DOI:
10.1039/C6RA26043H
(Paper)
RSC Adv., 2017,
7, 7640-7647
VUV/UV light inducing accelerated phenol degradation with a low electric input†
Received
30th October 2016
, Accepted 8th December 2016
First published on 23rd January 2017
Abstract
This study presents the first evidence for the accelerated degradation of phenol by Fenton's reagent in a mini-fluidic VUV/UV photoreaction system (MVPS). A low-pressure mercury lamp used in the MVPS led to a complete degradation of phenol within 4–6 min. The HO˙ and HO2˙ originating from both Fenton's reagent and VUV photolysis of water were identified with suitable radical scavengers. The effects of initial concentrations of phenol, H2O2 and Fe3+ as well as solution pH on phenol degradation kinetics were examined. Increasing the initial phenol concentration slowed down the phenol degradation, whereas increasing the initial H2O2 or Fe3+ concentration accelerated the phenol degradation. The optimal solution pH was 3.7. At both 254 and 185 nm, increasing phenol concentration enhanced its absorption for the incident photons. The reaction mechanism for the degradation of phenol was suggested consistent with the results obtained. This study indicates that the VUV/UV photo-Fenton process has potential applications in the treatment of industrial wastewater containing phenol and related aromatic pollutants.
Introduction
Industrial wastewater decontamination has been a research topic during the last few decades directed to abate toxic and recalcitrant pollutants of industrial origin.1–3 Phenol can only be degraded slowly by bacteria with low to moderate concentrations in mixtures of biodegradable and non-biodegradable pollutants in industrial wastewaters.4–8 Phenols, bi-phenols, chloro-phenols and nitro-phenols need at the present time a lower cost removal system working within an acceptable time than the biological and chemical methods currently available.9,10 Some advanced oxidation processes (AOPs) have used preferentially TiO2 as a photocatalyst for this purpose. TiO2 generates highly oxidative radicals under band-gap irradiation (band gap of 3.0–3.2 eV).11–14 Under UV or UV-Vis irradiation, TiO2 and doped-TiO2, either suspended in aqueous solutions or fixed on suitable substrates, have been applied to degrade phenol with relatively slow kinetics.15–20 Moreover, to increase the dispersion and light absorption of TiO2 and doped-TiO2, Raschig-rings have been used as supports to accelerate the phenol degradation kinetics.21,22
The degradation of phenol(s) by AOPs at current remains slow and inefficient in spite of recent improvements in reactor technology, lamp design and other engineering aspects. Fenton and photo-Fenton studies in homogeneous/heterogeneous systems leading to phenol degradation have increased in recent years. Esplugas et al. reported a time of ca. 10 min for phenol degradation.23 More recently, Zazo et al. reported a time of ca. 4 h for a complete degradation of phenol by Fenton's reagent.24 The authors also reported the phenol degradation intermediates: catechol, hydroquinone, p-benzoquinone and resorcinol, followed by short organic-acids branched or not before the final mineralization.24 Pontes et al. reported phenol degradation within 40 min in homogeneous solution,25 and by Friedrich et al. within 45 min.26 Heterogeneous Fenton and photo-Fenton using Fe–clinoptilolite led to a phenol reduction within 35 min,27 while clay pillared-Fe led to a phenol reduction within 120 min.28 Pillared montmorillonite Fe and clay/silica/zeolite reduced phenol within 60 and 120 min, respectively.29,30
The mechanism for the homogeneous catalysis of H2O2 leading to highly oxidative radicals in the dark has been reviewed.31–33 The fully hydrated Fe(H2O)63+ complex was reported as a catalytic species in the Fenton system at pH < 3.34 At pH ≤ 3.5, the complexes Fe(OH)(H2O)52+ and Fe(OH)2(H2O)4+ interact with H2O2 in the Fenton system, which has been comprehensively described.35,36 These Fe–aqua complexes have optical absorption up to 390 nm.7,8 The basic Fenton reactions are presented in eqn (1)–(4), whilst eqn (5) and (6) refer to the photo-Fenton reactions. Note that in eqn (4) and (5), Fe(OH)2+ represents a simplified form of Fe(OH)(H2O)52+.
| | |
Fe2+ + H2O2 → Fe3+ + HO˙ + OH−, k1 = 76 M−1 s−1
| (1) |
| | |
Fe3+ + H2O2 → FeOOH2+ + H+, k2 = 2 × 10−2 M−1 s−1
| (2) |
| | |
FeOOH2+ → Fe2+ + HO2˙
| (3) |
| | |
Fe3+ + H2O → Fe(OH)2+ + H+
| (4) |
| | |
Fe(OH)2+ + hν → Fe2+ + HO˙
| (5) |
The photo-Fenton reactions presented in eqn (5) and (6) generate additional HO˙ through the light activated reduction of Fe(OH)2+ and the direct photolysis of H2O2. This accelerates the degradation of organic compounds compared to the dark Fenton system.34 The HO˙ generated led in the first reaction step to an aromatic ring addition to the phenol, leading to the hydroxyl-cyclohexadienyl radical.37,38
The objective of this study was to achieve an accelerated degradation of phenol by the Fenton's reagent (Fe2+/3+/H2O2) in a more advanced photo-reactor by way of VUV/UV irradiation. Due to a short degradation time and a low electric input, this VUV/UV photo-Fenton process only required a low energy consumption. The experiments were carried out in a mini-fluidic VUV/UV photoreaction system (MVPS), which provided VUV/UV or UV irradiation with an accurate quantification of UV fluence. The light absorption by each of the solution components in the photo-reactor was determined as well as the identity of intermediate oxidative radicals. The effects of initial phenol concentration, initial H2O2 concentration, and solution pH on phenol degradation kinetics were examined in detail. The reaction mechanism leading to phenol degradation was also suggested.
