
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalpol prevents alteration of cholesterol homeostasis in non-alcoholic fatty liver disease via attenuating endoplasmic reticulum stress and NOX4 over-expression
Jiting Yan ,
Changyuan Wang,
Yue Jin,
Qiang Meng,
Qi Liu,
Zhihao Liu,
Kexin Liu and
Huijun Sun*
,
Changyuan Wang,
Yue Jin,
Qiang Meng,
Qi Liu,
Zhihao Liu,
Kexin Liu and
Huijun Sun*
Department of Clinical Pharmacology, College of Pharmacy, Dalian Medical University, 9 West Section, Lvshun South Road, Lvshunkou District, Dalian 116044, China. E-mail: sunhuijun@dlmedu.edu.cn; Fax: +86 411 8611 0407; Tel: +86 411 8611 0413
First published on 4th January 2017
Abstract
Catalpol is a natural product isolated from the root of Rehmannia glutinosa, and contributes to multiple pharmacological functions. Here, we showed the protective effects of catalpol against the alteration of cholesterol homeostasis in non-alcoholic fatty liver disease (NAFLD), and further investigated the role of endoplasmic reticulum stress (ER stress) and NOX4-mediated inhibition in alteration of cholesterol homeostasis by catalpol in NAFLD. NAFLD was induced in vivo in C57BL/6J mice by feeding with high-fat diet (HFD) for 12 weeks or in vitro in HepG2 cells by stimulating with palmitate (PA) for 24 hours. The results showed that catalpol reduced hepatic lipid accumulation, ER stress, NOX4-mediated oxidative stress and alteration of cholesterol homeostasis in HFD-induced C57BL/6J mice and PA-induced HepG2 cells. In addition, NOX4 over-expression was reduced by ER stress inhibitor TUDCA and GRP78 over-expression was down-regulated by NOX4 inhibitor DPI in PA-induced HepG2 cells. The results demonstrated that ER stress and NOX4 over-expression regulated each other in PA-induced NAFLD. Furthermore, administrating with ER stress inhibitor or NOX4 inhibitor attenuated lipid accumulation and alteration of cholesterol homeostasis in NAFLD induced by PA. Therefore, catalpol conferred prevention against alteration of cholesterol homeostasis in NAFLD at least partly through attenuating both ER stress and NOX4 over-expression.
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) is the most common chronic liver disease in the world.1–3 Metabolic syndromes such as obesity, type 2 diabetes and dyslipidemia are serious risk factors of NAFLD.4 NAFLD is characterized by hepatic intracellular lipid accumulation and can progress to non-alcoholic steatohepatitis (NASH), advanced fibrosis, cirrhosis and cancer.4–6 However, the mechanisms involved in the pathogenesis of NAFLD are not yet fully understood. Recent studies have indicated that endoplasmic reticulum stress (ER stress) and oxidative stress may play important roles in the pathogenesis of NAFLD.2,6–8 Furthermore, the alteration of cholesterol homeostasis leading to cholesterol accumulation has been found in NAFLD according to previous study.9 Thus, efforts to achieve a better understanding of the mechanisms that prevent hepatic lipid accumulation, ER stress, oxidative stress and alteration of cholesterol homeostasis in NAFLD are crucial to get new therapeutic strategies. It is well known that feeding with high-fat diet (HFD) could induce NAFLD in vivo and stimulating with palmitate (PA) which is the most abundant saturated fatty acid found in western diets and processed foods could induce NAFLD in vitro.10,11 Therefore, we fed C57BL/6J mice with HFD and stimulated HepG2 cells with PA as NAFLD models to explore new therapeutic agent of NAFLD.The progression of NAFLD involves pathological state that is attributed to the disruption of lipid metabolism homeostasis.12 It is well known that cholesterol is a kind of important lipid synthesized in the liver.9 The alteration of cholesterol homeostasis in NAFLD induced by feeding with HFD or stimulating with PA changes hepatic cholesterol metabolism of uptake, synthesis, efflux and elimination related indicators and then leads to cholesterol accumulation.9,13–15 Thus, efforts to maintain cholesterol homeostasis may be beneficial for treatment of NAFLD. Cholesterol homeostasis is closely regulated by a feedback mechanism.16,17 Both sterol regulatory element-binding protein-2 (SREBP-2) which belongs to sterol regulatory element-binding proteins (SREBPs) and SREBP cleavage activating protein (SCAP) predominantly regulate hepatic cholesterol uptake through low-density lipoprotein receptor (LDLr) and synthesis through HMGCoA reductase (HMGCoA-r) by a negative-feedback mechanism.17–19 Efflux of cholesterol is mainly regulated by adenosine triphosphate-binding cassette transporter A1 (ABCA1).20 The main route of hepatic cholesterol catabolism is through biliary excretion, either by conversion into bile acids or by directly transporting intact cholesterol to the bile in the liver. And cholesterol conversion into bile acids is mediated by the rate-limiting enzyme cholesterol-7a-hydroxylase (CYP7A1).21 CYP7A1 over-expression is contributed to prevent hepatic cholesterol excessive accumulation.22 Hepatic excess cholesterol accumulation and alteration of cholesterol homeostasis are associated with ER stress and oxidative stress, which aggravate NAFLD.23
ER stress is caused by the perturbation in ER homeostasis and leads to activate the unfold protein response (UPR).24 In response to ER stress, three main pathways including PKR-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6) are activated along with glucose regulated protein 78 (GRP78) expression and released, which in turn mediate the UPR.25 PERK autophosphorylation subsequently increases the expressions of the downstream makers including eukaryotic initiation factor-2α (eIF2α) autophosphorylation, activating transcription factor 4 (ATF4) and C/EBP homologous protein (CHOP) in the progress of ER stress.26,27 In PA induced HepG2 cells and HFD induced livers of mice, ER stress is believed to mainly activate PERK-eIF2α-CHOP pathway.10,28 Chronic exposure to ER stress could result in inflammation even apoptosis.27 ER stress plays a crucial role in NAFLD and reducing ER stress using pharmacological agents is believed to be a potential therapeutic target for NAFLD.6,29
Some studies have indicated that ER stress is linked to oxidative stress.6 Oxidative stress, which is generally existed in several liver diseases, plays a significant pathogenic role in HFD-induced NAFLD.7 NADPH oxidases (NOXs), which have been implicated to be the upstream of oxidative stress,31,32 are emerging as major sources of generating reactive oxidative species (ROS) in many pathological conditions.5,33 There are at least seven NOX homologue enzymes found in mammals (NOX1-5, DUOX1 and DUOX2), while NOX1, NOX2 and NOX4 are mainly expressed in hepatocytes.6,31 In particular, NOX4 is readily distinguished from other NOX homologue enzymes by its high level of expression in the pathogenesis of cardiovascular disease, diabetes and NASH.5,33,34 Therefore, the improvement of NAFLD may also relate to the attenuation of NOX4 expression.
Catalpol, an iridoid glucoside isolated from the root of Radix Rehmanniae, has been found to possess multiple pharmacological properties, including antioxidant, anti-inflammation, anti-apoptosis, anti-diabetic and neuroprotective properties in previous studies.35,36 Notably, catalpol might influence lipid metabolism in the liver,37–39 yet the role of catalpol in NAFLD is still unknown until now. Therefore, the purposes of the present study were to explore the therapeutic effects of catalpol on lipid accumulation, ER stress, NOX4-mediated oxidative stress and alteration of cholesterol homeostasis in NAFLD, and the possible molecular mechanism underlying the effects of catalpol protecting against lipid accumulation and alteration of cholesterol homeostasis in NAFLD.
2. Materials and methods
2.1. Reagents
Catalpol (98% purity) was purchased from Nanjing Jingzhu Biotechnology Co., Ltd (Jiangsu, China). Sodium palmitate and free fatty acid-free BSA were purchased from Sigma (St. Louis, MO, USA). The NOX inhibitor, diphenyleneiodonium (DPI), was purchased from Sigma (St. Louis, MO, USA). The ER stress inhibitor, tauroursodeoxycholic acid (TUDCA), was obtained from Dalian Meilun Biological Technology Co., Ltd (Dalian, China).2.2. Animals and treatment
Eight week-old male C57BL/6J mice weighting 20 ± 2 g were purchased from Liaoning changsheng Biotechnology Co., Ltd. (Liaoning, China). 40 healthy mice were acclimated for 1 week and then randomly distributed into five groups with 8 mice in each group: (1) control group, (2) single catalpol group: control + catalpol (100 mg kg−1 d−1) group, (3) HFD group, (4) HFD + catalpol (100 mg kg−1 d−1) group, and (5) HFD + catalpol (200 mg kg−1 d−1) group. Mice of control and single catalpol groups were received a standard diet. For induction of NAFLD, other mice were fed a 45% kcal fat diet (4.73 kcal g−1, MD12032, Medicience Ltd., Jiangsu, China; protein 24.0%, carbohydrate 41.0%, and fat 24.0%). Mice in catalpol treatment groups were gavaged for indicated concentrations of catalpol for 12 weeks and other mice were received normal saline, simultaneously fed with the corresponding diet. Mice were maintained on a temperature- and humidity-controlled environment with 12 h light and 12 h dark cycle. The mice were fed free access to sterile water and fortnightly weighted. At the end of the experiment period, blood was sampled by eyeball extracting and liver tissues were isolated for analysis. All experiments were approved by the Animal Care and Use Committee of Dalian Medical University, and the experimental procedures were strictly carried out in accordance with Legislation Regarding the Use and Care of Laboratory Animals of China.2.3. Cell culture and treatment
Human hepatocellular carcinoma cell line HepG2 was purchased from Nanjing KeyGEN Biotechnology Inc. (Jiangsu, China). The HepG2 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% (v/v) fetal bovine serum (FBS) (Gibco, MD, USA) in a humidified incubator with 5% CO2 at 37 °C. One day before treatment, the culture medium was changed to DMEM medium without FBS. Sodium palmitate was dissolved in PBS at 70 °C and then mixed with 10% BSA at 55 °C for 10 min to achieve a final PA concentration of 300 μM. The experiments were divided into six groups: control; induction with 25 μM catalpol; NAFLD induction with 300 μM PA; concomitant induction with 12.5, 25, 50 μM catalpol and 300 μM PA. Briefly, HepG2 cells were seeded on 6-well plates at a density of 1 × 106 per well and allowed to grow to desired confluence. Then the cells were treated with indicated concentrations of PA and catalpol for 24 hours and the cells or culture media were collected to analysis.2.4. Biochemical analyses
Blood samples were centrifuged (3000 rpm, 10 min, 4 °C) and serum was separated for analysis. Total cholesterol (TC), triglyceride (TG), alanine aminotransferase (ALT), aspartate aminotransferase (AST), superoxide dismutase (SOD), glutathione (GSH), malondialdehyde (MDA) and lactic dehydrogenase (LDH) levels of serum were measured using commercial kits purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Free fatty acid (FFA) level of serum was determined using mouse FFA ELISA kit from Shanghai Lengton Bioscience Co., LTD (Shanghai, China). SOD, GSH, MDA and LDH levels of HepG2 cells were determined with commercial kits from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). TC, TG and FFA of HepG2 cells were measured using human ELISA kits from Elabscience Biotechnology Co., Ltd (Hubei, China). All kits were used according to the corresponding manufacturers' instructions.2.5. Liver histological analysis
For pathological analysis, the isolated left lateral segment of the liver lobes was fixed into 4% paraformaldehyde solution. Then the fixed-livers were embedded in paraffin and sliced (5 μm sections). Finally, sections of liver tissues were stained with hematoxylin–eosin (H&E).2.6. Lipid accumulation analysis
The lipid accumulation in liver tissues or HepG2 cells was evaluated by Oil Red O staining. Briefly, the unfixed freshly frozen liver tissues were embedded in optimal cutting temperature (OCT) compound and sectioned at 10 μm frozen sections. HepG2 cells were fixed with 4% paraformaldehyde solution. After the operations, the samples were stained with Oil Red O and then counterstained with hematoxylin. The Oil Red O staining kits was purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China) and the experiments were performed according to the manufacturers' instructions.2.7. Electron microscopy
For ultrastructural analysis by electron microscopy, the liver samples of control, HFD and high-dose catalpol treatment group were fixed in 2.5% glutaraldehyde solution and embedded according to standard protocols. Briefly, the samples were rinsed in PBS and then postfixed in 1% osmium tetroxide. After rinsing in PBS for 45 min, the samples were dehydrated in a graded series of ethanol (70–100%) and then routinely embedded in epon. Then, embedded specimens were cut into ultrathin sections of 0.5 μm. Finally, ultrathin sections were stained with uranyl acetate and lead citrate.2.8. Western blot analysis and antibodies
Equal amounts of protein extracts from liver tissues or HepG2 cells were separated by 8–15% SDS-PAGE and transferred to polyvinyldifluoride (PVDF) membranes. These polyvinyldifluoride (PVDF) membranes were incubated with the appropriate primary antibodies after blocking and then incubated with goat anti-rabbit or goat anti-mouse HRP-labeled secondary antibodies. Finally, the singles were detected by ECL Western Blotting Detection Substrate (Biotool, Houston, USA).CYP7A1 polyclonal antibody, p-IRE1α polyclonal antibody and HMGCoA-r polyclonal antibody were purchased from Abcam Co., Ltd (Cambridge, UK). Polyclonal antibodies SCAP, SREBP-2, LDLr, NOX4, GRP78, ATF4, ATF6, CHOP, p65, Bcl-2, Caspase-3, Caspase-9 and Histone-H3 were purchased from Proteintech Group Inc. (Wuhan, China). Polyclonal antibodies PERK, p-PERK, IRE1α, eIF2α and p-eIF2α were purchased from Beijing Biosynthesis Biotechnology Co., Ltd (Beijing, China). Polyclonal antibodies β-actin, p22phox, p-p65 and ABCA1 were purchased from Bioworld Technology Co., Inc. (St. Louis Park, MN, USA).
2.9. Quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR)
According to manufacturer's instructions, total RNAs from liver tissues or HepG2 cells were isolated with TransZol Up (RNAiso Plus) (TransGen, Beijing, China) and the RNA was converted to cDNA using an all-in-one cDNA Synthesis SuperMix (Biotool, Houston, USA). Quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) amplification and detection were performed using the 7500 qPCR system (Applied Biosystems, USA) in a real-time PCR machine with 2× SYBR Green qPCR Master Mix (Low ROX) (Biotool, Houston, USA). We used the β-actin as an internal control gene and the relative expression levels were analyzed using the 2(−ΔΔCT) method. All the primers were purchased from Invitrogen Biotechnology Co., Ltd (Shanghai, China) (Tables 1 and 2).| Genes | Mouse primers | |
|---|---|---|
| β-Actin | Forward | 5′-ACTGCCGCATCCTCTTCCT-3′ |
| Reverse | 5′-TCAACGTCACACTTCATGATGGA-3′ | |
| SCAP | Forward | 5′-ACTGGACTGAAGGCAGGTCAA-3′ |
| Reverse | 5′-GCCTCTAGTCTAGGTCCAAAGAGTTG-3′ | |
| SREBP-2 | Forward | 5′-CATCCCTTGGGCCAGAAGTT-3′ |
| Reverse | 5′-TCCTTGGCTGCTGACTTGATC-3′ | |
| LDLr | Forward | 5′-TTGGGTTGATTCCAAACTCCAT-3′ |
| Reverse | 5′-CCGATTGCCCCCATTGA-3′ | |
| HMGCoA-r | Forward | 5′-CCTGGGCCCCACATTCA-3′ |
| Reverse | 5′-GACATGGTGCCAACTCCAATC-3′ | |
| ABCA1 | Forward | 5′-ACTTAGGGCACAATTCCACAAGA-3′ |
| Reverse | 5′-CTCCTGTGGTGTTTCTGGATGA-3′ | |
| CYP7A1 | Forward | 5′-AGCAATGAAAGCAGCCTCTGA-3′ |
| Reverse | 5′-GAGCCGCAGAGCCTCCTT-3′ | |
| NOX4 | Forward | 5′-TGTACTTTATCTGGGTGTGCAGAGA-3′ |
| Reverse | 5′-TCTTCTGAATCCCATCTGTTTGAC-3′ | |
| GRP78 | Forward | 5′-TCATCGGACGCACTTGGAA-3′ |
| Reverse | 5′-CAACCACCTTGAATGGCAAGA-3′ | |
| CHOP | Forward | 5′-GTCCCTAGCTTGGCTGACAGA-3′ |
| Reverse | 5′-TGGAGAGCGAGGGCTTTG-3′ | |
| ATF4 | Forward | 5′-CTCAGACAGTGAACCCAATTGG-3′ |
| Reverse | 5′-GGCAACCTGGTCGACTTTTATT-3′ |
| Genes | Human primers | |
|---|---|---|
| β-Actin | Forward | 5′-TGGCACCCAGCACAATGAA-3′ |
| Reverse | 5′-CTAAGTCATAGTCCGCCTAGAAGCA-3′ | |
| SCAP | Forward | 5′-GCTCACCAAGTCTGTGGTCTCAA-3′ |
| Reverse | 5′-GCACTAGGGTGAAGTAGCCGATG-3′ | |
| SREBP-2 | Forward | 5′-ATGCGGCTCTGGCCTATCAC-3′ |
| Reverse | 5′-CTGCACATTCAGCCAGGTTCA-3′ | |
| LDLr | Forward | 5′-GTGTCACAGCGGCGAATG-3′ |
| Reverse | 5′-CGCACTCTTTGATGGGTTCA-3′ | |
| HMGCoA-r | Forward | 5′-GGCCCAGTTGTGCGTCTT-3′ |
| Reverse | 5′-TTTCGAGCCAGGCTTTCACT-3′ | |
| ABCA1 | Forward | 5′-TTCCCGCATTATCTGGAAAGC-3′ |
| Reverse | 5′-CAAGGTCCATTTCTTGGCTGT-3′ | |
| CYP7A1 | Forward | 5′-AGAGAGCTTGAGGCACGAGAA-3′ |
| Reverse | 5′-AATGGTGTTTGCTTGCGATG-3′ | |
| NOX4 | Forward | 5′-GCTGCATCAGTCTTAACCGAAC-3′ |
| Reverse | 5′-GGCTCTTCCATACAAATCTTCACA-3′ | |
| GRP78 | Forward | 5′-CACAGTGGTGCCTACCAAGAAG-3′ |
| Reverse | 5′-AGCAGGAGGAATTCCAGTCAGA-3′ | |
| CHOP | Forward | 5′-GCTTCTCTGGCTTGGCTGACT-3′ |
| Reverse | 5′-CTGTTTCCGTTTCCTGGTTCTC-3′ | |
| ATF4 | Forward | 5′-CCAGCAAAGCACCGCAACA-3′ |
| Reverse | 5′-CCATCCACAGCCAGCCATT-3′ |
2.10. Statistical analysis
All statistical analysis was performed using SPSS 19.0 software. All data were expressed as means ± SD. Differences between the means of individual groups were analyzed by one-way ANOVA with LSD (least significant difference) post hoc test. Data was carried out at least three independent experiments. A significant difference was defined as P < 0.05.3. Results
3.1. Catalpol decreased body weights of mice
NAFLD presents a spectrum of liver abnormalities that is known to parallel with obesity.40 Therefore, we fortnightly measured body weights of mice. The body weights of mice were significantly increased in HFD group and catalpol decreased the body weights of HFD-fed mice (Fig. 1A). Concurrently, the body weights of mice in single catalpol group were clearly decreased compared to control group (Fig. 1A). These results showed that catalpol decreased body weights of mice feeding with or without HFD.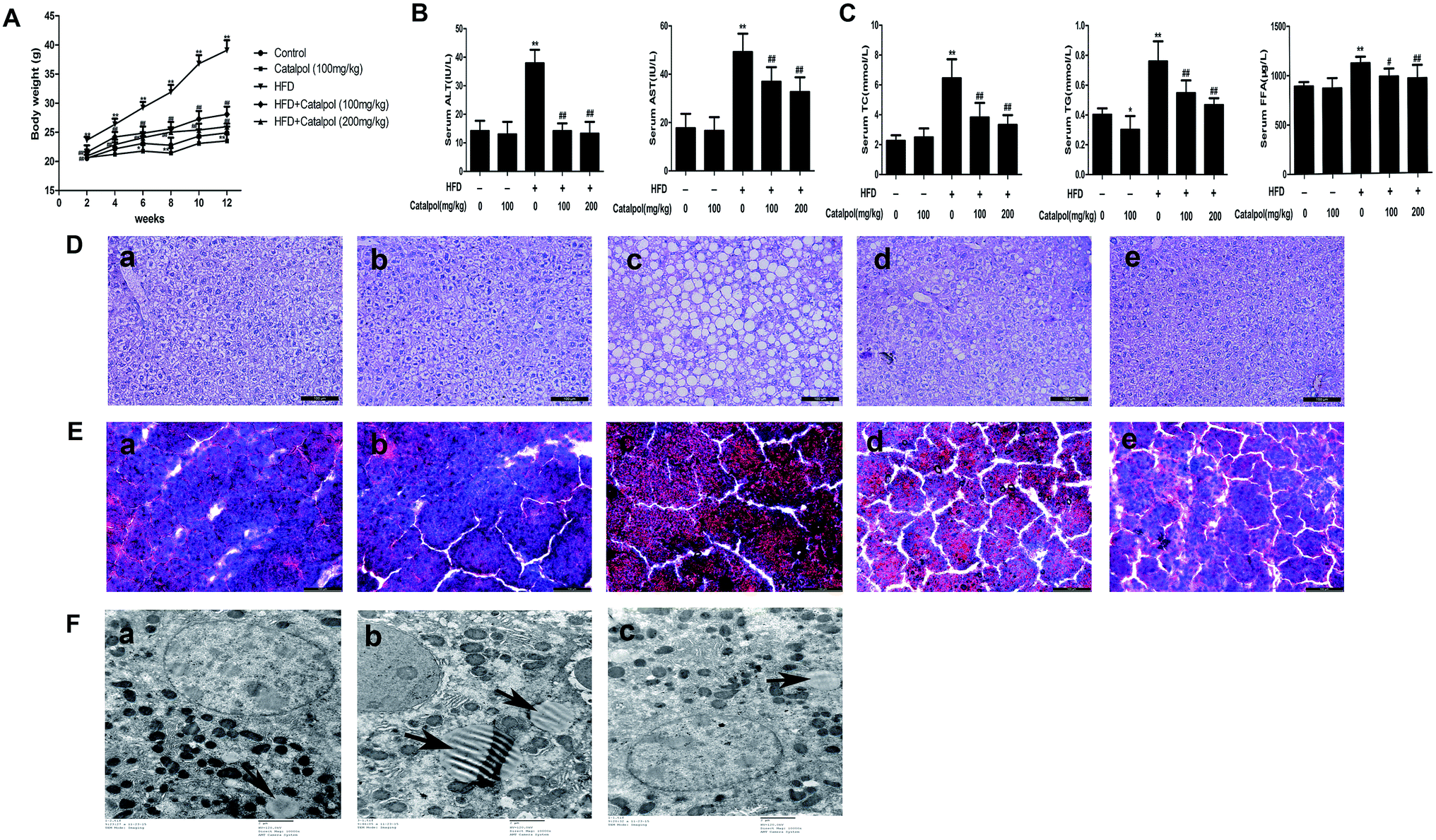 | ||
Fig. 1 Catalpol ameliorated body weights, hepatic injury and lipid accumulation in HFD-fed mice. (A) Body weights variation during 12 weeks period. (B) Serum levels of ALT and AST. (C) Serum levels of TC, TG and FFA. The results are the mean ± SD (n = 8), *P < 0.05 vs. control group, **P < 0.01 vs. control group, #P < 0.05 vs. HFD group, ##P < 0.01 vs. HFD group. (D) H&E staining of liver sections and (E) Oil Red O staining of liver sections: (a) control; (b) control + catalpol (100 mg kg−1); (c) HFD; (d) HFD + catalpol (100 mg kg−1); and (e) HFD + catalpol (200 mg kg−1). H&E- and Oil Red O-stained sections were photographed at 200× magnification. (F) Transmission electron microscopy of liver sections: (a) control; (b) HFD; and (c) HFD + catalpol (200 mg kg−1). The liver sections were photographed at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× magnification. The lipid droplets were indicated by arrows. 000× magnification. The lipid droplets were indicated by arrows. | ||
