
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of carbon-supported Pd–Co bimetallic catalysts templated by Co nanoparticles using the galvanic replacement method for selective hydrogenation
Takeharu Yoshiia,
Kazuki Nakatsukaa,
Yasutaka Kuwaharaab,
Kohsuke Mori abc and
Hiromi Yamashita*ab
abc and
Hiromi Yamashita*ab
aDivision of Materials and Manufacturing Science, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: yamashita@mat.eng.osaka-u.ac.jp; Fax: +81-6-6105-5029; Tel: +81-6-6879-7457
bElements Strategy Initiative for Catalysts Batteries (ESICB), Kyoto University, Katsura, Kyoto 615-8520, Japan
cJST, PRESTO, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 21st April 2017
Abstract
Pd–Co bimetallic catalysts were prepared by the controlled synthesis of carbon-supported Co catalysts, ranging from single-sites to nanoparticles, via structural transformation of a deposited Co(salen) complex precursor by heat treatment at different temperatures, followed by galvanic replacement with Pd ions. The catalysts were structurally characterized using XRD, TEM, HAADF-STEM, XAFS and XPS. Highly dispersed Pd-rich NPs containing a low concentration of Co species were formed and the original size of the Co NPs was retained. The electronic state of the Pd species was dependent on the size of the bimetallic NPs, and a catalyst with a mean diameter of 10.8 nm was found to be in the most electron-rich state. Not only the particle size, but also the electronic state of Pd play a crucial role in attaining high catalytic activity and selectivity in the selective hydrogenation of phenylacetylene. A Pd–Co catalyst based on a Co catalyst heat-treated at 600 °C showed 92% selectivity at 93% conversion. Furthermore, the Pd–Co catalysts synthesized by the galvanic replacement method showed superior performance to a monometallic Pd catalyst and a Pd–Co alloy catalyst.
Introduction
Bimetallic nanoparticles (NPs) have attracted much attention over the last several decades due to their potential applications as catalysts.1–6 Ligand and ensemble effects in bimetallic catalysts have been reported to dramatically enhance their catalytic activity and selectivity.7–9 These effects depend on the composition, size, morphology and structure of the bimetallic species,10–12 and therefore control of these properties is important in the design of heterogeneous catalysts for specific reactions. Bimetallic nanostructures are generally categorized by alloy structure, heterostructure, and core–shell structure, and different nanostructures have been reported to show unique catalytic performance.13,14 The preparation method is a crucial factor in determining the final structure of bimetallic NPs.15–17 Co-reduction of mixed metal ions is the most convenient method for preparing alloy NPs, whereas sequential deposition is typically used to prepare core–shell bimetallic NPs. A wide variety of additional strategies has been developed to synthesize bimetallic nanostructures.10,18The galvanic replacement (GR) method has been used over the last decade for the synthesis of bimetallic nanostructures.19,20 A pre-formed base metal, such as Co, Ni or Cu, and transition metal ions with a higher reduction potential, such as Pd2+, Pt2+ or Au3+, are used as precursors. The base metal is displaced by the transition metal ions due to the difference in ionization potential, and the second metal is redeposited onto the outer surface of the base metal. Various types of bimetallic nanostructures have been fabricated, including a core–shell structure, a hollow structure, and a porous structure, depending on the preparation conditions. A preparation technique based on GR has been developed for electro-catalysts, and various bimetallic catalysts show high electro-catalytic activities for oxygen reduction.21–27 The GR method has recently also been used to synthesize catalysts for other reactions, such as hydrogenation reaction,28–33 methane combustion34 and preferential oxidation reaction.35
In this study, a series of carbon-supported Co species with different structures, ranging from single-sites to NPs, was used for the synthesis of Pd–Co by the GR method. Bimetallic nanoparticle catalysts prepared through the GR process usually exhibit core–shell21,26,32–34 or hollow/porous20,23,28,35 structures. In contrast, the Pd–Co NPs fabricated in this study are Pd-rich NPs with a low Co content that retain the size of the original Co NPs. Thus, the size of Pd–Co NPs can be controlled by the heat treatment temperature during preparation of the carbon-supported Co species. The electronic state of the Pd species was varied by changing the size of the Pd–Co nanoparticles, and the electron-rich Pd species was found to be most active and selective for the hydrogenation of phenylacetylene. In addition, the Pd–Co catalysts prepared by the GR method exhibited performance superior to that of monometallic Pd catalyst and Pd–Co catalysts prepared by the conventional impregnation method.
