles demonstrate that these two intramolecular hydrogen bonds should strengthen in the S1 state. In fact, until now, it has been difficult to judge whether excited state proton transfer could occur, since the variations about bonds and angles are not very great. In addition, based on our calculations, the bond lengths and bond angles involved in the hydrogen bonds of stable YT–enol–keto and YT–keto–keto forms are also listed in Table 1. It can be found that there are also not obvious changes based on the photoexcitation from S0 to S1 states.
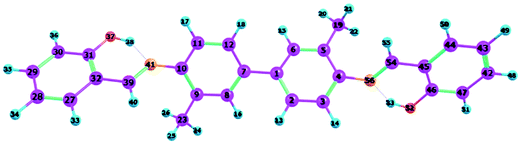 |
| | Scheme 1 The atomic numbers of the YT sensor. | |
Table 1 The primary bond lengths (Å) and bond angles (°) of YT–enol–enol, YT–enol–keto and YT–keto–keto forms in S0 and S1 states based on the DFT/TDDFT methods in DMSO solvent
| Electronic state |
YT–enol–enol |
YT–enol–keto |
YT–keto–keto |
| S0 |
S1 |
S0 |
S1 |
S0 |
S1 |
| O37–H38 |
1.000 |
1.003 |
1.726 |
1.723 |
1.723 |
1.703 |
| H38–N41 |
1.709 |
1.693 |
1.039 |
1.040 |
1.040 |
1.044 |
| O52–H53 |
1.000 |
1.003 |
1.000 |
1.001 |
1.723 |
1.703 |
| H53–N56 |
1.709 |
1.693 |
1.711 |
1.703 |
1.040 |
1.044 |
| δ(O37–H38–N41) |
148.1° |
150.2° |
139.4° |
143.7° |
139.6° |
143.7° |
| δ(O52–H53–N56) |
148.1° |
150.2° |
148.0° |
148.5° |
139.6° |
143.7° |
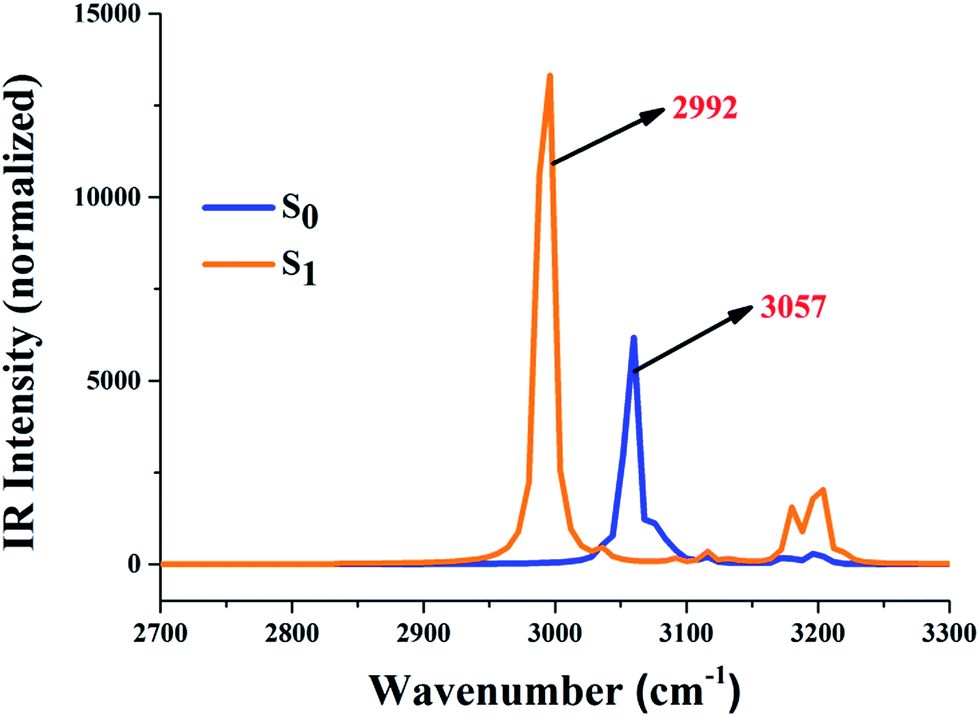
It is already known that the excited state dynamics of hydrogen bonds can be also provided by analyzing vibrational frequency.37–47 In this work, therefore, the IR vibration spectra of O37–H38 and O52–H53 moieties were calculated and are shown in Fig. 2. One should note that our theoretical stretching vibrational frequency in the S0 state is 3057 cm−1, while it changes to 2992 cm−1 in the S1 state. The apparent red-shift around 65 cm−1 in these two intramolecular hydrogen bonds (O37–H38⋯N41 and O52–H53⋯N56) shows that they are strengthened in the S1 state. Moreover, these changes about the excited state hydrogen bond could be also driven through the analysis of local charge distribution.
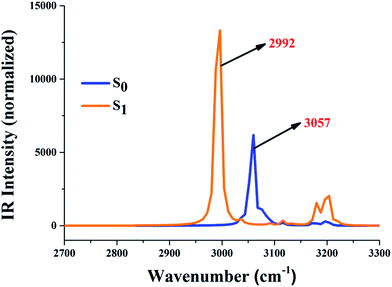 |
| | Fig. 2 The calculated IR vibrational spectra of the hydrogen bond moieties’ O37–H38 and O52–H53 stretching bands in the S0 and S1 state. | |
3.2 Frontier molecular orbitals and charge distribution
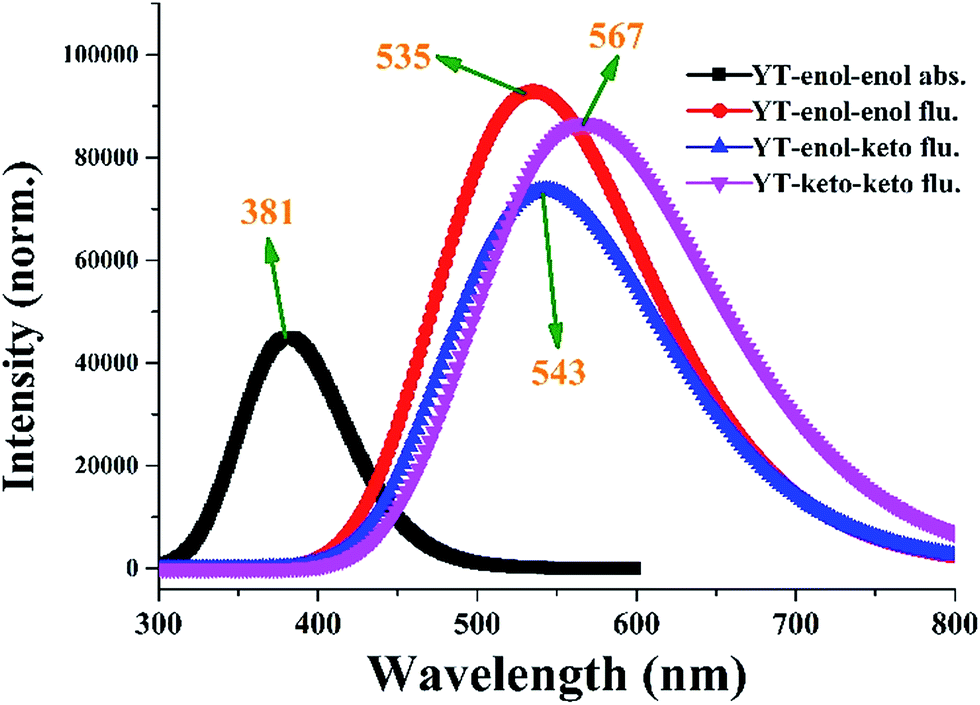
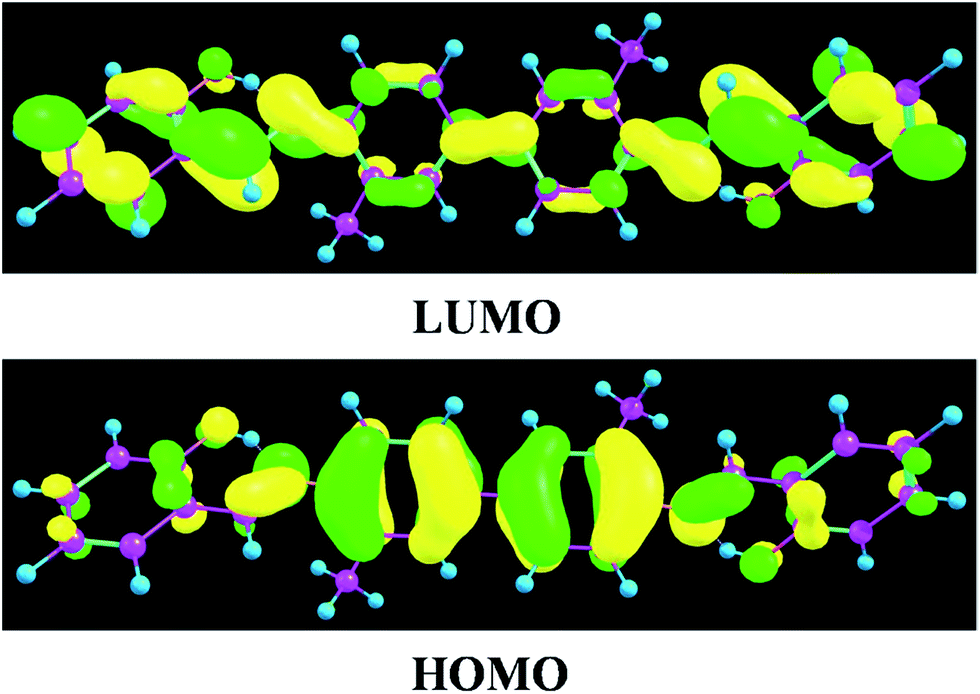
To inspect the influence of the photo-excitation process, the excitation energies of the six low-lying excited states of YT–enol–enol have been calculated in DMSO solvent (shown in Fig. 3). The theoretical results show that the absorption peak of YT–enol–enol is located at 381 nm, which is in good agreement with a previous experimental result (375 nm).36 Even though the previous experimental results did not provide the emission spectra of the YT tautomers, in this work, we calculated these fluorescence peaks which are shown in Fig. 3. For the sake of qualitative discussion about the changes of charge distribution in the excited state, the theoretical frontier molecular orbitals (MOs) of YT–enol–enol have been displayed to further explore the properties of the electronically excited state (shown in Fig. 4). Herein, based on the TDDFT/B3LYP/TZVP theoretical level, our calculated electronic transition energies, corresponding oscillator strengths and compositions are listed in Table 2. It is obvious that the first excited state mainly has ππ* character translating from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) with a very large 1.1096 oscillator strength. For this reason, we only show the HOMO and LUMO orbitals in this present work. In effect, different parts have been located from the HOMO to the LUMO, while we mainly focus on the shifted moieties involved in these two intramolecular hydrogen bonds (O37–H38⋯N41 and O52–H53⋯N56). Although the variations in the hydroxy and N atoms are not very apparent, the quantitative proportion of occupation are obviously different. Actually, the contribution of O37 and O52 of the molecular orbital drops from 12.58% to 9.92% based on the transition (HOMO to LUMO), while that of N41 and N56 increases from 14.86% to 17.28%, respectively.63 That is to say, the incremental charge density of N41 and N56 could enhance the intramolecular hydrogen bonds (O37–H38⋯N41 and O52–H53⋯N56) to some extent, which is consistent with the conclusion above. In general, this kind of charge transfer could promote the ESIPT process,37–47 while in the present work, it is hard to say whether ESIPT can occur since quantificational changed values are not so large. The specific situation depends on the height of the potential energy barriers, which we will discuss below.