3.2. Catalpol ameliorated hepatic injury and lipid accumulation in HFD-fed mice
Hepatic injury and lipid accumulation were frequently found in NAFLD.41 The results indicated that serum ALT and AST levels were increased in HFD-fed mice, and serum ALT and AST levels were decreased in catalpol-treated mice (Fig. 1B). Additionally, the TC, TG and FFA levels of serum were significantly increased in HFD-fed mice. However, catalpol clearly abrogated the increases of serum TC, TG and FFA in HFD-fed mice (Fig. 1C). Meanwhile, H&E staining of liver displayed significant macrovesicular steatosis in hepatocytes of HFD-fed mice, while steatosis was clearly reduced in hepatocytes of catalpol-treated mice (Fig. 1D). In agreement, the result of Oil Red O staining demonstrated a pronounced increase of hepatic lipid droplets in HFD-fed mice. However, hepatic lipid droplets accumulation was observably decreased in catalpol-treated groups compared with HFD-fed group (Fig. 1E), and it was concomitant with a significant reduction of hepatic intracellular lipid drops in high-dose catalpol treated mice compared with HFD-fed mice by transmission electron microscopy (Fig. 1F). Collectively, these results confirmed that catalpol ameliorated hepatic injury and lipid accumulation in HFD-fed mice.3.3. Catalpol prevented hepatic alteration of cholesterol homeostasis in HFD-fed mice
NAFLD is characterized with hepatic excess cholesterol accumulation and hepatic cholesterol abnormal metabolism is associated with NAFLD.9 The LDLr and HMGCoA-r, which respectively play pivotal roles in intracellular cholesterol uptake and synthesis, are predominantly regulated by SREBP-2 and SCAP expressions.9,17 The results showed that hepatic LDLr and HMGCoA-r mRNA and protein expressions were inhibited in HFD-fed mice (Fig. 2A and B). However, hepatic LDLr and HMGCoA-r mRNA and protein expressions were significantly increased in catalpol-treated mice compared to HFD-fed mice. As the regulators of LDLr and HMGCoA-r, SCAP and SREBP-2 mRNA and protein expressions were also clearly decreased in livers of HFD-fed mice, whereas catalpol significantly elevated hepatic SCAP and SREBP-2 mRNA and protein expressions in HFD-fed mice (Fig. 2C and D). ABCA1 is the key mediator for hepatic cholesterol efflux,42 and feeding with HFD to mice clearly up-regulated hepatic ABCA1 mRNA and protein expressions in the present study. However, hepatic ABCA1 mRNA and protein expressions were significantly down-regulated in catalpol-treated mice compared to HFD-fed mice (Fig. 2E). CYP7A1 is the limiting enzyme which mainly eliminates hepatic excessive cholesterol conversion to bile acids.22 In HFD-fed mice, the levels of hepatic CYP7A1 mRNA and protein expressions were markedly suppressed, whereas treatment with catalpol significantly reversed the suppressed CYP7A1 expressions (Fig. 2F). In conclusion, catalpol prevented hepatic alteration of cholesterol homeostasis in HFD-fed mice.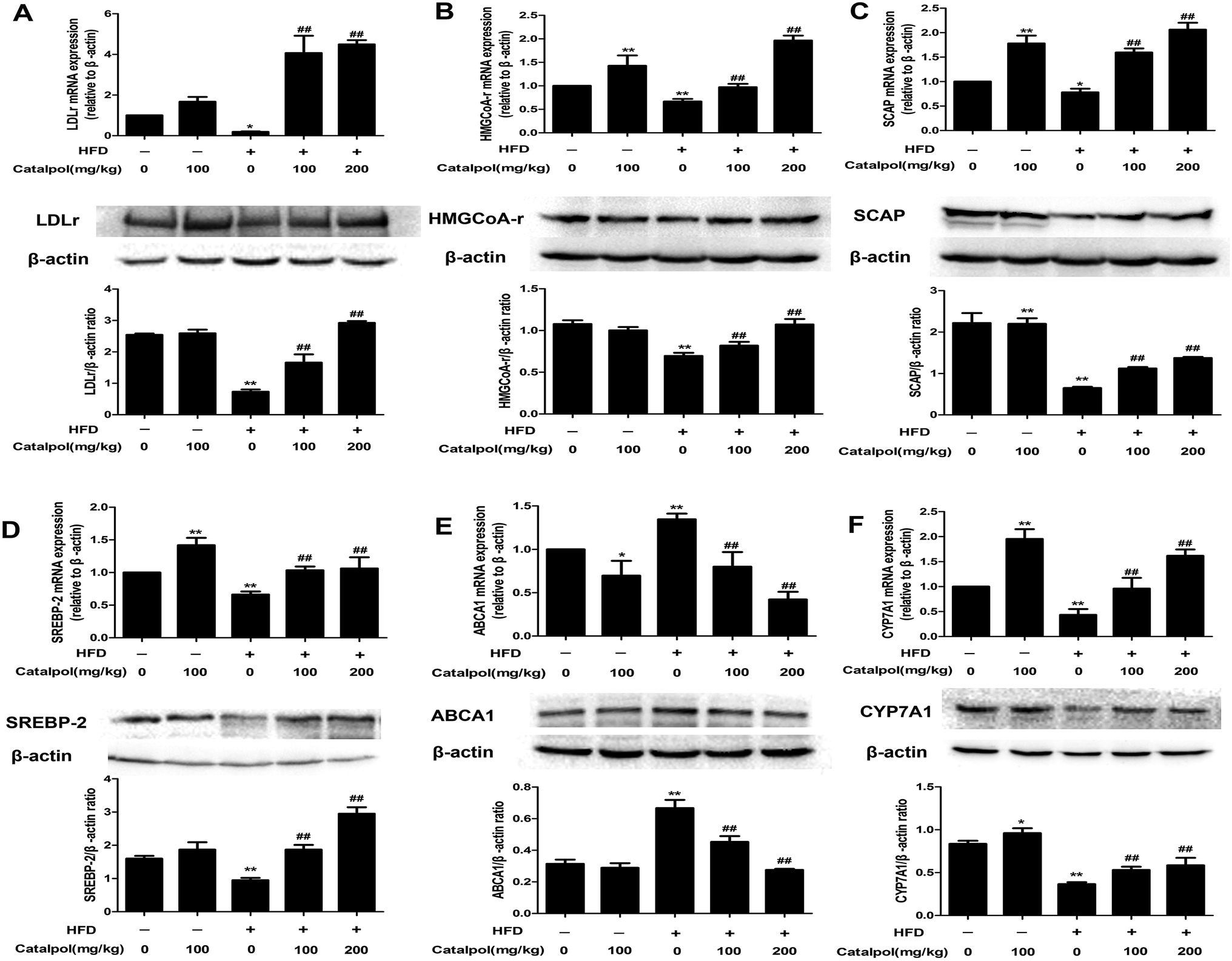 | ||
| Fig. 2 Catalpol prevented hepatic alteration of cholesterol homeostasis in HFD-fed mice. (A) Hepatic LDLr mRNA and protein expression levels. (B) Hepatic HMGCoA-r mRNA and protein expression levels. (C) Hepatic SCAP mRNA and protein expression levels. (D) Hepatic SREBP-2 mRNA and protein expression levels. (E) Hepatic ABCA1 mRNA and protein expression levels. (F) Hepatic CYP7A1 mRNA and protein expression levels. The results are mean ± SD (n = 3), *P < 0.05 vs. control group, **P < 0.01 vs. control group, ##P < 0.01 vs. HFD group. | ||
3.4. Catalpol inhibited hepatic ER stress in HFD-fed mice
ER stress plays a crucial role in NAFLD and thus could be a potential therapeutic target for NAFLD.43–45 GRP78, a key regulator of UPR and an ER resident chaperone protein, is followed to express and release in ER stress.29 The results showed that feeding with HFD to mice obviously up-regulated hepatic GRP78 mRNA and protein expressions. Reversely, catalpol significantly reduced the increases of hepatic GRP78 mRNA and protein expressions in HFD-fed mice (Fig. 3A). Following the release of GRP78, three ER stress sensors including PERK, IRE1 and ATF6 were activated.46 In HFD-fed mice, hepatic p-PERK, p-IRE1α, ATF6 protein expressions were clearly increased. In addition, hepatic protein expression of p-eIF2α and mRNA and protein expressions of ATF4 and CHOP were up-regulated in HFD-fed mice. However, catalpol significantly decreased hepatic protein over-expressions of p-PERK, p-IRE1α, ATF6 and p-eIF2α in HFD-fed mice and catalpol also reduced the increases of hepatic ATF4 and CHOP mRNA and protein expressions in HFD-fed mice (Fig. 3B–G). These results suggested that catalpol inhibited hepatic ER stress in HFD-fed mice.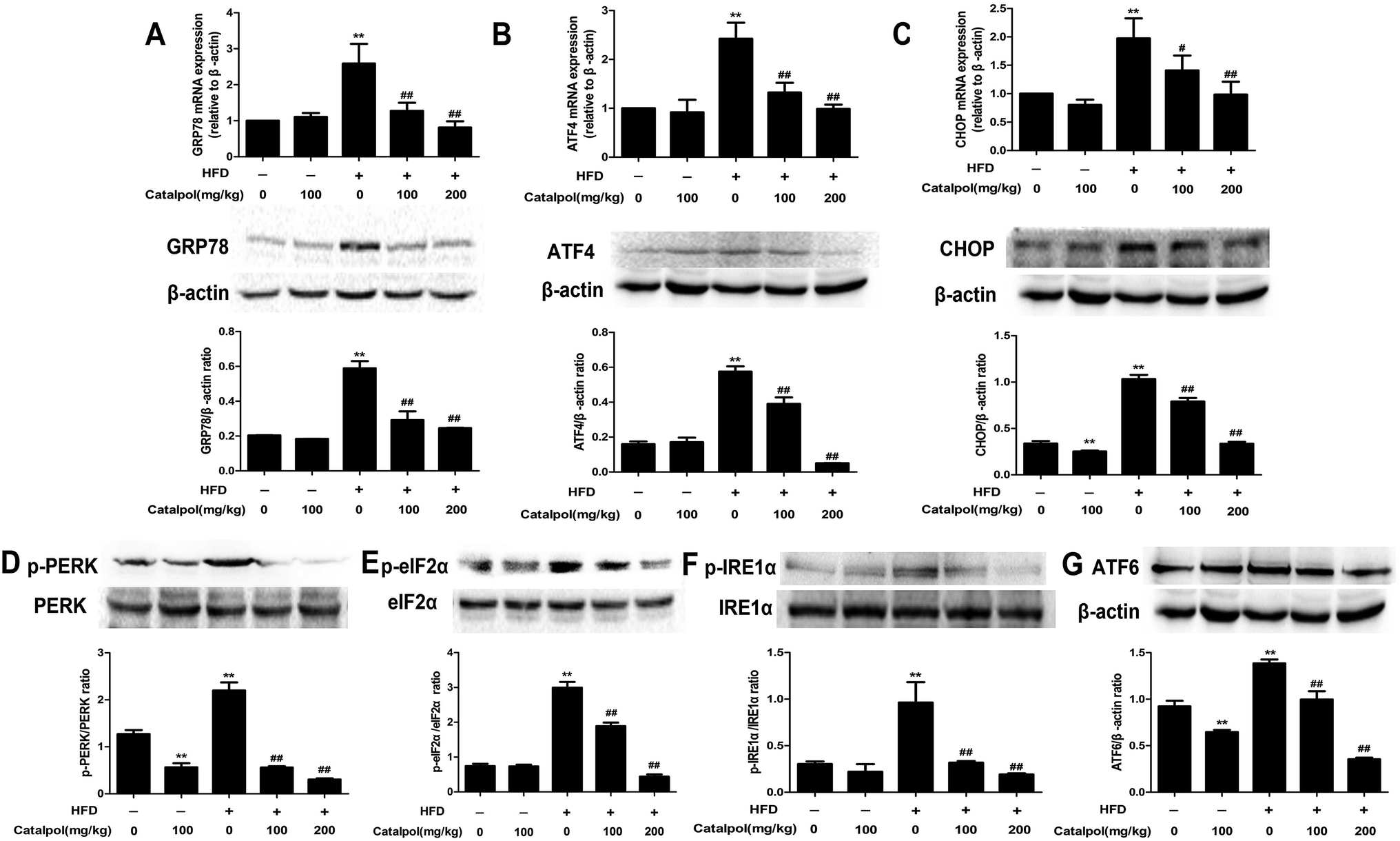 | ||
| Fig. 3 Catalpol inhibited hepatic ER stress in HFD-fed mice. (A) Hepatic GRP78 mRNA and protein expression levels. (B) Hepatic ATF4 mRNA and protein expression levels. (C) Hepatic CHOP mRNA and protein expression levels. (D) Hepatic p-PERK protein expression level. (E) Hepatic p-eIF2α protein expression level. (F) Hepatic p-IRE1α protein expression level. (G) Hepatic ATF6 protein expression level. The results are mean ± SD (n = 3), **P < 0.01 vs. control group, #P < 0.05 vs. HFD group, ##P < 0.01 vs. HFD group. | ||
3.5. Catalpol attenuated hepatic oxidative stress and NOX4 over-expression in HFD-fed mice
NAFLD patients were reported to increase hepatic oxidative stress which was an important factor in the progression of NAFLD.47,48 The results showed that the levels of serum LDH and lipid peroxidation products like MDA were increased, and the levels of serum anti-oxidants enzymes like SOD and anti-oxidants compounds such as GSH were decreased in HFD-fed mice. On the contrary, serum GSH and SOD levels were clearly increased and serum MDA and LDH levels were significantly reduced in catalpol-treated mice compared with HFD-fed mice (Fig. 4A–D). NOX4 is one of the homologue enzymes of NADPH oxidases (NOXs) which regulate oxidative stress.34,49 The results demonstrated that feeding with HFD to mice induced hepatic NOX4 mRNA and protein over-expressions were greatly attenuated by treating with catalpol (Fig. 4E and F). The protein expression of p22phox, which is required for all NADPH oxidase isoforms as a catalytic subunit of NADPH oxidases,50 was increased in livers of HFD-fed mice. However, catalpol attenuated p22phox protein over-expression in HFD-fed mice (Fig. 4G). Overall, these findings further proved that catalpol attenuated hepatic oxidative stress and NOX4 over-expression in HFD-fed mice.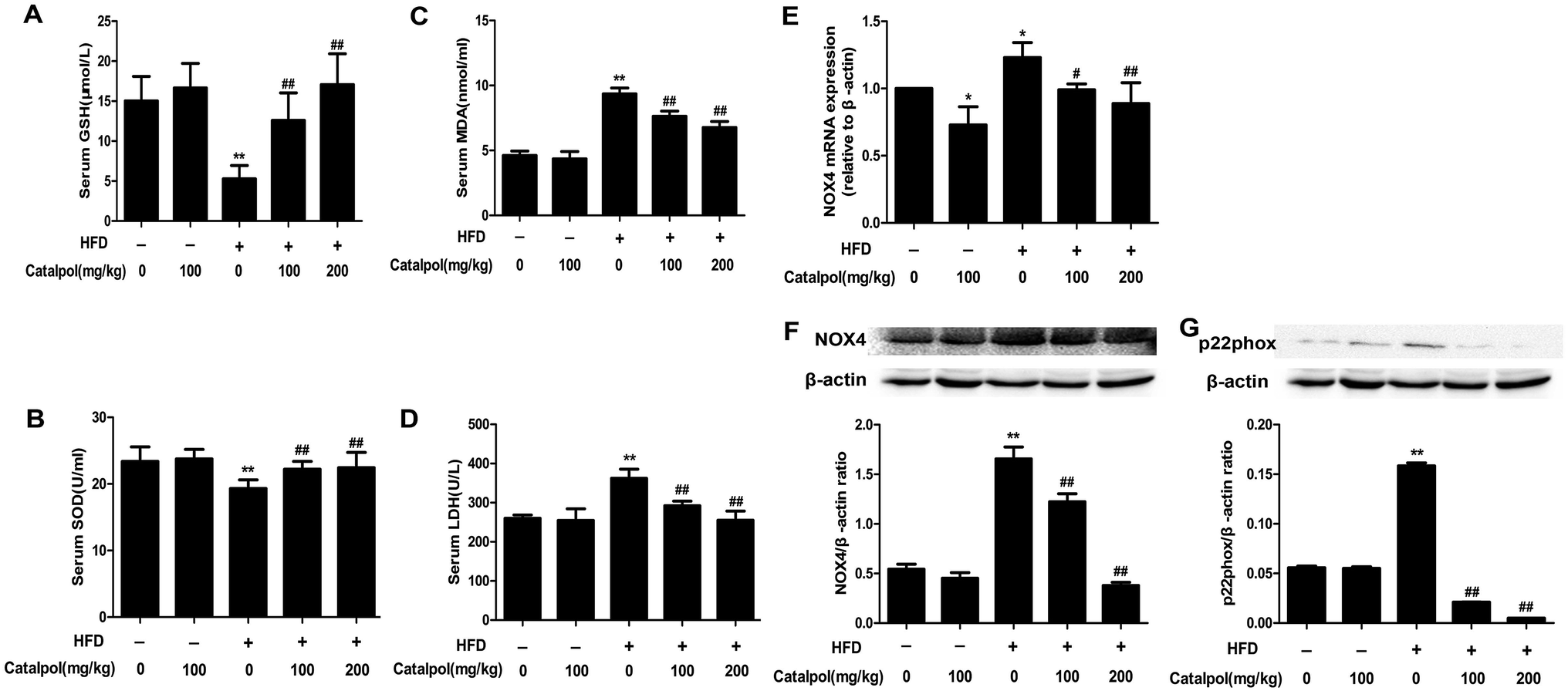 | ||
| Fig. 4 Catalpol attenuated hepatic oxidative stress and NOX4 over-expression in HFD-fed mice. (A) Serum level of GSH. (B) Serum level of SOD. (C) Serum level of MDA. (D) Serum level of LDH. The results are the mean ± SD (n = 8), **P < 0.01 vs. control group, ##P < 0.01 vs. HFD group. (E) Hepatic NOX4 mRNA expression level. (F) Hepatic NOX4 protein expression level. (G) Hepatic p22phox protein expression level. The results are the mean ± SD (n = 3), *P < 0.05 vs. control group, **P < 0.01 vs. control group, #P < 0.05 vs. HFD group, ##P < 0.01 vs. HFD group. | ||
3.6. Catalpol inhibited hepatic activation of NF-κB pathway and apoptosis in HFD-fed mice
Some studies considered that ER stress and oxidative stress could activate inflammation effects such as activation of NF-κB pathway.30,51,52 The results showed that feeding with HFD up-regulated hepatic protein expressions of nucleus p65 and total p-p65, and the hepatic protein expressions of nucleus p65 and total p-p65 were significantly down-regulated in catalpol-treated mice (Fig. 5A and B). Therefore, catalpol inhibited hepatic NF-κB pathway activation in HFD-fed mice.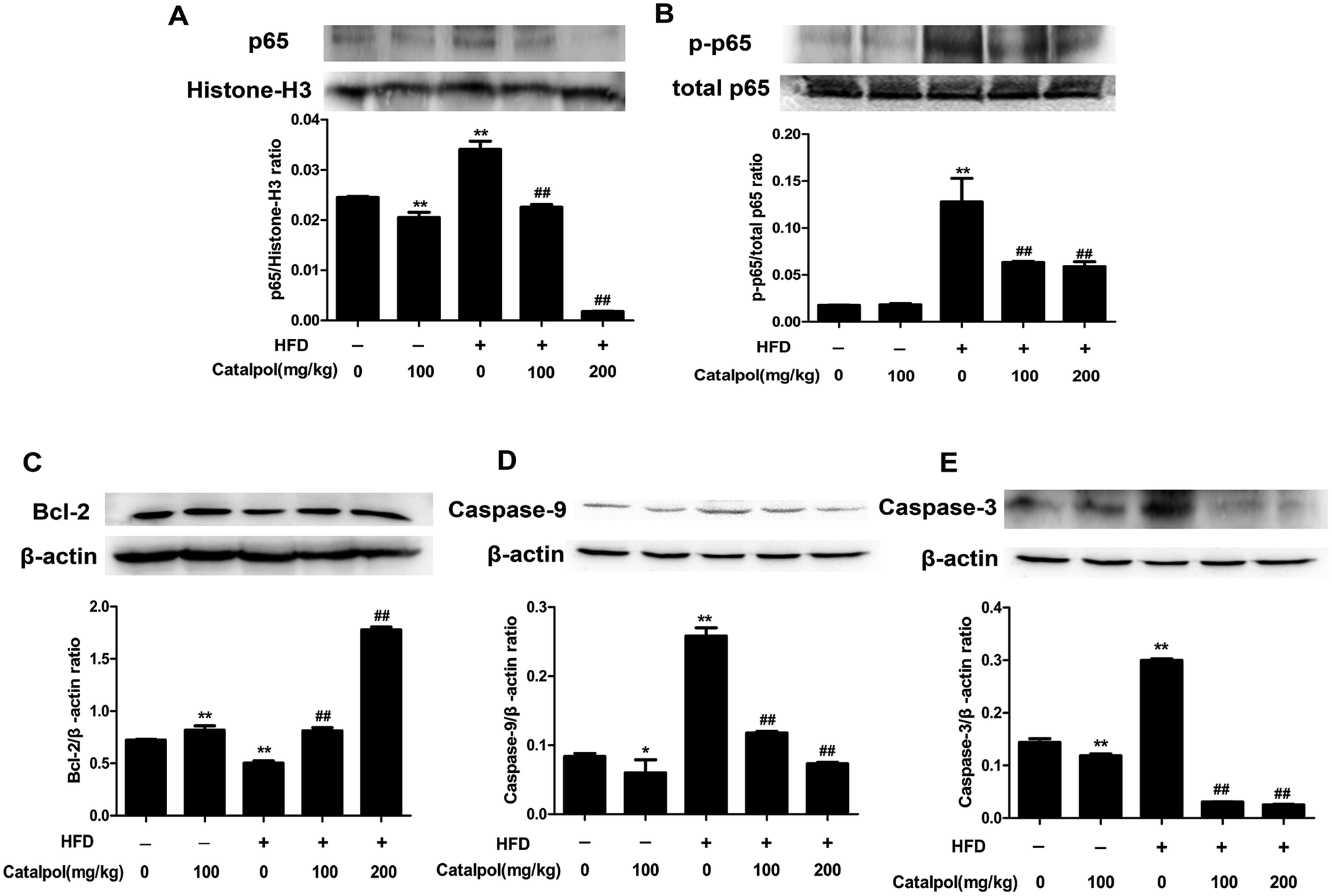 | ||
| Fig. 5 Catalpol inhibited hepatic activation of NF-κB pathway and apoptosis in HFD-fed mice. (A) Hepatic nucleus p65 protein expression level. (B) Hepatic p-p65 protein expression level. (C) Hepatic Bcl-2 protein expression level. (D) Hepatic Caspase-9 protein expression level. (E) Hepatic Caspase-3 protein expression level. The results are the mean ± SD (n = 3), *P < 0.05 vs. control group, **P < 0.01 vs. control group, ##P < 0.01 vs. HFD group. | ||
Hepatic cells apoptosis was reported to be activated by prolonging ER stress.53 The results showed that feeding with HFD to mice could induce significant increases of hepatic caspase protein expressions such as Caspase-9 and Caspase-3. Moreover, the anti-apoptotic protein Bcl-2 was clearly down-regulated in livers of HFD-fed mice. Whereas, catalpol obviously decreased hepatic Caspase-3 and Caspase-9 as well as elevated Bcl-2 protein expression levels in HFD-fed mice (Fig. 5C–E). Thus, catalpol inhibited hepatic apoptosis in HFD-fed mice.
3.7. Catalpol ameliorated lipid accumulation in PA-induced HepG2 cells
The results demonstrated that catalpol dose-dependently decreased the increases of TC, TG and FFA levels in HepG2 cells induced by PA (Fig. 6A). Additionally, Oil Red O staining demonstrated more lipid droplets in HepG2 cells induced by PA, however, less lipid droplets was found in HepG2 cells of catalpol-treated groups compared with PA group (Fig. 6B). Thus, these results confirmed that catalpol ameliorated lipid accumulation in PA-induced HepG2 cells.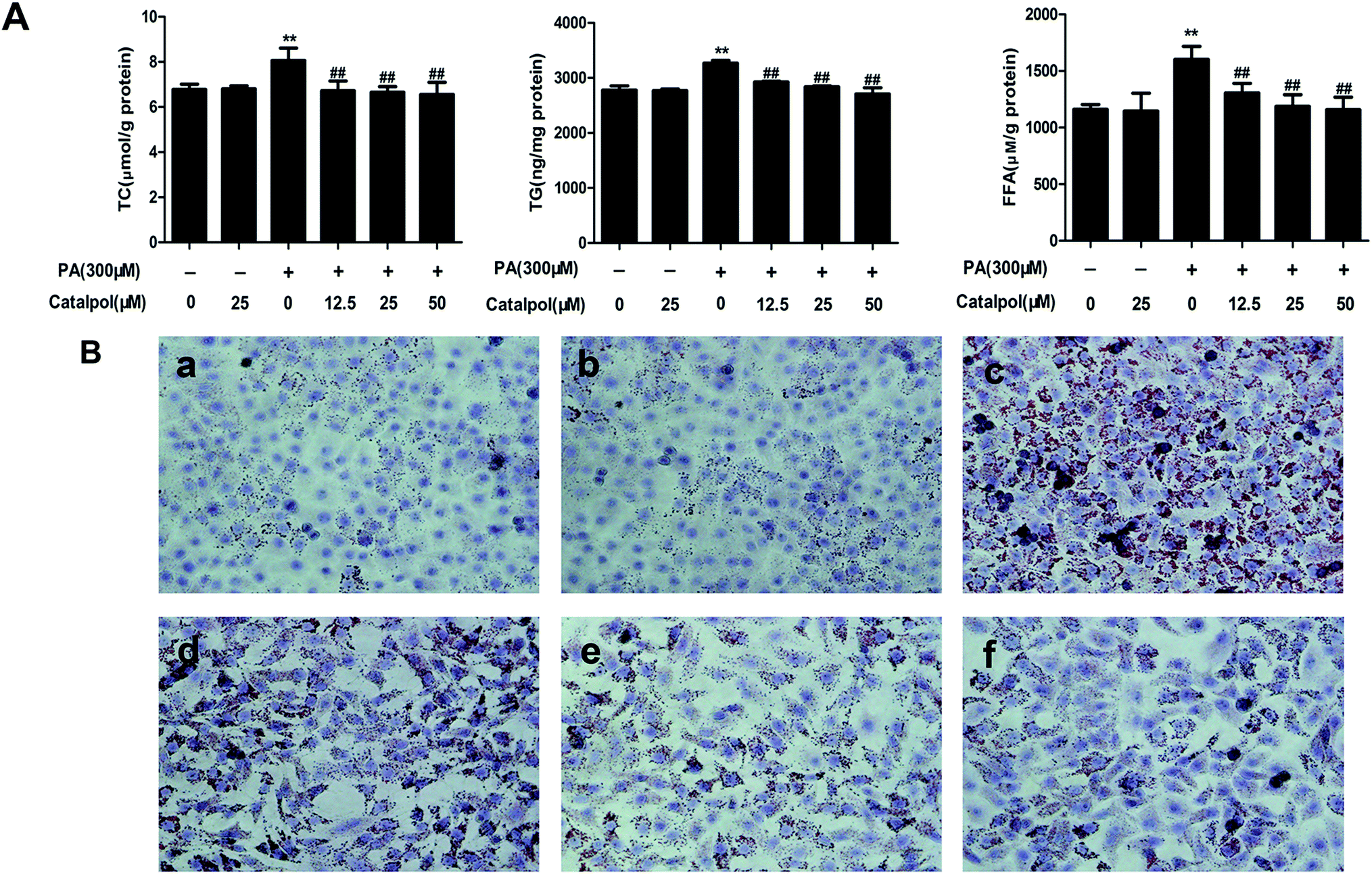 | ||
| Fig. 6 Catalpol ameliorated lipid accumulation in PA-induced HepG2 cells. (A) The levels of TC, TG and FFA. The results are the mean ± SD (n = 3), **P < 0.01 vs. control group, ##P < 0.01 vs. PA group. (B) Oil Red O staining of HepG2 cells: (a) control; (b) catalpol (25 μM); (c) PA (300 μM); (d) PA + catalpol (12.5 μM); (e) PA + catalpol (25 μM); and (f) PA + catalpol (50 μM). Oil Red O staining of HepG2 cells were photographed at 400× magnifications. | ||
3.8. Catalpol prevented alteration of cholesterol homeostasis in PA-induced HepG2 cells
As the results indicated, PA obviously inhibited HepG2 cells LDLr, HMGCoA-r, SCAP and SREBP-2 mRNA and protein expressions. However, catalpol significantly up-regulated LDLr, HMGCoA-r, SCAP and SREBP-2 mRNA and protein expressions in a dose-dependent manner in PA-induced HepG2 cells compared with PA group (Fig. 7A–D). In addition, catalpol dose-dependently repressed PA induced elevations of ABCA1 mRNA and protein expressions in HepG2 cells (Fig. 7E). As expected, CYP7A1 expressions of mRNA and protein in HepG2 cells were decreased by PA, yet catalpol significantly increased the mRNA and protein expressions of CYP7A1 in a dose-dependent manner in PA-induced HepG2 cells (Fig. 7F). In conclusion, catalpol prevented alteration of cholesterol homeostasis in PA-induced HepG2 cells.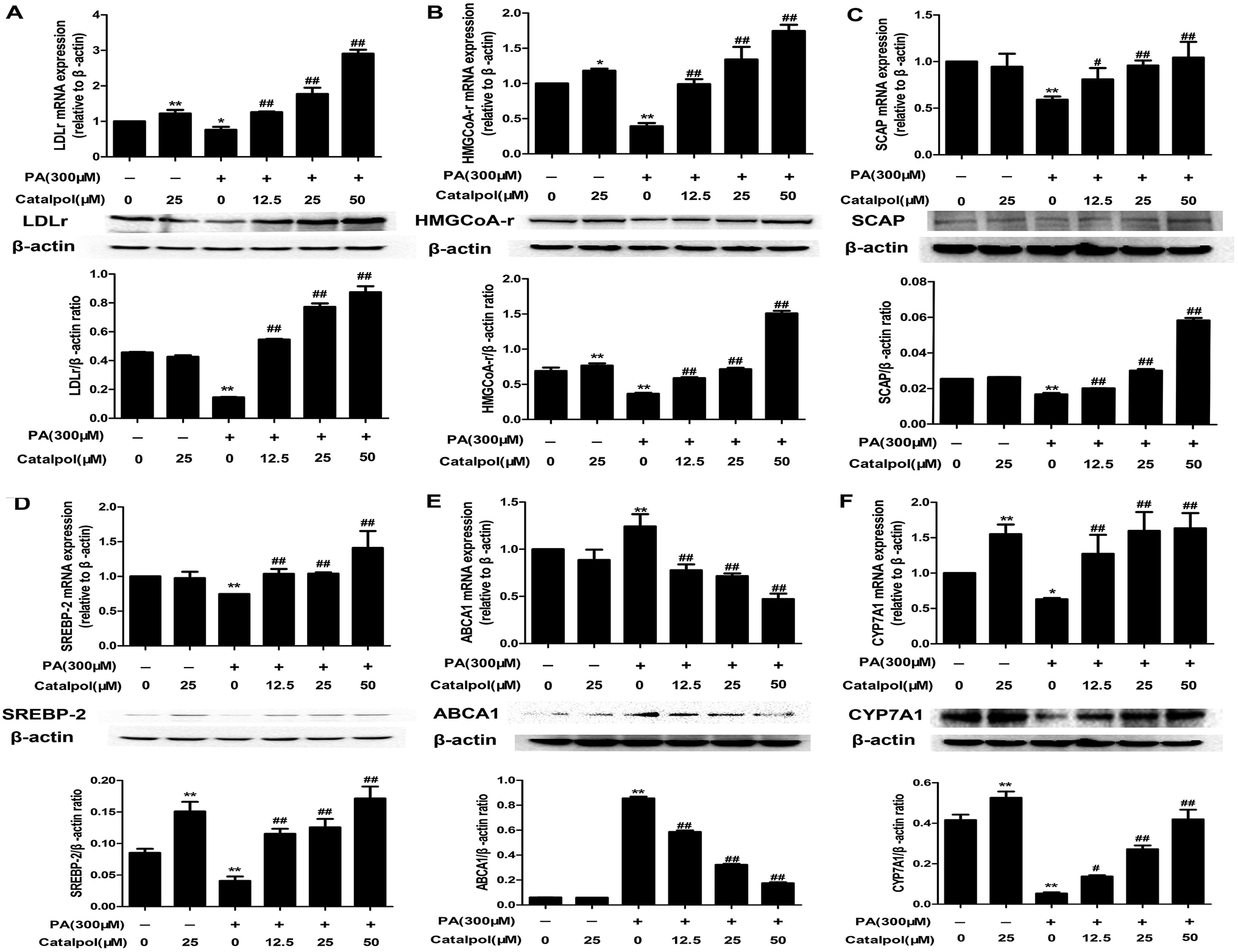 | ||
| Fig. 7 Catalpol prevented the alteration of cholesterol homeostasis in PA-induced HepG2 cells. (A) LDLr mRNA and protein expression levels. (B) HMGCoA-r mRNA and protein expression levels. (C) SCAP mRNA and protein expression levels. (D) SREBP-2 mRNA and protein expression levels. (E) ABCA1 mRNA and protein expression levels. (F) CYP7A1 mRNA and protein expression levels. The results are mean ± SD (n = 3), *P < 0.05 vs. control group, **P < 0.01 vs. control group, #P < 0.05 vs. PA group, ##P < 0.01 vs. PA group. | ||
3.9. Catalpol inhibited ER stress in PA-induced HepG2 cells
The results demonstrated that GRP78, ATF4 and CHOP mRNA and protein expressions were clearly elevated in HepG2 cells induced by PA, whereas treatment with catalpol could significantly decreased GRP78, ATF4 and CHOP mRNA and protein expressions in a dose-dependent manner (Fig. 8A–C). Additionally, PA markedly elevated protein expression levels of p-PERK, p-IRE1α, ATF6 and p-eIF2α in HepG2 cells, however, the expressions of p-PERK, p-IRE1α, ATF6 and p-eIF2α were significantly inhibited by treating with catalpol in a dose-dependent manner in PA-induced HepG2 cells (Fig. 8D–G). These data indicated that catalpol inhibited ER stress in PA-induced HepG2 cells.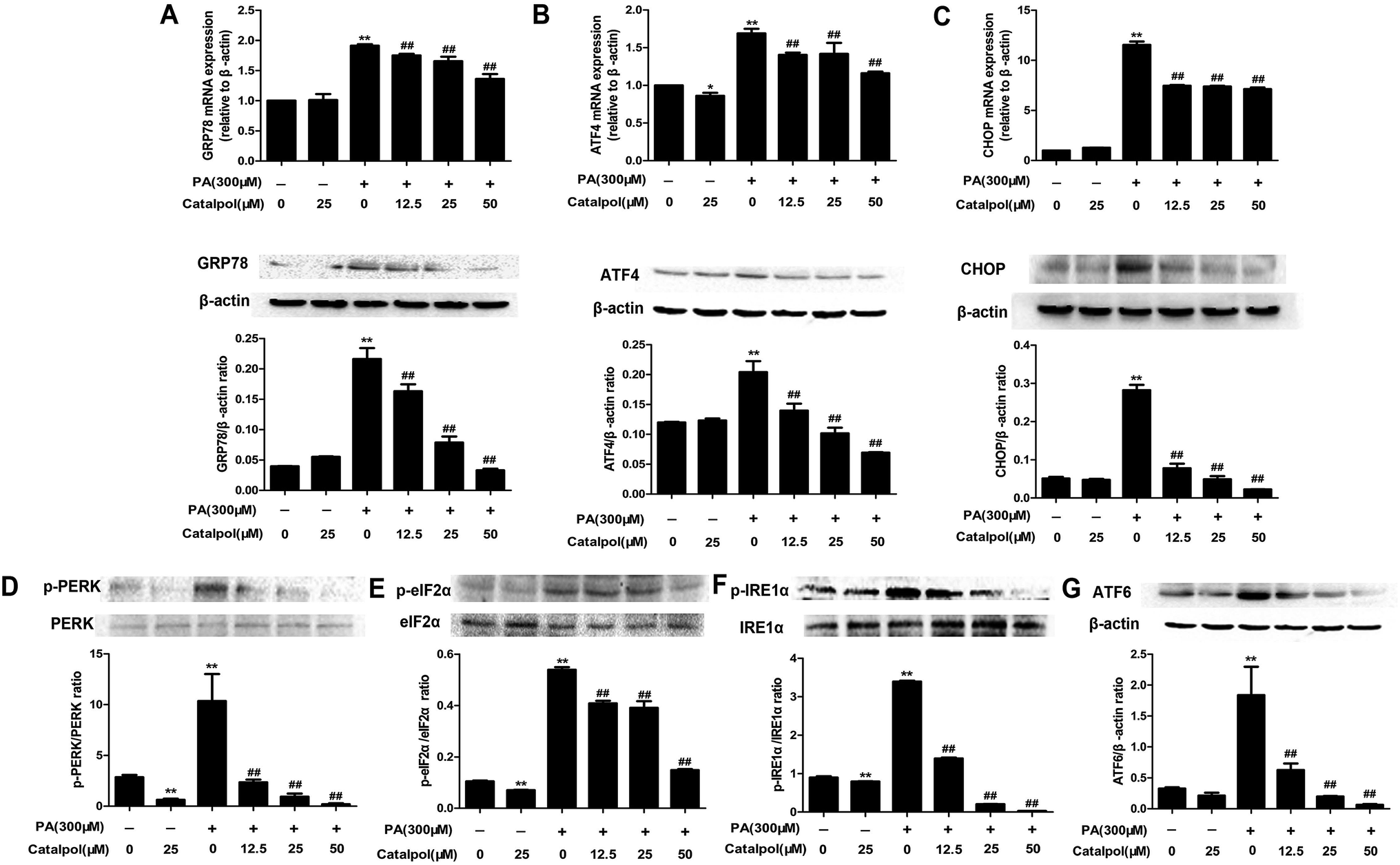 | ||
| Fig. 8 Catalpol attenuated ER stress in PA-induced HepG2 cells. (A) GRP78 mRNA and protein expression levels. (B) ATF4 mRNA and protein expression levels. (C) CHOP mRNA and protein expression levels. (D) Level of p-PERK protein expression. (E) Level of p-eIF2α protein expression. (F) Level of p-IRE1α protein expression. (G) ATF6 protein expression level. The results are mean ± SD (n = 3), *P < 0.05 vs. control group, **P < 0.01 vs. control group, ##P < 0.01 vs. PA group. | ||
3.10. Catalpol attenuated oxidative stress and NOX4 over-expression in PA-induced HepG2 cells
The results showed that catalpol significantly increased GSH level and SOD activity, yet reduced MDA level in a dose-dependent manner in PA-induced HepG2 cells (Fig. 9A–C). The production of LDH in the culture supernatant was up-regulated by PA, whereas catalpol treatment resulted in the down-regulation of LDH dose-dependently (Fig. 9D). Additionally, the attenuating effects of catalpol on NOX4 over-expressions of mRNA and protein dose-dependently were also observed in PA-induced HepG2 cells (Fig. 9E and F). The results also showed catalpol significantly decreased PA induced p22phox protein over-expression in PA-induced HepG2 cells (Fig. 9G). Overall, these findings further proved that catalpol attenuated oxidative stress and NOX4 over-expression in PA-induced HepG2 cells.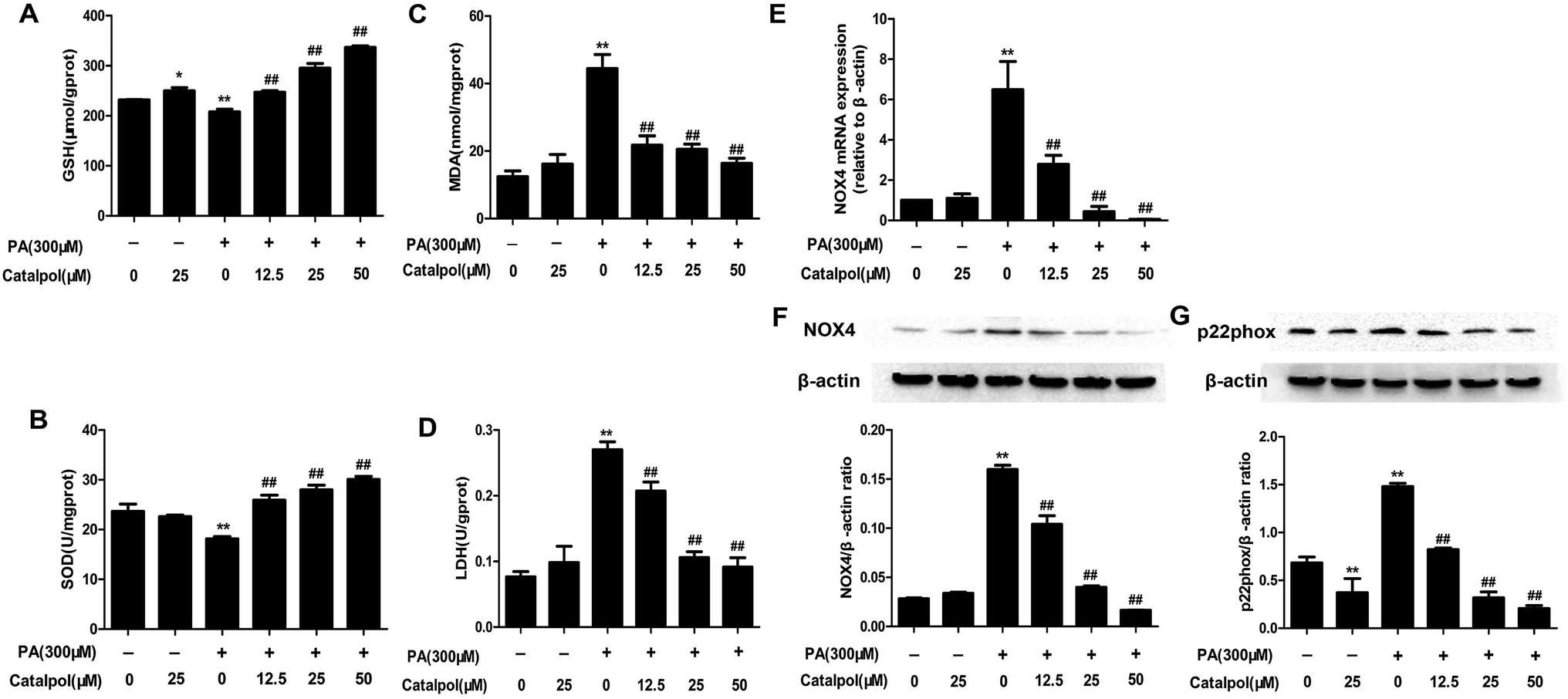 | ||
| Fig. 9 Catalpol inhibited oxidative stress and NOX4 over-expression in PA-induced HepG2 cells. (A) The level of GSH. (B) The level of SOD. (C) The level of MDA. (D) The level of LDH. (E) NOX4 mRNA expression level. (F) NOX4 protein expression level. (G) Level of p22phox protein expression. The results are the mean ± SD (n = 3), *P < 0.05 vs. control group, **P < 0.01 vs. control group, ##P < 0.01 vs. PA group. | ||
3.11. NOX4 over-expression and ER stress regulated each other in PA-induced NAFLD
To determine the interaction between PA-induced NOX4 over-expression and ER stress in HepG2 cells, we treated PA-induced HepG2 cells with the ER stress inhibitor TUDCA or the NOX inhibitor DPI to analyze NOX4 or GRP78 protein expressions, respectively. TUDCA inhibited PA induced NOX4 protein over-expression and DPI reduced PA induced GRP78 protein over-expression (Fig. 10A and B). These results provided ample evidence that NOX4 over-expression and ER stress regulated each other in PA-induced NAFLD.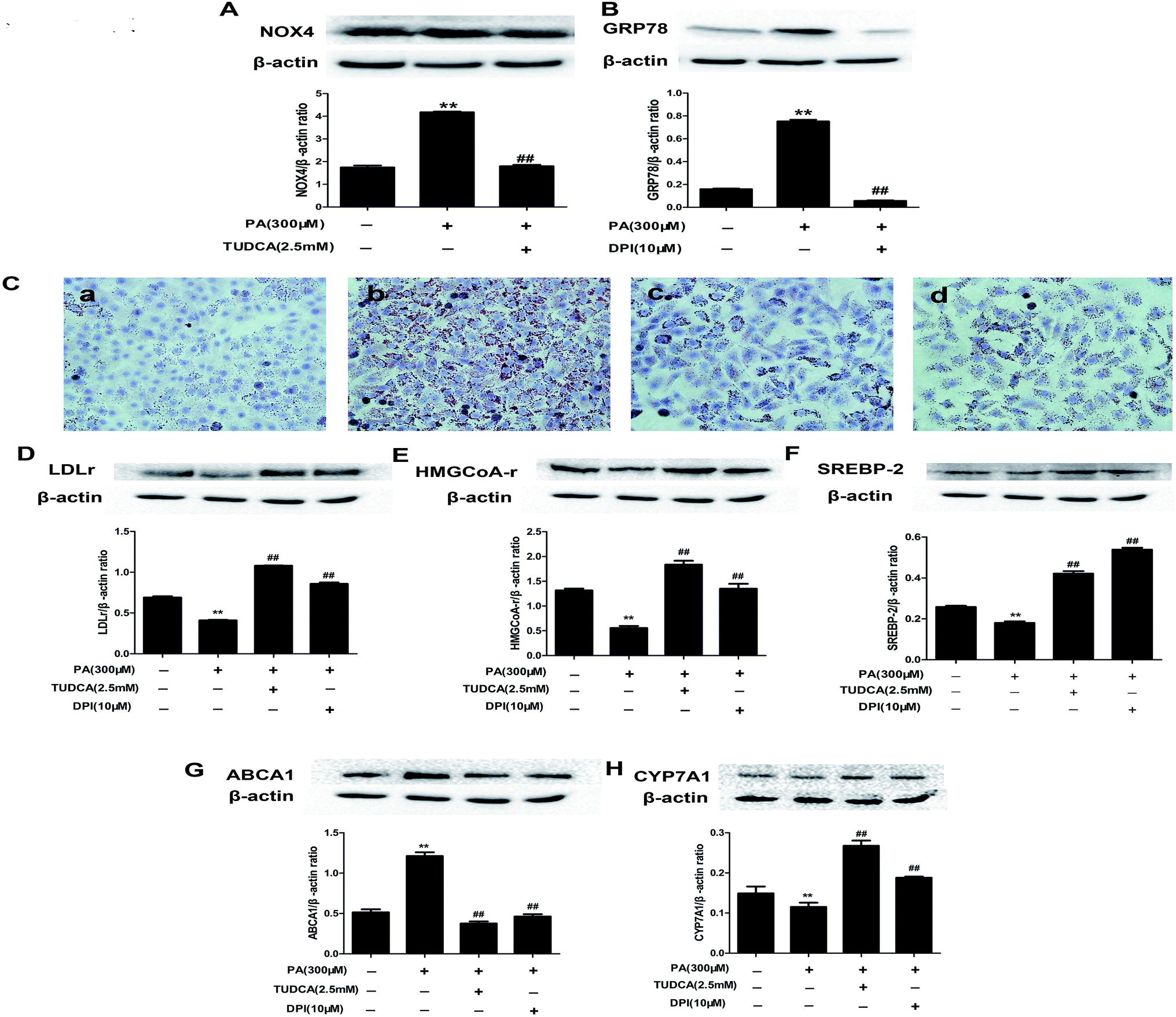 | ||
| Fig. 10 NOX4 over-expression and ER stress regulated each other and catalpol prevented lipid accumulation and alteration of cholesterol homeostasis via attenuating ER stress and NOX4 over-expression in PA-induced HepG2 cells. (A) The effect of TUDCA on NOX4 protein expression. (B) The effect of DPI on GRP78 protein expression. The results are the mean ± SD (n = 3), **P < 0.01 vs. control group, ##P < 0.01 vs. PA group. (C) Oil Red O staining of HepG2 cells: (a) control; (b) PA (300 μM); (c) PA + TUDCA (2.5 mM); (d) PA + DPI (10 μM). Oil Red O staining of HepG2 cells were photographed at 400× magnifications. (D) The effect of TUDCA or DPI on LDLr protein expression. (E) The effect of TUDCA or DPI on HMGCoA-r protein expression. (F) The effect of TUDCA or DPI on SREBP-2 protein expression. (G) The effect of TUDCA or DPI on ABCA1 protein expression. (H) The effect of TUDCA or DPI on CYP7A1 protein expression. The results are the mean ± SD (n = 3), **P < 0.01 vs. control group, ##P < 0.01 vs. PA group. | ||
3.12. Catalpol prevented PA induced lipid accumulation and alteration of cholesterol homeostasis via attenuating ER stress and NOX4 over-expression in HepG2 cells
To determine whether attenuating ER stress and NOX4 over-expression were the mechanisms of catalpol preventing PA-induced lipid accumulation of NAFLD, we respectively adopted TUDCA or DPI to investigate lipid accumulation in HepG2 cells by using staining of Oil Red O. Consistent with the effect of catalpol reducing intracellular lipid droplets in PA-induced HepG2 cells, the effect of PA increasing the number of intracellular lipid droplets was respectively abolished when ER stress or NOX4 was blocked by TUDCA or DPI (Fig. 10C). Additionally, the results showed that the protein levels of SREBP-2, LDLr, HMGCoA-r and CYP7A1 were attenuated and ABCA1 protein expression was elevated in PA-induced HepG2 cells. In line with catalpol, these protein levels were clearly reversed by administrating with TUDCA or DPI (Fig. 10D–H). Collectively, these results confirmed that catalpol prevented PA induced lipid accumulation and alteration of cholesterol homeostasis in NAFLD partly via attenuating ER stress and NOX4 over-expression in HepG2 cells.3.13. Catalpol inhibited activation of NF-κB pathway and apoptosis in PA-induced HepG2 cells
As the results, catalpol dose-dependently decreased PA induced increases of nucleus p65 and total p-p65 protein expressions in HepG2 cells (Fig. 11A and B). The findings suggested that catalpol inhibited activation of NF-κB pathway in PA-induced HepG2 cells.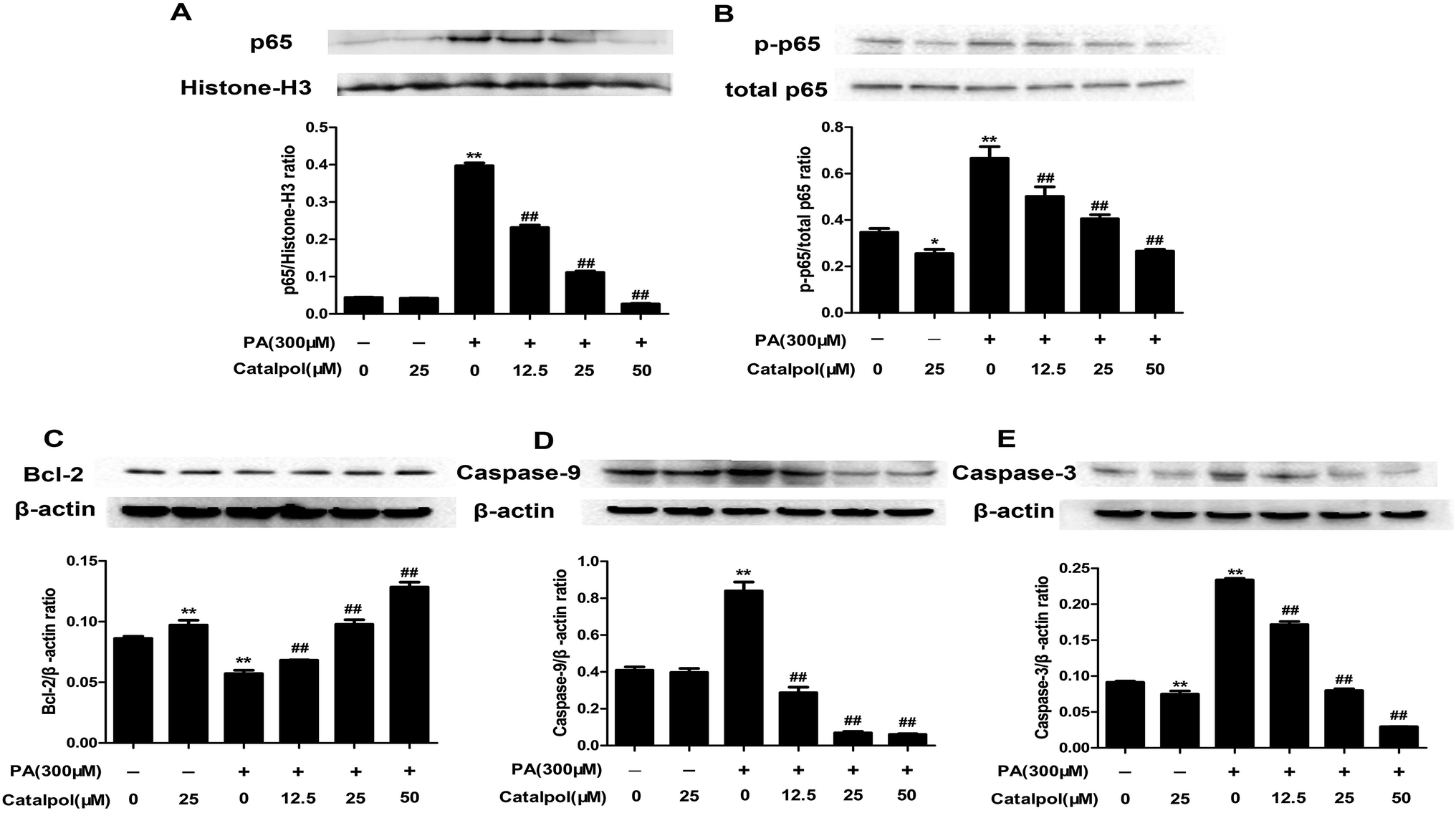 | ||
| Fig. 11 Catalpol inhibited activation of NF-κB pathway and apoptosis in PA-induced HepG2 cells. (A) Nucleus p65 protein expression level. (B) Level of p-p65 protein expression. (C) Bcl-2 protein expression level. (D) Caspase-9 protein expression level. (E) Caspase-3 protein expression level. The results are the mean ± SD (n = 3), *P < 0.05 vs. control group, **P < 0.01 vs. control group, ##P < 0.01 vs. PA group. | ||
Apoptosis could be induced by PA.54 The results showed that catalpol dose-dependently elevated Bcl-2 and decreased Caspase-9 and Caspase-3 protein expressions in PA group (Fig. 11C–E). The results indicated that catalpol inhibited apoptosis in PA-induced HepG2 cells.