Experimental
Materials
Ethanol, methanol, 1,4-dioxane, acetone, styrene(monomer), ethylbenzene, biphenyl and sodium borohydride [NaBH4] were purchased from Nakalai Tesque, Inc. (CH3COO)2Co·4H2O and Pd(II) nitrate solution were obtained from Wako Pure Chemical Ind. Co., Ltd. N,N′-bis(salicylidene)-ethylenediamine (H2(salen)) and ethynylbenzene phenylacetylene were purchased from Tokyo Chemical Industry Co., Ltd. Activated carbon (Shirasagi, M644) was obtained from Osaka Gas Chemicals Co., Ltd.Synthesis of Co(salen) complex
The Co(salen) complex was prepared by a typical procedure.36 H2(salen) (805 mg) was added into Ar-saturated ethanol (10 mL) and the solution was heated up to 90 °C in an oil bath. (CH3COO)2Co·4H2O (375 mg) as a Co precursor was added to the solution. After stirring for 24 h, the solution was cooled, filtered and washed for removing the remaining H2(salen) and Co2+ ions, followed by drying under vacuum for a day.Synthesis of Co/AC-T
A series of carbon-supported Co catalysts were synthesized thorough the process we previously reported.37 Co(salen) complex (50 mg) was dissolved into acetone (100 mL) and the activated carbon (AC, 450 mg) was added to the solution. After stirring for 1 h, the solvent was removed under vacuum with a rotary evaporator, and heat-treated at different temperatures for 2 h under a 100 mL min−1 nitrogen flow. The obtained powder was denoted as Co/AC-T (Co: 1.8 wt%), in which the T indicates the heat treatment temperature.Synthesis of PdCo/AC-T-GR
PdCo/AC-T-GR was prepared using GR method. Co/AC-T (100 mg) was dispersed in 20 mL of ethanol/H2O and an aqueous solution of NaBH4 (4 mL, 75 mM) was added dropwise under vigorous stirring at 30 °C to reduce Co2+ to Co0. After 30 min stirring, the suspension was centrifuged and washed thoroughly with Ar-saturated ethanol/H2O. The product was dispersed in 20 mL of Ar-saturated ethanol/H2O, and Pd(NO3)2 aqueous solution (140 μL, Pd 5 mg mL−1) was added dropwise under continuous stirring at 30 °C for 30 min. Then, the reaction of Pd2+ + Co0 → Pd0 + Co2+ occurred, and Pd NPs were formed by replacing Co species. Finally, the suspension was filtered and dried under vacuum for a day to obtain PdCo/AC-T-GR catalysts.Synthesis of Pd/AC
The reference catalyst of Pd/AC was synthesized by the following method. Activated carbon (100 mg) was dispersed in 20 mL of the ethanol/H2O and Pd(NO3)2 aqueous solution (140 μL, Pd 5 mg mL−1) was added dropwise under continuous stirring. After string for 1 h, the solvent was removed under vacuum with a rotary evaporator. The obtained powder was dried up for 1 day under vacuum and denoted as Pd/AC.Synthesis of PdCo/AC-600-imp
The reference catalyst of PdCo/AC-600-imp was prepared using impregnation method. Co(salen) complex (10 mg) was dissolved into acetone (20 mL) and the activated carbon (90 mg) was added to the solution. After stirring for 1 h, the solvent was removed under vacuum with a rotary evaporator. The obtained powder was dispersed into ethanol/H2O, and Pd(NO3)2 aqueous solution (140 μL, Pd 5 mg mL−1) was added. After string for 1 h, the solvent was removed under vacuum with the rotary evaporator. The obtained powder was heat-treated at 600 °C for 2 h under a 100 mL min−1 nitrogen flow, and will be referred as PdCo/AC-600-imp.Characterization
Powder X-ray diffraction patterns (XRD) were measured using a Rigaku Ultima IV diffractometer with a semiconductor analyzer. The Pd K-edge X-ray absorption fine structure analysis (XAFS) spectra were recorded at the BL-01 at SPring-8, JASRI, Harima, Japan (Pro. No. 2016B1104 and 2016A1095). In XAFS measurements, a Si(111) double crystal was used to monochromatize the X-rays from the 2.5 GeV electron storage ring. In the XAFS measurement, a sample was pretreated with hydrogen at 200 °C for 1 h, loaded into the in situ cell with plastic windows and measured in fluorescence mode. The EXAFS data was examined using the EXAFS analysis program, Rigaku EXAFS. Transmission electron microscopy (TEM) images were obtained using a field emission-transmission electron microscope (FE-TEM; Hitachi Hf-2000) operated at 200 kV. Scanning transmission electron microscopy (STEM) images and elemental mapping were obtained using JEOL-ARM 200F equipped with a Kvex energy-dispersive X-ray detector (JED-2300T) operated at 200 kV. X-ray photoelectron spectroscopy (XPS) was performed with a Shimadzu XPS system, ESCA-3400, using the Mg Kα line (1253.6 eV). All samples were measured after hydrogen reduction at 200 °C for 1 h, and the designated closed container was utilized for sample setting to prevent reoxidation. Binding energy was calibrated using the C 1s photoelectronic peak at 284.5 eV.Catalytic test