Experimental section
Chemical reagents and analytical methods
All solutions were prepared in ultrapure water produced from a Milli-Q system (Advantage A10, Millipore, USA). Chemicals used were of at least reagent grade and purchased from Sigma-Aldrich (St. Louis, MO, USA) or Thermo Fisher Scientific (Fair Lawn, NJ, USA). Fenton experiments were conducted employing FeCl3 and H2O2 (∼35% by weight). The initial pH was adjusted by NaOH and HCl solutions. 1,4-Benzoquinone (BQ) and tert-butanol (TBA) were used as O2˙− and HO˙ scavengers, respectively. Uridine (0.12 mM) and methanol (0.10 mM) were utilized as chemical actinometers to determine the UV fluence rate output from the MVPS.
Phenol concentration was determined by using a high-performance liquid chromatograph coupled with a photodiode-array detector (HPLC/DAD, Agilent Technologies, US). Methanol concentration was determined by a gas chromatograph equipped with a flame ionization detector (GC/FID, Shimadzu, Japan). Uridine concentration was measured by light absorption at 664 nm on a UV-Vis spectrophotometer (2600, Shimadzu, Japan) directly linked to the MVPS. H2O2 concentration was measured by Titanium(IV) oxysulfate (TiOSO4) with a detection limit of 0.01 mg L−1.39 Total organic carbon (TOC) was analyzed with a TOC analyzer (TOC-VCPH, Shimadzu, Japan).
VUV/UV and UV irradiations in the MVPS
To overcome the limitations of traditional batch VUV/UV photo-reactors, an innovative design of a bench-scale MVPS was developed in our previous study (see Fig. S1†).39,40 The photo-reactor chamber was cylindrical (length = 300 mm, outer diameter = 40 mm) with a jacketed quartz-wall. An 8 W low-pressure mercury lamp (arc length = 200 mm, Wanhua Co., Zhejiang, China) manufactured by synthetic quartz was housed in the center of the photo-reactor, which could emit both 185 (VUV) and 254 nm (UV) beams. Cooling water was recirculated through the outer chamber at a constant temperature to maintain stable VUV and UV outputs from the LP lamp. A synthetic quartz tube (i.e., VUV/UV tube, high VUV transmittance) and a Ti-doped silica tube (i.e., UV tube, no VUV transmittance) were installed parallel to the lamp in the photo-reactor chamber with the same distance (ca. 5 mm) to the lamp surface. The inner diameter and length of both tubes were 2 and 100 mm, respectively. The reaction solution flowing through the VUV/UV and UV tubes could receive the 185/254 nm and the 254 nm irradiations, respectively. Nearly same UV (at 254 nm) fluence rate (FR) could be obtained in the VUV/UV and UV tubes because both tubes had a very high 254 nm transmittance and were positioned at the same distance to the lamp surface. Nitrogen gas was flushed through the inner chamber of the photo-reactor, to eliminate the air absorption (primarily O2) of VUV and thus maximize the VUV light reaching the reaction solution. By using the chemical actinometers of uridine and methanol, the VUV and UV FRs (i.e., Eo,VUV and Eo,UV) were determined to be 1.7 and 14.5 mW cm−2, respectively. An UV-Vis spectrophotometer (2600, Shimadzu, Japan) was installed in the MVPS to provide online absorbance measurements.
Experimental procedures and reduction equivalent exposure time
After a 15 min lamp equilibration period, the phenol solution of 50 mL was circulated in the MVPS by a peristaltic pump. Samples of 1 mL each were taken out at preselected times for chemical analysis. During the MVPS runs, the reaction solution was recirculated sequentially through the exposure section (i.e., the VUV/UV or UV tube) and the dark section until a desired fluence was reached. The reduction equivalent exposure time (tree, s) is defined as the total reaction time (t, s) multiplied by the ratio of the exposure volume of the operation tube (πr2h, m3) to the total sample volume (V, m3):| |
 | (7) |
where r and h are the radius and the length of the VUV/UV (or UV) tube, respectively. In other words, tree represents the exposure time when the whole reaction solution receives the exposure under the same exposure conditions (i.e., the same Eo,UV and Eo,VUV) in the relevant tube. The VUV (FVUV, mJ cm−2) or UV (FUV, mJ cm−2) fluences can be estimated as follows:
Results and discussion
Phenol absorption spectrum and degradation kinetics and TOC reduction in the MVPS
Fig. 1 shows the absorption spectra of H2O2, FeCl3 and phenol. Note that the mercury resonance lines at 185/254 nm (i.e., in the VUV/UV tube) or 254 nm (i.e., in the UV tube) irradiations were used throughout this study. The molar absorption coefficients of H2O2, phenol and Fe(III) increased towards shorter wavelengths. The light absorption of H2O2 was so low that it could not absorb a significant amount of VUV/UV and UV photons at the low concentrations (i.e., mostly 0.147 mM) used in the MVPS. The Fe (d–d) transitions take place in the nanosecond time scale, and the intensity of these transitions giving rise to the Fe(III)-spectrum depends on the counter-ion, pH and Fe(III) concentration.41,42
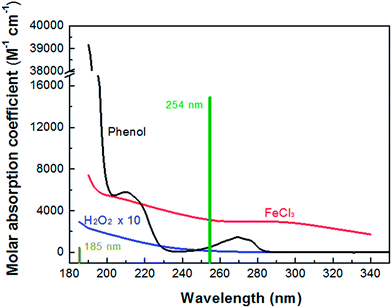 |
| | Fig. 1 Absorption spectra of H2O2, FeCl3 and phenol and mercury resonance lines at 254 and 185 nm of the low-pressure mercury lamp. | |
Fig. 2a shows the phenol degradation by the dark Fenton, UV or VUV/UV photolysis, UV photo-Fenton, and VUV/UV photo-Fenton processes at an initial pH of 3.7. The VUV/UV in the absence of the Fenton's reagent (Fe2+/3+/H2O2) induced phenol degradation due to the VUV (185 nm) light generating HO˙ from the photolysis of water.43,44 Without UV and VUV irradiation, little phenol degradation was found although both Fe3+ and H2O2 were present. UV and VUV/UV photo-Fenton processes led to a complete phenol degradation within 6–8 min. The photochemical behavior of the Fe(III)–aqua complexes plays a determining role in pollutant degradation.35,36 At pH 3.7 in this study, Fe(H2O)63+ was partially transformed into Fe(III)(OH)(H2O)52+, which was able to undergo ligand-to-metal-charge-transfer. This transformation depends on the excitation wavelength and involves inner sphere photo-induced electron transfer leading to the Fe(II)–aqua complex (i.e., Fe(II)(H2O)62+) and HO˙, as shown in eqn (5) above.
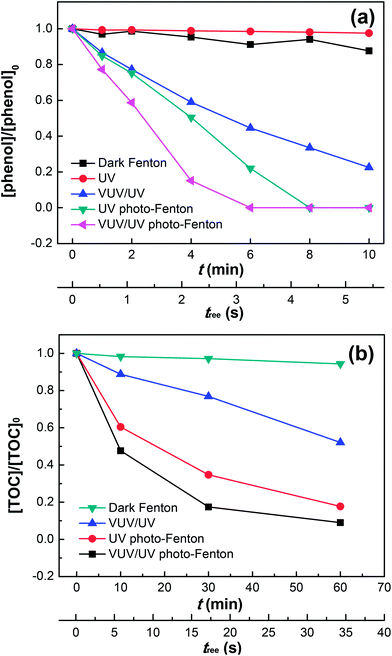 |
| | Fig. 2 Phenol degradation (a) and TOC removal (b) in the dark Fenton, UV, VUV/UV, UV photo-Fenton and VUV/UV photo-Fenton processes at pH0 = 3.7. Experimental conditions: (a) [phenol]0 = 0.011 mM, [H2O2]0 = 0.147 mM, [Fe3+]0 = 0.05 mM; and (b) [phenol]0 = 0.055 mM, [H2O2]0 = 0.735 mM, [Fe3+]0 = 0.25 mM. | |
The HO˙ leading to phenol degradation was generated by the VUV (185 nm) photolysis of water and the Fe3+ photo-reduction. In addition, VUV also generates H˙ and eaq− in aqueous solutions due to homolysis and water ionization.43,44 Fig. 2a shows that UV (254 nm) by itself did not lead to phenol degradation. The VUV (185 nm) photons preferentially react with water (concentration of 55.6 M) compared to phenol (0.011 mM). Hence, the VUV (185 nm) photon absorption by phenol only contributed marginally to the phenol degradation (see Table 1). When H2O2 and Fe3+ were added during phenol degradation under UV or VUV/UV irradiation, the higher efficiency of phenol degradation could be explained by the effective Fe(II)/Fe(III) inter-conversion and subsequently the higher generation of oxidative radicals. The Fe(OH)(H2O)52+ may also react with dissolved oxygen in the reaction solution to form O2˙−, thus enhancing the phenol degradation. It has been reported that dissolved oxygen is able to enhance Fenton/photo-Fenton reactions.37,38
Table 1 Solution parameters determining the light absorption during phenol degradation in the MVPSa
| Reactant |
C (mM) |
ε254 (M−1 cm−1) |
ε185 (M−1 cm−1) |
A254 |
A185 |
P254 (%) |
P185 (%) |
| C is the molar concentration of each solution component; ε254 and ε185 are the molar absorption coefficient of each solution component at 254 and 185 nm, respectively; A254 and A185 are the absorbance of each solution component for 1 cm optical path length at 254 and 185 nm, respectively (A254 = ε254 × C and A185 = ε185 × C); P254 is the ratio of A254 of a specific solution component to the sum of A254 of all solution components (i.e., total A254 = 0.175); and P185 is the ratio of A185 of a specific solution component to the sum of A185 for all solution components (i.e., total A185 = 2.644). |
| H2O |
55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 600 |
0.0002 |
0.03 |
0.01 |
1.8 |
5.7 |
68.1 |
| Phenol |
0.011 |
472 |
39178 |
0.005 |
0.431 |
3.0 |
16.3 |
| H2O2 |
0.147 |
19 |
341 |
0.003 |
0.050 |
1.6 |
1.9 |
| FeCl3 |
0.05 |
3140 |
7250 |
0.157 |
0.363 |
89.7 |
13.7 |
| Total |
|
|
|
0.175 |
2.644 |
100 |
100 |
Fig. 2b presents the mineralization of phenol under different irradiation conditions up to 60 min. Only 90% of the initial phenol was converted into CO2 in the VUV/UV photo-Fenton process. This suggests the existence of long-lived organic intermediates in the reaction solution, such as catechol, hydroquinone, p-benzoquinone and resorcinol as reported by Zazo et al.,24 which probably accounts for the tail after 30 min. Note that a higher concentration of phenol, H2O2 and Fe3+ was used here than that in Fig. 2a, because TOC could only be measured accurately at a higher phenol concentration (i.e., 0.055 mM). The total mineralization reaction of phenol can be written as follows:
| | |
C6H5OH + 14H2O2 → 6CO2 + 17H2O
| (10) |
The fractions of the incident photons absorbed by each solution component (i.e., Fe3+, H2O2, phenol, and H2O) during the TOC removal (Fig. 2b) is shown Fig. S2 (ESI†).
Effects of initial phenol, H2O2 and Fe3+ concentrations on phenol degradation kinetics
Fig. 3a shows phenol degradation as a function of the initial phenol concentration in the range of 0.011–0.266 mM. High concentrations of phenol decreased the degradation rate, due to the competition for the oxidative radicals in solution. In addition, the increasing phenol concentration would induce a higher fraction of UV (254 nm) photons absorbed by phenol, leaving less photons for the photo-reduction of Fe3+ (see eqn (5)). Similarly, this would also lower the amount of HO˙ generated by the VUV photolysis of water. Therefore, at high concentrations of phenol, the radicals generated by the Fenton's reagent (Fe2+/3+/H2O2) would not be enough to degrade effectively an increased amount of phenol. Although secondary reactions due to these oxidative radicals still led to the degradation of phenol, only a low efficiency was observed.5,7,48
 |
| | Fig. 3 Effects of initial concentrations of phenol (a) and H2O2 (b) on phenol degradation as well as H2O2 decomposition (c) in the VUV/UV photo-Fenton process. Experimental conditions: [phenol]0 = 0.011 mM, [H2O2]0 = 0.147 mM, [Fe3+]0 = 0.05 mM, and pH0 = 3.7. | |
Fig. 3b shows that the degradation rate of phenol increased with increasing H2O2 concentration, which implies that the interaction of H2O2 with phenol was significant. This can be explained by the photolysis of the intermediate complex/exciplex [Fe3+⋯phenol⋯H2O2] in solution.23–27 H2O2 could also scavenge HO˙ in solution,11–14 but this was not significant at H2O2 concentrations used in this study (i.e., ≤2.941 mM). Hence, no inhibition of phenol degradation was seen in Fig. 3b. Moreover, the increasing Fe3+ concentration (up to 0.5 mM) could also enhance the phenol degradation (see Fig. S3, ESI†), which allowed Fe3+ to absorb more 254 and 185 nm photons (see eqn (5)), thus enhancing the HO˙ generation. Concomitantly, this would also decrease the fraction of photons absorbed by H2O2 and subsequently the HO˙ generation through H2O2 photolysis (see eqn (6)). Comparatively, the negative effect of the decreased H2O2 photolysis was less significant. The Cl− ions added with FeCl3 could substitute water in the first coordination sphere of Fe(OH)(H2O)52+ to yield mixed ligand complexes, such as FeCl(H2O)52+ and FeCl2(H2O)4+.41,45,46
Fig. 3c shows the H2O2 consumption during phenol degradation. It is readily seen that using the Fenton's reagent in the dark led to a small H2O2 consumption due to the slow radical generation (i.e., HO˙ and HO2˙) (see eqn (1) and (3)). A higher H2O2 consumption was observed when UV irradiation was applied. The reason is that more H2O2 was consumed by Fe2+, which was effectively photo-generated from Fe3+ (see eqn (4) and (5)), and by HO˙ in the radical scavenging process as well. Under VUV/UV irradiation, a significant increase in H2O2 consumption was observed compared to that under UV irradiation, because of the enhanced Fe3+ reduction, water photolysis,43,44 and H2O2 photolysis by the VUV light.
The VUV light generates radicals and ions in water as reported by previous researchers.43,44 VUV photolysis of water leads to both radicals HO˙, HO2˙, O2˙−, H˙ and stable species such as H2O2 and O2. In this study, the kinetics of phenol degradation depended on the absorbers in the UV and VUV regions (see Table 1). Even though the H2O2/UV process presents similarities with the VUV/UV process, there are substantial differences: (a) in the H2O2/UV process, the photolysis of H2O2 only generates HO˙; whereas in the VUV/UV process, the photolysis of water generates ˙OH, H˙ and eaq− (H2O + hν (185 nm) → ˙OH + H+ + eaq−), which modifies the observed radical reactions.43,44 The direct addition of eaq− to H2O2 during VUV irradiation (i.e., H2O2 → HO2− + H+) was not possible in the Marcus inverse region,8 because the activation energy necessary for this reaction was not available.
The decomposition of H2O2 produced the oxidative radicals HO˙ (in the first place) and HO2˙ (to a lesser degree), as demonstrated by the scavenging experiments with TBA and BQ (see Fig. 4). The decomposition of H2O2 involves eqn (1), (2) and (6) as well as additionally the reactions reported in literature:31,37,47
| | |
H2O2 + HO˙ → H2O + HO2˙
| (11) |
| | |
H2O2 + HO2˙ → H2O + O2 + HO˙
| (12) |
| | |
Fe2+ + HO2˙ → Fe3+ + HO2−
| (13) |
| | |
Fe3+ + HO2˙ → Fe2+ + H+ + O2
| (14) |
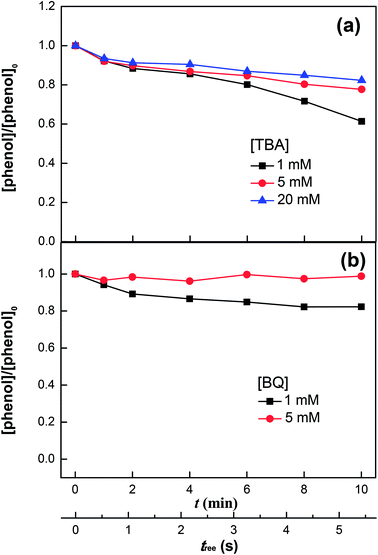 |
| | Fig. 4 Phenol degradation in the VUV/UV photo-Fenton process in the presence of tert-butanol (TBA) (a) and benzoquinone (BQ) (b). Experimental conditions: [phenol]0 = 0.011 mM, [H2O2]0 = 0.147 mM, [Fe3+]0 = 0.05 mM, and pH0 = 3.7. | |
Eqn (11) and (15) denote the scavenging and recombination of HO˙, respectively. As the H2O2 concentration increased, both reactions fostered the decomposition of H2O2.
Identification of oxidative radicals produced in the VUV/UV photo-Fenton process
Fig. 4a shows the phenol degradation under VUV/UV irradiation with TBA used as a HO˙ scavenger. HO˙ is scavenged by TBA with a reaction rate of 1.9 × 109 M−1 s−1 and reacts with phenol at a diffusion controlled reaction rate of 9.6 × 109 M−1 s−1.47 It is readily seen that increasing the TBA concentration inhibited phenol degradation due to the quenching of the HO˙.
Fig. 4b shows the phenol degradation in the presence of BQ, an HO2˙ scavenger. The phenol degradation slowed down at higher BQ concentrations. BQ scavenges HO2˙ with a reaction rate of 9.6 × 108 M−1 s−1 according to the reaction:
| | |
BQ + HO2˙ → BQ˙− + H+ + O2
| (16) |
Mechanism for phenol degradation by Fenton's reagent under VUV/UV irradiation
Fig. 5a shows the effect of initial pH on phenol degradation in the VUV/UV photo-Fenton process. Both Fe(OH)2+ reduction and H2O2 hydrolysis under UVC light (see eqn (5) and (6)), generated additional HO˙ compared to the dark Fenton system.31,34,46 The Fe(III)–aqua complexes present different molar absorption coefficients and optical absorption wavelength ranges as a function of the solution pH.35,36 This in turn induced a different rate of phenol degradation and quantum yield as a function of pH. The highest phenol degradation rate was found at pH 3.7. At this pH, the initial fully coordinated Fe3+ ion under strongly acidic conditions, Fe(H2O)63+, could incorporate an OH− group in its structure leading to the formation of Fe(OH)(H2O)52+.35,36,42 Under UVC light irradiation, Fe(OH)(H2O)52+ led to the formation of HO˙ (see eqn (5)). The relatively slower phenol degradation rates at pHs 2.1 and 3.1 were probably due to the acidic Fe–aqua complexes substituted by Cl− in the first coordination sphere such as FeCl(H2O)52+ and FeCl2(H2O)42+.47 At pH 5.2, the Fe–aqua complex, Fe(OH)2(H2O)4+, incorporated progressively one more OH− group slowing down the phenol degradation rate.48 The degradation of other organic pollutants by photo-Fenton processes have been reported recently.49,50
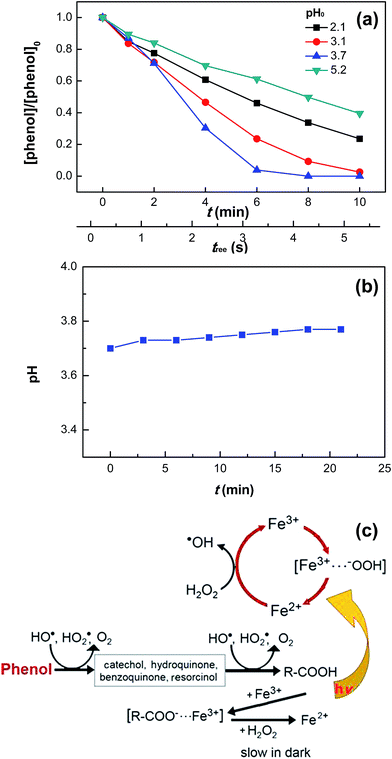 |
| | Fig. 5 Phenol degradation at various initial pH values (a) and pH variation (pH0 = 3.7) (b) in the VUV/UV photo-Fenton process as well as proposed reaction mechanisms for the dark Fenton and VUV/UV photo-Fenton processes (c). Experimental conditions: [phenol]0 = 0.011 mM, [H2O2]0 = 0.147 mM, and [Fe3+]0 = 0.05 mM. | |
Fig. 5b shows that during phenol degradation under VUV/UV irradiation, the solution pH moved slightly to more basic values. This suggests that the phenol degradation was mainly due to the HO˙/OH− conversion. Fenton reactions induced the hydroxylation of the aromatic ring of phenol through the hydroxy-cyclohexadienyl radical by the addition of HO˙, which subsequently led to the formation of quinones and other intermediates shown in Fig. 5c and also found during phenol degradation by the Fenton's reagent by Zazo et al.24 A mechanistic cycle for phenol degradation in the dark and by a photo-Fenton system under UVC irradiation is suggested in Fig. 5c.
Light absorption by each solution component during phenol degradation with various initial concentrations
Although the effect of initial phenol concentration on its degradation kinetics was already examined in Fig. 3a, the specific contribution of the highly oxidative radical species leading to phenol degradation has not been completely elucidated. This remains a controversial issue. Fig. 6a shows the fraction variation of the 185 nm photons absorbed by each solution component as a function of the phenol concentration. For the degradation of low-concentration phenol (i.e., [phenol]0 = 0.011 mM) by the VUV/UV photo-Fenton process, the fraction of 185 nm photons absorbed by water was 68.1%, while those by phenol and FeCl3 were 16.3% and 13.7%, respectively, indicating that the effect of the additional VUV irradiation was the HO˙ generation from VUV photolysis of water as compared to that of the UV photo-Fenton process. The enhancement for Fe3+ reduction and direct photolysis of phenol by the VUV (185 nm) light was rather small. When the phenol concentration increased to 0.266 mM, phenol could absorb 82.5% of the 185 nm photons. In this case, the HO˙ generation through VUV photolysis of water became much less important.
 |
| | Fig. 6 Fractions of 185 (a) and 254 nm (b) photons absorbed by each solution component as a function of initial phenol concentration in the VUV/UV photo-Fenton process. Experimental conditions: [H2O2]0 = 0.735 mM, [Fe3+]0 = 0.25 mM, and pH0 = 3.7. | |
Fig. 6b shows the fraction of the 254 nm photons absorbed by each component in the solution as a function of phenol concentration. Because of the low molar absorption coefficient of water at 254 nm (see Table 1), the photon absorption by water was not significant. For a low phenol concentration, Fe3+ could absorb most of the 254 nm photons. However, when the phenol concentration increased from 0.011 to 0.266 mM, the fraction of the 254 nm photons absorbed by phenol increased from 3.0% to 42.5%, which considerably decreased the generation of HO˙ through eqn (4)–(6).
The results of photon absorption by the components in solution show that under UV and VUV irradiations, the 254 and 185 nm photons absorbed by phenol itself played an important role in phenol degradation, especially when the phenol concentration was high. The ionization of phenol requires 8.50 eV and may go through either a mono-photonic or bi-photonic absorption process. The 185 nm light is made up of 6.70 eV photons; therefore, it might induce a mono-photonic ionization of phenol. For the 254 nm photons (4.87 eV), a bi-photonic ionization of phenol was likely to occur. The ionization reactions are presented in eqn (17)–(19). These results demonstrate that the phenol concentration strongly affected its degradation kinetics and the VUV/UV photo-Fenton process was most suitable for the treatment of dilute phenol solutions.
| | |
Phenol + hν → phenol+ + eaq−
| (17) |
| | |
Phenol + eaq− → phenol−
| (18) |
| | |
eaq− + O2 + H+ → HO2˙
| (19) |
Conclusions
This study addressed the optimization of the solution parameters leading to the enhanced abatement of phenol in a mini-fluidic VUV/UV photoreaction system. The phenol degradation required a reduced consumption of electricity because the VUV light was made full use. By the VUV/UV mercury resonant lines (185/254 nm), water and H2O2 generated the highly oxidative radicals (HO˙ and HO2˙) leading to phenol degradation. The generation of these oxidative radicals was further increased in the presence of Fe3+ ions in the photo-Fenton processes. The oxidative radicals which intervened in the phenol degradation were identified. In addition, the fraction of the incident photons absorbed by each solution component was quantified. For a dilute phenol solution (0.011 mM), the fraction absorbed by phenol was 16.3% for the VUV photons (185 nm) and 3.0% for the UV photons (254 nm), while for a high-concentration phenol solution (0.266 mM), the fraction increased to 82.5% (185 nm photons) and to 42.5% (254 nm photons). Due to a relatively low electrical energy used to generate the oxidative radicals to abate phenol, the VUV/UV photo-Fenton process has potential applications in the treatment of industrial wastewater containing phenol and related aromatic pollutants.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21590814, 51408592, 51611540344), National Geographic Air and Water Conservation Fund (GEFC23-15), and the People Program (Marie Curie Actions) of the European Union's Seventh Program FP7/2007–2013 under a REA grant (Agreement No. 318926).
References
- S. Malato, P. Fernández-Ibáñez, M. I. Maldonado, J. Blanco and W. Gernjak, Catal. Today, 2009, 147, 1–59 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Y. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- H. Roques, Chemical Water Treatment: Principles and Practice, VCH Publishers Inc., New York, NY, 1996 Search PubMed.
- N. P. Cheremissinoff, Handbook of Water and Waste Water Treatment Technologies, Butterworth-Heinemann, Woburn, MA, 2002 Search PubMed.
- J. P. Scott and D. F. Ollis, Environ. Prog., 1995, 14, 88–103 CrossRef CAS.
- O. Legrini, E. Oliveros and A. M. Braun, Chem. Rev., 1993, 93, 671–698 CrossRef CAS.
- Aquatic and Surface Photochemistry, ed. G. R. Helz, R. G. Sepp and D. G. Crosby, Lewis Publishers, Boca Raton, FL, 1994 Search PubMed.
- W. Stumm and J. J. Morgan, Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters, John Wiley & Sons, Inc., New York, NY, 3rd edn, 1996 Search PubMed.
- A. Machulek Jr, S. C. Oliveira, M. E. Osugi, V. S. Ferreira, F. H. Quina, R. F. Dantas, S. L. Oliveira, G. A. Casagrande, F. J. Anaissi, V. O. Silva, R. P. Cavalcante, F. Gozzi, D. D. Ramos, A. P. P. da Rosa, A. P. F. Santos, D. C. de Castro and J. A. Nogueira, Application of Different Advanced Oxidation Processes for the Degradation of Organic Pollutants, InTech, Sao Paulo, Brazil, 2013 Search PubMed.
- M. Stefan, Advanced Oxidation Processes for Water Treatment: Fundamental and Applications, IWA Publishing, Alliance House, London UK, 2017, in press Search PubMed.
- A. Fujishima, X. T. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. M. Dunlop, J. W. J. Hamilton, J. A. Byrne, K. O'Shea, M. H. Entezari and D. D. Dionysiou, Appl. Catal., B, 2012, 125, 331–349 CrossRef CAS.
- S. Banerjee, D. D. Dionysiou and S. C. Pillai, Appl. Catal., B, 2015, 176, 396–428 CrossRef.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. L. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- R. W. Matthews, J. Catal., 1988, 111, 264–272 CrossRef CAS.
- Environmental Health and Toxicology; https://sis.nlm.nih.gov, National Library of Medicine, USA, 2005.
- G. Balasubramanian, D. D. Dionysiou, M. T. Suidan, Y. Subramanian, I. Baudin and J. M. Laine, J. Mater. Sci., 2003, 38, 823–831 CrossRef CAS.
- M. R. Wiesner, G. V. Lowry, P. Alvarez, D. Dionysiou and P. Biswas, Environ. Sci. Technol., 2006, 40, 4336–4345 CrossRef CAS PubMed.
- J. M. Rosas, F. Vicente, A. Santos and A. Romero, Chem. Eng. J., 2013, 220, 125–132 CrossRef CAS.
- G. Veréb, L. Manczinger, A. Oszkó, A. Sienkiewicz, L. Forró, K. Mogyorósi, A. Dombi and K. Hernádi, Appl. Catal., B, 2013, 129, 194–201 CrossRef.
- P. Raja, M. Bensimon, A. Kulik, R. Foschia, D. Laub, P. Albers, R. Renganathan and J. Kiwi, J. Mol. Catal. A: Chem., 2005, 237, 215–223 CrossRef CAS.
- M. J. Sampaio, C. G. Silva, A. M. T. Silva, V. J. P. Vilar, R. A. R. Boaventura and J. L. Faria, Chem. Eng. J., 2013, 224, 32–38 CrossRef CAS.
- S. Esplugas, J. Giménez, S. Contreras, E. Pascual and M. Rodríguez, Water Res., 2002, 36, 1034–1042 CrossRef CAS PubMed.
- J. A. Zazo, J. A. Casas, A. F. Mohedano, M. A. Gilarranz and J. J. Rodríguez, Environ. Sci. Technol., 2005, 39, 9295–9302 CrossRef CAS PubMed.
- R. F. F. Pontes, J. E. F. Moraes, A. Machulek Jr and J. M. Pinto, J. Hazard. Mater., 2010, 176, 402–413 CrossRef CAS PubMed.
- L. C. Friedrich, C. L. P. S. Zanta, A. Machulek Jr, V. D. Silva and F. H. Quina, Sci. Agric., 2012, 69, 347–351 CrossRef CAS.
- M. Bayat, M. Sohrabi and S. J. Royaee, J. Ind. Eng. Chem., 2012, 18, 957–962 CrossRef CAS.
- C. Catrinescu, D. Arsene, P. Apopei and C. Teodosiu, Appl. Clay Sci., 2012, 58, 96–101 CrossRef CAS.
- O. B. Ayodele, J. K. Lim and B. H. Hameed, Appl. Catal., A, 2012, 413–414, 301–309 CrossRef CAS.
- S. Navalon, M. Alvaro and H. Garcia, Appl. Catal., B, 2010, 99, 1–26 CrossRef CAS.
- A. Y. Sychiov and V. G. Isac, Usp. Khim., 1995, 64, 1183–1209 Search PubMed.
- P. Cieśla, P. Kocot, P. Mytych and Z. Stasicka, J. Mol. Catal. A: Chem., 2004, 224, 17–33 CrossRef.
- J. Sima and J. Makanova, Coord. Chem. Rev., 1997, 160, 161–189 CrossRef CAS.
- G. Ruppert, R. Bauer and G. Heisler, J. Photochem. Photobiol., A, 1993, 73, 75–78 CrossRef CAS.
- A. Safarzadeh-Amiri, J. R. Bolton and S. R. Cater, Sol. Energy, 1996, 56, 439–443 CrossRef CAS.
- D. Wang, T. Oppenländer, M. G. El-Din and J. R. Bolton, Photochem. Photobiol., 2010, 86, 176–181 CrossRef CAS PubMed.
- C. Walling, Acc. Chem. Res., 1975, 8, 125–131 CrossRef CAS.
- S. Ito, A. Mitarai, K. Hikino, M. Hirama and K. Sasaki, J. Org. Chem., 1992, 57, 6937–6941 CrossRef CAS.
- M. K. Li, Z. M. Qiang, P. Hou, J. R. Bolton, J. H. Qu, P. Li and C. Wang, Environ. Sci. Technol., 2016, 50, 5849–5856 CrossRef CAS PubMed.
- M. K. Li, Z. M. Qiang, C. Pulgarin and J. Kiwi, Appl. Catal., B, 2016, 187, 83–89 CrossRef CAS.
- W. P. Kwan and B. M. Voelker, Environ. Sci. Technol., 2003, 37, 1150–1158 CrossRef CAS PubMed.
- V. Balzani and V. Carassiti, Photochemistry of Coordination Compounds, Academic Press, London, 1970 Search PubMed.
- S. Robl, M. Worner, D. Maier and A. M. Braun, Photochem. Photobiol. Sci., 2012, 11, 1041–1050 CAS.
- M. Bagheri and M. Mohseni, J. Hazard. Mater., 2015, 294, 1–8 CrossRef CAS PubMed.
- Inorganic Chemistry: Towards the 21st Century, ACS Symposium Series, ed. M. H. Chisholm, American Chemical Society, Washington, DC, 1983, vol. 211 Search PubMed.
- V. Nadtochenko and J. Kiwi, Environ. Sci. Technol., 1998, 32, 3273–3281 CrossRef CAS.
- P. Wardman, J. Phys. Chem. Ref. Data, 1989, 18, 1637–1755 CrossRef CAS.
- L. M. Dorfman and G. E. Adams, National Standard Reference Data System, National Bureau of Standards 46, Washington, DC, 1973 Search PubMed.
- X. L. Yang, W. Chen, J. F. Huang, Y. Zhou, Y. H. Zhu and C. Z. Li, Sci. Rep., 2015, 5, 10632 CrossRef PubMed.
- W. Shi, D. Du, B. Shen, C. F. Cui, L. J. Lu, L. Z. Wang and J. L. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 20831–20838 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26043h |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *b
*b