4. Discussion
In recent years, the incidence of NAFLD was increasingly high.1–3 However available therapeutic approaches of NAFLD were still limited.2 Therefore, the new treatment strategies of NAFLD are required. Catalpol is a natural product present in the roots of Rehmannia glutinosa with multiple pharmacological effects,35,36 and recent studies indicated that catalpol might influence lipid metabolism in the liver.39 In our study, we firstly attempted to demonstrate that (1) catalpol confers the prevention against hepatic lipid accumulation, ER stress, NOX4-mediated oxidative stress and alteration of cholesterol homeostasis in HFD- and PA-induced NAFLD; (2) ER stress and NOX4 over-expression regulate each other in PA-induced NAFLD; and (3) catalpol prevents against PA induced hepatic lipid accumulation and alteration of cholesterol homeostasis by attenuating ER stress and NOX4 over-expression in HepG2 cells.NAFLD, which is characterized by hepatic intracellular lipid accumulation, is paralleled with obesity.40 In the present study, we found catalpol decreased body weights of HFD-fed mice. Furthermore, we found the role of catalpol ameliorating hepatic lipid accumulation and injury in HFD- and PA-induced NAFLD. Catalpol decreased lipid levels such as TC, TG and FFA in serum of HFD-induced mice and in PA-induced HepG2 cells. In addition, Oil Red O and H&E staining and transmission electron microscopy demonstrated that catalpol reduced hepatic massive lipid accumulation in HFD-induced mice. Consistent with experiments in mice, Oil Red O staining showed that catalpol reduced the increase of lipid accumulation in PA-induced HepG2 cells. The results also showed catalpol decreased ALT and AST levels of serum in HFD-induced mice. Therefore, catalpol has remarkable favorable functions for treating NAFLD.
ER stress is an important feature of several liver diseases especially HFD and saturated fatty acid such as PA induced NAFLD.10,27,45,54 The activated components of ER stress induced by feeding with HFD and stimulating with PA have previously been thought to be primarily confined to PERK-dependent pathway.10,28 In the present study, the up-regulated expressions of signs of ER stress such as GRP78, p-PERK, p-eIF2α and ATF4 were found in livers of HFD-fed mice and in PA-induced HepG2 cells. Furthermore, we also found ATF6 and p-IRE1α expressions were up-regulated in livers of HFD-fed mice and in PA-induced HepG2 cells, suggested that not only the PERK pathway but also ATF6 and IRE1 pathways of ER stress might be activated by feeding with HFD and stimulating with PA. Interestingly, these signs of ER stress were down-regulated after treating with catalpol in livers of HFD-fed mice and HepG2 cells induced by PA, indicating catalpol could prevent the three pathways of ER stress in HFD- and PA-induced NAFLD.
Hepatic apoptosis has been shown in the process of NAFLD and unresolved ER stress could induce apoptosis.29,55 Several apoptosis mediators such as transcription factor CHOP, Bcl-2 family proteins and caspase activation are implicated in ER stress-associated cell apoptosis.29 The transcription factor CHOP is the most well characterized proapoptotic pathway that operates as a downstream component of ER-stress pathways converging the IRE1, PERK and ATF6 pathways and activated by ATF4, ATF6 and XBP1.27,56 As shown in the results, the increased of CHOP, Caspase-9 and Caspase-3 and the decreased of Bcl-2 expression in liver of HFD-fed mice and HepG2 cells induced by PA were significantly reversed by catalpol in the present study. Therefore, catalpol could effectively prevent hepatic apoptosis in HFD- and PA-induced NAFLD.
NF-κB plays a crucial role in the transformation from simple steatosis to steatohepatitis.57 And in many models of diseases in vivo and in vitro, NF-κB pathway has been proved to be associated with ER stress and oxidative stress.30,58–60 In the present study, catalpol decreased both p-p65 and nucleus p65 expressions in livers of HFD-fed mice and in HepG2 cells induced by PA, suggesting catalpol inhibited hepatic NF-κB pathway activation in HFD- and PA-induced NAFLD.
Oxidative stress, which generally leads to several liver diseases, was demonstrated to play a critical role in the pathogenesis of NAFLD.7,61 Excessive lipid accumulation increases oxidative stress and consequently developing into metabolic syndrome.62,63 In the present study, catalpol exhibited beneficial effects against oxidative stress in NAFLD by increasing GSH content and SOD activity but decreasing levels of MDA and LDH in serum in HFD-fed mice and in PA-induced HepG2 cells. NADPH oxidases, as the regulators of oxidative stress, have been implicated to be the major sources of ROS generation in many pathological conditions and play important roles in liver injury and fibrosis.5,34 In the liver, NOX1, NOX2 and NOX4 are the main ROS producers.49 Previous study reported that fa/fa rats fed with HFD increased hepatic activities of NADPH oxidases.64 In addition, the expression of NOX4 was significantly increased in livers of NASH5 and in livers of db/db mice fed with a western diet.34 In the present study, we demonstrated that catalpol down-regulated NOX4 and p22phox expressions in livers of HFD-fed mice and in PA-induced HepG2 cells. Therefore, catalpol might attenuate hepatic NOX4-mediated oxidative stress in HFD- and PA-induced NAFLD.
It is well known that ER stress associates with oxidative stress and recent study reported ER stress is related to NOX4 over-expression.6,66 Consistent evidence demonstrated that the NOX4 mRNA and protein expressions were increased during ER stress induced by distinct stimuli.66 However, the exact relationship of ER stress and NOX4 over-expression stimulated by PA has not been described. In the present study, the NOX4 was down-regulated by ER stress inhibitor TUDCA and GRP78 was down-regulated by NOX4 inhibitor DPI in PA-induced HepG2 cells, suggesting that ER stress and NOX4 over-expression regulated each other in PA-induced HepG2 cells. Recent findings raised the possibility that ER stress is a contributing factor to the development of hepatic steatosis.67 Unfolded protein response (UPR) was originally identified to maintain essential ER homeostasis and various studies indicated its important role in maintaining hepatic lipid metabolism.6 More specifically, PERK siRNA has been shown to abrogate lipid accumulation in immortalized embryonic fibroblasts.68 It is possibly that decreased ER stress may improve hepatic lipid accumulation. In addition, recent studies reported NOX4 as a regulator of metabolic homeostasis and treatment with NADPH oxidases inhibitor in obese mice attenuated hepatic steatosis.65 In the present study, DPI or TUDCA respectively blocked the increased lipid accumulation in PA-induced HepG2 cells by Oil Red O staining. Thus, catalpol prevented PA induced hepatic lipid accumulation of NAFLD at least partly via attenuating NOX4 over-expression and ER stress.
Experimental inductions of hepatic alteration of cholesterol homeostasis are relevant to the pathogenesis of NAFLD.9,13 The main rate-limiting enzyme of hepatic cholesterol conversion into bile acids is CYP7A1.21 Previous study showed that gene expression and the activity of CYP7A1 were reduced in livers of HFD-feeding mice.14 Additionally, over-expressing of CYP7A1 could be a resistance to accumulation of cholesterol in the liver of transgenic mice.22,69 Therefore, the reduction of CYP7A1 may be a crucial reason of cholesterol accumulation and up-regulated CYP7A1 is possibly associated with the improvement of hepatic cholesterol level in NAFLD. In the present study, we found that hepatic CYP7A1 expressions were down-regulated by feeding with HFD and stimulating with PA, yet catalpol up-regulated CYP7A1 expressions. The liver LDLr which is the most important receptor regulating LDL-cholesterol uptake and HMGCoA-r which is the rate-limiting enzyme in cholesterol synthesis are predominantly regulated by SREBP-2 and SCAP in a negative-feedback mechanism.9,17 Previous studies demonstrated that the suppression of hepatic SREBP-2, LDLr and HMGCoA-r induced by feeding with HFD.9,70–72 Interestingly, over-expression of CYP7A1 in hepatoma cells activated SREBP-mediated LDL receptor gene expression73 and constitutive expression of CYP7A1 transgene in mice could cause the reduction in diet-induced hypercholesterolemia and the increased expressions in SREBP-mediated LDLr and HMGCoA-r in liver.74 In the present study, catalpol increased the expressions of SCAP, SREBP-2, LDLr and HMGCoA-r in livers of HFD-fed mice and PA-induced HepG2 cells. ABCA1 whose over-expression in NAFLD has been demonstrated to regulate hepatic cholesterol efflux.75 Furthermore, the expression of ABCA1 in primary hepatocytes was increased after 16 h or 40 h incubation with PA.15 In the present study, catalpol inhibited ABCA1 over-expression in livers of HFD-fed mice and PA-induced HepG2 cells. In brief, catalpol could prevent against alteration of cholesterol homeostasis in HFD- and PA-induced NAFLD.
In the present study, catalpol prevented PA induced hepatic lipid accumulation of NAFLD at least partly via attenuating ER stress and NOX4 over-expression. Thus, the possibility is rising that the mechanism of catalpol preventing alteration of cholesterol homeostasis was also via attenuating ER stress and NOX4 over-expression. As expected, the inhibited expressions of SREBP-2, LDLr, HMGCoA-r and CYP7A1 and elevated expression of ABCA1 in PA-induced HepG2 cells were abolished by administrating the inhibitor of ER stress TUDCA or the inhibitor of NOX4 DPI. Thus, catalpol conferred prevention against alteration of cholesterol homeostasis in NAFLD at least partly through attenuating ER stress and NOX4 over-expression.
5. Conclusions
In summary, the present study is the first to display that administration with catalpol has protective effects against hepatic lipid accumulation and alteration of cholesterol homeostasis in HFD- and PA-induced NAFLD via inhibiting ER stress and NOX4 over-expression. In addition, we also demonstrate ER stress and NOX4 over-expression regulated each other in PA-induced NAFLD. It should be noted that there are still some limitations in our work and further study whether catalpol can reverse symptoms of NAFLD after feeding HFD should be carried out to investigate the treating effects of catalpol on NAFLD. Although more work is needed, the findings in the present study threw light that catalpol provides new insights toward the development of therapeutic agents of NAFLD.Conflicts of interest
The authors declare that this report content has no conflict of interest.Abbreviations
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| SCAP | SREBP cleavage activating protein |
| SREBP-2 | Sterol regulatory element-binding protein-2 |
| LDLr | Low-density lipoprotein receptor |
| HMGCoA-r | HMGCoA reductase |
| ABCA1 | Adenosine triphosphate-binding cassette transporter A1 |
| CYP7A1 | Cholesterol-7a-hydroxylase |
| ER stress | Endoplasmic reticulum stress |
| UPR | Unfolded protein response |
| GRP78 | Glucose regulated protein 78 |
| PERK | PKR-like ER kinase |
| ATF6 | Activating transcription factor 6 |
| IRE1α | Inositol-requiring enzyme 1α |
| eIF2α | Eukaryotic translation initiation factor-2α |
| ATF4 | Activating transcription factor 4 |
| CHOP | C/EBP homologous protein |
| NOX | NADPH oxidase |
| SOD | Superoxide dismutase |
| GSH | Glutathione |
| MDA | Malondialdehyde |
| LDH | Lactic dehydrogenase |
| TG | Triglyceride |
| TC | Total cholesterol |
| FFA | Free fatty acid |
| ALT | Alanine aminotransferase |
| AST | Aspartate aminotransferase |
| PA | Palmitate |
| HFD | High-fat diet |
| TUDCA | Tauroursodeoxycholic acid |
| DPI | Diphenyleneiodonium |
Acknowledgements
This study was supported in part by Grants from the National Natural Science Foundation of China (No. 81273508).References
- B. N. Bohinc and A. M. Diehl, Clin. Liver. Dis., 2012, 16, 549–565 CrossRef PubMed.
- H. C. Masuoka and N. Chalasani, Ann. N. Y. Acad. Sci., 2013, 1281, 106–122 CrossRef CAS PubMed.
- G. C. Farrell and C. Z. Larter, Hepatology, 2006, 43, S99–S112 CrossRef CAS PubMed.
- P. Angulo, N. Engl. J. Med., 2002, 346, 1221–1231 CrossRef CAS PubMed.
- A. Bettaieb, J. X. Jiang, Y. Sasaki, T. I. Chao, Z. Kiss, X. Chen, J. Tian, M. Katsuyama, C. Yabe-Nishimura, Y. Xi, C. Szyndralewiez, K. Schroder, A. Shah, R. P. Brandes, F. G. Haj and N. J. Torok, Gastroenterology, 2015, 149, 468–480 CrossRef CAS PubMed.
- N. U. Ashraf and T. A. Sheikh, Free Radical Res., 2015, 49, 1405–1418 CrossRef CAS PubMed.
- G. H. Heeba and M. A. Morsy, Environ. Toxicol. Pharmacol., 2015, 40, 907–914 CrossRef CAS PubMed.
- C. D. Fuchs, T. Claudel, P. Kumari, G. Haemmerle, M. J. Pollheimer, T. Stojakovic, H. Scharnagl, E. Halilbasic, J. Gumhold, D. Silbert, H. Koefeler and M. Trauner, Hepatology, 2012, 56, 270–280 CrossRef CAS PubMed.
- L. Zhao, Y. Chen, R. Tang, Y. Chen, Q. Li, J. Gong, A. Huang, Z. Varghese, J. F. Moorhead and X. Z. Ruan, J. Gastroenterol. Hepatol., 2011, 26, 875–883 CrossRef CAS PubMed.
- H. Y. Lee, G. H. Lee, M. R. Lee, H. K. Kim, N. Y. Kim, S. H. Kim, Y. C. Lee, H. R. Kim and H. J. Chae, PLoS One, 2013, 8, e81349 Search PubMed.
- L. Martinez, S. Torres, A. Baulies, C. Alarcon-Vila, M. Elena, G. Fabrias, J. Casas, J. Caballeria, J. C. Fernandez-Checa and C. Garcia-Ruiz, Oncotarget, 2015, 6, 41479–41496 Search PubMed.