The catalytic performance was evaluated in the selective hydrogenation of phenylacetylene. Prior to the catalytic reaction, the catalyst were pretreated with hydrogen at 200 °C for 1 h. Next, the suspension containing methanol (5.0 mL), 1,4-dioxane (5.0 mL), phenylacetylene (1.0 mmol), biphenyl (0.5 mmol) as an internal standard and the catalyst (10 mg) were added into a glass reactor. The reactor was placed in an oil bath at 30 °C, and the reaction was started by 10 mL min−1 hydrogen bubbling through the suspension under continuous stirring. The samples were examined by gas chromatography (Shimadzu, GC-2014 with TC-1 column).Results and discussion
Characterization
The crystallinity of samples was investigated by XRD measurements. Fig. 1(A) shows XRD patterns for a series of Co/AC-T precursors (T = 400, 500, 600 and 800). The broad peak at 40–50° for all samples originates from the amorphous structure of activated carbon. Co/AC-400 exhibited no significant peaks associated with Co species. In contrast, Co/AC-500, -600 and -800 showed clear peaks corresponding to metallic Co at 44.1° or to CoO at 42.5°, and the intensity of the peaks increased with increasing heat treatment temperature, indicating the formation and aggregation of Co NPs above 500 °C. XRD patterns for PdCo/AC-T-GR and PdCo/AC-600-imp are shown in Fig. 1(B). All samples were measured after pretreatment with hydrogen at 200 °C for 1 h. PdCo/AC-T-GR (T = 400, 500, 600 and 800) provided no peaks arising from Co species, unlike their Co/AC-T counterparts, and no significant peaks derived from metallic Pd structure were observed (typically shown at 40.0° and 46.6°).38 These results suggest that the crystallinity of Co NPs is reduced probably due to the atomic-scale replacement with Pd to form amorphous Pd–Co NPs. In contrast, PdCo/AC-600-imp showed a peak at around 41.5°, which is 1.5° larger than that for the Pd face centered cubic (fcc) structure and 3.0° lower than that for the Co fcc structure. This peak shift indicates the formation of a Pd–Co bimetallic alloy structure in PdCo/AC-600-imp.39,40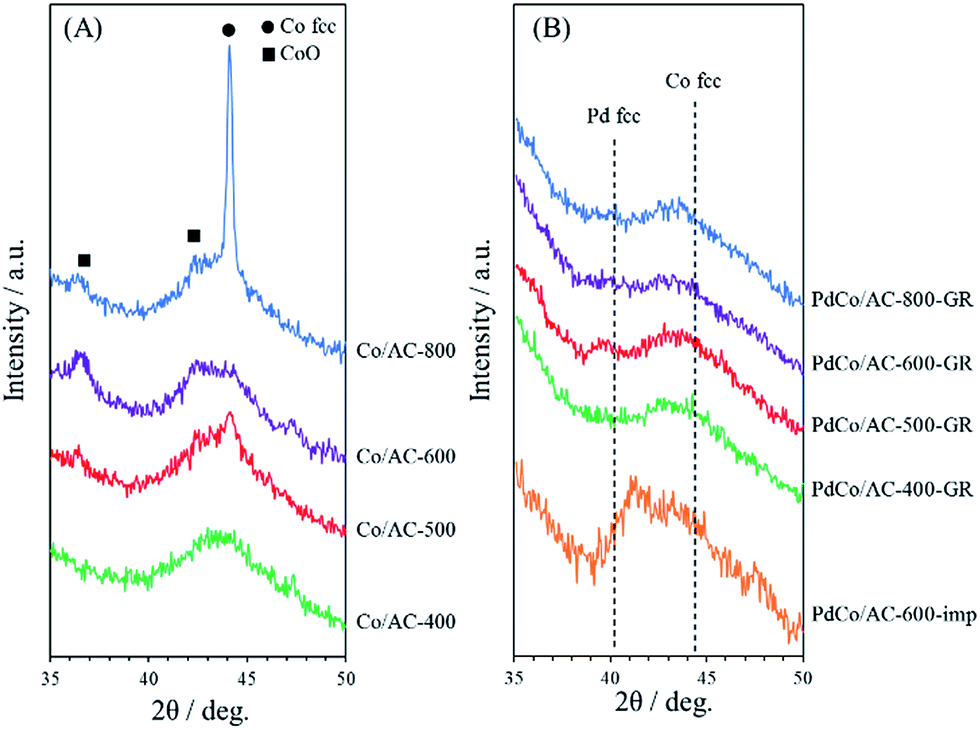 | ||
| Fig. 1 XRD patterns of (A) Co/AC-T (T = 400, 500, 600, 800) and (B) PdCo/AC-T-GR (T = 400, 500, 600, 800) and PdCo/AC-600-imp. | ||
Fig. 2(A) shows transmission electron microscopy (TEM) images of Co/AC-T. No NPs are observed in Co/AC-400, suggesting the formation of single-site Co species on the support, as we previously demonstrated.37 In contrast, Co/AC-500 contained distinct NPs with a mean diameter of 5.9 nm, and the Co/AC-600 and Co/AC-800 samples contained NPs with a mean diameter of 13.0 nm and 18.8 nm, respectively. These TEM images, together with the XRD results, indicate that NPs are produced above 500 °C and that the diameter of the NPs increases as the heat treatment temperature increases. TEM images of PdCo/AC-T-GR and PdCo/AC-600-imp subjected to hydrogen reduction at 200 °C for 1 h are shown in Fig. 2(B). PdCo/AC-400-GR contained clear NPs with a mean diameter of 3.4 nm, in contrast to Co/AC-400, which suggests the formation of Pd or Pd–Co NPs by the GR method, followed by hydrogen reduction. PdCo/AC-500, -600 and -800 also contained distinct NPs with a mean diameter of 5.9 nm, 10.8 nm and 14.8 nm, respectively. The size of the NPs in PdCo/AC-500-GR was similar to that of Co/AC-500, whereas the PdCo/AC-600-GR and -800-GR NPs were slightly smaller than those of their Co/AC-T counterparts. This finding suggests that Co NPs are replaced by Pd species while maintaining the size of the original Co NPs throughout the GR process. Thus, the size of the NPs in PdCo/AC-T-GR is primarily determined by the heat treatment temperature during Co/AC-T synthesis. PdCo/AC-600-imp as a reference sample provided NPs with a mean diameter of 14.2 nm.
 | ||
| Fig. 2 TEM images of (A) Co/AC-T (T = (a) 400, (b) 500, (c) 600, (d) 800) and (B) PdCo/AC-T-GR (T = (a) 400, (b) 500, (c) 600, (d) 800) and (e) PdCo/AC-600-imp. | ||
The crystallinity and composition of the NPs in PdCo/AC-600-GR were investigated by scanning transmission electron microscopy (STEM). Fig. 3 shows a high-angle annular dark-field (HAADF) STEM image and EDX elemental mapping images of PdCo/AC-600-GR. Regularly aligned fringes of the Pd fcc structure are partly visible in the NPs in the HAADF-STEM image, but fringes are not discernible in most areas of the NPs, indicating their low degree of crystallization, consistent with the XRD results. The EDX elemental mapping images indicate that the NPs are mainly composed of Pd species. Co species are segregated in the vicinity of the NPs and are also highly dispersed over the surface of the carbon support.
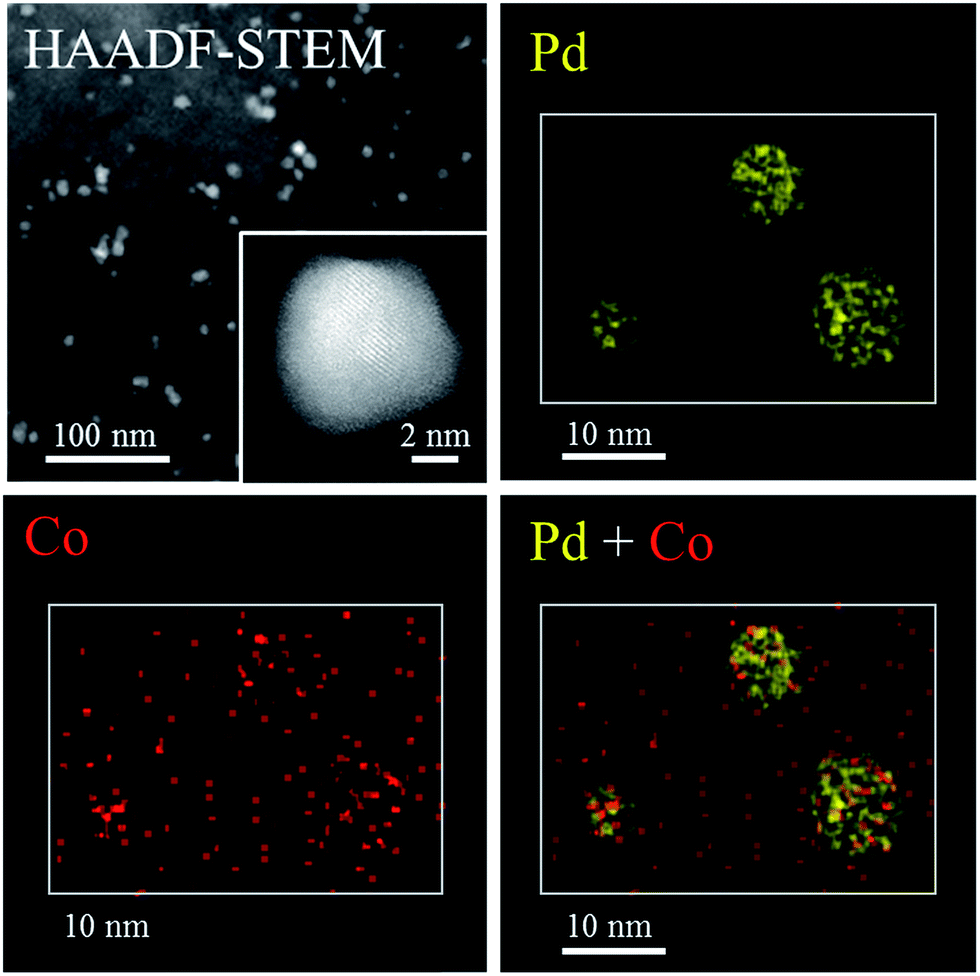 | ||
| Fig. 3 HAADF-STEM image and EDX mapping of PdCo/AC-600-GR. | ||
X-ray absorption fine structure (XAFS) measurements were performed to investigate the more detailed local structure of the Pd species. Fig. 4 shows the Pd K-edge X-ray absorption near-edge structure (XANES) and Fourier transformed extended X-ray absorption fine structure (FT-EXAFS) spectra of PdCo/AC-T-GR, PdCo/AC-600-imp, and reference samples. The peak shapes in the XANES spectra of a series of PdCo/AC-T-GR are similar to those for Pd foil. The FT-EXAFS spectra of all PdCo/AC-T-GR samples exhibit a strong peak at around 2.6 Å, attributed to Pd–Pd bonds, revealing the presence of Pd NPs. No clear peak attributed to the Pd–Co bond (typically seen at 2.2 Å)26 is observed in any PdCo/AC-T-GR sample. On the other hand, PdCo/AC-600-imp shows two strong peaks, at 2.2 Å and 2.6 Å, which can be attributed to Pd–Co and Pd–Pd bonds, respectively. This supports the formation of a Pd–Co bimetallic alloy, consistent with the XRD result.
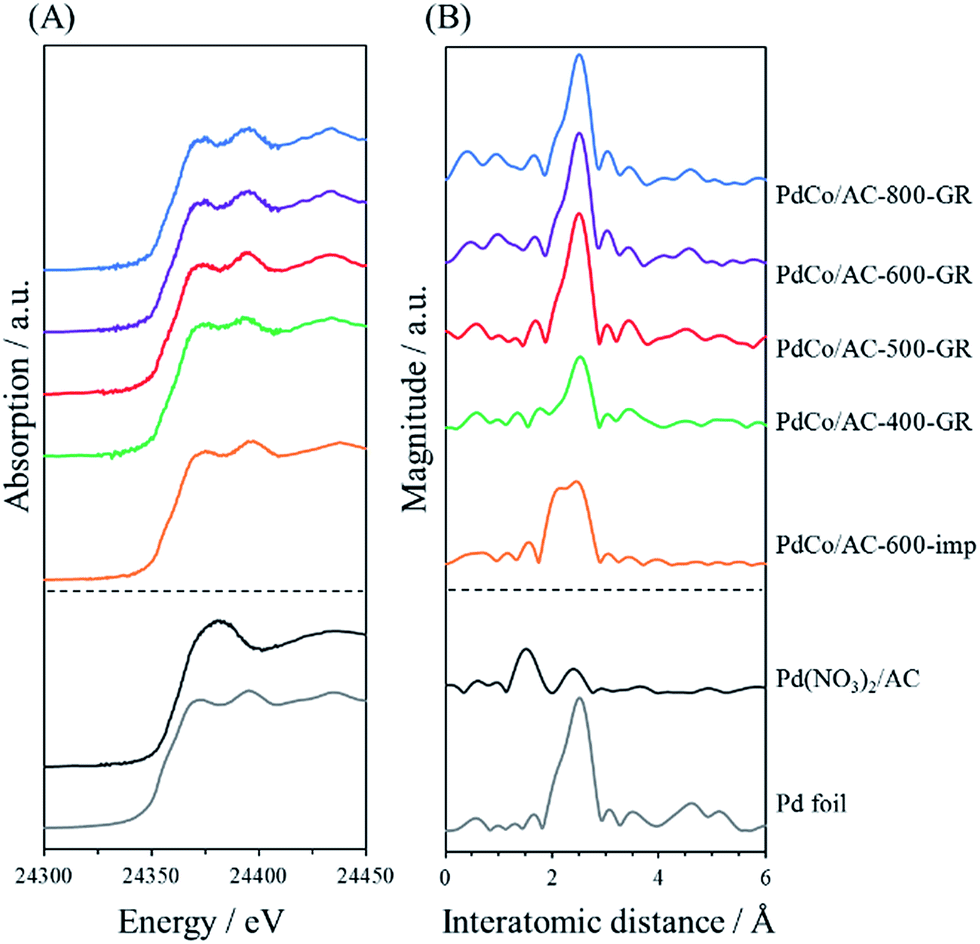 | ||
| Fig. 4 Pd K-edge (A) XANES spectra and (B) FT-EXAFS spectra of PdCo/AC-T-GR, PdCo/AC-600-imp and reference samples. | ||
The above characterization results indicate that Pd2+ ions displace Co in Co/AC-T during the GR process, followed by the formation of partially crystallized Pd NPs containing a low concentration of Co in all samples of PdCo/AC-T-GR. The size of the original Co NPs does not change significantly during the GR process. At the same time, highly dispersed displaced Co species are deposited on the surface of the carbon support. Previous use of the GR method for catalyst preparation indicated that the Pd species was replaced on the surface of the Co NPs with high dispersion, resulting mainly in the formation of Co@Pd core–shell catalysts.32–34 In contrast, in the present preparation, Co NPs are almost fully displaced by Pd species in all samples, and Pd-rich NPs are produced rather than Co@Pd core–shell NPs, probably due to the small size of the original Co NPs.
Catalytic test
The catalytic performance of PdCo/AC-T-GR was examined in the hydrogenation of phenylacetylene. This is a sequential reaction in which styrene, produced from phenylacetylene, is successively hydrogenated into ethylbenzene. Fig. 5 shows the catalytic performance of PdCo/AC-T-GR (T = 400, 500, 600 and 800), evaluated by the conversion of phenylacetylene and the selectivity for styrene. As the heat treatment temperature during Co/AC-T preparation increased from 400 to 600 °C, the activity and selectivity increased, whereas PdCo/AC-800-GR exhibited lower activity and selectivity than PdCo/AC-600-GR. Activity and selectivity were altered by the heat treatment temperature of PdCo/AC-T-GR, and PdCo/AC-600-GR showed the highest conversion and selectivity among the PdCo/AC-GR-T samples: 92% selectivity and 93% conversion after 75 min reaction.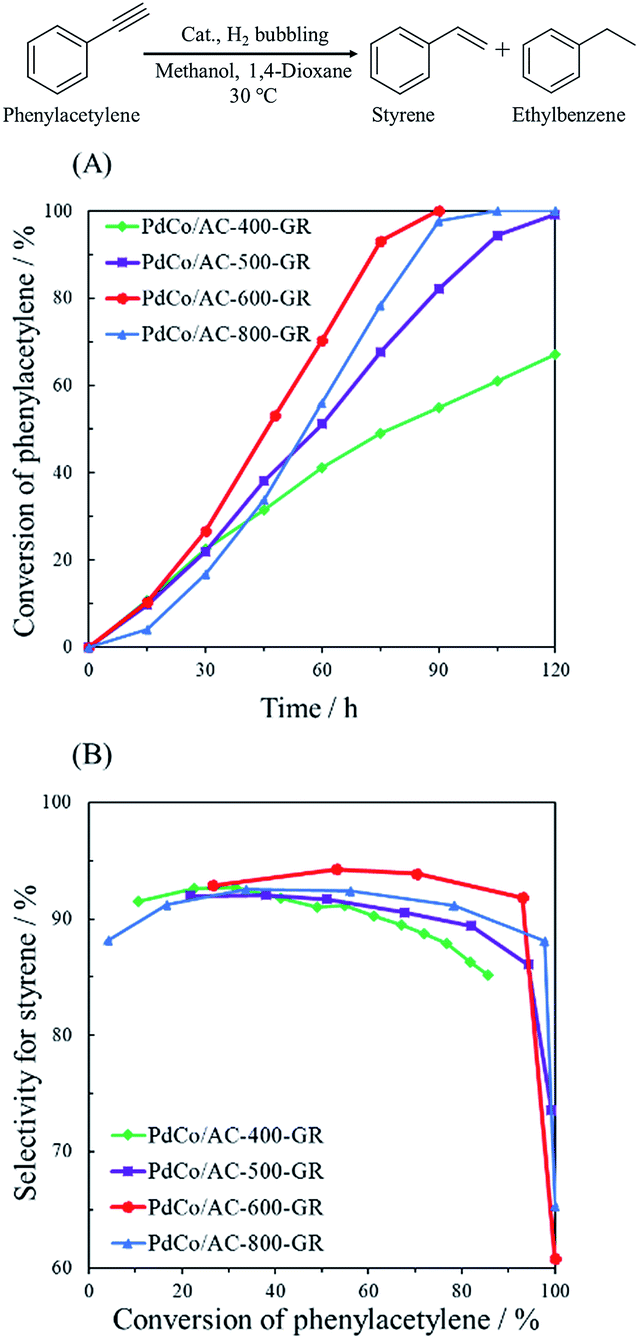 | ||
| Fig. 5 (A) Reaction profiles and (B) conversion–selectivity relationship in the hydrogenation of phenylacetylene over PdCo/AC-T-GR (T = 400, 500, 600, 800). | ||
In order to examine the reason for the different catalytic performance in a series of PdCo/AC-T-GR, the electronic properties of the Pd species in the catalysts were analyzed by XPS measurements. The Pd 3d spectra of PdCo/AC-T-GR are shown in Fig. 6. PdCo/AC-400-GR showed a binding energy (BE) peak of Pd 3d5/2 at 336.0 eV. As the heat treatment temperature increased, the BE of PdCo/AC-500-GR and PdCo/AC-600-GR decreased (335.4 and 335.1 eV, respectively), indicating that the Pd species in PdCo/AC-600 were in a more electron-rich state compared with the case for PdCo/AC-400. On the other hand, the BE for PdCo/AC-800 was 335.3 eV, which is higher than that of PdCo/AC-600. These electronic states of a series of PdCo/AC-T-GR were probably influenced by the remaining Co species incorporated in NPs. Fig. 7 shows the relationship between the electronic state (represented by the BE peak of the Pd 3d2/5 spectra of PdCo/AC-T-GR (Fig. 6)) and catalytic activity and selectivity. A clear correlation between the electronic properties and catalytic performance was observed, and catalysts with more electron-rich Pd exhibited higher catalytic activity and selectivity in the selective hydrogenation reaction. It is suggested that the Pd–Co ensembles with electron-rich Pd act as suitable active sites for selective hydrogenation of phenylacetylene. The electron richness of Pd would promote the charge transfer into the π* orbitals and destabilization of triple bond of phenylacetylene, which accelerate the hydrogenation of phenylacetylene to yield styrene.41 On the other hand, the interaction between electron-rich π-bond of styrene and the electron-rich Pd species is not as strong as that of the electron-deficient Pd species. Therefore, the electron-rich Pd species exhibit a lower adsorption energy toward styrene, which probably suppresses the sequential hydrogenation of styrene to yield ethylbenzene.31 This is the probable reason for the high activity and selectivity of PdCo/AC-600-GR possessing electron-rich Pd species.
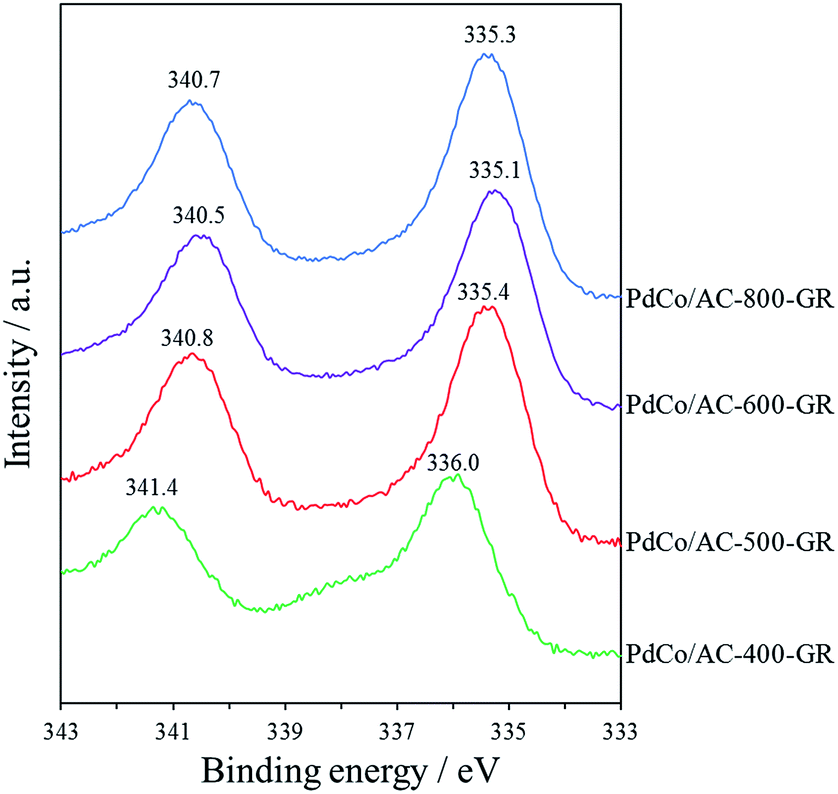 | ||
| Fig. 6 Pd 3d XPS spectra of PdCo/AC-T-GR, PdCo/AC-600-imp. | ||
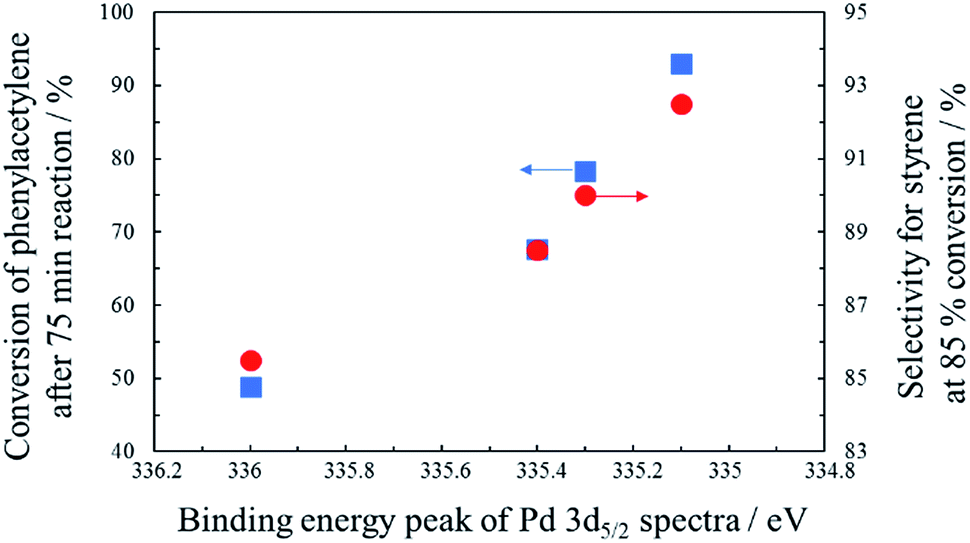 | ||
| Fig. 7 The relationship between the electronic state of Pd and conversion of phenylacetylene/selectivity for styrene. | ||
Fig. 8 shows the catalytic performance of PdCo/AC-600-GR and the reference catalysts. Co/AC-600 exhibited no catalytic activity in this reaction, whereas Pd/AC showed 80% selectivity and 90% conversion after 105 min reaction, which is about 12% lower selectivity than that of PdCo/AC-600-GR. These results reveal that Pd is active site for the hydrogenation of phenylacetylene in PdCo/AC-600-GR, while selectivity is enhanced by the addition of Co species. PdCo/AC-600-imp showed nearly the same selectivity as PdCo/AC-600-GR, but lower activity than PdCo/AC-600-GR, probably due to the low concentration of surface Pd species.
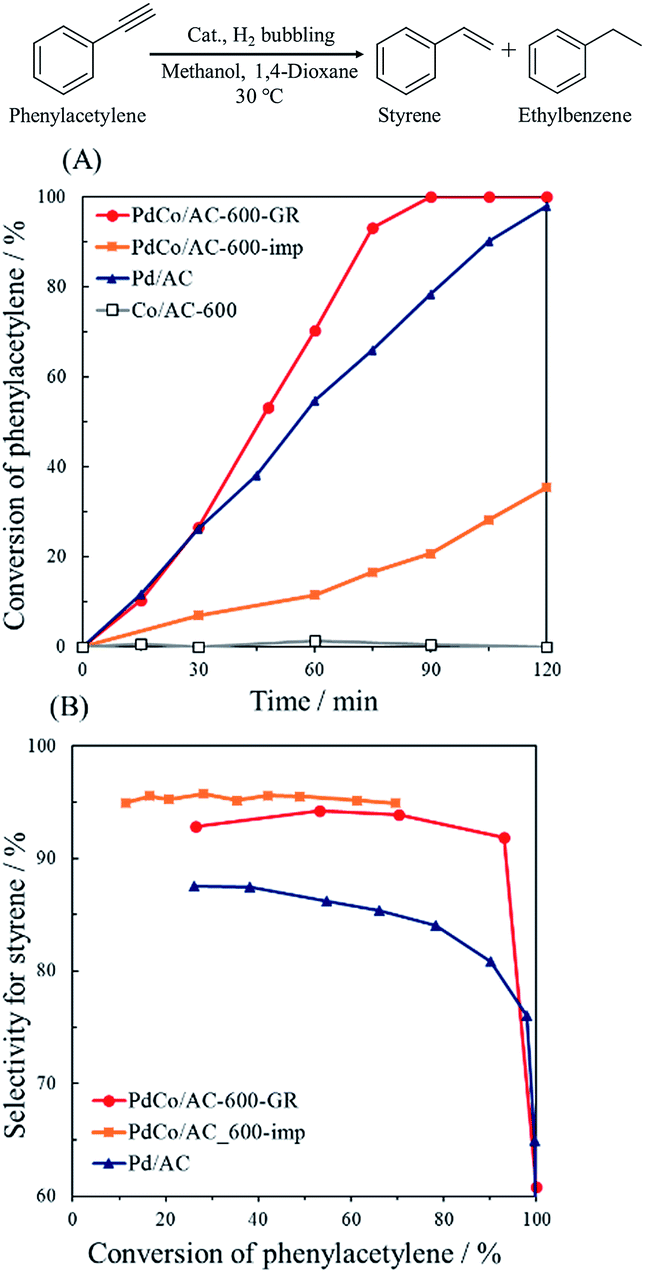 | ||
| Fig. 8 (A) Reaction profiles and (B) conversion–selectivity relationship in the hydrogenation of phenylacetylene over PdCo/AC-600-GR, PdCo/AC-600-imp, Pd/AC and Co/AC-600. | ||
Conclusions
We adopted the GR method for the synthesis of Pd–Co NP catalysts using Co NPs with various sizes and tested their catalytic performance for the selective hydrogenation of phenylacetylene. Carbon-supported Co species ranging from single-site to NPs were prepared by simply varying heat treatment temperature, and subsequently replaced with Pd2+ ions via the GR process. In contrast to previously reported results, the formed NPs were mainly composed of Pd species, with Co species present at low concentration. The size of the NPs did not change significantly during the GR process, as judged from the TEM images. The electronic properties of the Pd species were dependent on the size of the Pd–Co NPs, as determined from the XPS results. We found a correlation between the electronic state of the Pd species and catalytic activity/selectivity in the selective hydrogenation of phenylacetylene: PdCo/AC-600-GR, possessing the most electron-rich Pd species, showed the best catalytic performance, and provided 92% selectivity at 93% conversion. Moreover, the selectivity of PdCo/AC-600-GR was higher than that of pure Pd catalyst and its activity was higher than that of a Pd–Co alloy catalyst.Acknowledgements
The present work was supported by the Grants-in-Aid for Scientific Research (KAKENHI, No. 26220911 and 26620194) from the Japan Society for the Promotion of Science (JSPS).Notes and references
- N. Toshima and T. Yonezawa, New J. Chem., 1998, 22, 1179 RSC.
- L. Peng, E. Ringe, R. P. Van Duyne and L. D. Marks, Phys. Chem. Chem. Phys., 2015, 17, 27940 RSC.
- W. Xu, X. Liu, J. Ren, P. Zhang, Y. Wang, Y. Guo, Y. Guo and G. Lu, Catal. Commun., 2010, 11, 721 CrossRef CAS.
- W. Yu, M. D. Porosoff and J. G. Chen, Chem. Rev., 2012, 112, 5780 CrossRef CAS PubMed.
- K. Fuku, T. Sakano, T. Kamegawa, K. Mori and H. Yamashita, J. Mater. Chem., 2012, 22, 16243 RSC.
- K. Mori, K. Miyawaki and H. Yamashita, ACS Catal., 2016, 6, 3128 CrossRef CAS.
- F. Gao and D. W. Goodman, Chem. Soc. Rev., 2012, 41, 8009 RSC.
- B. Coq and F. Figueras, J. Mol. Catal. A: Chem., 2001, 173, 117 CrossRef CAS.
- C. Huang, H. Zhang, Y. Zhao, S. Chen and Z. Liu, J. Colloid Interface Sci., 2012, 386, 60 CrossRef CAS PubMed.
- X. Liu, D. Wang and Y. Li, Nano Today, 2012, 7, 448 CrossRef CAS.
- M. Oezaslan, M. Heggen and P. Strasser, J. Am. Chem. Soc., 2012, 134, 514 CrossRef CAS PubMed.
- Z. Wang, L. Yang, R. Zhang, L. Li, Z. Cheng and Z. Zhou, Catal. Today, 2016, 264, 37 CrossRef CAS.
- Y. Xu, L. Chen, X. Wang, W. Yao and Q. Zhang, Nanoscale, 2015, 7, 10559 RSC.
- A. Villa, D. Wang, D. S. Su and L. Prati, Catal. Sci. Technol., 2015, 5, 55 CAS.
- Z. M. Adriana, M. Martyna, D. Magdalena and G. Ewelina, Adv. Colloid Interface Sci., 2016, 229, 80 CrossRef PubMed.
- A. Dehghan Banadaki and A. Kajbafvala, J. Nanomater., 2014, 2014, 985948 Search PubMed.
- W. G. Menezes, L. Altmann, V. Zielasek, K. Thiel and M. Bäumer, J. Catal., 2013, 300, 125 CrossRef CAS.
- L. Zhang, Z. Xie and J. Gong, Chem. Soc. Rev., 2016, 45, 3916 RSC.
- S. M. Alia, Y. Yan and B. Pivovar, Catal. Sci. Technol., 2014, 4, 3589 CAS.
- X. Xia, Y. Wang, A. Ruditskiy and Y. Xia, Adv. Mater., 2013, 25, 6313 CrossRef CAS PubMed.
- L. Jiang, A. Hsu, D. Chu and R. Chen, Electrochim. Acta, 2010, 55, 4506 CrossRef CAS.
- C. L. Lee and C. M. Tseng, J. Phys. Chem. C, 2008, 112, 13342 CAS.
- C. L. Lee, C. M. Tseng, R. B. Wu, C. C. Wu and S. C. Syu, Electrochim. Acta, 2009, 54, 5544 CrossRef CAS.
- S. M. Alia, K. Duong, T. Liu, K. Jensen and Y. Yan, ChemSusChem, 2014, 7, 1739 CrossRef CAS PubMed.
- S. M. Alia, B. S. Pivovar and Y. Yan, J. Am. Chem. Soc., 2013, 135, 13473 CrossRef CAS PubMed.
- M. Shao, K. Sasaki, N. S. Marinkovic, L. Zhang and R. R. Adzic, Electrochem. Commun., 2007, 9, 2848 CrossRef CAS.
- S. M. Alia, K. O. Jensen, B. S. Pivovar and Y. Yan, ACS Catal., 2012, 2, 858 CrossRef CAS.
- H. Li, H. Lin, Y. Hu, H. Li, P. Li and X. Zhou, J. Mater. Chem., 2011, 21, 18447 RSC.
- M. B. Boucher, B. Zugic, G. Cladaras, J. Kammert, M. D. Marcinkowski, T. J. Lawton, E. C. H. Sykes and M. Flytzani-Stephanopoulos, Phys. Chem. Chem. Phys., 2013, 15, 12187 RSC.
- S. Okada, T. Kamegawa, K. Mori and H. Yamashita, Catal. Today, 2012, 185, 109 CrossRef CAS.
- Y. Zhang, W. Diao, J. R. Monnier and C. T. Williams, Catal. Sci. Technol., 2015, 5, 4123 CAS.
- K. Shen, L. Chen, J. Long, W. Zhong and Y. Li, ACS Catal., 2015, 5, 5264 CrossRef CAS.
- J. Ma, L. Xu, L. Xu, H. Wang, S. Xu, H. Li, S. Xie and H. Li, ACS Catal., 2013, 3, 985 CrossRef CAS.
- Y. Mahara, J. Ohyama, T. Tojo, K. Murata, H. Ishikawa and A. Satsuma, Catal. Sci. Technol., 2016, 6, 4773 CAS.
- H. Zhang, M. Jin, H. Liu, J. Wang, M. J. Kim, D. Yang, Z. Xie, J. Liu and Y. Xia, ACS Nano, 2011, 5, 8212 CrossRef CAS PubMed.
- T. Fukuda and T. Katsuki, Tetrahedron, 1997, 53, 7201 CrossRef CAS.
- K. Nakatsuka, T. Yoshii, Y. Kuwahara, K. Mori and H. Yamashita, Phys. Chem. Chem. Phys., 2017, 19, 4967 RSC.
- S. Navaladian, B. Viswanathan, T. K. Varadarajan and R. P. Viswanath, Nanoscale Res. Lett., 2009, 4, 181 CrossRef CAS PubMed.
- L. Zhang, K. Lee and J. Zhang, Electrochim. Acta, 2007, 52, 3088 CrossRef CAS.
- R. N. Singh and C. S. Sharma, Eng. Tech. Appl. Sci. Res., 2012, 2, 295 Search PubMed.
- G. X. Pei, X. Y. Liu, A. Wang, A. F. Lee, M. A. Isaacs, L. Li, X. Pan, X. Yang, X. Wang, Z. Tai, K. Wilson and T. Zhang, ACS Catal., 2015, 5, 3717 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |