
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQM/MM studies of the type II isopentenyl diphosphate–dimethylallyl diphosphate isomerase demonstrate a novel role for the flavin coenzyme†
Qianqian Hou *a,
Kang Wanga,
Feng Xua,
Wenshen Zhanga,
Kejian Ji*a and
Yongjun Liub
*a,
Kang Wanga,
Feng Xua,
Wenshen Zhanga,
Kejian Ji*a and
Yongjun Liub
aShandong Non-Metallic Materials Institute, Jinan, Shandong 250100, China. E-mail: houqianqian53@163.com; Fax: +86-531-85062524; Tel: +86-531-85878054
bSchool of Chemistry and Chemical Engineering, Shandong University, Jinan, Shandong 250100, China
First published on 21st April 2017
Abstract
The type II isopentenyl diphosphate:dimethylallyl diphosphate isomerase (IDI-2) catalyzes the reversible isomerization of isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP). Although a growing body of experiments have suggested that the flavin coenzyme of IDI-2 serves a novel function as an acid–base catalyst, the detailed reaction mechanism of IDI-2 is still unknown. In this paper, a combined quantum-mechanical/molecular-mechanical (QM/MM) approach has been applied to investigate the detailed reaction mechanism of IDI-2. The one-base mechanism in which the N-5 nitrogen of the zwitterionic form of reduced FMN acts as the acid–base catalyst has been supported by our computational results, and a IPP-FMN adduct is also proposed for the first time. The mechanistic details including the fundamental reaction pathways, the complete energy profiles of the whole catalytic cycle, and the specific role of the coenzyme and key residues are all obtained. It is proved that IDI-2 employs novel flavin chemistry with the coenzyme acting as a general acid–base catalyst.
1. Introduction
Isoprenoids are a large and structurally diverse class of metabolites which are essential to life.1–3 Sterols, carotenoids, dolichols, ubiquinones, and the lipophilic side chains of prenylated proteins are included in this class, which are utilized in a variety of ways by living organisms, functioning as hormones, vitamins, visual pigments, pheromones, toxins, and components of cell membranes. All isoprenoids derive from the same five-carbon building blocks, isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP). Isopentenyl diphosphate:dimethylallyl diphosphate isomerases (IDI) catalyze the reversible isomerization of IPP and DMAPP,4 as described in Scheme 1.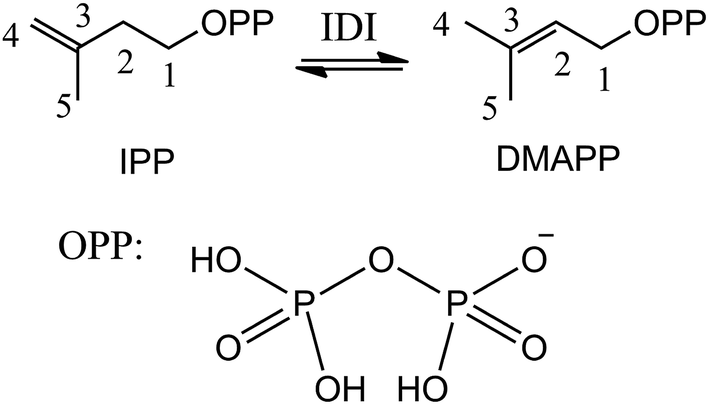 | ||
| Scheme 1 Reversible isomerization of IPP and DMAPP. | ||
Two structurally distinct IDIs have been identified. The structure and mechanism of type I IDI (IDI-1) have been extensively studied. IDI-1 requires two divalent metal ions (Mg2+ and Zn2+) for activity.5–9 The Mg2+ ion promotes substrate binding, while the Zn2+ ion binds to a hexacoordinate site in the enzyme that contains an essential glutamate residue. This glutamate, in conjunction with a nearby tyrosine residue, acts as a catalytic acid, protonating the double bond of IPP to generate a carbocation intermediate. Subsequent removal of the pro-R C2 proton by a conserved cysteine residue completes the isomerization reaction. In contrast to IDI-1, the type II enzyme (IDI-2) requires both a divalent metal ion and reduced flavin mononucleotide (FMN) coenzyme.10–13 However, mechanistic studies on IDI-2 remain spare, and it is still unclear what role the reduced flavin coenzyme plays in this isomerization reaction without net redox change.
To date, several IDI-2 homologues have been purified and characterized to different extents.14,15 But most of reported studies have focused on the biochemical characterization of the purified proteins. IDI-2 is a member of the (S)-α-hydroxy acid dehydrogenase superfamily, which has been purified from several pathogenic bacteria including methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus faecalis. The proteins crystallized as typical α8β8 triosephosphate isomerase (TIM) barrels, and one molecule of FMN was bound per monomer and located in the standard phosphate binding (SPB) region of the TIM barrel.
Based on the crystal structures and experimental studies, several proposals for the role of the flavin have been discussed for IDI-2,16–19 as described in Scheme 2. For the inactivity of apoenzyme reconstituted with cofactor analogues such as 5-deaza-FMN, a radical-mediated mechanism was initially proposed.16,17 However, the radical substrate intermediates were not detected in sopped-flow studies under single turn over conditions. Alternatively, a protonation–deprotonation mechanism was suggested for IDI-2.19 The conserved glutamine residue (Gln154 in S. aureus or Gln160 in S. shibatae IDI-2) is optimally positioned (at a distance of 3.4–4.5 Å) to mediate the proton transfer at C4 position of IPP/DMAPP. While glutamine is not a typical acid/base catalyst, it could be part of a proton relay consisting of conserved amino acid residues (specifically His11 and Glu229 in S. shibatae). Indeed, site-directed mutagenesis of this glutamine residue led to a mutant enzyme with a substantially lowered kcat value and an unperturbed KM value for IPP. Another possibility is that the reduced FMN directly participates in acid–base catalysis, either with the assistance of an amino acid group or by serving as both an acid and a base via FMNH2 or its zwitterionic tautomer.18,20 Considering the short distance between N5 of FMN and C4 of IPP, it is conceivable that N5 is the catalyst for both proton transfers at C2 and C4. In the studies of Thermus thermophilus IDI-2, C. Dale Poulter discovered that the olefinic hydrogens of a vinyl thiomethyl analogue of isopentenyl diphosphate exchanged with solvent when the enzyme was incubated with D2O without concomitant isomerization of the double bond. These results suggest the enzyme-catalyzed isomerization reaction is not concerted. A substantial accumulation of positive charge on the reduced FMN during turnover as indicated by the kinetic linear free energy relationship (LFER) studies is also consistent with the role of N5 as a putative general acid/base catalyst. The latter observation is likely due to the transfer of the pro-R C2–H of IPP to the N5 atom of the reduced FMN coenzyme, which is at least partially rate limiting in steady state IDI-2 turnover based on substrate deuterium kinetic isotope effect (KIE) studies.20,21
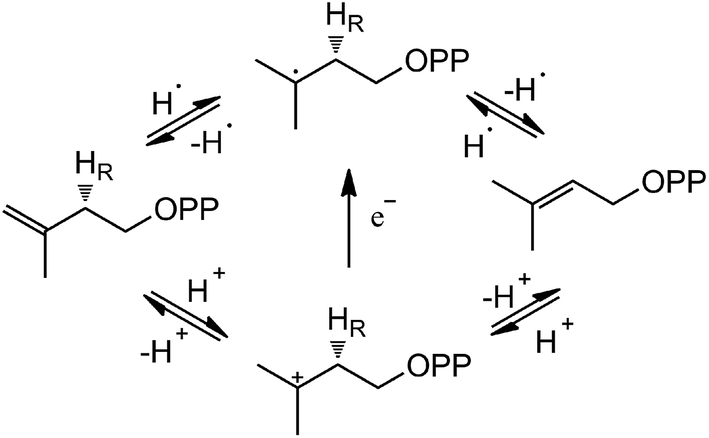 | ||
| Scheme 2 Potential IDI-2 chemical mechanisms. | ||
As described above, the reaction mechanism of IDI-2 is still controversial. In this paper, a combined quantum mechanics and molecular mechanics (QM/MM) study on IDI-2 is presented and different pathways are devised. The mechanistic details of the reaction, including the detailed energetic profile, structures of the intermediates, transition states along the reaction pathways and the role of FMN are illustrated.
2. Computational methods
The crystal structure of IDI in complex with reduced FMN and IPP (PDB ID:2ZRY)5 was used to construct the computational model. The molecular dynamics simulations were done with CHARMM27 force field as implemented in the CHARMM program.22 The protonation states of the titratable residues were determined according to the experimental condition and the PROPKA method.23,24 As for FMN, its different protonation states will be discussed in the following section. The missing hydrogen atoms were added using the HBUILD facility. Each system was neutralized by adding Na+ ion at random position, and solvated into a water sphere of radius centered on the C1 atom of the substrate. To relax the system, preliminary structural optimization was firstly carried out, then a 8 ns simulation was performed to equilibrate the system. A relative smooth RMSD (the root-mean-squared deviation) curve to represent equilibration of protein backbone (shown in Fig. S1†) was obtained. It is shown that the dynamics trajectory was basically stable after 6 ns, and there would be no major fluctuations for the protein backbone unless undergoing reaction.A representative initial structure selected from MD trajectory is important for obtaining a meaningful energy barrier. In our calculations, a total of 11 snapshots were extracted from the MD trajectory for the following QM/MM calculations. The superposition of QM region from 11 obtained structures was displayed in Fig. S2.† To find a representative initial configuration, the RMSDs of 11 structures relative to the average one were firstly calculated (Fig. S2†), then a presentative structure derived from 5.6 ns of MD trajectory was selected for the following step.
In QM/MM calculations, the QM subsystem contains the substrate (IPP), coenzyme FMN, an Mg2+, one water molecule and the sidechains of Lys193, Ser195, and His11 (depicted in Fig. 2) which directly participate in the catalysis or make a significant impact on the distribution of electron density in the active site. MM subsystem includes all the other atoms and solvent. The QM/MM calculations were performed with the ChemShell package25 which integrates TURBOMOLE26 and DL-PLOY programs.27 The B3LYP functional and 6-31G (d, p) basis set were used for the QM part, and the CHARMM22 force field for the MM part. An electronic embedding scheme28 which incorporates the MM charges into the one-electron Hamiltonian of the QM treatment and hydrogen link atoms with charge shift model for the QM/MM boundary were adopted in the QM/MM treatment. During the geometry optimization, the QM subsystem and the MM atoms within a distance of 12 Å of N atom of IPP were allowed to relax, whereas the remaining MM atoms were kept frozen. The hybrid delocalized internal coordinates (HDLC) optimizer29 in ChemShell was employed for geometry optimizations. A quasi-Newton limited memory Broyden–Fletcher–Goldfarb–Shanno (L-BFGS) algorithm29 was used for minima search. In the transition state search, the highest point on the potential energy profile along the reaction coordinate was chosen to be optimized with partitioned rational function optimization (P-RFO) method29 and characterized by a single negative eigenvalue. The electronic embedding scheme and the hydrogen link atoms with charge shift model for QM/MM boundary were applied in the QM/MM treatment. Finally, a high-level single point electronic energy calculation was performed at a larger basis set 6-311++G (d, p) to obtain accurate energies. All the energies reported herein were corrected by zero-point vibrational effects.
3. Results and discussion
3.1 Structure of enzyme–substrate complex
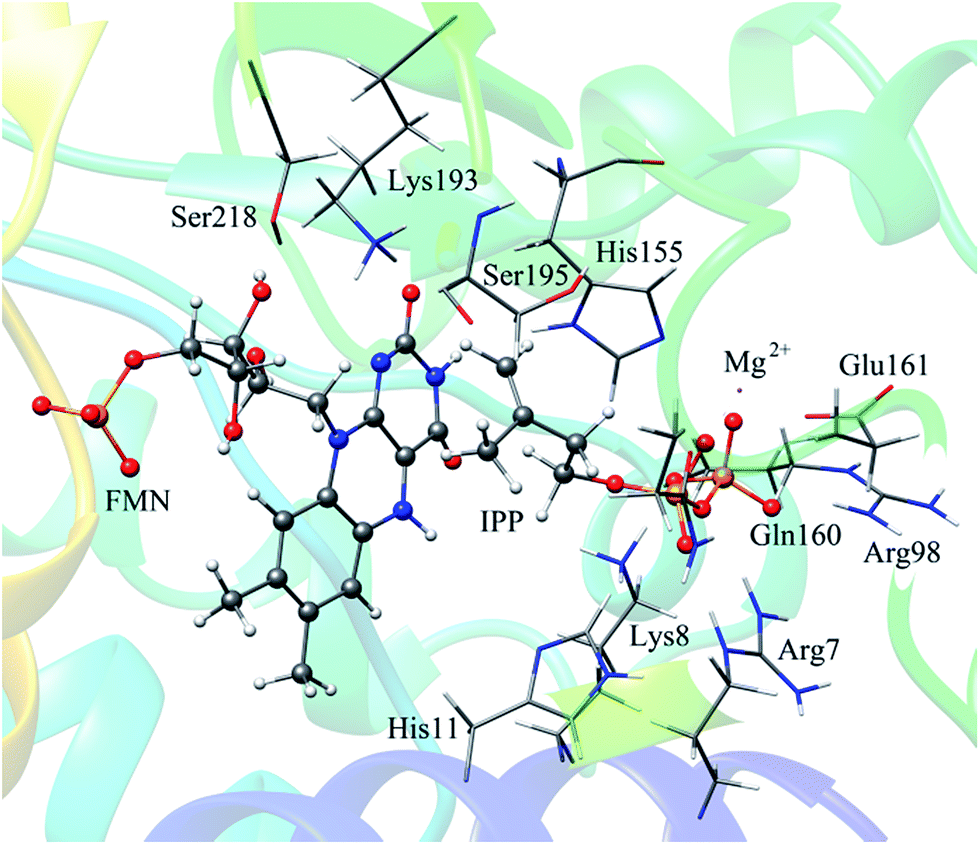
The reported crystal structure of IDI-2 and the active pocket with the key surrounding residues are shown in Fig. 1. In the crystal structure, the asymmetric unit contains four monomers that are related by a non-crystallographic 4-fold rotation, and each monomer contains a regular triose-phosphate isomerase barrel structure. The substrate-binding sites surrounded by helices, loops and the isoalloxazine ring of FMN are located on the top of the triose-phosphate isomerase barrel. Via hydrophobic and electrostatic interactions, IPP directly interacts with the side chains of Arg7, Lys8, Ser96 Arg98, His155, Gln160 and the isoalloxazine ring of FMN. The above residues are not included in the QM region, because they mainly play role of binding to the diphosphate moiety of IPP. Additionally, an Mg2+, which is supported by Glu161 and a water molecule, also binds to the diphosphate moiety of the substrate. The ionic and hydrogen bonds formed among the diphosphate group, Mg2+ ion, water molecules and surrounding amino acid resides, contribute to stabilize substrate binding. The tight binding confronts the isopentenyl or dimethylallyl plane of the substrate with the si-face of the isoalloxazine ring of FMN.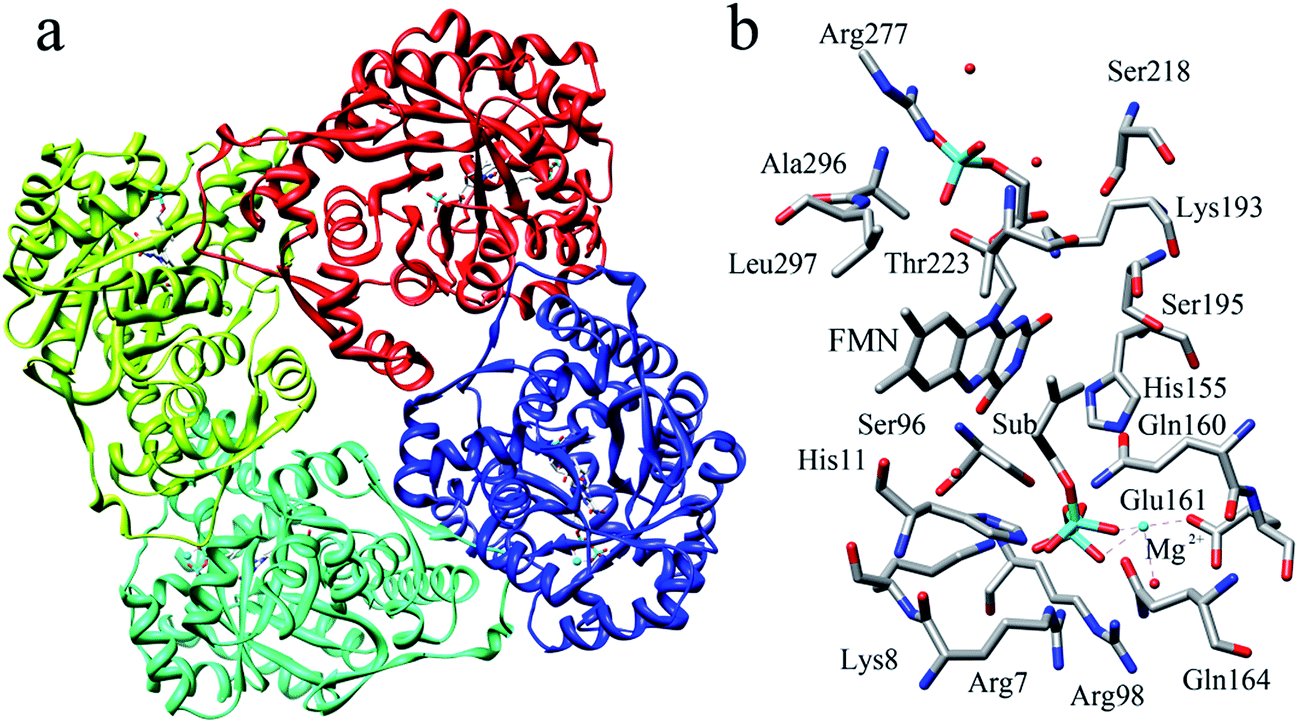 | ||
| Fig. 1 (a) Crystal structure of IDI-2 in complex with IPP (PDB code: 2ZRY), and (b) a active site structure of IDI-2. | ||
3.2 Reaction paths
In our QM/MM calculations, three models (model A, B and C) based on the experiments were examined as stated in Scheme 3. UV-visible spectral analysis of IDI-2 reaction has revealed the accumulation of an anionic reduced FMNH− or a protonated reduced flavin (FMNH2) when the reduced enzyme was incubated with IPP or DMAPP.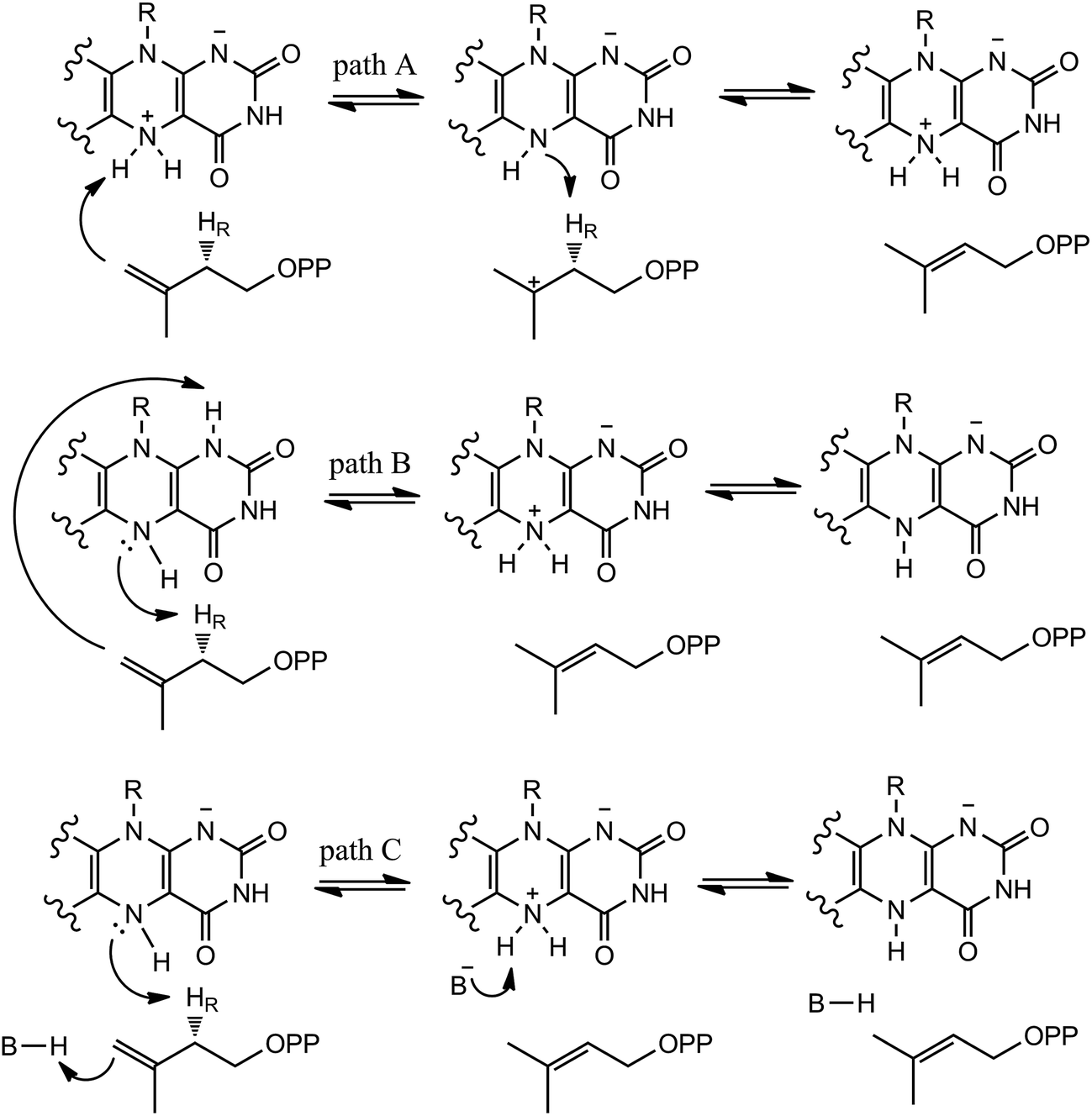 | ||
| Scheme 3 Hypothetical mechanisms of IDI-2. | ||
The reduced FMN may play three possible roles in IDI-2: (1) as shown in the one-base mechanism, the N-5 nitrogen of the zwitterionic form of FMN acts as the acid–base catalyst (path A); (2) the N-1 nitrogen and N-5 nitrogen of reduced FMN catalyze the protonation addition/elimination (path B); and (3) protonation and deprotonation are catalyzed by amino acid residues, and reduced FMN stabilizes the intermediate (path C).
If the N-5 nitrogen acts as the acid–base catalyst in the reaction, probably by generating the zwitterionic flavin species, it is considered to monofunctionally catalyze (1,3)-suprafacial proton addition/elimination. The stationary points involved in path A are shown in Fig. 3. In the reactant complex (ReactA), the C2–C3 bond of IPP has rotated 180° related to the binding conformation of the substrate in the crystal, which is supposed to facilitate the proton transfer. The double bond of IPP strongly interacts with the isoalloxazine ring in FMN by π interactions. The distance between H(N5) and C4(IPP) is 3.44 Å, and the C3 atom of IPP positions a distance of 3.69 Å from the C4A atom of FMNH2. In the first step, the proton transfer of H(N5) is concerted with the formation C4A(FMNH2)–C3(IPP) bond, forming the FMN-IPP adduct. In the transition state (TS1A), the distances of N5–H and H–C4(IPP) is 1.40 Å and 1.34 Å, respectively, and the C4A(FMNH2)–C3(IPP) bond changes from 3.69 Å in the reactant to 3.10 Å in TS1A, which indicate this step to be a concerted but asynchronous reaction, i.e., the proton transfer is little earlier than the C–C bond formation. The dihedral angle O5–C2 increases to 156.5°. After passing the transition state, a FMN-IPP adduct is generated. The C4A(FMNH2)–C3(IPP) bond is formed with a distance of 1.67 Å in Int1A. The calculated energy barrier is 18.0 kcal mol−1. In the second step, the enzyme undergoes process of the hydrogen transfer and C4A(FMNH2)–C3(IPP) bond breakage concertedly. In TS2A, the distances of H(N5) and pro-R H–C2(IPP) is 1.52 Å and 1.36 Å, respectively, and the C4A(FMNH2)–C3(IPP) bond is 3.55 Å, suggesting the completion of the cleavage of C4A(FMNH2)–C3(IPP) bond. Finally, the product DMAP is formed. The energy barrier for this step is calculated to be 18.8 kcal mol−1. The corresponding energy profile along the reaction path is presented in Fig. 4. The energy values displayed in the Fig. 4 are taken relative to the reactant, and the energy barriers are calculated relative to the preceding intermediate. Another round of catalysis will start when the zwitterionic form of reduced FMN transforms to the protonated reduced FMN.
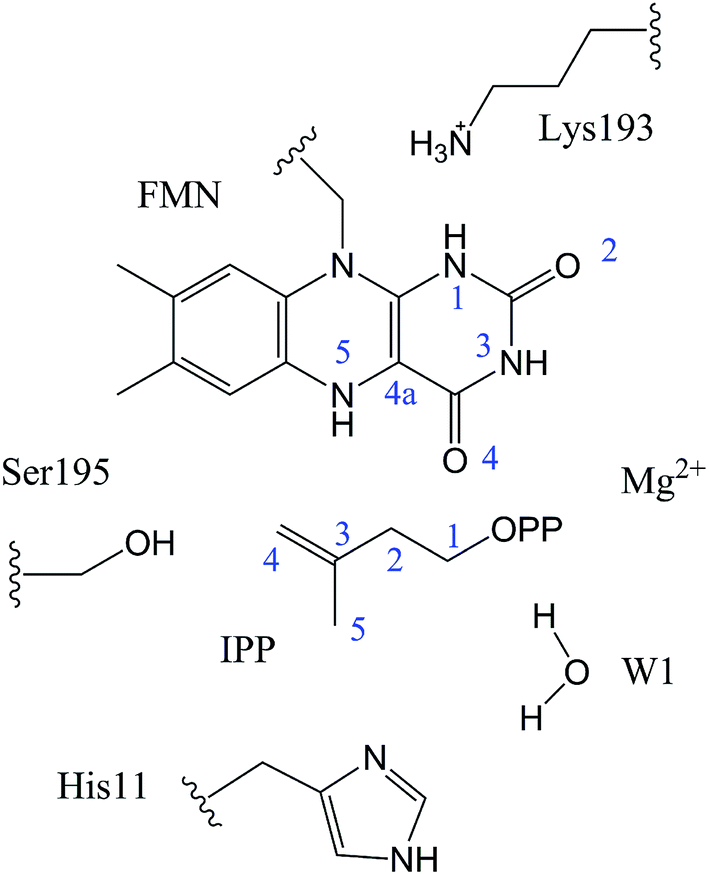 | ||
| Fig. 2 The selected QM region in our QM/MM calculations. | ||
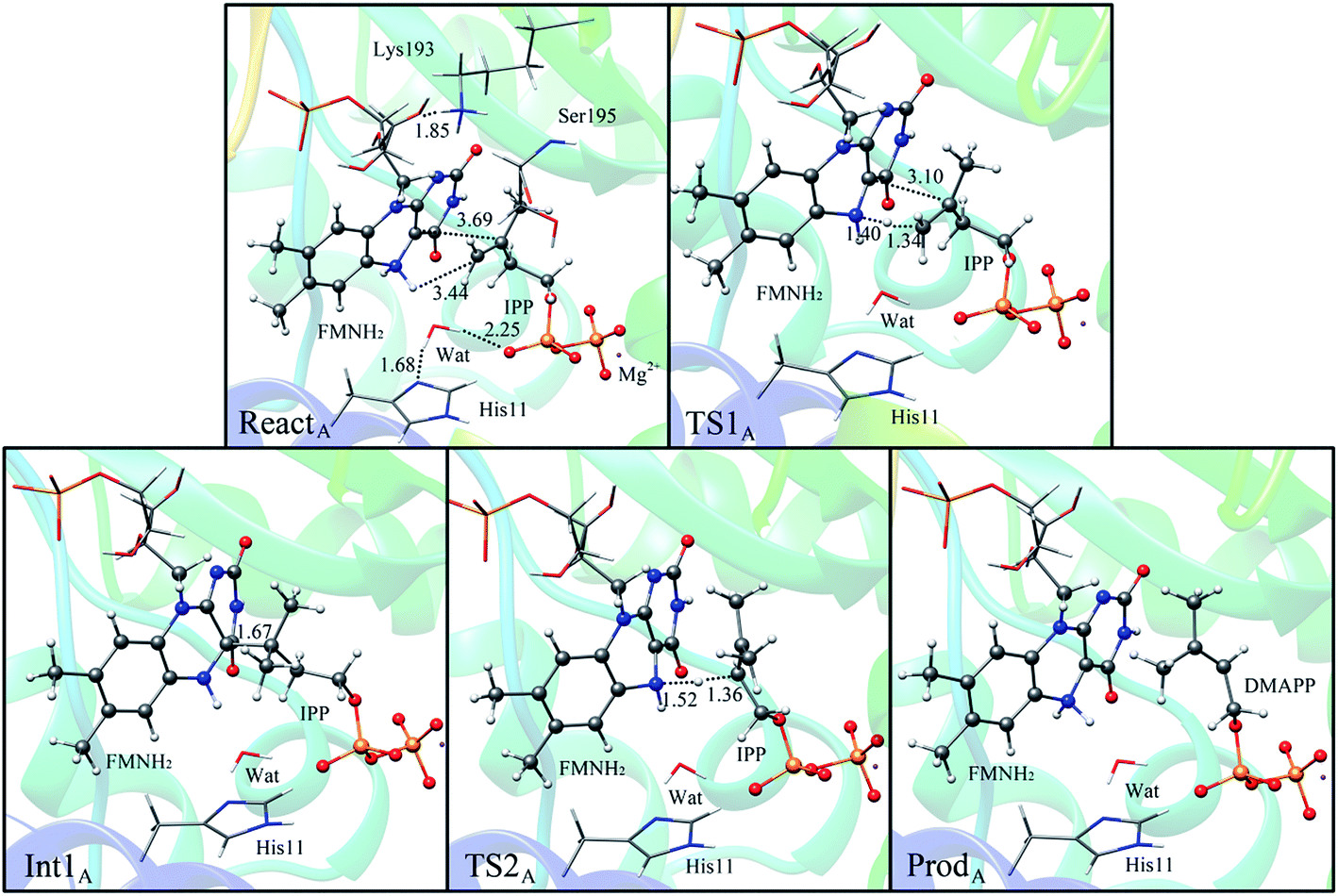 | ||
| Fig. 3 Optimized structures of reactant, transition states, intermediate, and product for model A. Distances are given in Å. | ||
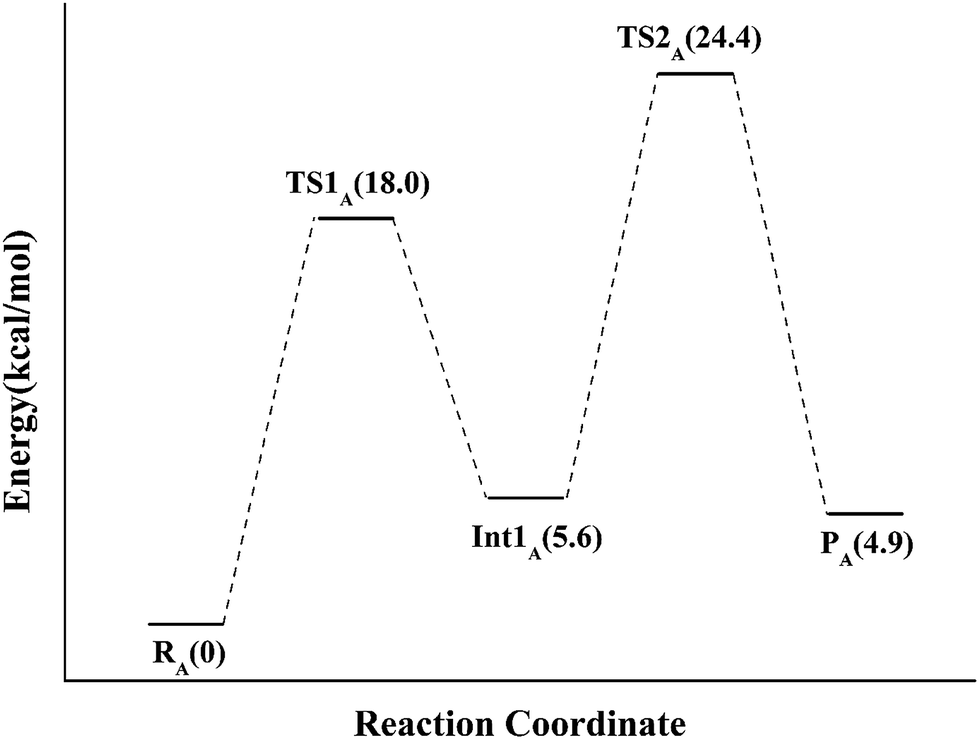 | ||
| Fig. 4 The QM/MM energy profile for model A. | ||
Alternatively, the N-1 nitrogen and N-5 nitrogen are likely to act as a pair of acid/base, along with the tautomeric rearrangement of reduced flavin. The stationary points concerning path B are shown in Fig. 5. In the reactant complex (ReactB), the distance between H and C4 is 3.95 Å, and the C2 atom of IPP positions a distance of 3.81 Å from the C4A atom of FMNH2. In the first step, the proton transfer and formation of bond C4A(FMNH2)–C3(IPP) are considered to be a concerted mechanism. In the transition state (TS1B), the distances of N1–H and H–C4(IPP) is 1.52 Å and 1.26 Å, respectively. Besides, the C4A(FMNH2)–C3(IPP) bond changes to 3.68 Å in TS1B. After passing the transition state, the system reaches a FMN-IPP adduct. The C4A(FMNH2)–C3(IPP) bond is formed with a distance of 1.64 Å in Int1B. The calculated energy barrier is 33.8 kcal mol−1. Notably, it is 15 kcal mol−1 higher than the rate-limiting step in path A. In the second step, the enzyme undergoes process of the hydrogen transfer and C4A(FMNH2)–C3(IPP) bond breakage concertedly. In TS2B, the distances of N5–H and H–C2(IPP) is 1.39 Å and 1.50 Å, respectively, and the C4A(FMNH2)–C3(IPP) bond is 2.93 Å. It is suggested that C4A(FMNH2)–C3(IPP) bond is already broken. Finally, the product DMAP is formed. The calculated energy barrier for this step is 21.3 kcal mol−1. The corresponding energy profile along the reaction path is presented in Fig. 6. The energy values displayed in the Fig. 6 are taken relative to the reactant, and the energy barriers are calculated relative to the preceding intermediate.
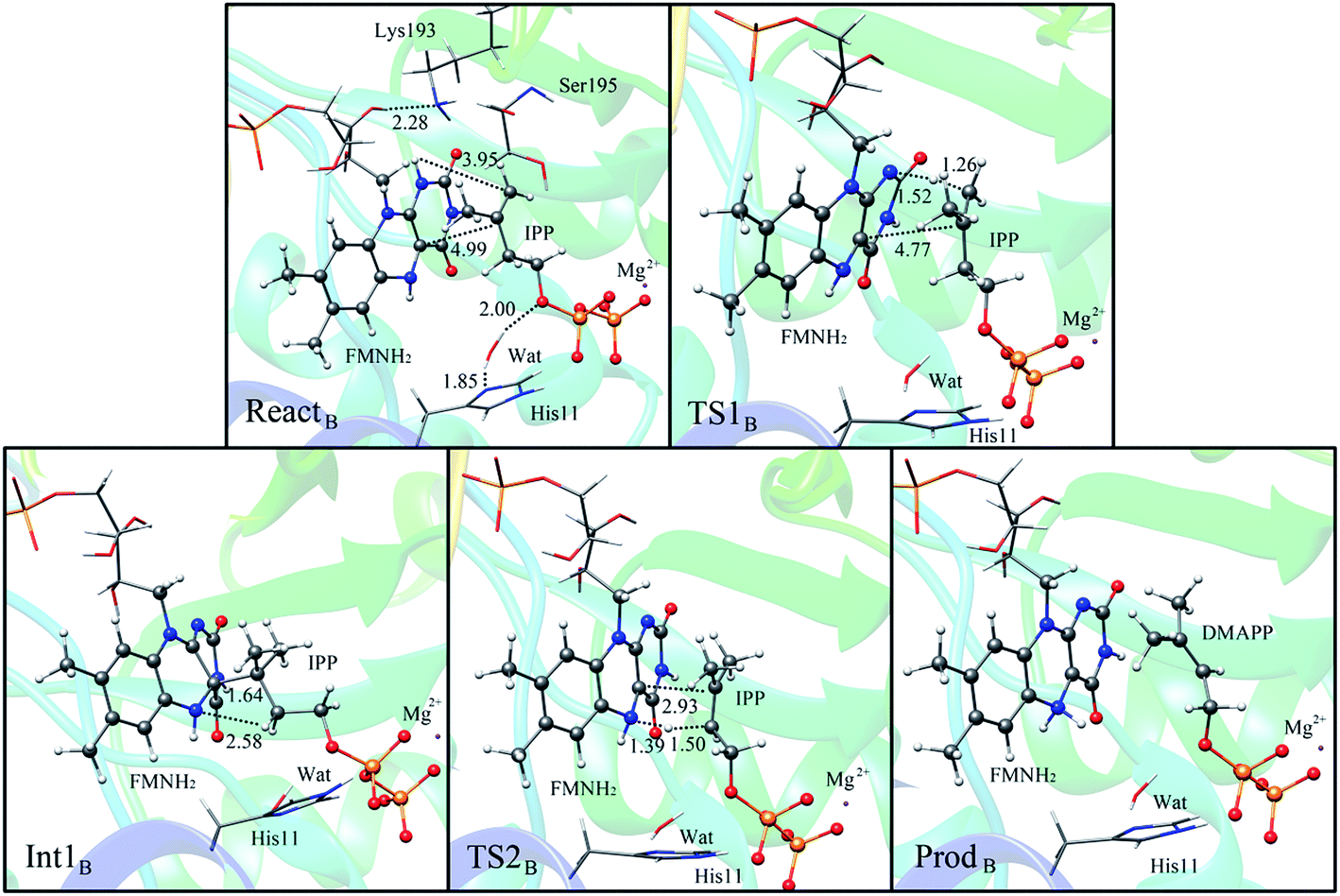 | ||
| Fig. 5 Optimized structures of reactant, transition states, intermediate, and product for model B. Distances are given in Å. | ||
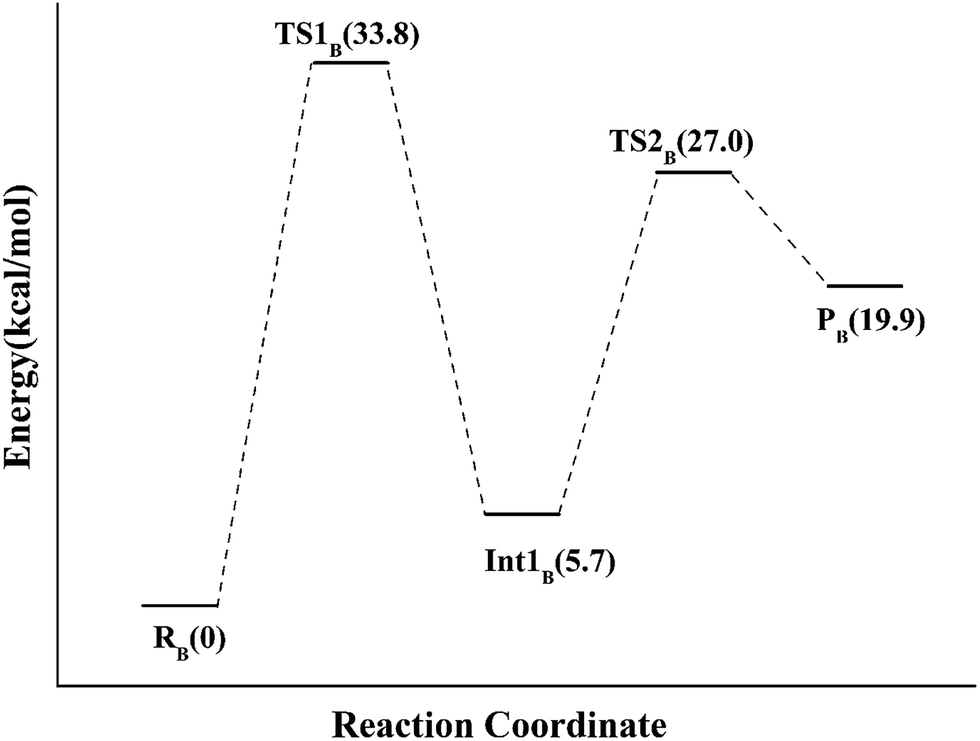 | ||
| Fig. 6 The QM/MM energy profile for model B. | ||
We also explored another possibility that the anionic reduced FMNH− participates in acid–base catalysis with the assistance of an amino acid group. The optimized structure taken from the reactive pocket of IDI is shown in Fig. 7. Compared with the crystal structure, the active pocket of model B is located at the same position, but the binding mode is a little bit different. The C2–C3 bond of the substrate has rotated about 180°. As described in the Scheme 3, when the N-5 of FMNH− attracts a proton from the C2 of IPP, a proton from an amino acid transfers to the double bond of IPP concertedly. Notably, there are several charged and polar residues surrounding the double bond of IPP, including Lys193, His155, and Ser195. Lys193 is far away from IPP with the distance from C-4 of the substrate being 5.04 Å. Obviously, His155 and Ser 195 cannot act as a general acid–base catalyst for C-4 of IPP because they cannot access the reaction site with the pro-R specificity. Unfortunately, no putative proton donor and acceptor can be identified to support this mechanism.
 | ||
| Fig. 7 Optimized structure of model C. | ||
Our QM/MM results have provided much mechanistic information for the unusual flavoenzyme IDI-2. The mechanistic details support the mechanism of model A. Because of the ability of FMN to accept and donate either one or two protons according to the nitrogen atoms of the flavin ring, the coenzyme of FMN is supposed to participate in the enzymatic reaction directly. The N5 atom of FMN is properly positioned to perform a similar role as an active site acid–base catalyst. The N5 atom loses a proton to the C4 atom of IPP in the first step, and then attracts a proton from the C2 atom of IPP. FMN-IPP adduct is formed as an intermediate during the catalysis. Interestingly, Eguchi and co-workers30 have recently found that an epoxide analogue of IPP binds inclose proximity to the flavin ring and suggests that electron density is capable of accumulating at N5. Our results support that the substrate and the flavin ring are easily form a cavalent adduct according to C4A–C3 bond. Our results are also consistent with the linear free energy relationships (LFER) studies which suggest that the IDI-2 chemical mechanism involves general base catalysis by the N5 atom of the reduced flavin coenzyme. The fact that IDI-1 and IDI-2 catalyze isomerization by totally different mechanisms has also been demonstrated by these results.
Actually, flavin coenzymes play multiple roles in biological systems. In the catalytic mechanism of IDI-2, the role of N5 acting as a putative general acid/base catalyst has been approved by our QM/MM calculations. Meanwhile, an adduct is firstly proposed and obtained in the catalytic mechanism. Though the involvement of a formal reduced N5 anion in acid/base chemistry has never been demonstrated in flavin-dependent enzyme catalysis, such as a species could both electrostatically stabilize a developing substrate carbocation and act as a general base. Acyl-CoA dehydrogenases are capable of catalyzing the one and two-electron oxidation/reduction reactions.31–33 While in CDP-6-deoxy-L-threo-D-glycero-4-hexulose-3-dehydrase reductase, flavin acts as a redox mediator between two-electron and one-electron transfer reactions.34 In some other flavin coenzymes, flavin mononucleotide or flavin adenine dinucleotide are not involved in redox chemistry, they may electrostatically stabilize the reaction intermediates.35,36 In UDP-galactopyranose mutase, flavin acts as a covalent catalyst, facilitating the ring contraction through the formation of an N5 adduct. Excitingly, the behavior of the flavin coenzyme during IDI-2 catalysis is quite different to that observed in the reactions catalyzed by these flavoenzymes.
4. Conclusion
Despite its extreme importance, the mechanism of IDI-2 has not yet been determined. In this study, QM/MM method was firstly employed to investigate the detailed mechanism of IDI-2 by three models. Path A is supported to be the minimum energy pathway by our QM/MM calculations. In this path, IDI-2 employs novel flavin chemistry, with the N5 atom of FMN acting as a general acid/base catalyst. In the first step, the proton transfer of H(N5) and the formation C4A(FMNH2)–C3(IPP) bond occur concertedly to form the FMN-IPP adduct with an energy barrier of 18.0 kcal mol−1. It is notably that the intermediate of FMN-IPP adduct with the C4A–C3 bond is found for the first time. In the second step, the enzyme undergoes process of the hydrogen transfer and C4A(FMNH2)–C3(IPP) bond breakage concertedly, corresponding to an energy barrier of 18.8 kcal mol−1. Path B has been identified to be a high-energy pathway. The rate-limiting step of formation of IPP-FMN adduct undergoes an energy barrier of 33.8 kcal mol−1. In path C, there is no putative proton donor and acceptor can be identified to support this mechanism. It is clear that our study elucidated the reversible isomerization of IPP and DMAPP catalyzed by this unusual enzyme at the atomistic level, which could further provide useful information for the development of new antibiotics.Acknowledgements
This work was supported by the Natural Science Foundation of China (21403201).References
- N. Qureshi and J. W. Porter, in Biosynthesis of Isoprenoid Compounds, ed. J. W. Porter and S. L. Spurgeon, John Wiley, New York, 1981, vol. 1, pp. 47–94 Search PubMed.
- T. Koyama and K. Ogura, in Comprehensive Natural Products Chemistry, ed. D. E. Cane, Elsevier, Amsterdam, 1999, vol. 2, pp. 69–96 Search PubMed.
- B. A. Kellogg and C. D. Poulter, Chain elongation in the isoprenoid biosynthetic pathway, Curr. Opin. Chem. Biol., 1997, 1, 570–578 CrossRef CAS PubMed.
- A. C. Ramos-Valdiva, R. van der Heijden and R. Verpoorte, Isopentenyl diphosphate isomerase: a core enzyme in isoprenoid biosynthesis. A review of its biochemistry and function, Nat. Prod. Rep., 1997, 14, 591–603 RSC.
- S. Lee and C. D. Poulter, Escherichia coli type I isopentenyl diphosphate isomerase: structural and catalytic roles for divalent metals, J. Am. Chem. Soc., 2006, 128, 11545–11550 CrossRef CAS PubMed.
- J. Wouters, Y. Oudjama, S. Ghosh, V. Stalon, L. Droogmans and E. Oldfield, Structure and mechanism of action of isopentenyl pyrophosphate–dimethylallyl pyrophosphate isomerase, J. Am. Chem. Soc., 2003, 125, 3198–3199 CrossRef CAS PubMed.
- J. Wouters, Y. Oudjama, S. J. Barkley, C. Tricot, V. Stalon, L. Droogmans and C. D. Poulter, Catalytic mechanism of Escherichia coli isopentenyl diphosphate isomerase involves Cys-67, Glu-116, and Tyr-104 as suggested by crystal structures of complexes with transition state analogues and irreversible inhibitors, J. Biol. Chem., 2003, 278, 11903–11908 CrossRef CAS PubMed.
- V. Durbecq, G. Sainz, Y. Oudjama, B. Clantin, C. Bompard-Gilles, C. Tricot, J. Caillet, V. Stalon, L. Droogmans and V. Villeret, Crystal structure of isopentenyl diphosphate:dim-ethylallyl diphosphate isomerase, EMBO J., 2001, 20, 1530–1537 CrossRef CAS PubMed.
- R. B. Silverman, K. A. Bichler and A. J. Leon, Unusual mechanistic difference in the inactivation of r-aminobutyric acid aminotransferase by (E)- and (Z)-4-amino-6-fluoro-5-hexenoic acid, J. Am. Chem. Soc., 1996, 118, 1253–1261 CrossRef CAS.
- K. Kaneda, T. Kuzuyama, M. Takagi, Y. Hayakawa and H. Seto, An unusual isopentenyl diphosphate isomerase found in the mevalonate pathway gene cluster from Streptomyces sp. strain CL190, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 932–937 CrossRef CAS.
- H. Hemmi, Y. Ikeda, S. Yamashita, T. Nakayama and T. Nishino, Catalytic mechanism of type 2 isopentenyl diphosphate:dimethylallyl diphosphate isomerase: verification of a redox role of the flavin cofactor in a reaction with no net redox change, Biochem. Biophys. Res. Commun., 2004, 322, 905–910 CrossRef CAS PubMed.
- T. Kuzuyama and H. Seto, Diversity of the biosynthesis of the isoprene units, Nat. Prod. Rep., 2003, 20, 171–183 RSC.
- C. J. Thibodeaux, W.-c. Chang and H.-w. Liu, Linear free energy relationships demonstrate a catalytic role for the flavin mononucleotide coenzyme of the type II isopentenyl diphosphate:dimethylallyl diphosphate isomerase, J. Am. Chem. Soc., 2010, 132, 9994–9996 CrossRef CAS PubMed.
- J. de Ruyck, J. Pouyez, S. C. Rothman, D. Poulter and J. Wouters, Crystal Structure of Type 2 Isopentenyl Diphosphate Isomerase from Thermus thermophilus in Complex with Inorganic Pyrophosphate, Biochemistry, 2008, 47, 9051–9053 CrossRef CAS PubMed.
- S. C. Rothman, J. B. Johnston, S. Lee, J. R. Walker, C. D. Poulter, S. C. Rothman, J. B. Johnston, S. Lee, J. R. Walker and C. D. Poulter, Type II isopentenyl diphosphate isomerase: irreversible inactivation by covalent modification of flavin, J. Am. Chem. Soc., 2008, 130, 4906–4913 CrossRef CAS PubMed.
- S. Rothman, T. R. Helm and C. D. Poulter, Kinetic and spectroscopic characterization of type II isopentenyl diphosphate isomerase from thermus thermophilus: evidence for formation of substrate-induced flavin species, Biochemistry, 2007, 46, 5437–5445 CrossRef CAS PubMed.
- C. J. Thibodeaux, S. O. Mansoorabadi, W. Kittleman, W. C. Chang and H. W. Liu, Evidence for the involvement of acid/base chemistry in the reaction catalyzed by the type II isopentenyl diphosphate/dimethylallyl diphosphate isomerase from Staphylococcus aureus, Biochemistry, 2008, 47, 2547–2558 CrossRef CAS PubMed.
- S. O. Mansoorabadi, C. J. Thibodeaux and H. W. Liu, The diverse roles of flavin coenzymes-nature's most versatile thespians, J. Org. Chem., 2007, 72, 6329–6342 CrossRef CAS PubMed.
- J. B. Johnston, J. R. Walker, S. C. Rothman and C. D. Poulter, Type-2 isopentenyl diphosphate isomerase. mechanistic studies with cyclopropyl and epoxy analogs, J. Am. Chem. Soc., 2007, 129, 7740–7741 CrossRef CAS PubMed.
- H. Unno, S. Yamashita, Y. Ikeda, S. Sekiguchi, N. Yoshida, T. Yoshimura, M. Kusunoki, T. Nakayama, T. Nishino and H. Hemmi, New role of flavin as a general acid–base catalyst with no redox function in type 2 isopentenyl-diphosphate isomerase, J. Biol. Chem., 2009, 284, 9160–9167 CrossRef CAS PubMed.
- S. C. Rothman, J. B. Johnston, S. Lee, J. R. Walker and C. D. Poulter, Type II isopentenyl diphosphate isomerase: irreversible inactivation by covalent modification of flavin, J. Am. Chem. Soc., 2008, 130, 4906–4913 CrossRef CAS PubMed.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, CHARMM: a program for macromolecular energy, minimization, and dynamics calculations, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- C. R. Sondergaard, M. H. Olsson, M. Rostkowski and J. H. Jensen, Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values, J. Chem. Theory Comput., 2011, 7, 2284–2295 CrossRef CAS PubMed.
- M. H. Olsson, C. R. Sondergaard, M. Rostkowski and J. H. Jensen, PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS PubMed.
- P. Sherwood, A. H. de Vries, M. F. Guest, G. Schreckenbach, C. R. A. Catlow, S. A. French, A. A. Sokol, S. T. Bromley, W. Thiel, A. J. Turner, S. Billeter, F. Terstegen, S. Thiel, J. Kendrick, S. C. Rogers, J. Casci, M. Watson, F. King, E. Karlsen, M. Sjovoll, A. Fahmi, A. Schäfer and C. Lennartz, QUASI: a general purpose implementation of the QM/MM approach and its application to problems in catalysis, J. Mol. Struct.: THEOCHEM, 2003, 632, 1–3 CrossRef CAS.
- R. Ahlrichs, M. Bar, M. Haser, H. Horn and C. Kolmel, Electronic structure calculations on workstation computers: the program system turbomole, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- W. Smith and T. R. Forester, DL_POLY_2.0: a general-purpose parallel molecular dynamics simulation package, J. Mol. Graphics Modell., 1996, 14, 136–141 CrossRef CAS.
- D. Bakowies and W. Thiel, Hybrid models for combined quantum mechanical and molecular mechanical approaches, J. Phys. Chem., 1996, 100, 10580–10594 CrossRef CAS.
- S. R. Billeter, A. J. Turner and W. Thiel, Linear scaling geometry optimisation and transition state search in hybrid delocalised internal coordinates, Phys. Chem. Chem. Phys., 2000, 2, 2177–2186 RSC.
- T. Hoshino, H. Tamegai, K. Kakinuma and T. Eguchi, Bioorg. Med. Chem., 2006, 14, 6555–6559 CrossRef CAS PubMed.
- C. Thorpe, R. G. Matthews and C. H. Williams Jr, Acyl-coenzyme A dehydrogenase from pig kidney. Purification and properties, Biochemistry, 1979, 18, 331–337 CrossRef CAS PubMed.
- B. Pohl, T. Raichle and S. Ghisla, Studies on the reaction mechanism of general acyl-CoA dehydrogenase. Determination of selective isotope effects in the dehydrogenation of butyryl-CoA, Eur. J. Biochem., 1986, 160, 109–115 CrossRef CAS PubMed.
- S. Ghisla, C. Thorpe and V. Massey, Mechanistic studies with general acyl-CoA dehydrogenase and butyryl-CoA dehydrogenase: evidence for the transfer of the beta-hydrogen to the flavin N(5)-position as a hydride, Biochemistry, 1984, 23, 3154–3161 CrossRef CAS PubMed.
- H. Grisebach, Biosynthesis of sugar components of antibiotic substances, Adv. Carbohydr. Chem. Biochem., 1978, 35, 81–126 CrossRef CAS.
- P. M. Nassau, S. L. Martin, R. E. Brown, A. Weston, D. Monsey, M. R. McNeil and K. Duncan, Galactofuranose biosynthesis in Escherichia coli K-12: identification and cloning of UDP-galactofuranose mutase, J. Bacteriol., 1996, 178, 1047–1052 CrossRef CAS PubMed.
- Q. Zhang and H. W. Liu, Mechanistic investigation of UDP-galactopyranose mutase from Escherichis coli using 2- and 3-fluorinated UDP-galactofuranose as probes, J. Am. Chem. Soc., 2001, 123, 6756–6766 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26397f |
| This journal is © The Royal Society of Chemistry 2017 |