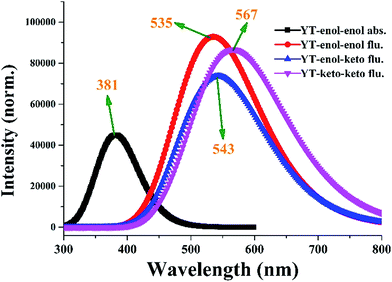 |
| | Fig. 3 The calculated absorption and fluorescence spectra of YT–enol–enol, YT–enol–keto and YT–keto–keto in DMSO solvent. | |
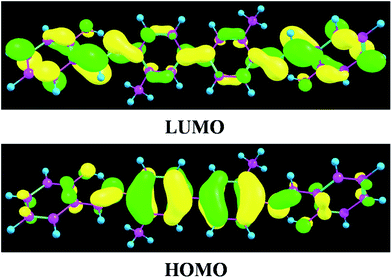 |
| | Fig. 4 View of frontier molecular orbitals (HOMO and LUMO) of YT–enol–enol. | |
Table 2 Electronic excitation energy (nm), corresponding oscillator strengths and the corresponding compositions for the YT–enol–enol chemosensor based on the TDDFT method
| |
Transition |
λ (nm) |
f |
Composition |
CI (%) |
| YT–enol–enol |
S0–S1 |
381 |
1.1096 |
H → L |
94.5% |
| S0–S2 |
356 |
0.0017 |
H → L+1 |
91.8% |
| S0–S3 |
320 |
0.0083 |
H−1 → L |
77.1% |
3.3 Potential energy surfaces
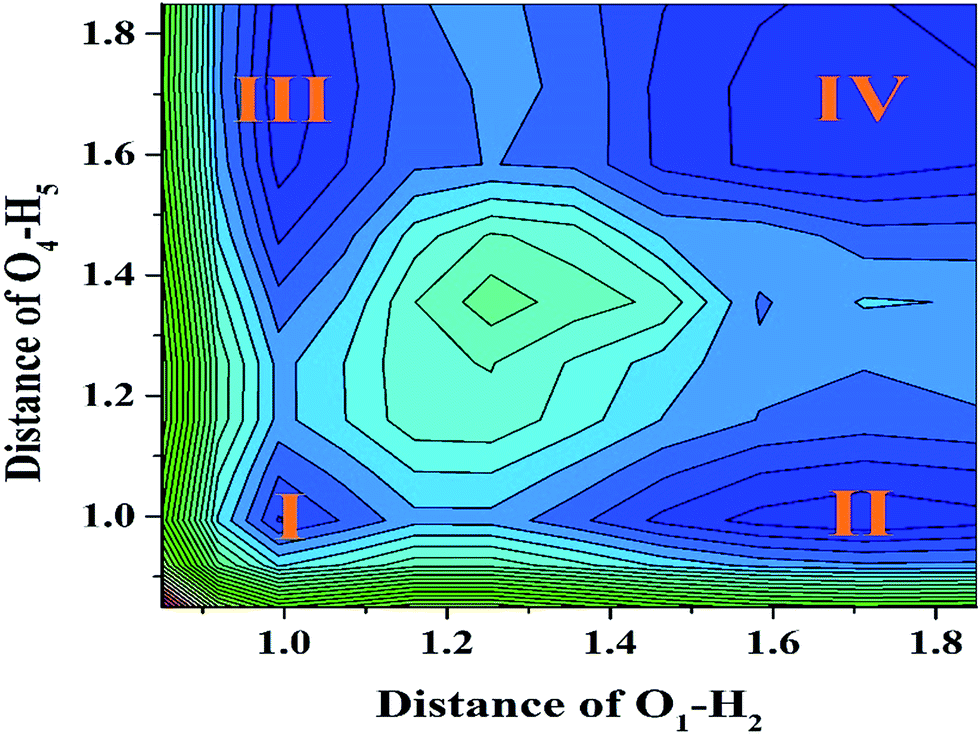
To further reveal the detailed mechanism about whether the ESIPT process could occur for a YT sensor in DMSO solvent, we constructed the potential energy surfaces (PESs) of both the S0 and S1 states with both fixed O37–H38 and O52–H53 bond lengths based on the DFT/TDDFT/B3LYP/TZVP theoretical level. Specifically, our calculated S1-state PES through fixed O37–H38 and O52–H53 ranged from 0.85 Å to 1.85 Å in steps of 0.1 Å (shown in Fig. 5), respectively. We believe it is particularly worth mentioning that the process of constructing S1 stated PES is very time-consuming. Moreover, even though previous work has mentioned that the TDDFT theoretical level may not be expected to be sufficiently accurate to yield the correct ordering of closely spaced excited states, previous theoretical results have shown that this method can reliably offer qualitative energetic pathways for the intra- or inter-molecular proton transfer processes.64–68 Hence it is not disputed for our theoretical PESs. From Fig. 5, it can be clearly seen that four stable structures exist in the S1 state (i.e. I (YT–enol–enol), II (YT–enol–keto), III (YT–enol–keto) and IV (YT–keto–keto)) PES. In addition, this PES is symmetrical along the diagonal (i.e. I–IV). Our calculated results show that a low potential barrier (2.066 kcal mol−1) exists from I to II or III form, which supports an excited state single proton transfer reaction. Then, we find that 2.859 kcal mol−1 separates II or III from structure IV, which shows that a single-proton transfer structure can also transfer another proton forming double-proton transfer form (YT–keto–keto). In addition, we also consider the third excited state path (i.e. transferring the double protons simultaneously from I to IV). In this way, our results indicate a low barrier of 4.814 kcal mol−1. Also it is reasonable for transferring protons in the excited state. Therefore, up to now, we can confirm that the stepwise ESDPT reaction and the synchronous ESDPT is concomitant in the S1 state for the YT system. Since the barriers among these four stable structures are low enough to finish the ESPT process, we thus believe that a competitive mechanism exists for both stepwise synchronous ESDPT processes.
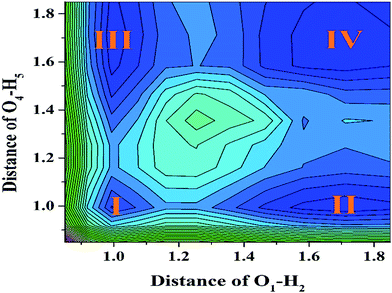 |
| | Fig. 5 The S1-state projective plane with four stable points (I, II, III and IV) of YT–enol–enol. | |
4. Conclusions
In summary, in this present work, based on DFT and TDDFT methods the excited state dynamics of a new fluorescent chemosensor YT have been investigated in detail. Our calculated electronic spectra for YT–enol–enol have been able to reproduce previous experimental results through our optimized geometries of both S0 and S1 states. Through the study of changes in bond lengths and bond angles, we find that these two intramolecular hydrogen bonds (O37–H38⋯N41 and O52–H53⋯N56) should be strengthened in the S1 state, which may provide the possibility for excited state proton transfer. In addition, based on IR spectra analyses about O37–H38 and O52–H53, we further confirm the conclusion of hydrogen bonds strengthening. In order to investigate the situations about charge distribution, the relative frontier molecular orbitals (HOMO and LUMO) have been analyzed through charge changes over specific atoms (O37, O52, N41 and N56) involved in these two hydrogen bonds. It cannot be denied that upon excitation, the charge distributions are really vicissitudinous, however, these changes are not large. That is to say, from these variations it is difficult to judge whether the ESIPT process could occur, as only the tendency of proton transfer could be found. The specific proton transferred situation was estimated through the potential energy barriers. Our constructed potential energy surfaces of both the S0 and S1 states reveal a competitive mechanism existing for both stepwise synchronous ESDPT processes.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 11304095, 11404112 and 11604333), the Science and Technology Research Key Project of Education Department of Henan Province of China (Grant No. 13A140690) and the Key Scientific Research Project of Colleges and Universities of Henan Province of China (Grant No. 16B140002).
References
- W. H. Chen, Y. Xing and Y. Pang, Org. Lett., 2011, 13, 1362 CrossRef CAS PubMed.
- J. F. Zhao, H. N. Yao, J. Y. Liu and M. R. Hoffmann, J. Phys. Chem. A, 2015, 119, 681 CrossRef CAS PubMed.
- D. B. Bucher, A. Schlueter, T. Carell and W. Zinth, Angew. Chem., Int. Ed., 2014, 53, 11366 CrossRef CAS PubMed.
- D. Yang, Y. Yang and Y. Liu, Spectrochim. Acta, Part A, 2014, 117, 379 CrossRef CAS PubMed.
- D. Yang, J. Zhao, R. Zheng, Y. Wang and J. Lv, Spectrochim. Acta, Part A, 2015, 151, 368 CrossRef CAS PubMed.
- J. Zhao, S. Ji, Y. Chen, H. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8003 Search PubMed.
- J. Zhao, J. Chen, J. Liu and M. R. Hoffmann, Phys. Chem. Chem. Phys., 2015, 17, 11990 RSC.
- P. Song and F. C. Ma, Int. Rev. Phys. Chem., 2013, 32, 589 CrossRef CAS.
- G. Y. Li and T. S. Chu, Phys. Chem. Chem. Phys., 2011, 13, 20766 RSC.
- Y. H. Liu, S. C. Lan, C. Y. Zhu and S. H. Lin, J. Phys. Chem. A, 2015, 119, 6269 CrossRef CAS PubMed.
- C. C. Hsieh, C. M. Jiang and P. T. Chou, Acc. Chem. Res., 2010, 43, 1364 CrossRef CAS PubMed.
- K. C. Tang, M. J. Chang, T. Y. Lin, H. A. Pan, T. C. Fang, K. Y. Chen, W. Y. Huang, Y. H. Hsu and P. T. Chou, J. Am. Chem. Soc., 2011, 133, 17738 CrossRef CAS PubMed.
- Y. G. Yang, Y. F. Liu, D. P. Yang, H. Li, K. Jiang and J. F. Sun, Phys. Chem. Chem. Phys., 2015, 17, 32132 RSC.
- J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483 RSC.
- Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2012, 113, 192 CrossRef PubMed.
- F. B. Yu, P. Li, B. S. Wang and K. L. Han, J. Am. Chem. Soc., 2013, 135, 7674 CrossRef CAS PubMed.
- F. B. Yu, P. Li, G. J. Zhao, T. S. Chu and K. L. Han, J. Am. Chem. Soc., 2011, 133, 11030 CrossRef CAS PubMed.
- S. J. Lim, J. Seo and S. Y. Park, J. Am. Chem. Soc., 2006, 128, 14542 CrossRef CAS PubMed.
- H. Shono, T. Ohkawa, H. Tomoda, T. Mutai and K. Araki, ACS Appl. Mater. Interfaces, 2011, 3, 654 CAS.
- P. T. Chou, M. L. Martinez, W. C. Cooper and C. P. Chang, Appl. Spectrosc., 1994, 48, 604 CrossRef CAS.
- P. T. Chou, M. L. Martinez and S. L. Studer, Appl. Spectrosc., 1994, 45, 918 CrossRef.
- P. T. Chou, S. L. Studer and M. L. Martinez, Appl. Spectrosc., 1994, 45, 513 CrossRef.
- I. Masahiro, Chem. Rev., 2000, 100, 1685 CrossRef.
- G. Y. Li, G. J. Zhao, Y. H. Liu, K. L. Han and G. Z. He, J. Comput. Chem., 2010, 31, 1759 CrossRef CAS PubMed.
- M. Zhou, J. Zhao, Y. Cui, Q. Wang, Y. Dai, P. Song and L. Xia, J. Lumin., 2015, 161, 1 CrossRef CAS.
- Y. Yang, Y. Liu, D. Yang, H. Li, K. Jiang and J. Sun, Spectrochim. Acta, Part A, 2015, 151, 814 CrossRef CAS PubMed.
- Y. Dai, J. Zhao, Y. Cui, Q. Wang, P. Song, F. Ma and Y. Zhao, Spectrochim. Acta, Part A, 2015, 144, 76 CrossRef CAS PubMed.
- Y. Li, J. S. Chen and T. S. Chu, J. Lumin., 2016, 179, 203 CrossRef CAS.
- S. C. Lan and Y. H. Liu, Spectrochim. Acta, Part A, 2015, 139, 49 CrossRef CAS PubMed.
- D. Yang, F. Zhao, R. Zheng and J. Lv, J. Alloys Compd., 2016, 659, 82 CrossRef CAS.
- Y. H. Liu, M. S. Mehata and S. C. Lan, Spectrochim. Acta, Part A, 2014, 128, 280 CrossRef CAS PubMed.
-
(a) K. C. Tang, C. L. Chen, H. H. Chuang, J. L. Chen, Y. J. Chen, Y. C. Lin, J. Y. Shen, W. P. Hu and P. T. Chou, J. Phys. Chem. Lett., 2011, 2, 3063 CrossRef CAS;
(b) V. I. Tomin, A. P. Demchenko and P. T. Chou, J. Photochem. Photobiol., C, 2015, 22, 1 CrossRef CAS.
- R. Crespo-Otero, N. Kungwan and M. Barbatti, Chem. Sci., 2015, 6, 5762 RSC.
- S. Mandal, S. Ghosh, G. Banerjee, J. Kuchlyan and N. Sarkar, J. Phys. Chem. B, 2013, 117, 12212 CrossRef CAS PubMed.
- P. T. Chou, G. R. Wu, C. Y. Wei, C. C. Cheng, C. P. Chang and F. T. Hungs, J. Phys. Chem. B, 1999, 103, 10042 CrossRef CAS.
- G. Yan, H. Li, Y. R. Zhu, B. B. Shi, W. Qu, Q. Lin, H. Yao, Y. Zhang and T. B. Wei, New J. Chem., 2015, 39, 8797 RSC.
- G. J. Zhao and K. L. Han, J. Phys. Chem. A, 2007, 111, 2469 CrossRef CAS PubMed.
- G. J. Zhao and K. L. Han, J. Phys. Chem. A, 2007, 111, 9218 CrossRef CAS PubMed.
- J. Zhao and Y. Yang, J. Mol. Liq., 2016, 220, 735 CrossRef CAS.
- J. F. Zhao, J. S. Chen, Y. L. Cui, J. Wang, L. X. Xia, Y. M. Dai, P. Song and F. C. Ma, Phys. Chem. Chem. Phys., 2015, 17, 1142 RSC.
- G. J. Zhao, B. H. Northrop, P. J. Stang and K. L. Han, J. Phys. Chem. A, 2010, 114, 3418 CrossRef CAS PubMed.
- G. J. Zhao, B. H. Northrop, K. L. Han and P. J. Stang, J. Phys. Chem. A, 2010, 114, 9007 CrossRef CAS PubMed.
- G. J. Zhao, J. Y. Liu, L. C. Zhou and K. L. Han, J. Phys. Chem. B, 2007, 111, 8940 CrossRef CAS PubMed.
- J. F. Zhao and P. Li, RSC Adv., 2015, 5, 73619 RSC.
- G. J. Zhao and K. L. Han, Acc. Chem. Res., 2012, 45, 404 CrossRef CAS PubMed.
- G. J. Zhao and K. L. Han, ChemPhysChem, 2008, 9, 1842 CrossRef CAS PubMed.
- J. F. Zhao, P. Song, Y. L. Cui, X. M. Liu, S. W. Sun, S. Y. Hou and F. C. Ma, Spectrochim. Acta, Part A, 2014, 131, 282 CrossRef CAS PubMed.
- Y. Wang, H. Yin, Y. Shi, M. Jin and D. Ding, New J. Chem., 2014, 38, 4458 RSC.
- H. Li, Y. Shi, H. Yin, Y. Wang, L. Cong, M. Jin and D. Ding, Spectrochim. Acta, Part A, 2015, 141, 211 CrossRef CAS PubMed.
- H. Yin, H. Li, G. Xia, C. Ruan, Y. Shi, H. Wang, M. Jin and D. Ding, Sci. Rep., 2016, 6, 19774 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS.
- B. Miehlic, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200 CrossRef.
- D. J. Feller, J. Comput. Chem., 1996, 17, 1571 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision C.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- M. A. Marques and E. Gross, Annu. Rev. Phys. Chem., 2004, 55, 427 CrossRef CAS PubMed.
- C. Adamo and D. Jacquemin, Chem. Soc. Rev., 2013, 42, 845 RSC.
- R. Cammi and J. Tomasi, J. Comput. Chem., 1995, 16, 1449 CrossRef CAS.
- E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef CAS.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef CAS.
- M. Caricato, B. Mennucci, J. Tomasi, F. Ingrosso, R. Cammi, S. Corni and G. Salmani, J. Chem. Phys., 2006, 124, 124520 CrossRef PubMed.
- M. X. Zhang, Q. Zhou, C. Du, Y. Ding and P. Song, RSC Adv., 2016, 6, 59389 RSC.
- Y. Saga, Y. Shibata and H. Tamiaki, J. Photochem. Photobiol., C, 2010, 11, 15 CrossRef CAS.
- A. L. Sobolewski and W. Domcke, Phys. Chem. Chem. Phys., 1999, 1, 3065 RSC.
- L. Serrano-Andres and M. Merchan, J. Photochem. Photobiol., C, 2009, 10, 21 CrossRef CAS.
- Q. Chu, D. A. Medvetz and Y. Pang, Chem. Mater., 2007, 19, 6421 CrossRef CAS.
- K. Furukawa, K. Hino, N. Yamamoto, K. Awasthi, T. Nakabayashi, N. Ohta and H. Sekiya, J. Phys. Chem. A, 2015, 119, 9599 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a