- S. Parekh and F. A. Anania, Gastroenterology, 2007, 132, 2191–2207 CrossRef CAS PubMed.
- E. Y. Park, H. Choi, J. Y. Yoon, I. Y. Lee, Y. Seo, H. S. Moon, J. H. Hwang and H. S. Jun, Mar. Drugs, 2015, 13, 6866–6883 CrossRef CAS PubMed.
- A. Leikin-Frenkel, P. Parini, F. M. Konikoff, L. Benthin, D. Leikin-Gobbi, I. Goldiner, C. Einarsson and T. Gilat, Arch. Biochem. Biophys., 2008, 471, 63–71 CrossRef CAS PubMed.
- K. Konstantynowicz-Nowicka, E. Harasim, M. Baranowski and A. Chabowski, PLoS One, 2015, 10, e0116858 Search PubMed.
- P. Simonen, A. Kotronen, M. Hallikainen, K. Sevastianova, J. Makkonen, A. Hakkarainen, N. Lundbom, T. A. Miettinen, H. Gylling and H. Yki-Jarvinen, J. Hepatol., 2011, 54, 153–159 CrossRef CAS PubMed.
- A. Nohturfft, R. A. DeBose-Boyd, S. Scheek, J. L. Goldstein and M. S. Brown, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11235–11240 CrossRef CAS.
- J. Sakai and R. B. Rawson, Curr. Opin. Lipidol., 2001, 12, 261–266 CrossRef CAS PubMed.
- S. M. Vallett, H. B. Sanchez, J. M. Rosenfeld and T. F. Osborne, J. Biol. Chem., 1996, 271, 12247–12253 CrossRef CAS PubMed.
- J. M. Timmins, J. Y. Lee, E. Boudyguina, K. D. Kluckman, L. R. Brunham, A. Mulya, A. K. Gebre, J. M. Coutinho, P. L. Colvin, T. L. Smith, M. R. Hayden, N. Maeda and J. S. Parks, J. Clin. Invest., 2005, 115, 1333–1342 CrossRef CAS PubMed.
- D. W. Russell and K. D. Setchell, Biochemistry, 1992, 31, 4737–4749 CrossRef CAS PubMed.
- K. E. Andersson, U. Axling, J. Xu, K. Sward, S. Ahrne, G. Molin, C. Holm and P. Hellstrand, Eur. J. Nutr., 2013, 52, 1755–1769 CrossRef CAS PubMed.
- A. Bashiri, G. Tavallaee, L. Li and D. S. Ng, Curr. Opin. Lipidol., 2013, 24, 275–276 CrossRef CAS PubMed.
- J. Lee and U. Ozcan, J. Biol. Chem., 2014, 289, 1203–1211 CrossRef CAS PubMed.
- K. Zhang and R. J. Kaufman, Nature, 2008, 454, 455–462 CrossRef CAS PubMed.
- E. Szegezdi, S. E. Logue, A. M. Gorman and A. Samali, EMBO Rep., 2006, 7, 880–885 CrossRef CAS PubMed.
- I. Kim, W. Xu and J. C. Reed, Nat. Rev. Drug Discovery, 2008, 7, 1013–1030 CrossRef CAS PubMed.
- U. Ozcan, Q. Cao, E. Yilmaz, A. H. Lee, N. N. Iwakoshi, E. Ozdelen, G. Tuncman, C. Gorgun, L. H. Glimcher and G. S. Hotamisligil, Science, 2004, 306, 457–461 CrossRef PubMed.
- H. Malhi and R. J. Kaufman, J. Hepatol., 2011, 54, 795–809 CrossRef CAS PubMed.
- S. Reuter, S. C. Gupta, M. M. Chaturvedi and B. B. Aggarwal, Free Radical Biol. Med., 2010, 49, 1603–1616 CrossRef CAS PubMed.
- N. S. de Mochel, S. Seronello, S. H. Wang, C. Ito, J. X. Zheng, T. J. Liang, J. D. Lambeth and J. Choi, Hepatology, 2010, 52, 47–59 CrossRef PubMed.
- W. Li, C. Wang, J. Peng, J. Liang, Y. Jin, Q. Liu, Q. Meng, K. Liu and H. Sun, Curr. Pharm. Biotechnol., 2014, 15, 1173–1182 CAS.
- E. Maloney, I. R. Sweet, D. M. Hockenbery, M. Pham, N. O. Rizzo, S. Tateya, P. Handa, M. W. Schwartz and F. Kim, Arterioscler., Thromb., Vasc. Biol., 2009, 29, 1370–1375 CrossRef CAS PubMed.
- M. Fukuda, T. Nakamura, K. Kataoka, H. Nako, Y. Tokutomi, Y. F. Dong, O. Yasuda, H. Ogawa and S. Kim-Mitsuyama, J. Pharmacol. Exp. Ther., 2010, 335, 70–75 CrossRef CAS PubMed.
- B. Jiang, R. F. Shen, J. Bi, X. S. Tian, T. Hinchliffe and Y. Xia, Curr. Med. Chem., 2015, 22, 1278–1291 CrossRef CAS PubMed.
- J. Zhou, G. Xu, S. Ma, F. Li, M. Yuan, H. Xu and K. Huang, Biochem. Biophys. Res. Commun., 2015, 467, 853–858 CrossRef CAS PubMed.
- W. J. Huang, H. S. Niu, M. H. Lin, J. T. Cheng and F. L. Hsu, J. Nat. Prod., 2010, 73, 1170–1172 CrossRef CAS PubMed.
- J. P. Shieh, K. C. Cheng, H. H. Chung, Y. F. Kerh, C. H. Yeh and J. T. Cheng, J. Agric. Food Chem., 2011, 59, 3747–3753 CrossRef CAS PubMed.
- Z. Xu, L. Zhang, X. Li, Z. Jiang, L. Sun, G. Zhao, G. Zhou, H. Zhang, J. Shang and T. Wang, Acta Biochim. Biophys. Sin., 2015, 47, 730–740 CrossRef PubMed.
- E. Fabbrini, S. Sullivan and S. Klein, Hepatology, 2010, 51, 679–689 CrossRef CAS PubMed.
- W. Zeng, W. Shan, L. Gao, D. Gao, Y. Hu, G. Wang, N. Zhang, Z. Li, X. Tian, W. Xu, J. Peng, X. Ma and J. Yao, Sci. Rep., 2015, 5, 16013 CrossRef CAS PubMed.
- K. L. Ma, X. Z. Ruan, S. H. Powis, Y. Chen, J. F. Moorhead and Z. Varghese, Hepatology, 2008, 48, 770–781 CrossRef CAS PubMed.
- M. J. Pagliassotti, Annu. Rev. Nutr., 2012, 32, 17–33 CrossRef CAS PubMed.
- C. L. Gentile, M. A. Frye and M. J. Pagliassotti, BioFactors, 2011, 37, 8–16 CrossRef CAS PubMed.
- M. Hamano, H. Ezaki, S. Kiso, K. Furuta, M. Egawa, T. Kizu, N. Chatani, Y. Kamada, Y. Yoshida and T. Takehara, J. Gastroenterol., 2014, 49, 305–316 CrossRef CAS PubMed.
- X. Cai, L. Chen and J. F. McGinnis, Exp. Eye Res., 2015, 140, 130–138 CrossRef CAS PubMed.
- Z. Yesilova, H. Yaman, C. Oktenli, A. Ozcan, A. Uygun, E. Cakir, S. Y. Sanisoglu, A. Erdil, Y. Ates, M. Aslan, U. Musabak, M. K. Erbil, N. Karaeren and K. Dagalp, Am. J. Gastroenterol., 2005, 100, 850–855 CrossRef CAS PubMed.
- Y. Sumida, T. Nakashima, T. Yoh, M. Furutani, A. Hirohama, Y. Kakisaka, Y. Nakajima, H. Ishikawa, H. Mitsuyoshi, T. Okanoue, K. Kashima, H. Nakamura and J. Yodoi, J. Hepatol., 2003, 38, 32–38 CrossRef CAS PubMed.
- Y. H. Paik, J. Kim, T. Aoyama, S. De Minicis, R. Bataller and D. A. Brenner, Antioxid. Redox Signaling, 2014, 20, 2854–2872 CrossRef CAS PubMed.
- R. K. Ambasta, P. Kumar, K. K. Griendling, H. H. Schmidt, R. Busse and R. P. Brandes, J. Biol. Chem., 2004, 279, 45935–45941 CrossRef CAS PubMed.
- B. Sun and M. Karin, J. Hepatol., 2012, 56, 704–713 CrossRef CAS PubMed.
- S. Fu, S. M. Watkins and G. S. Hotamisligil, Cell Metab., 2012, 15, 623–634 CrossRef CAS PubMed.
- J. D. Mosinski, M. R. Pagadala, A. Mulya, H. Huang, O. Dan, H. Shimizu, E. Batayyah, R. K. Pai, P. R. Schauer, S. A. Brethauer and J. P. Kirwan, Acta Physiol., 2016, 217, 141–151 CrossRef CAS PubMed.
- J. Cao, D. L. Dai, L. Yao, H. H. Yu, B. Ning, Q. Zhang, J. Chen, W. H. Cheng, W. Shen and Z. X. Yang, Mol. Cell. Biochem., 2012, 364, 115–129 CrossRef CAS PubMed.
- J. D. Mosinski, M. R. Pagadala, A. Mulya, H. Huang, O. Dan, H. Shimizu, E. Batayyah, R. K. Pai, P. R. Schauer, S. A. Brethauer and J. P. Kirwan, Acta Physiol., 2016, 217, 141–151 CrossRef CAS PubMed.
- Y. Ma, J. W. Brewer, J. A. Diehl and L. M. Hendershot, J. Mol. Biol., 2002, 318, 1351–1365 CrossRef CAS PubMed.
- W. Chen, X. Wang, L. I. Huang and B. O. Liu, Exp. Ther. Med., 2016, 11, 73–76 CAS.
- M. Kaneko, Y. Niinuma and Y. Nomura, Biol. Pharm. Bull., 2003, 26, 931–935 CAS.
- P. Hu, Z. Han, A. D. Couvillon, R. J. Kaufman and J. H. Exton, Mol. Cell. Biol., 2006, 26, 3071–3084 CrossRef CAS PubMed.
- F. Urano, X. Wang, A. Bertolotti, Y. Zhang, P. Chung, H. P. Harding and D. Ron, Science, 2000, 287, 664–666 CrossRef CAS PubMed.
- S. Spahis, E. Delvin, J. M. Borys and E. Levy, Antioxid. Redox Signaling, 2016 DOI:10.1089/ars.2016.6776.
- Q. M. Anstee, D. Concas, H. Kudo, A. Levene, J. Pollard, P. Charlton, H. C. Thomas, M. R. Thursz and R. D. Goldin, J. Hepatol., 2010, 53, 542–550 CrossRef CAS PubMed.
- U. Nair, H. Bartsch and J. Nair, Free Radical Biol. Med., 2007, 43, 1109–1120 CrossRef CAS PubMed.
- M. Carmiel-Haggai, A. I. Cederbaum and N. Nieto, FASEB J., 2005, 19, 136–138 CAS.
- S. Furukawa, T. Fujita, M. Shimabukuro, M. Iwaki, Y. Yamada, Y. Nakajima, O. Nakayama, M. Makishima, M. Matsuda and I. Shimomura, J. Clin. Invest., 2004, 114, 1752–1761 CAS.
- F. R. Laurindo, T. L. Araujo and T. B. Abrahao, Antioxid. Redox Signaling, 2014, 20, 2755–2775 CrossRef CAS PubMed.
- D. T. Rutkowski, J. Wu, S. H. Back, M. U. Callaghan, S. P. Ferris, J. Iqbal, R. Clark, H. Miao, J. R. Hassler, J. Fornek, M. G. Katze, M. M. Hussain, B. Song, J. Swathirajan, J. Wang, G. D. Yau and R. J. Kaufman, Dev. Cell, 2008, 15, 829–840 CrossRef CAS PubMed.
- E. Bobrovnikova-Marjon, G. Hatzivassiliou, C. Grigoriadou, M. Romero, D. R. Cavener, C. B. Thompson and J. A. Diehl, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16314–16319 CrossRef CAS PubMed.
- E. P. Ratliff, A. Gutierrez and R. A. Davis, J. Lipid Res., 2006, 47, 1513–1520 CrossRef CAS PubMed.
- Z. Huang, X. Zhou, A. C. Nicholson, A. M. Gotto Jr, D. P. Hajjar and J. Han, Br. J. Pharmacol., 2008, 155, 596–605 CrossRef CAS PubMed.
- Y. J. Jia, R. X. Xu, J. Sun, Y. Tang and J. J. Li, J. Transl. Med., 2014, 12, 103 CrossRef PubMed.
- M. S. Brown and J. L. Goldstein, Cell, 1997, 89, 331–340 CrossRef CAS PubMed.
- S. L. Wang, E. Z. Du, T. D. Martin and R. A. Davis, J. Biol. Chem., 1997, 272, 19351–19358 CrossRef CAS PubMed.
- J. H. Miyake, X. T. Duong-Polk, J. M. Taylor, E. Z. Du, L. W. Castellani, A. J. Lusis and R. A. Davis, Arterioscler., Thromb., Vasc. Biol., 2002, 22, 121–126 CrossRef CAS.
- S. B. Ahn, K. Jang, D. W. Jun, B. H. Lee and K. J. Shin, Dig. Dis. Sci., 2014, 59, 2975–2982 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |