
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the photophysical properties of BODIPY dyes through extended aromatic pyrroles
Shawn Swavey*a,
John Quinna,
Michael Coladipietroa,
Kegan G. Coxb and
M. Kyle Brennamanb
aDepartment of Chemistry, University of Dayton, 300 College Park, Dayton, OH 45469-2357, USA. E-mail: sswavey1@udayton.edu; Web: https://www.udayton.edu/directory/artssciences/chemistry/swavey_shawn.php Tel: +1-937-229-3145
bDepartment of Chemistry, University of North Carolina, Chapel Hill, NC 27599, USA
First published on 22nd December 2016
Abstract
Three new BODIPY dyes have been synthesized by a two-step synthetic route. This expands the series to nine different BODIPY dyes by this method. Naphtha[1,2-c]pyrrole was combined with 1-pyrenecarboxaldehyde to give a symmetric dipyrrin followed by reaction with boron trifluoride to give a symmetric highly conjugated BODIPY dye. Expanding this synthetic route to a more conjugated pyrrole fluorantho[2,3-c]pyrrole was combined with 1-pyrenecarboxaldehyde followed by reaction with boron trifluoride to give the asymmetric BODIPY dye (9). Dyes with the more highly conjugated fluoranthopyrrole resulted in a bathochromic shift of ca. 50 nm in the electronic absorption and showed greater stability of the LUMO energy, as determined by electrochemical measurements, compared to their naphthapyrrole analogs. All of the dyes synthesized by this method display molar absorptivities greater than 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 with photoluminescence quantum efficiencies of 0.8–1.0. Excited state lifetimes of the dyes in dichloromethane are modest, ranging from 3.2 ns to 4.3 ns.
000 M−1 cm−1 with photoluminescence quantum efficiencies of 0.8–1.0. Excited state lifetimes of the dyes in dichloromethane are modest, ranging from 3.2 ns to 4.3 ns.
Introduction
Over the past decade, the interest in BODIPY (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene) dyes has generated hundreds of new compounds. Although much of this work has focused on improving the lasing properties of these compounds, other applications have emerged.1–5 In bioimaging, for example, tailoring of the photophysical properties of BODIPY dyes through synthetic modification has led to increased contrast and spatial imaging in living cells.6–8 Appropriate meso substitution enables these probes to be directed to particular regions of cells allowing insight into specific bio-reactions in real time.9–11 Other suitable applications for BODIPY dyes include dye sensitized solar cells (DSSCs) and opto-electronics, due in part to their near-unity luminescence quantum efficiency and NIR light absorption originally afforded by substitution at the meso-position of the dipyrrin core.12,13 This strategy to achieve absorption in the NIR region of the electromagnetic spectrum, though effective, is somewhat limited. Fortunately, an alternative strategy has been found which involves introducing aromatically fused BODIPY cores to achieve sizable absorption red shifts.12–20 Notably, bathochromic shifts in the absorption spectrum have been achieved through β-substitution leading to fused ring systems incorporating biphenyl21 (673 nm), indole22 (607 nm), carbazole23 (508 nm), and benzothieno24 (658 nm). Luminescence quantum efficiencies near unity were achieved with shorter-wavelength-absorbing BODIPY dyes; however, a significant decrease in quantum efficiency was observed for the biphenyl- and benzothieno-fused BODIPY dyes, 0.51 and 0.05 respectively. In addition to lower quantum efficiencies, the synthetic route to these fused BODIPY dyes requires multiple synthetic steps, harsh chemicals, and expensive catalysts. Rurack et al. has achieved luminescence quantum efficiencies near unity with absorption wavelengths between 621 nm and 642 nm from phenanthrene-fused BODIPY dyes with various meso-substituents.25We have recently described a two-step route toward BODIPY dyes26 in which formation of the dipyrrin was accomplished in the absence of solvent. Using naphthyl-pyrrole and a variety of aromatic aldehydes, a series of new BODIPY dyes were generated with long-wavelength absorption properties ranging from ca. 580 nm to 620 nm and near unity quantum efficiencies. It was argued that by judicious choice of aromatic aldehyde, i.e. sterically hindered versus non-sterically hindered, the BODIPY dyes generated would be either asymmetric or symmetric respectively, Scheme 1. To continue this study in an attempt to advance this new synthetic route as well as shift light absorption closer to the NIR, herein we report new BODIPY dyes based on fluoranthene pyrroles, Scheme 1 (framed).
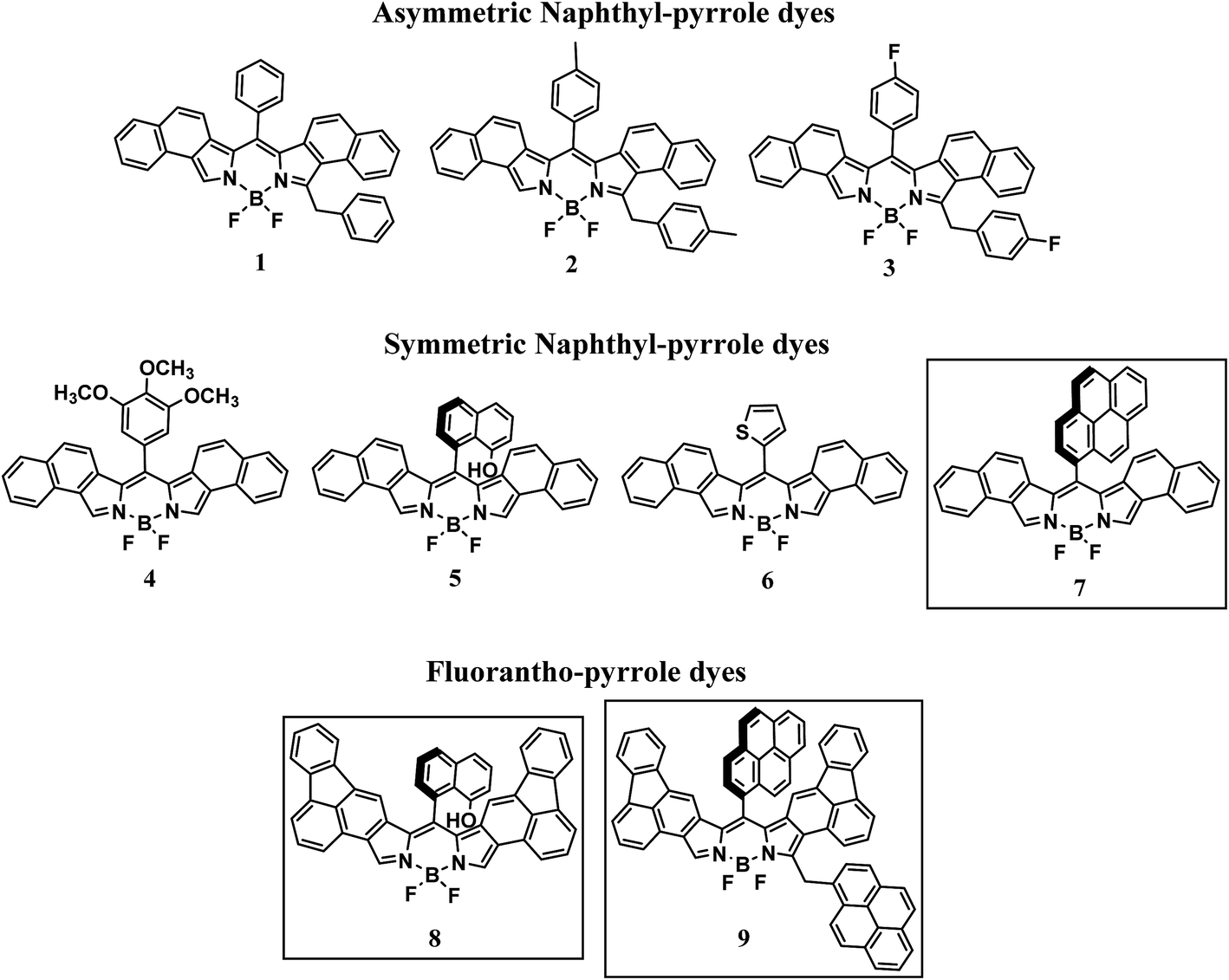 | ||
| Scheme 1 Molecular structures of BODIPY dyes. | ||
Experimental
All chemicals were reagent grade and used without further purification. Naphtha[1,2-c]pyrrole,27 fluorantho[2,3-c]pyrrole28 and BODIPY dyes 1–6 (ref. 26) were synthesized as previously described. Chromatography was performed on a Teledyne Combiflash Rf+ equipped with UV detection. High resolution mass spectral analysis was performed at the Mass Spectrometry and Proteomics facility at the Ohio State University. Elemental analysis was performed at Atlantic Microlabs Inc. Norcross, Georgia. 1H NMR spectra were recorded on a Bruker 300 MHz NMR spectrophotometer at 298 K. Electronic absorption spectra were recorded at room temperature using an HP8453 photodiode array spectrophotometer with 2 nm resolution. All spectra were recorded at 298 K. Room temperature luminescence spectra in a 1 cm quartz spectrophotometer fluorescence cell (Starna) in DCM were run on a Cary Eclipse fluorescence spectrophotometer. Cyclic voltammograms were recorded under a nitrogen atmosphere using a one-compartment, three electrode cell, CH-Instruments, equipped with a platinum wire auxiliary electrode. The working electrode was a 2.0 mm diameter glassy carbon disk from CH-Instruments, which was polished first using 0.30 μm followed by 0.05 μm alumina polish (Buehler) and then sonicated for 10 s prior to use. Potentials were referenced to a Ag/Ag+ non-aqueous electrode with ferrocene as an internal standard, CH-Instruments. The supporting electrolyte was 0.1 M tetrabutylammonium hexafluorophosphate (Bu4NPF6) and the measurements were made in dry DCM. Luminescence quantum yields were determined at room temperature in HPLC-grade DCM relative to Rhodamine 6G as the reference (ϕ = 0.95, in ethanol).29 The quantum yields were obtained using the following equation:| ϕs = ϕr[Arηs2Ds/Asηr2Dr] |
Time-resolved emission measurements
Excited-state lifetimes were measured by the time-correlated single photon counting (TCSPC) technique implemented by using an Edinburgh Instruments FLS920 photoluminescence spectrophotometer. The FLS920 was outfitted with a Hamamatsu 2658P photomultiplier tube (PMT) mounted in a PMT housing cooled to −22 °C. Laser excitation was provided by a 484 nm diode laser (Edinburgh Instruments EPL-485, 93 ps FWHM pulse width, 2 mm beam diameter) operated at 20 MHz. Time-resolved emission for compounds 1–9 was monitored at 608, 612, 612, 612, 610, 620, 605, 650 and 650 nm, respectively with a bandwidth of 5–10 nm. A 515 nm long-pass filter was placed before the entrance of the emission monochromator to remove laser scatter. Methylene chloride solutions of each compound were prepared with maximum visible absorbance values less than 0.2 OD. For each compound, at least two measurements were performed using air-saturated solutions at 22 ± 2 °C. Each transient decay was recorded on a 50 ns time range (12.2 ps per channel) to achieve 10000 peak counts. An instrument response function (IRF), with FWHM of 1.32 ns, was obtained by tuning the monochromator to 484 nm and collecting the laser light scattered by an aqueous Ludox solution. Emission decays were treated in Edinburgh L900 software (v7.1.1) with a reconvolution fit incorporating the Ludox-based IRF.
General synthetic procedure
To a 25 mL round bottom flask 50.0 mg (0.30 mmol) of naphtha[1,2-c]pyrrole or (0.21 mmol) of fluorantho[2,3-c]pyrrole and a molar equivalent of the appropriate aldehyde were combined. Approximately 1 mL of dichloromethane (other solvents i.e. methanol, acetone, chloroform also work) was added to give a homogenous solution. The solvent was then removed under reduced pressure and the flask was placed in a warm water bath preset at 70 °C for 15 min. Temperatures were increased until the reactant mixture turned to a purple paste. In the case of the fluorantho[2,3-c]pyrrole reactions, the mixture was placed in an oven and heated to 140 °C. The resulting purple paste was chromatographed on silica gel using DCM (to remove any polymers formed) collecting the first deep red band. The solvent was removed under reduced pressure and the flask was degassed with nitrogen. Approximately 4 mL of dry DCM was added followed by 100 μL of triethylamine and then 200 μL boron trifluoride etherate. The solution was stirred at room temperature under nitrogen overnight. The reaction mixture was washed with distilled water, dried over magnesium sulfate and chromatographed on silica gel using 90/10 chloroform/hexanes as eluent. The first band was collected and the solvent removed.Results and discussion
Synthesis
In light of the recent report on the two-step synthetic route of highly conjugated BODIPY dyes26 we have attempted to extend this synthetic route. By heating naphtha[1,2-c]pyrrole and 1-pyrenecarboxaldehyde at 80 °C for 15 min followed by chromatography on silica gel, the red dipyrrin was obtained. The dipyrrin was converted to the BODIPY dye 7 (Scheme 2) by reaction with boron trifluoride in the presence of triethylamine and dry DCM as previously described. After workup and purification, 7 was obtained as a golden solid. Characterization by high resolution mass spectrometry and 1H NMR confirmed the structure.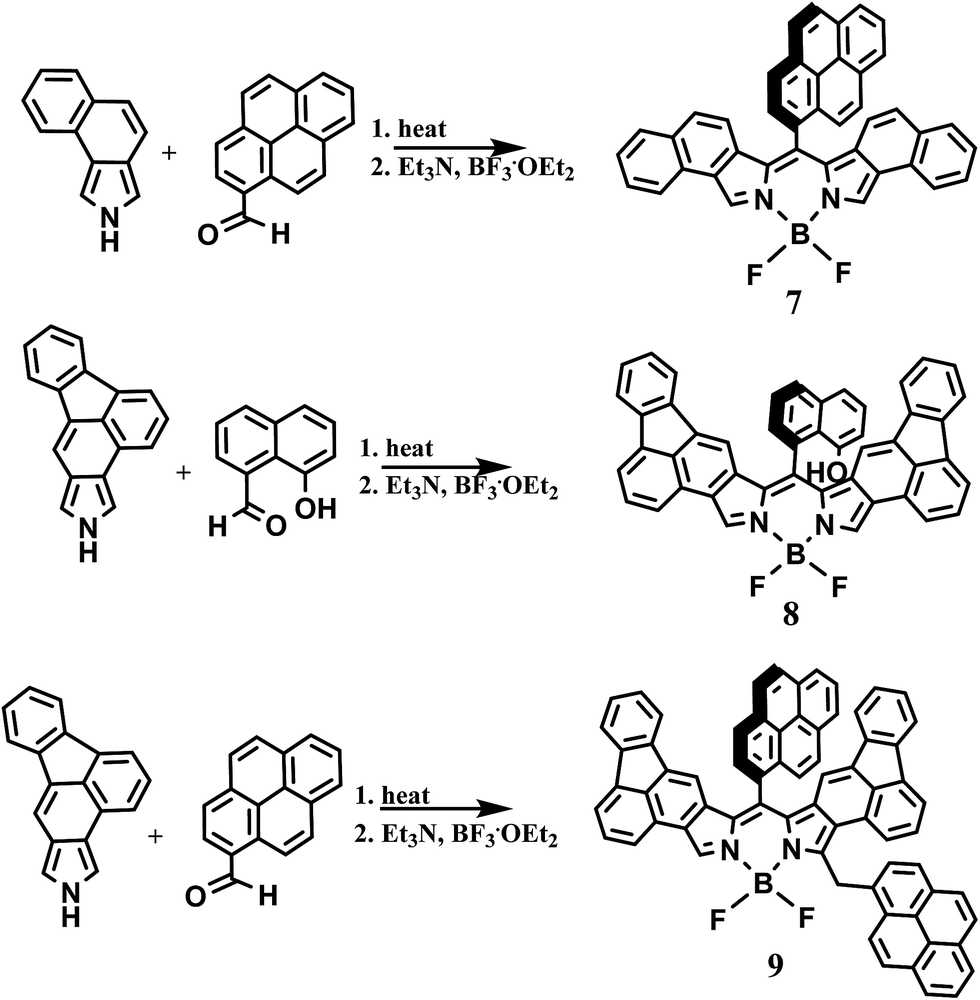 | ||
| Scheme 2 Synthetic route of BODIPY dyes. | ||
To further extend this procedure to more highly conjugated pyrroles, the readily available fluorantho[2,3-c]pyrrole was chosen.28 To obtain the dipyrrins based on this pyrrole; however, required heating at 140 °C, considerably higher than the napthapyrrole based dipyrrins. After purification, the dipyrrins were converted to the BODIPY dyes 8 and 9 as previously described, Scheme 2. Due to their complex proton NMR spectra, these dyes were characterized by high-resolution mass spectrometry and their purity determined by elemental analysis.
For naphtha[1,2-c]pyrrole, it was suggested that the steric bulk of the aldehyde determined the symmetry of the dipyrrin with the symmetric dipyrrin favored by highly substituted aldehydes. This does not seem to be the case with the fluorantho[2,3-c]pyrrole. This is evidenced by the formation of the symmetric BODIPY dye 8 while the bulkier 1-pyrenecarboxaldehyde favors the asymmetric BODIPY dye 9. In addition, attempts to form BODIPY dyes from 3,4,5-trimethoxybenzaldehyde and 2-thiophenecarboxaldehyde, for comparison with our earlier study, resulted in only trace amounts of the desired products.
Spectroscopic properties
Electronic absorption and emission spectroscopy was performed for 7–9 in dry DCM at room temperature, Table 1 and Fig. 1–3. BODIPY 5, from our previous study, was included for comparison. Fig. 1A illustrates the electronic absorption spectra of 5 and 8. In general, electronic excitations of BODIPY dyes are the result of HOMO to LUMO transitions localized on the dipyrrin core.1 Both 5 and 8 exhibit sharp electronic absorption features, with the maximum absorption of 5 at 596 nm (ε = 158000 M−1 cm−1) and of 8 at 642 nm (ε = 110000 M−1 cm−1). The more highly conjugated BODIPY dye 8 shows a bathochromic shift of 46 nm compared to 5.
| Dyes | λabs (nm) | λem (nm) | ε, M−1 cm−1 | τ (ns) | ϕa | LUMO (eV) | HOMO (eV) |
|---|---|---|---|---|---|---|---|
| a Fluorescence quantum yields were obtained using Rhodamine 6G as a reference (ϕ = 0.94 in ethanol).29 | |||||||
| 1 | 600 | 608 | 107000 |
4.2 | 1.0 | −3.6 | −5.3 |
| 2 | 600 | 612 | 152000 |
4.3 | 0.8 | −3.5 | −5.3 |
| 3 | 602 | 612 | 140000 |
4.0 | 1.0 | −3.5 | −5.3 |
| 4 | 596 | 604 | 158000 |
4.1 | 0.9 | −3.6 | −5.4 |
| 5 | 603 | 612 | 176000 |
4.2 | 1.0 | −3.7 | −5.4 |
| 6 | 612 | 619 | 194000 |
3.2 | 0.5 | −3.8 | −5.4 |
| 7 | 599 | 613 | 138000 |
4.0 | 1.0 | −3.7 | −5.4 |
| 8 | 642 | 654 | 110000 |
3.5 | 0.8 | −4.0 | −5.4 |
| 9 | 645 | 657 | 124000 |
3.5 | 0.8 | −3.9 | −5.4 |
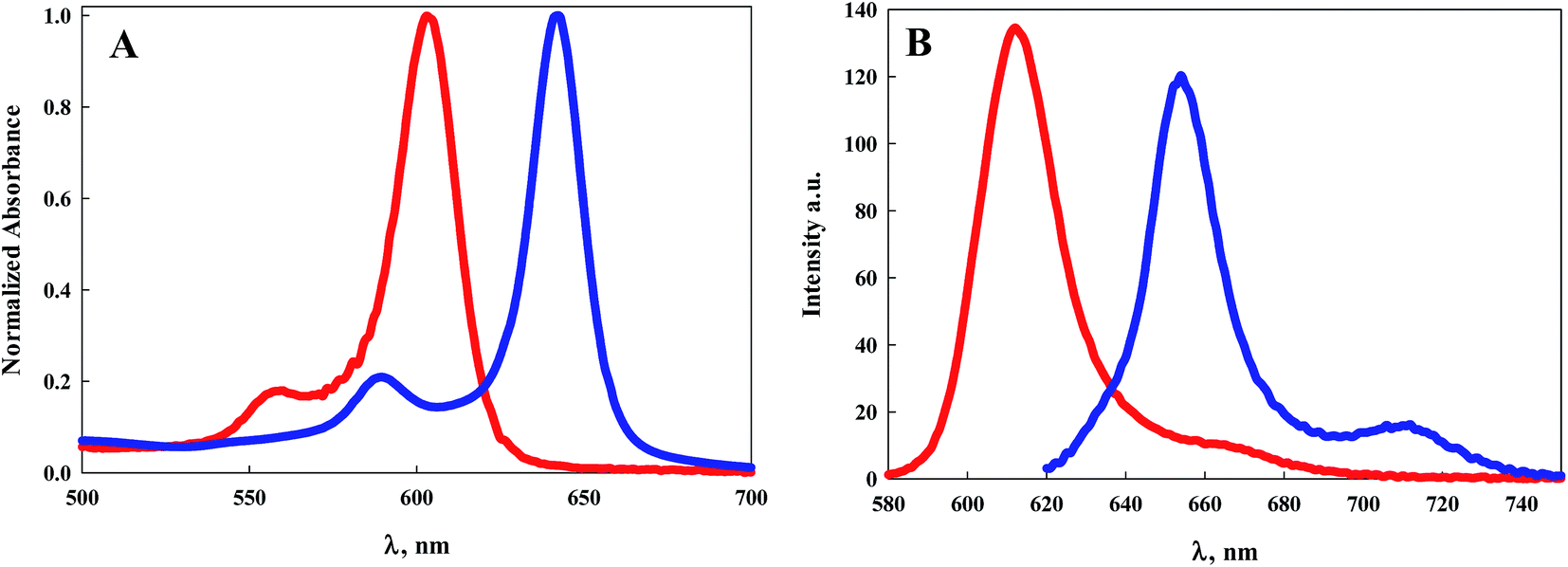 | ||
| Fig. 1 (A) Electronic absorption spectra of 5 (red line) and 8 (blue line) in DCM; (B) emission spectra of 5 (red line) and 8 (blue line) when excited at their maximum absorption wavelength. | ||
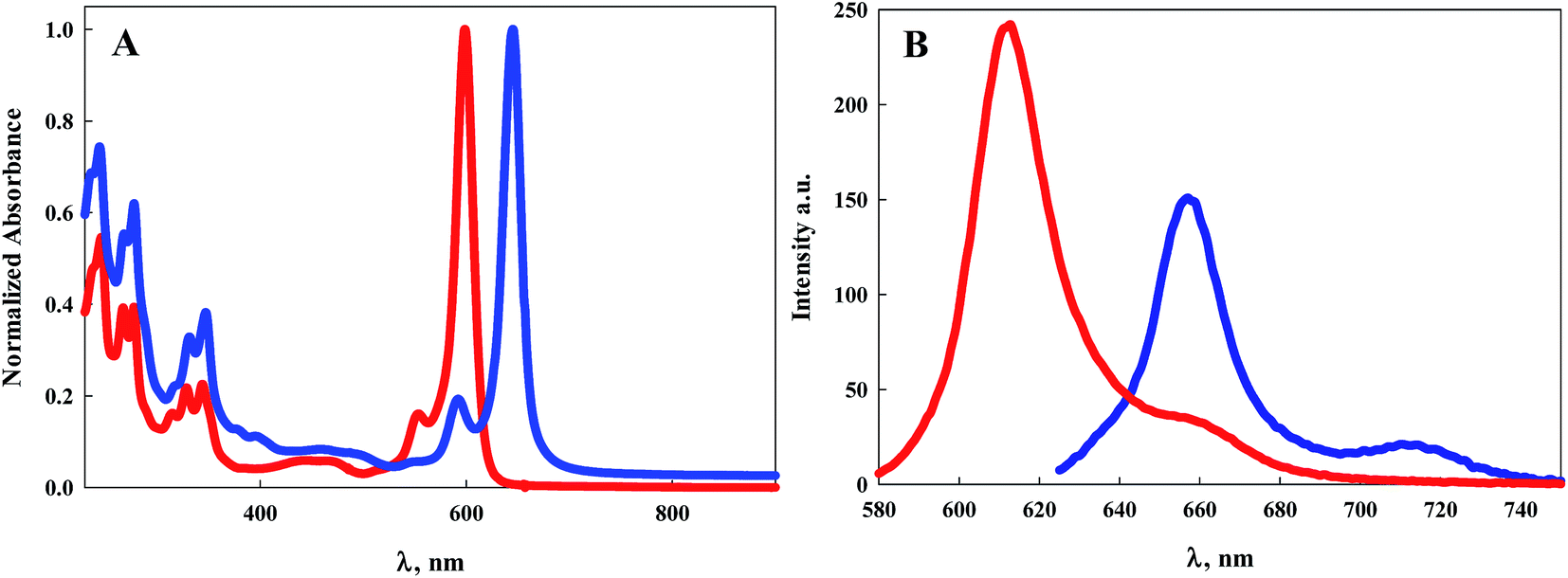 | ||
| Fig. 2 (A) Electronic absorption spectra of 7 (red line) and 9 (blue line) in DCM; (B) emission spectra of 7 (red line) and 9 (blue line) when excited at their maximum absorption wavelength. | ||
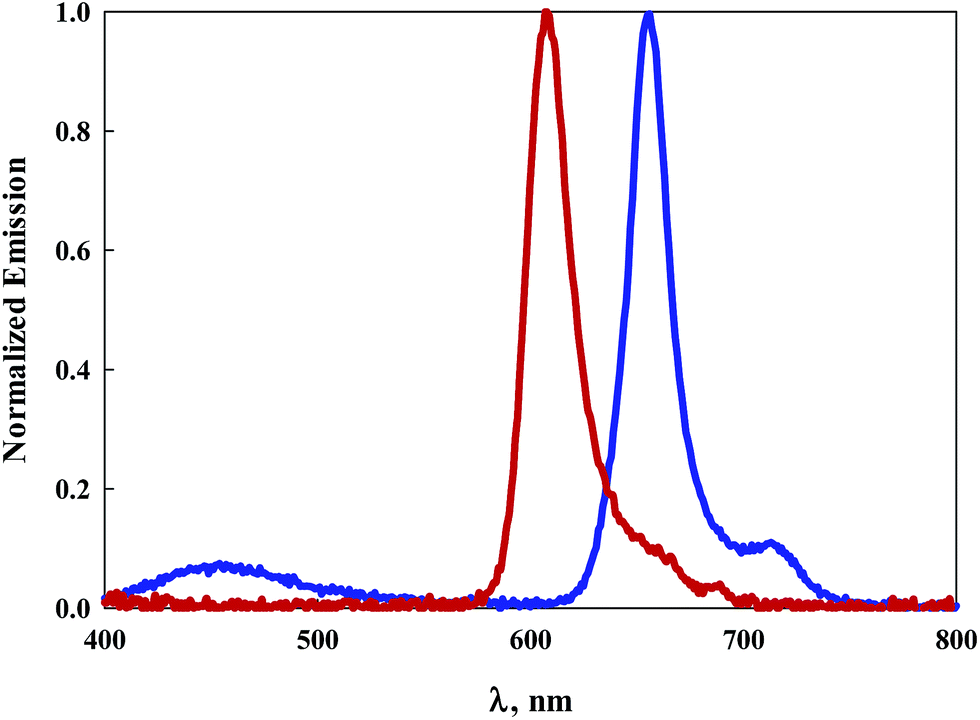 | ||
| Fig. 3 Emission spectra of 7 (red line) and 9 (blue line) in DCM upon excitation at 344 nm. | ||
The emission spectra of 5 and 8 are illustrated in Fig. 1B. The emission peak for 5 appears at 604 nm with a Stokes shift of 223 cm−1. The emission peak for 8 appears at 654 nm with a slightly larger Stokes shift of 285 cm−1. The luminescence quantum efficiency for 5 was determined previously to be 0.9.26 Typically BODIPY dyes with emission maxima greater than 600 nm exhibit quantum efficiencies well below unity. Notably, the quantum efficiency of 8 is 0.8, considerably higher than that observed for similar dyes with emission also peaking near 650 nm.1
Normalized electronic absorption spectra of BODIPY dyes 7 and 9 are illustrated in Fig. 2A. Despite their structural differences with 7 symmetric and 9 asymmetric, both exhibit intense absorption peaks at wavelengths shorter than 400 nm indicative of the pyrene moieties. It is noteworthy that 9, which contains an extra pyrene arm compared to 7, displays similar but more intense absorption within this region of the spectrum. Similar to the other BODIPY dyes, both 7 and 9 display sharp, intense, absorption bands with absorption peaks of 599 nm (ε = 138000 M−1 cm−1) for 7 and 645 nm (ε = 124000 M−1 cm−1) for 9. This represents a 46 nm bathochromic shift for the more conjugated pyrrole, Fig. 2A and Table 1. Emission of 7 is observed with a maximum at 613 nm with a Stokes shift of 381 cm−1, Fig. 2B. Dye 9 displays an emission peak at 657 nm with a Stokes shift of 283 cm−1, Fig. 2B. Luminescence quantum efficiencies for both 7 and 9 are at or near unity, Table 1.
It has been previously established that due to a near-orthogonal arrangement between the BODIPY core and its meso-substituent, there is typically no overlap of the π-electron clouds.30 Indeed bulky meso-substituents limited by their size to freely rotate allow for extended lifetimes of the excited dyes by eliminating non-radiative decay mechanisms.31 Lifetime measurements of dyes 1–9 in DCM range from 3.2 ns to 4.3 ns suggesting the ability for non-radiative deactivation via some rotation at the meso positions. Surprisingly, dyes 8 and 9 exhibit the shortest lifetimes even though, structurally, they represent the most resistance to free rotation at the meso position. This suggests that another mechanism for non-radiative deactivation is likely responsible for the shorter excited-state lifetimes. Of the dyes in this study, only the pyrene-substituted BODIPY compounds 7 and 9 display significant absorption in the UV region. For both 7 and 9, photoexcitation of the pyrene substituents at 344 nm results in emission peaking at 608 nm and 656 nm respectively, Fig. 3. A small emission peak is observed for the asymmetric, more highly conjugated dye 9 at ∼450 nm associated with the pyrene moiety. The observation that excitation of the pyrene generates emission from the BODIPY core suggests either a through-bond or through-space intramolecular excitation energy transfer process.30
Electrochemical properties
Cyclic voltammetry experiments were performed for 7–9 in DCM with TBAPF6 as the supporting electrolyte. A one compartment three electrode cell was employed with a platinum wire auxiliary electrode and glassy carbon working electrode. The cell was equipped with a non-aqueous Ag+ reference electrode and ferrocene as the internal standard. All experiments were performed at room temperature under a nitrogen atmosphere.Scanning in the anodic direction, a quasi-reversible redox couple is observed for the BODIPY dye 7 with E1/2 = 0.68 V (ΔE = 120 mV), Fig. 4A.
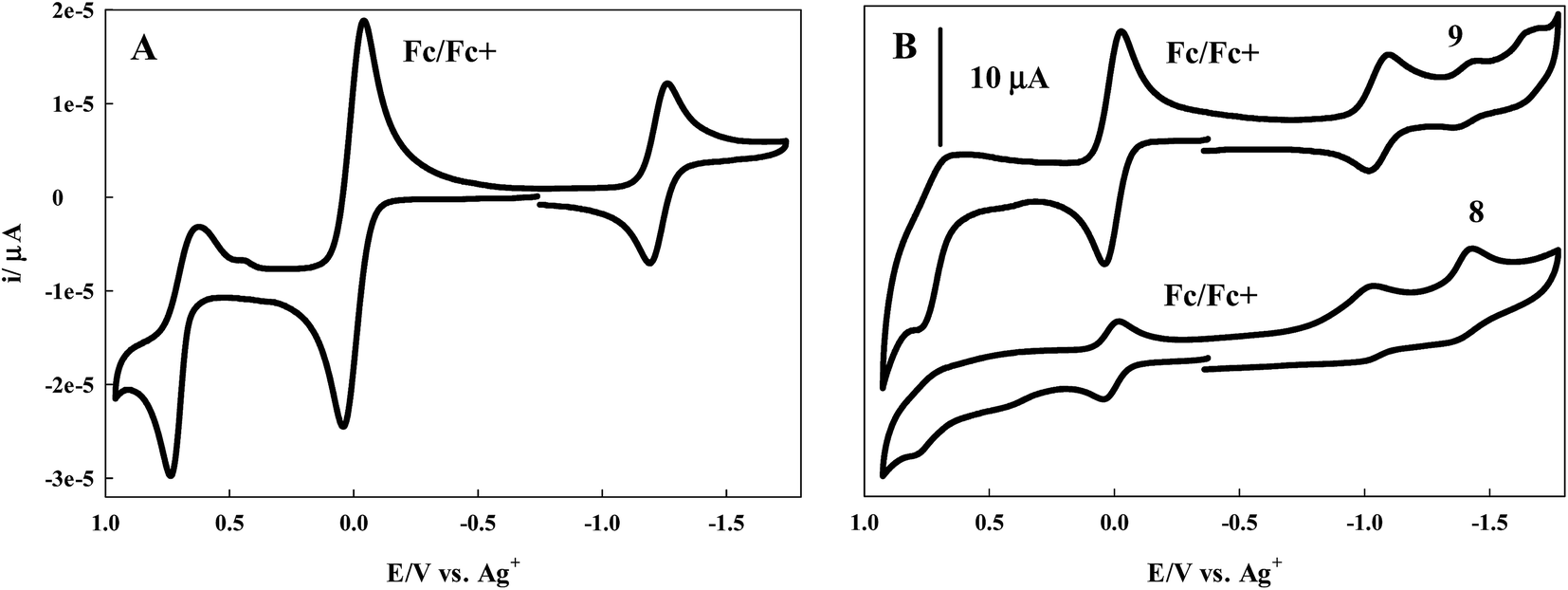 | ||
| Fig. 4 (A) Cyclic voltammogram of 7 in DCM, (B) CV of 8 and 9 in DCM containing 0.1 M TBAPF6 as the supporting electrolyte at room temperature, a glassy carbon working electrode, platinum wire auxilliary, Ag/Ag+ reference electrode, and ferrocene as internal standard. Scan rate = 100 mV s−1. | ||
A reversible couple with E1/2 = −1.23 V (ΔE = 60 mV) is revealed upon scanning in the cathodic direction. The cyclic voltammograms of the more conjugated BODIPY dyes 8 and 9 are less well defined, though redox activity is observed. In the case of 8, a broad, irreversible oxidation is observed in the anodic region of the voltammogram with a peak potential of 0.77 V. When scanned in the cathodic direction, two irreversible reductions with peak potentials of −1.03 V and −1.43 V are observed, Fig. 4B. For 9, Fig. 4B, an irreversible oxidation with peak potential of 0.75 V is followed by a quasi-reversible redox couple in the cathodic region with E1/2 = −1.06 V (ΔE = 70 mV). Scanning further in the cathodic direction reveals two irreversible reduction waves with peak potentials of −1.45 V and −1.67 V.
From the onset of the oxidation and reduction waves, the HOMO and LUMO energies can be estimated.32 Scheme 3 depicts a Jablonski diagram of these energies for the BODIPY dyes of Scheme 1.
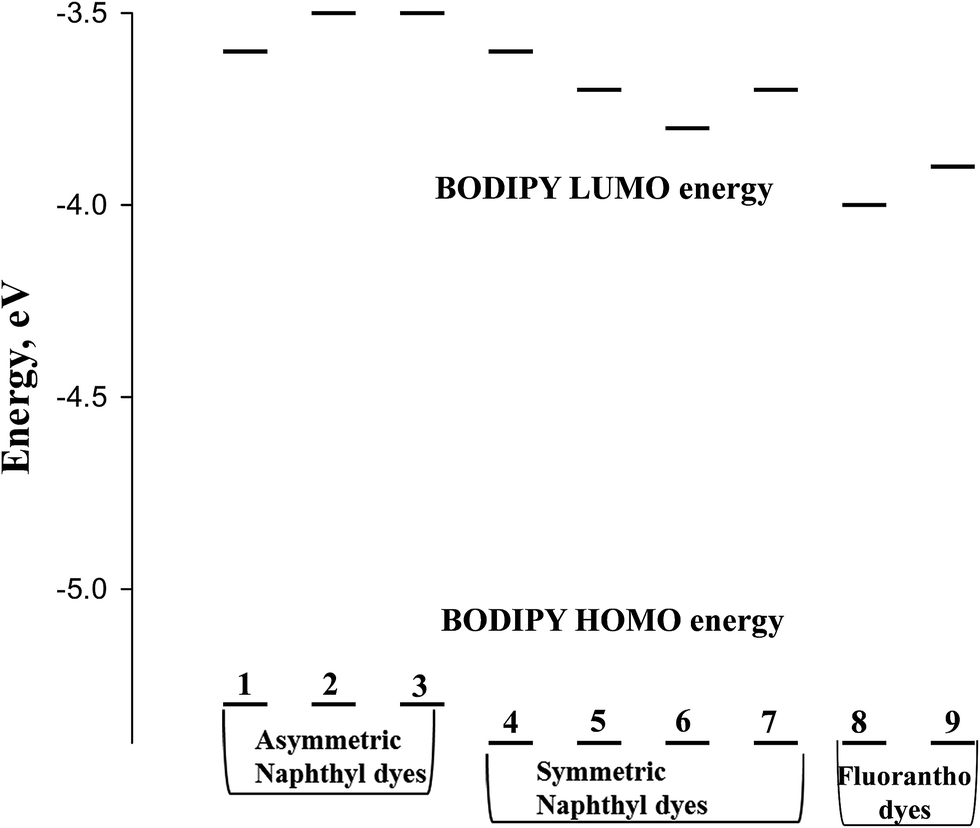 | ||
| Scheme 3 Jablonski diagram for the BODIPY dyes illustrated in Scheme 1. | ||
It has been established that coordination of the BF2 moiety to the dipyrrin serves to bring the π-orbitals of the dipyrrin into alignment, resulting in a sharper, more intense, bathochromically shifted electronic transition.1 Yet, theoretical calculations indicate that neither the HOMO nor LUMO electron density is located on the boron, but instead resides on the dipyrrin core.33 It is reasonable to assume then that any shifts in energy of the electronic transition of the BODIPY dyes is related to the pyrrole or the meso-substituent of the dipyrrin. From the Jablonski diagram of Scheme 3, it appears that with the naphtha[1,2-c]pyrrole-based dyes, the HOMO energy is unaffected by the composition of the meso-substituent. Despite this, the symmetric dyes show greater stabilization of the HOMO energy compared to the asymmetric dyes. In the case of the fluorantho[2,3-c]pyrrole-based dyes, the HOMO energy is the same as that for the symmetric naphtha[1,2-c]pyrrole dyes and independent of the symmetry of the dye.
It is well established that meso-substituents of BODIPY dyes are typically orthogonal to the BODIPY core thereby preventing overlap of the π-cloud associated with the BODIPY core and the π-cloud of the aromatic meso-substituent. Theoretical studies have indicated that electron-withdrawing meso-substituents stabilize the LUMO energy, while electron-donating meso-substituents destabilize the LUMO energy.34 Considering this, comparison of the asymmetric napthyl-pyrrole dyes (1–3) shows that the LUMO energy is destabilized in the dyes with the electron donating para-fluoro and para-methyl phenyl meso-substituents compared to the meso-phenyl dye, Scheme 3. For the symmetric napthyl-pyrrole dyes (4–7), the LUMO energy exhibiting the greatest destabilization is associated with the meso-trimethoxyphenyl-substituted dye (4) while the most stabilized of this group belongs to the meso-thienyl substituted dye (6). The LUMO energy for the meso-substituted hydroxy-naphthyl (5) and pyrene (7) dyes is identical. This is surprising because, in the case of the fluorantho-pyrrole dyes, the pyrene-substituted dye (9) shows greater LUMO destabilization compared to the hydroxy-naphthyl fluorantho dye (8). However, this difference in energy may be associated with the symmetry or lack of symmetry since the asymmetric dyes destabilize the LUMO energy more than the symmetric dyes.
Conclusions
A simple two-step synthetic route to new BODIPY dyes has been established for a variety of aromatic aldehydes and π-conjugated pyrroles. These dyes display significant photophysical properties that can be tuned by both the choice of aldehyde as well as the choice of π-conjugated pyrrole. The role of the aldehydes plays less of a role in tuning the photophysical properties. Nevertheless, the versatility displayed by these aldehydes may allow for structural adaptations which could prove advantages for numerous applications. It is clear that the greatest effect on the photophysical properties lies with the conjugation associated with the pyrroles. Extending this conjugation further may lead to efficient BODIPY dyes capable of light absorption and emission properties well into the near-IR region of the electromagnetic spectrum.Acknowledgements
Time-resolved emission data was collected on an emission spectrophotometer (Edinburgh Instrument, FLS920) housed in the UNC EFRC Instrumentation Facility established by the UNC EFRC Center for Solar Fuels, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award DE-SC0001011.Notes and references
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- A. Mishra, M. K. R. Fischer and P. Bauerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed.
- S. Kolemen, M. Isik, G. M. Kim, D. Kim, H. Geng, M. Buyuktemiz, T. Karatas, X.-F. Zhang, Y. Dede, J. Yoon and E. U. Akkaya, Angew. Chem., Int. Ed., 2015, 54, 5340–5344 CrossRef CAS PubMed.
- (a) A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC; (b) S. G. Awuah and Y. You, RSC Adv., 2012, 2, 11169–11183 RSC; (c) Z. Wang, X. Hong, S. Zong, C. Tang, Y. Cui and Q. Zeng, Sci. Rep., 2015, 5, 12602 CrossRef CAS PubMed; (d) X. Hong, Z. Wang, J. Yang, Q. Zheng, S. Zong, Y. Sheng, D. Zhu, C. Tanga and Y. Cuic, Analyst, 2012, 137, 4140–4149 RSC.
- T. Gayathri, A. K. Barui, S. Prashanthi, C. R. Patra and S. P. Singh, RSC Adv., 2014, 4, 47409–47413 RSC.
- A. M. Courtis, S. A. Santos, Y. Guan, J. A. Hendricks, B. Ghosh, D. M. Szantai-Kris, S. A. Reis, J. V. Shah and R. Mazitschek, Bioconjugate Chem., 2014, 25, 1043–1051 CrossRef CAS PubMed.
- L. Yang, Y. Liu, W. Liu, C. Ma, C. Zhang and Y. Li, Bioorg. Med. Chem. Lett., 2015, 25, 5716–5719 CrossRef CAS PubMed.
- S. D. Topel, O. Topel, R. B. Bostancioglu and A. T. Koparal, Colloids Surf., B, 2015, 128, 245–253 CrossRef CAS PubMed.
- G. Fan, L. Yang and Z. Chen, Front. Chem. Sci. Eng., 2014, 8, 405–417 CrossRef CAS.
- B. Sui, S. Tang, T. Liu, B. Kim and K. D. Belfield, ACS Appl. Mater. Interfaces, 2014, 6, 18408–18412 CAS.
- S. P. Singh and T. Gayathri, Eur. J. Org. Chem., 2014, 22, 4689–4707 CrossRef.
- S. Debnath, S. Singh, A. Bedi, K. Krishnamoorthy and S. S. Zade, J. Phys. Chem. C, 2015, 119, 15859–15867 CAS.
- S. Madhu and M. Ravikanth, Inorg. Chem., 2012, 51, 4285–4292 CrossRef CAS PubMed.
- T. Kowada, S. Yamaguchi and K. Ohe, Org. Lett., 2010, 12, 296–299 CrossRef CAS PubMed.
- C. Yu, L. Jiao, P. Zhang, Z. Feng, C. Cheng, Y. Wei, X. Mu and E. Hao, Org. Lett., 2014, 16, 3048–3051 CrossRef CAS PubMed.
- C. Cheng, N. Gao, C. Yu, Z. Wang, J. Wang, E. Hao, Y. Wei, X. Mu, Y. Tian, C. Ran and L. Jiao, Org. Lett., 2015, 17, 278–281 CrossRef CAS PubMed.
- A. B. Descalzo, H.-J. Xu, Z.-L. Xue, K. Hoffmann, Z. Shen, M. G. Weller, X.-Z. You and K. Rurack, Org. Lett., 2008, 10, 1581–1584 CrossRef CAS PubMed.
- Y. Kubo, K. Watanabe, R. Nishiyabu, R. Hata, A. Murakami, T. Shoda and H. Ota, Org. Lett., 2011, 13, 4574–4577 CrossRef CAS PubMed.
- S. Yamazawa, M. Nakashima, Y. Suda, R. Nishiyabu and Y. Kubo, J. Org. Chem., 2016, 81, 1310–1315 CrossRef CAS PubMed.
- Y. Hayashi, N. Obata, M. Tamaru, S. Yamaguchi, Y. Matsuo, A. Saeki, S. Seki, Y. Kureishi, S. Saito, S. Yamaguchi and H. Shinokubo, Org. Lett., 2012, 14, 866–869 CrossRef CAS PubMed.
- L. Luo, D. Wu, W. Li, S. Zhang, Y. Ma, S. Yan and J. You, Org. Lett., 2014, 16, 6080–6083 CrossRef CAS PubMed.
- C. Maeda, T. Todaka and T. Ema, Org. Lett., 2015, 17, 3090–3093 CrossRef CAS PubMed.
- Z.-B. Sun, M. Guo and C.-H. Zhao, J. Org. Chem., 2015, 229–237 Search PubMed.
- Y.-W. Wang, A. B. Descalzo, Z. Shen, X.-Z. You and K. Rurack, Chem.–Eur. J., 2010, 16, 2887–2903 CrossRef CAS PubMed.
- S. Swavey, M. Coladipietro, A. Burbayea and J. A. Krause, Eur. J. Org. Chem., 2016, 4429–4435 CrossRef CAS.
- T. D. Lash, M. L. Thompson, T. M. Werner and J. D. Spence, Synlett, 2000, 2, 213–216 CrossRef.
- T. D. Lash, T. M. Werner, M. L. Thompson and J. M. Manley, J. Org. Chem., 2001, 66, 3152–3159 CrossRef CAS PubMed.
- H. Sunahara, Y. rano, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2007, 129, 5597–5605 CrossRef CAS PubMed.
- J. Banuelos, I. J. Arroyo-Cordoba, I. Valois-Escamilla, A. Alvarez-Hernandez, E. Pena-Cabrera, R. Hu, B. Z. Tang, I. Esnal, V. Martinez and I. L. Arbeloa, RSC Adv., 2011, 1, 677–684 RSC.
- R. Ziessel, C. Goze, G. Ulrich, M. Cesario, P. Retailleau, A. Harriman and J. P. Rostron, Chem.–Eur. J., 2005, 11, 7366–7378 CrossRef CAS PubMed.
- LUMO (eV) = −(Ered + 4.8); HOMO (eV) = −(Eox + 4.8).
- J. Banuelos Prieto, F. Lopez Arbeloa, V. Martinez Martinez and I. Lopez Arbeloa, Chem. Phys., 2004, 296, 13–22 CrossRef CAS.
- J. K. Petrushenko and K. B. Petrushenko, Spectrochim. Acta, Part A, 2015, 138, 623–627 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2017 |