
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The binding of free and copper-complexed fluoroquinolones to OmpF porins: an experimental and molecular docking study†
Sousa C. F. a,
Coimbra J. T. S.a,
Gomes I.bc,
Franco R.b,
Fernandes P. A.*a and
Gameiro P.*a
a,
Coimbra J. T. S.a,
Gomes I.bc,
Franco R.b,
Fernandes P. A.*a and
Gameiro P.*a
aRequimte, UCIBIO, Departamento de Química e Bioquímica, Faculdade de Ciências, Universidade do Porto, Porto, Portugal. E-mail: agsantos@fc.up.pt; pafernan@fc.up.pt
bRequimte, UCIBIO, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova Lisboa, Caparica, Portugal
cInstituto de Medicina Molecular, Faculdade de Medicina da Universidade de Lisboa, Lisboa, Portugal
First published on 3rd February 2017
Abstract
Bacterial resistance is a critical public health issue and the development of alternative antibiotics to counteract this problem is an urgent matter. Fluoroquinolones are widely used antibiotics and numerous cases of bacterial resistance to these drugs have already been reported. One important mechanism of resistance is the decrease of the permeability of the bacterial membrane to antibiotics by mutation of the OmpF porin. Fluoroquinolone complexation with copper and a nitrogen-donor heterocyclic ligand is an approach developed to counteract this mechanism of bacterial resistance. The binding of antibiotics to the constriction zone residues of porins has been shown to be essential for their influx across the channel. In the present work, the interaction of sparfloxacin, ciprofloxacin and their respective copper ternary complexes with the outer-membrane protein F (OmpF) porin was studied to further explore the channel selectivity for each of these molecules. Surface plasmon resonance was used to determine the association constants between the drugs and the OmpF protein and molecular docking was used to further analyze the interaction between them. The results support a higher affinity of free fluoroquinolones to the constriction zone of the OmpF channel, when compared to the respective copper-complexes. The findings of this work provide insights that will be helpful to proceed with the study of metal-complexes as alternatives to free fluoroquinolones in resistant infections, avoiding the high-cost and time-consuming processes of discovering and designing new antibiotics.
Introduction
Antimicrobial resistance is currently a global public health threat and the misuse of antibiotics in the last decades is one of the key factors that led to this problem. Among the antibiotics currently in use, fluoroquinolones (FQs) are a family with over twenty members in the market, that are widely used against Gram-negative and some Gram-positive bacteria. Bacterial resistance to FQs is an emergent and serious public health problem, especially regarding Gram-negative bacteria. FQs derive from quinolones and are characterized by having a fluorine at position 6 of the quinolone ring, resulting in improved antibacterial activity and pharmacokinetic properties.1,2 These antibiotics act in the bacterial cell by binding and inhibiting topoisomerase enzymes that are fundamental for DNA replication. The internalization of FQs in the bacterial cell is, therefore, a key step to their anti-bacterial activity.3 The bacterial outer-membrane is known as the major permeability barrier of Gram-negative bacteria. Hydrophobic FQs can partially diffuse through the lipid bilayer but most of the FQs are hydrophilic and thus their ability to cross membranes by diffusion is compromised. Porins, especially the outer-membrane protein F (OmpF), become then the main pathway for the influx of these FQs.4–6The OmpF porin is a homo-trimer, composed by three hydrophilic channels that transport small cationic molecules (≤600 Da). The channel size is mainly determined by loop 3, which is particularly long and folds into the barrel, forming the constricted area of the channel. The constriction area is formed by D113 and E117 from loop 3 and K16 and R132, R82 and R42 from the opposing barrel wall. As these aminoacids have opposite charges in opposite sides of the barrel, the constriction zone is characterized by a strong transversal electric field.4,7,8 OmpF is a general diffusion porin and thus, in its normal function, there is no affinity-enhanced or substrate-specific uptake. However, some antibiotics can bind to specific residues, thus favouring their influx through the channel.9–12 The constriction zone residues, due to their electrostatic properties, constitute a very favourable binding motif for zwitterionic antibiotics. This specific binding is known to be very relevant to facilitate drug translocation through the channel, compensating for the entropic and desolvation costs of crossing this narrow region.9–12 Furthermore, a recent study has highlighted that distinct pore characteristics over different OmpF proteins, have an impact on the physiological functions and type of molecules that they transport.13
Porin mutation is an important mechanism of bacterial resistance to FQs and their complexation with divalent metals is an approach that has been developed to counteract this type of bacterial resistance.14–17 Ternary complexes (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) with one copper(II) ion, one FQ and one nitrogen-donor heterocyclic ligand, such as 1,10-phenathroline (Phen), are stable under biological conditions and have shown an anti-bacterial potential comparable to the free drug.17 They also appear to have a porin-independent route of entry in the bacterial cell that should be promoted by favourable drug–lipid interactions.17–21 Due to their high nuclease activity, ternary complexes of drugs with copper and phen have also been studied as anticancer agents. In this context, their cytotoxicity in healthy and in cancer cell lines was evaluated and compared. Overall, both the copper-complexes, and copper(II) ions and phen exhibit low cytotoxicity for human healthy cells.22–24
:1:1) with one copper(II) ion, one FQ and one nitrogen-donor heterocyclic ligand, such as 1,10-phenathroline (Phen), are stable under biological conditions and have shown an anti-bacterial potential comparable to the free drug.17 They also appear to have a porin-independent route of entry in the bacterial cell that should be promoted by favourable drug–lipid interactions.17–21 Due to their high nuclease activity, ternary complexes of drugs with copper and phen have also been studied as anticancer agents. In this context, their cytotoxicity in healthy and in cancer cell lines was evaluated and compared. Overall, both the copper-complexes, and copper(II) ions and phen exhibit low cytotoxicity for human healthy cells.22–24
Recent studies evaluating the interaction of two different FQs, sparfloxacin (Spx) and ciprofloxacin (Cpx), and their respective metalloantibiotics (CuFQPhen) with Gram-negative membrane models, showed that, while Cpx has a small partition coefficient and its influx through the bacterial outer membrane should be OmpF-dependent; Spx shows a more hydrophobic behaviour, being likely capable of passing this membrane by passive diffusion.25,26 Previous microbiological studies also showed that while Cpx antibacterial activity is decreased in an Escherichia coli (E. coli) strain with deleted OmpF, Spx's activity remains similar.17 It is thus expected that Spx and Cpx have different affinities for OmpF and abilities to cross the OmpF protein channel. On the other hand, both metalloantibiotics showed large partition coefficients with Gram-negative membrane models but, microbiological studies showed that CuCpxPhen internalization in the bacterial cell can be partially OmpF-dependent, since its antibacterial activity is marginally affected by porin deletion.17,25,26
Following these results, we aim to study the interaction of Spx, Cpx and their respective copper ternary complexes with OmpF, to further explore the channel's selectivity for each of these molecules. For this purpose, the interactions of the drugs with the OmpF porin are studied using a combination of experimental and theoretical techniques. Experimental studies were accomplished using Surface Plasmon Resonance (SPR), a technique that has been greatly employed in the last years to study the binding between two molecules.27,28 SPR detects a binding interaction between a molecule immobilized on a sensor chip and another in a flowed solution by using an optical method that measures the changes in the refractive index at the sensor surface upon binding. A great advantage of this technique is the possibility of assessing the binding between ligands and membrane proteins in a lipid environment that mimics the bacterial membrane.29,30 However, despite being a highly sensible technique to calculate binding constants between two molecules, SPR is not able to provide information about the location nor the residues that are involved in the binding. Therefore, molecular docking was performed to get atomic detail of the binding of FQs to OmpF's pore residues.31 Molecular docking is able to provide structural information about the binding like the location of the ligand within the target protein and the residues that are involved in the binding. It also analyses the affinity of the resulting complex. This technique is particularly important in this study, as the transport of the antibiotics across the OmpF channel protein is strongly dependent on its ability to interact with the residues at the constriction zone.9–12
The findings of this work are important to proceed with the study of CuFQPhen complexes as an alternative to free FQs in resistant bacteria with mutated OmpF porins.
Experimental
E. coli total lipid extract (PE 57.5%; PG 15.1%; cardiolipin 9.8%; unknown lipids 17.6%) (Cas number 1240502-50-4) was obtained from AVANTI Polar Lipids. Sparfloxacin (Spx) and ciprofloxacin (Cpx) (>98.0%) were purchased from SIGMA-ALDRICH. Ternary FQ complexes with Cu(II) and phen were synthesized as described before. Briefly, stock solutions of the FQ, Cu(NO3)2 and phen were prepared and mixed in the proportion 1:1:1 to achieve complex formation in solution.17 The stability constants were determined and complexes characterization by UV-visible spectroscopy, Infra-Red spectroscopy and X-ray diffraction was already published by Feio et al.17 All experiments described were carried out at 37 °C and at pH 7.4 using HEPES buffer solution (10 mmol dm−3 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid hemisodium salt (purity ≥ 99.0%), 0.1 mol dm−3 NaCl) provided by SIGMA-ALDRICH. All other chemicals were from MERCK or SIGMA-ALDRICH (grade, p.a.).
OmpF purification
OmpF was purified from E. coli, strain BL21 (DE3) Omp8, following published procedures with some changes performed to improve the purification yield and described below.32An isolated colony was added to Luria Bertani Broth (LB) medium with ampicillin 40 μg cm−3 and allowed to incubate for 16 h in an orbital incubator at 37 °C, 180 rpm. This culture was inoculated into a new LB medium and incubated (37 °C, 180 rpm) until it reaches and optical density in the range 0.75–1.1 at 600 nm. Induction was performed with 0.4 mmol dm−3 isopropyl β-D-1-thiogalactopyranoside (IPTG), followed by further 6 h of incubation, and centrifugation at 8000× g, 4 °C, and for 20 min. The cell-containing pellet was suspended in 20 mmol dm−3 Tris buffer, pH 8 with 2% SDS, a protease inhibitor, phenylmethylsulfonyl fluoride (PMSF) 50 g dm−3 and DNase 50 μg cm−3. The suspended E. coli cell pellet was lysed in a French-press cell (THERMO, Germany) with a 2000 psi gauge pressure, followed by incubation for 90 min at 50 °C in an orbital incubator (150 rpm). The lysed cells solution was centrifuged at 11000× g, 25 °C for 45 min. The pellet was suspended in 20 mmol dm−3 sodium phosphate buffer pH 7, containing 0.125% n-octylpolyoxyethylene (o-POE) and PMSF 50 g dm−3. After a further passage through the French press for additional cell lysis, followed by a 45 min incubation (37 °C, 150 rpm), the solution was ultracentrifuged for 40 min at 219331× g, 4 °C. The ultracentrifugation pellet was suspended in 20 mmol dm−3 sodium phosphate buffer pH 7, 3% o-POE and PMSF 50 g dm−3, and the supernatant was collected. This supernatant contains the pure protein and was concentrated by ultrafiltration using a 30 kDa-pore membrane (MILIPORE), and the buffer was exchanged to 20 mmol dm−3 sodium phosphate buffer pH 7, with 1% o-POE.
Protein concentration was determined by the bicinchoninic acid assay (BCA) assay (SIGMA-ALDRICH), and protein purity was assessed by SDS-PAGE (10%).
Liposome preparation
E. coli total lipid extract liposomes were prepared by evaporation to dryness of the lipid solution in chloroform, using a stream of argon. The films were left under vacuum for 3 h to remove all traces of the organic solvent. The resulting dried lipid films were dispersed with HEPES buffer and the mixture was vortexed above the phase transition temperature of the lipid (70 °C) to produce multilamellar large vesicles (MLVs). The MLVs were processed by freezing in liquid nitrogen and thawing the sample in a boiling water bath. This process was repeated five times. Lipid suspensions were then equilibrated above the phase transition temperature of the lipid for 30 min and extruded ten times through 100 nm polycarbonate filters to produce large unilamellar vesicles (LUVs). Extrusion of the liposomes was performed using a LIPEX Biomembranes extruder attached to a CLIFTON thermostatic circulating water bath. The size distributions of the LUVs were determined by dynamic light scattering analysis using a MALVERN Instruments Zeta SizerNano ZS and the mean particle size was 130 ± 0.3 nm.OmpF reconstitution in liposomes
OmpF was reconstituted in pre-formed E. coli total lipid extract LUVs, with detergent (o-POE) concentration below the critical micelle concentration of formation of lipid–detergent mixed micelles.33 The insertion of OmpF protein in the liposomes was performed by direct incorporation to a final molar ratio OmpF:lipid of 1:1000, using a well-established methodology.34,35 OmpF protein (1% o-POE) was mixed with the LUVs suspension in HEPES buffer to obtain a final concentration of 2 mmol dm−3 liposomes and 2 μmol dm−3 OmpF. After homogenization, the mixture was incubated at room temperature for 20 min, with gentle stirring, followed by a 1 h incubation into an ice bath with a temperature shock with boiling water after 30 min of incubation. Firstly, the detergent was adsorbed with SM2-Bio-Beads at a concentration of 75 mg cm−3, by gentle shaking the suspension during a period of 3 h at room temperature. After this time, the supernatant was placed in a new vial with fresh Bio-Beads and incubated overnight, in ice, with gentle stirring. At the end of this period, proteoliposomes were gently removed by decanting the Bio-Beads. Five cycles of freezing with liquid nitrogen and thawing in a boiling water bath were then performed. Extrusion and final size distribution measurements were performed as described before for the liposomes.
Surface plasmon resonance (SPR) measurements
SPR experiments were carried out using a Biacore X100 analytical system with a L1 sensor chip (GE Healthcare, United Kingdom). Prior to use, the L1 chip surface was washed with a conditioning cycle consisting on the injection of 3-[(3-cholamidopropyl)dimethylammonium]-1-propanesulfonate (CHAPS), 20 mmol dm−3 followed by running buffer (HEPES). E. coli total lipid extract liposomes vesicles, 2 mmol dm−3 were immobilized in channel 1, at a flow rate of 2 μl min−1 for 45 min. Stabilization of the signal was achieved by four successive injections of running buffer (50 μl min−1 for 100 s), interposed by an NaOH injection (1 mmol dm−3) (50 μl min−1 for 60 s) and finished with two injections of running buffer at 10 μl min−1 for 500 s. Subsequently, proteoliposomes of OmpF/E. coli total lipid extract (2 μmol dm−3/2 mmol dm−3) were immobilized on channel 2, following the same approach described above. After immobilization of liposomes and proteoliposomes, the drug injection cycles were initiated. Each drug injection cycle comprised a first injection of buffer (5 μl min−1 for 500 s), followed by drug solution injection at a flow rate of 30 μl min−1, association during 180 s and dissociation during 10 min and finalized by buffer injection (10 μl min−1 for 230 s) to ensure the complete removal of the drug. To begin with, three drug injection cycles using buffer as drug solution were performed as start-ups. Then, a range of six different drug concentrations were tested, from 0 to 1 mmol dm−3, comprising a repetition with a drug solution of intermediate concentration. In the end, liposomes and proteoliposomes were removed by a double injection of CHAPS and a double injection of running buffer, both at a flow rate of 10 μl min−1 for 150 s. Equilibrium steady-state and kinetic dissociation constants were calculated using 1:1 binding equations.
Molecular docking
Molecular docking studies were conducted on the OmpF protein channel with two FQs and their respective copper ternary complexes: (i) Spx and CuSpxPhen; and (ii) Cpx and CuCpxPhen. The two FQs were studied using the forms in which they are likely to be present at physiological pH (pH 7.4). For Spx, the neutral (FQ), zwitterionic (FQ±) and negative (FQ−) forms were tested, since the pKa of the amide group is pKa = 7.43 and the ratio [FQ±]/[FQ] is 20.0. Cpx, was only tested in the zwitterionic and neutral forms, since its pKa is significantly higher than the physiological pH (pKa = 8.95). The neutral form was also tested since a small percentage could still be present in solution: despite the [FQ±]/[FQ] ratio being significantly higher than the ratio for Spx (≈631).17,36 For the copper complexes, the [Cu(FQ±)Phen]2+ form was studied, considering that for both molecules this is the predominant form at physiological pH. Molecular structures of Cpx, CuCpxPhen, Spx and CuSpxPhen and the different protonation states used are represented in Fig. 1.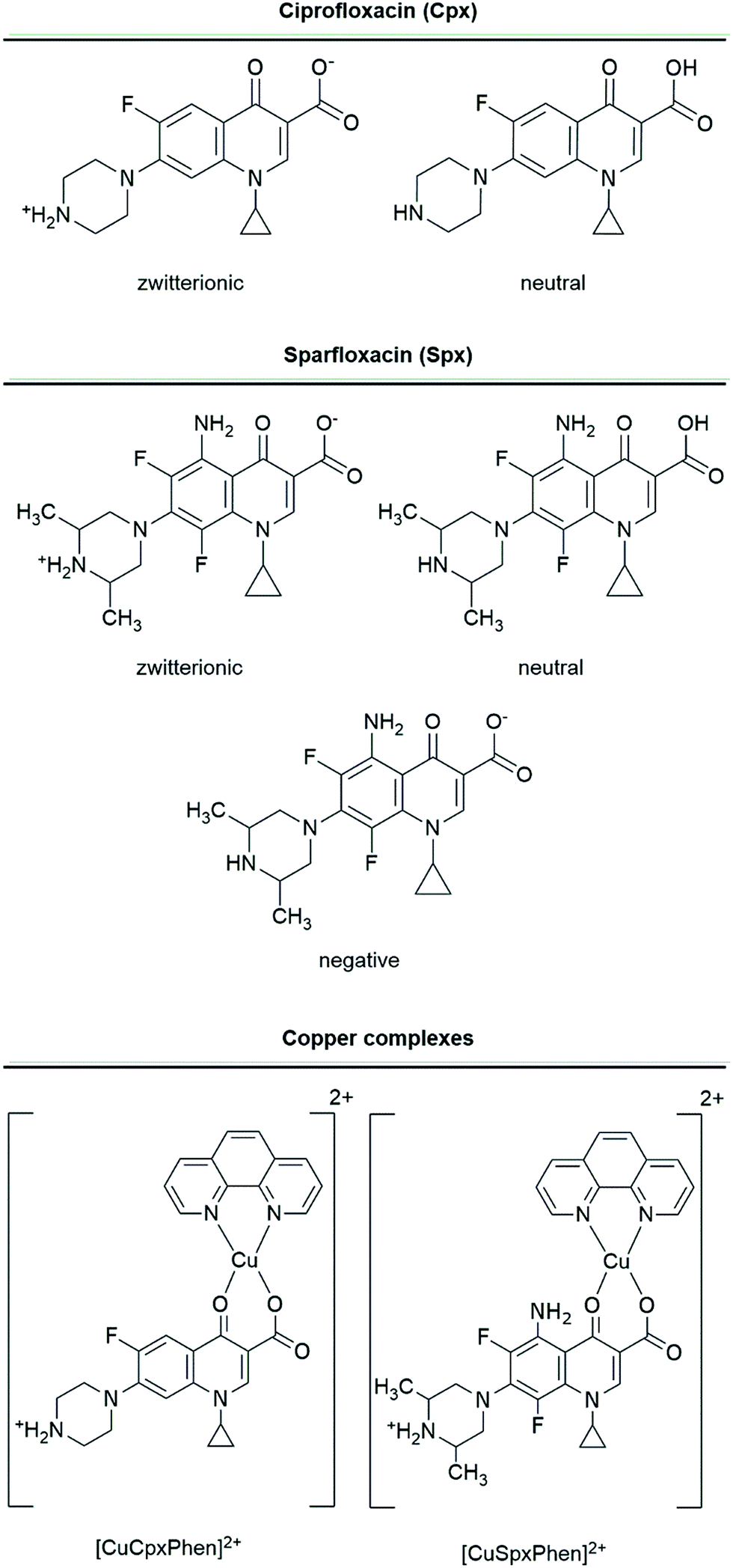 | ||
| Fig. 1 Molecular representation of the studied molecules and the different ionization states considered. | ||
For the comparison of docking results between the different forms of the free FQs, a distribution of charges calculated by the Open Babel software was used.37 However, for the copper complexes a different charge distribution was needed to account for the copper(II) atom charge. Hence, a Mulliken charge distribution of the copper complexes and the zwitterionic forms of Spx and Cpx was also calculated using the Gaussian 09 software.38 The structures were prior optimized using a B3LYP/6-31G(d) default optimization protocol and the Mulliken charges were determined at this level of theory. This protocol considers the charge distribution between copper and its ligands while the protocols often employed in docking evaluations use a unitary metal charge.
Two different OmpF protein structures were used: 2OMF (DOI: 10.2210/pdb2omf/pdb) and 4KRA (DOI: 10.2210/pdb4kra/pdb). The 4KRA structure was used in this study because it has the advantage of having been co-crystalized with Cpx. However, this protein structure has some disadvantages: (i) it has been purified from Salmonella enterica and we intended to study the protein from E. coli; (ii) it presents a low resolution (3.32 Å) and (iii) there is no evidence of being crystallized in a lipid-mimetic matrix. Furthermore, a recent study comparing the pore residues of an OmpF porin from E. coli with one from Salmonella typhi suggested that their interaction with antibiotics should be different and unique for each species.39 Given that, we decided to also test and compare an alternative OmpF X-ray structure, 2OMF, purified from E. coli. This protein structure is not co-crystalized with Cpx but it has a higher resolution (2.40 Å), and it was crystalized in an (hidroxyethyloxy)tri(ethyloxy)octane matrix.
The 2OMF and 4KRA protein sequences were compared using the BLAST program and they present a sequence identity of 57%, which means that the 57% of their amino acid sequences have the same residues at exactly the same position. Sequence similarity, also calculated using BLAST, showed that 72% of the proteins' sequence is similar and related.40,41 The root-mean-square deviation (RMSD) value for the aligned structures was 1.91 Å (considering all the residues of both protein structures), and 0.73 Å with outlier rejection.
Docking-defined procedure
Docking was performed using the AutoDock 4.0 software. AutoDock 4.0 combines a semi-empirical free energy force field with a Lamarckian genetic algorithm, predicting bound conformations and free energies of association.31 The Lamarckian genetic algorithm was used with 50 runs and a population size of 50. A maximum of 1.5 million energy evaluations was applied. Free energy of binding was predicted using the AutoDock 4.0 semi-empirical free energy force field that is based on a thermodynamic model that evaluates enthalpic and entropic contributions accounting with the intramolecular energy and incorporating a full desolvation model.42 Default Autodock 4.0 docking parameters were employed. The results were clustered using a tolerance of 1.5 Å. For the docking considering the constriction zone, the docking-site was defined using the localization of Cpx in the 4KRA crystal structure and the docking grid-box size was set to (32, 16, 32 Å). For the full-channel docking, a docking grid-box was created to include the entire protein channel and its size was set to (34, 44, 34 Å). Flexible docking was performed allowing the torsion of all the rotatable bonds of the residues on the constriction zone.31 Flexibilized residues were E117, D113, R132, R82, R42 and K16 for 2OMF and E116, D108, R130, R77, R60 and K16 for 4KRA.Results and discussion
Association constants by surface plasmon resonance (SPR)
The association constants of Cpx and Spx, both in free or copper-complexed forms, with proteoliposomes of E. coli total lipid extract with reconstituted OmpF protein were determined using SPR. The proteoliposomes were immobilized on the active channel surface of the SPR sensor ship (channel 2), whereas liposomes without protein were immobilized in the reference channel (channel 1) in order to subtract for the non-specific binding of the drugs to the lipids. Fig. 2 shows an example of a SPR sensorgram presenting the binding of Cpx and CuCpxPhen complex to protein OmpF through time (results reflect the Response Units (R.U.) of the difference between channel 2 and channel 1). From t = 0 s until t = 180 s an association period is taking place, while the drug is flowing in the system; from t = 180 s until t = 780 s a dissociation period is occurring, with buffer running in the system to induce the dissociation of the drug from the protein.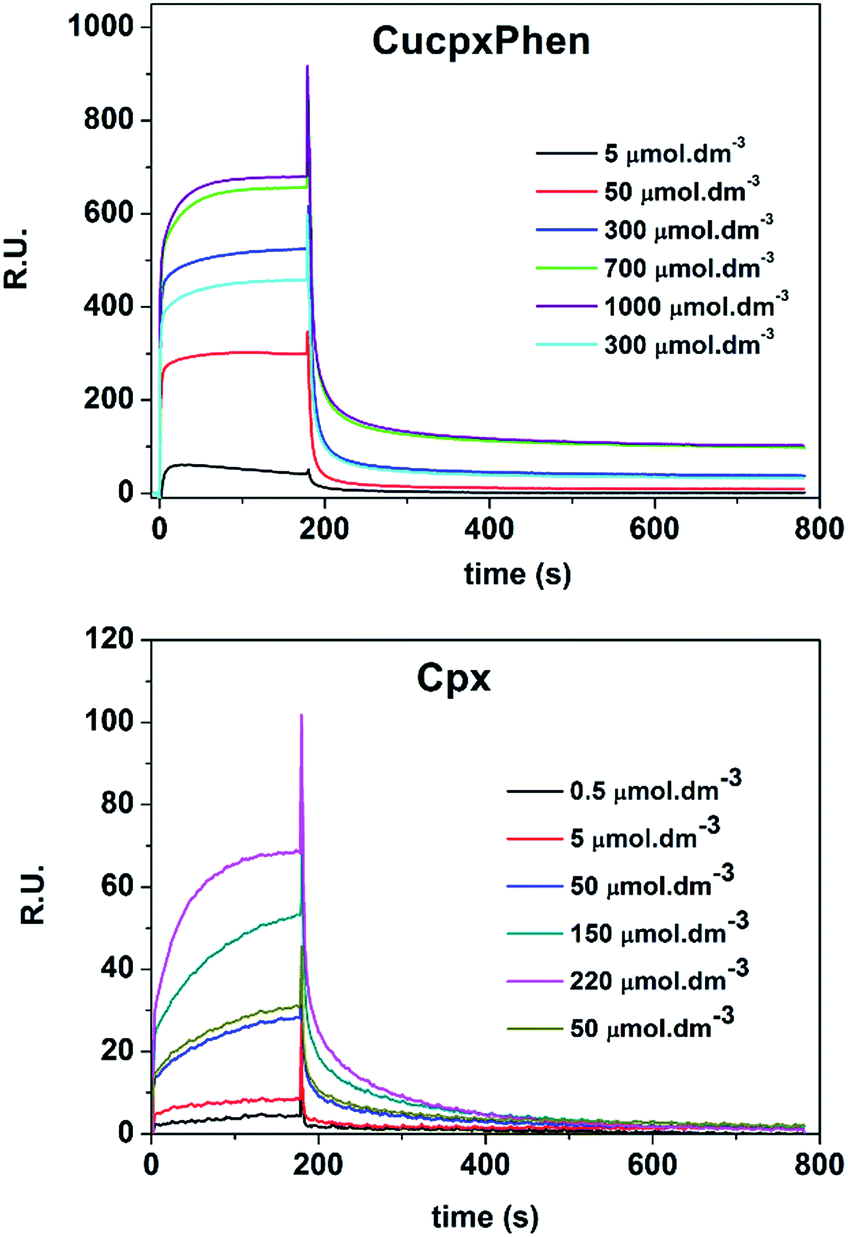 | ||
| Fig. 2 SPR sensorgram showing the association and dissociation of CuCpxPhen and Cpx with the OmpF protein at different concentrations. Response Units (R.U.) reflect the value of subtracting channel 1 results from channel 2. Association period occurs from t = 0 s until t = 180 s and dissociation from t = 180 s until t = 780 s. | ||
The equilibrium steady-state dissociation constants were calculated using the value of R.U. at the equilibrium (t = 175 s) for each concentration used. Fig. 3 shows the plots for the determination of the dissociation constant (Kd) for each compound used, calculated by fitting the one-site binding equation to the results:
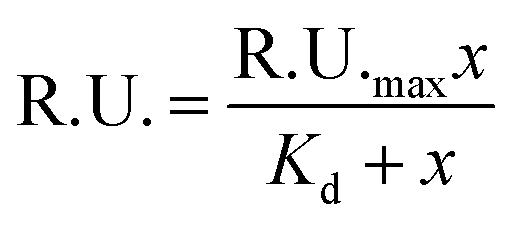
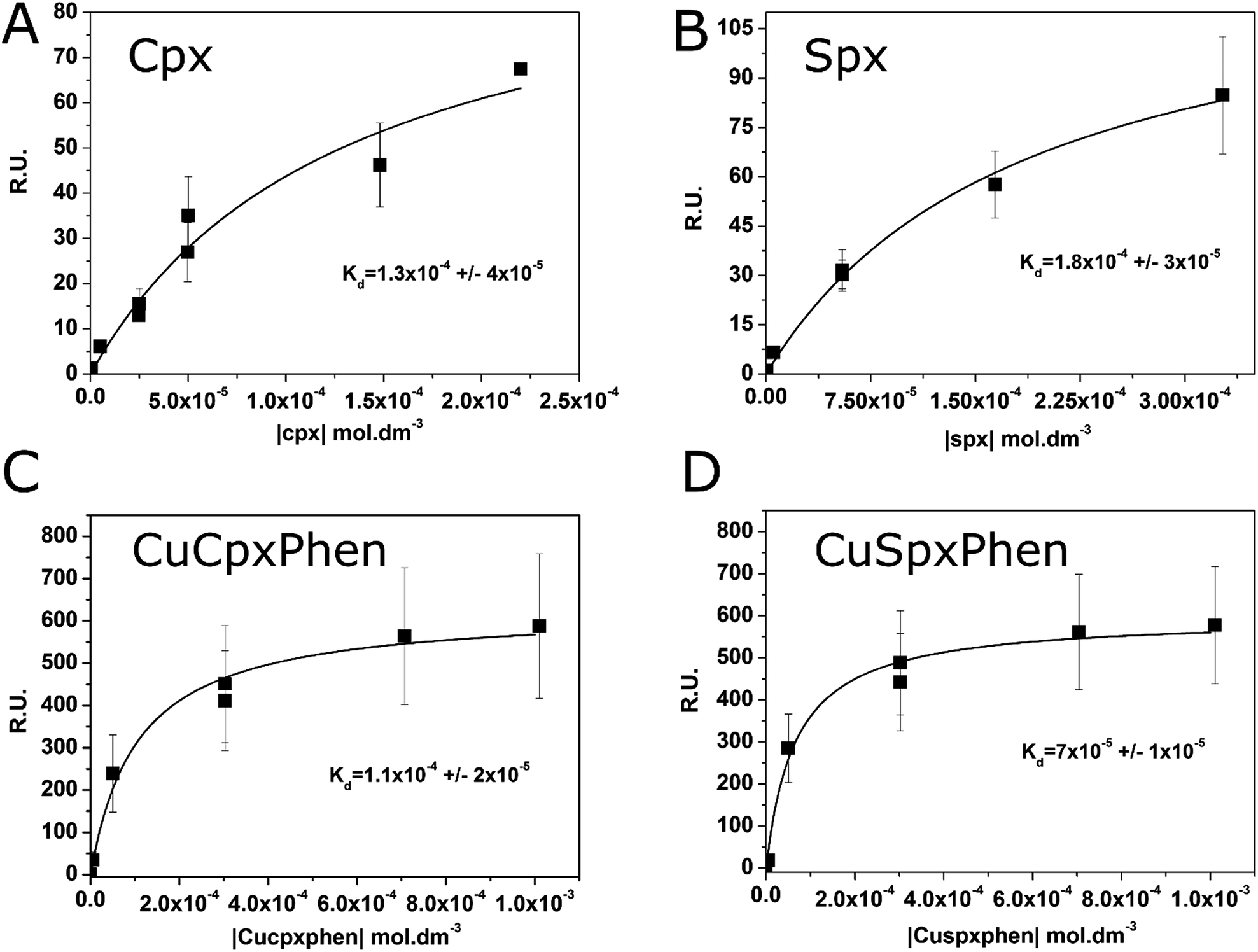 | ||
| Fig. 3 Plots of the Response Units (R.U.) at the equilibrium (t = 175 s) versus the concentration of Cpx (A), Spx (B), CuCpxPhen (C) and CuSpxPhen (D). Kd was calculated with the non-linear fit of the one-site direct binding equation. Results are a mean of at least three individual experiments. | ||
For Cpx and Spx the kinetic dissociation constant was also calculated, along with the on-rate association (kon) and dissociation (koff) constants:
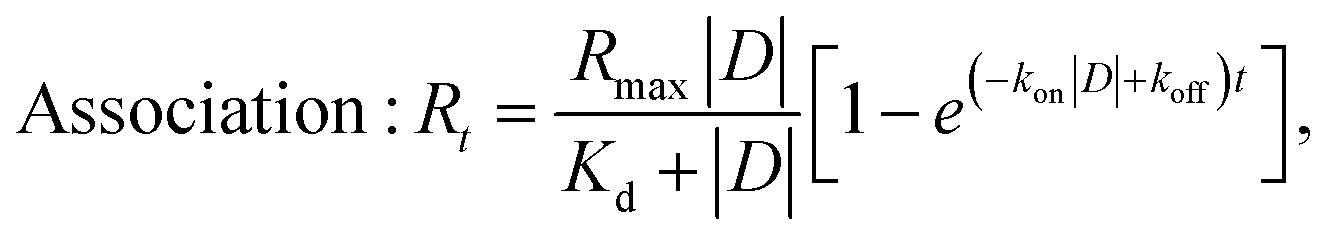
| Dissociation: Rt = R0e−kofft, |
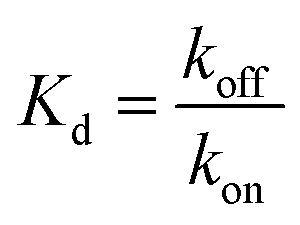
It is also important to mention that, for the copper complexes, the determination of the kinetic parameters was not possible, since the binding of these complexes is too fast to solve the binding kinetics. By comparing the sensorgrams presented in Fig. 2 it is possible to verify that the binding curves of Cpx increase gradually with time until they reach the equilibrium, while the binding curves for CuCpxPhen increase dramatically in the first seconds of the association period, reaching the equilibrium after only a few seconds.
The association constants (Kass) were calculated using the inverse of Kd and the values are listed on Table 1. Results show similar binding affinities for Cpx and CuCpxPhen and slightly smaller steady-state binding affinities for Spx when compared to CuSpxPhen.
| Compound | Steady-state | Kinetics | ||
|---|---|---|---|---|
| log(Kass)/mol dm−3 | kon/mol−1 dm3 s−1 | koff/s−1 | log(Kass)/mol dm−3 | |
| Cpx | 3.90 ± 0.10 | 238.4 | 0.026 | 3.97 |
| CuCpxPhen | 3.96 ± 0.08 | — | — | — |
| Spx | 3.74 ± 0.07 | 319.7 | 0.064 | 3.70 |
| CuSpxPhen | 4.15 ± 0.06 | — | — | — |
This equilibrium steady-state binding affinity however, does not necessarily express transition across the protein channel. It is not possible to decipher, by SPR, if the determined binding affinity represents an association to constriction zone residues or just binding to other regions of the OmpF porin. Therefore, these results can be just showing a high and fast binding of the copper complexes to random residues of the OmpF porin.26 In this context, since it is known that the binding of antibiotics to constriction zone residues is important to antibiotic flux through the OmpF porin, the high binding constant calculated for the copper complexes by SPR should not express a higher affinity to cross the channel.10 This is also supported by the fast association and dissociation of the copper complexes when compared to the free-drugs. This fast binding does not allow the calculation of the kinetic parameters, possibly because it is mainly controlled by electrostatic interactions outside the constriction zone of the OmpF channel.
For Spx and Cpx, similar Kass values were obtained from equilibrium and kinetic analysis. Spx has an on-rate and off-rate higher than Cpx, by 34% and 46% respectively. The reason behind the faster binding/unbinding kinetics is unclear, given the similarity between the two molecules. This faster kinetics is consistent with the lower affinity of Spx to the channel, as a tighter binding is usually associated with a slower unbinding. This difference may then result in an easier traffic of Cpx on both the entry and exit chambers of the channel. Further studies would be needed to clarify this aspect.
Molecular docking
By using molecular docking, we intend to corroborate and get further atomic-level structural information over the OmpF channel selectivity between the different FQs, contemplating both free and copper-complexed forms. For this purpose, we employ docking calculations, which are very adequate to get a picture about the affinity of the drugs for different, specific regions of the channel.Free-energy sampling methods, such as umbrella sampling molecular dynamics simulations, are sometimes used in systems as the one explored here, to calculate free energy profiles for drug translocation, and obtain kinetic information about drug permeation. Here, however, the purpose is to obtain structural information about the binding and the selectivity of the channel for different drugs, and not to study the permeation of the drugs across the channel.43–45 As it is clear, and preciously demonstrated, that the FQ transport across the OmpF porin is strongly dependent on its interaction and binding to the constriction zone residues, molecular docking results will give us relevant insights on OmpF selectivity for each molecule.9–12
One of our first assumptions, was to consider a static protein channel, assuming that the protein residues are already in the most energetically favourable conformation. This static docking protocol was defined as protocol 1. The assumption that we would get a meaningful description of the interaction energies and poses over a single conformation of the constriction zone of the pore is a reasonable simplification of the entire process and should be more relevant for the 4KRA protein structure (since it was co-crystalized with Cpx), although it should be less true when considering the 2OMF protein structure. Hence, a second protocol was defined, in which relevant residues were flexibilized during the docking routines. This protocol, defined as protocol 2, would grant us in principle a necessary level of flexibility that could have an impact in the binding energetics of FQs. Subsequently, we will describe the results for each protocol (1 and 2) in detail.
Protocol 1 (rigid docking) – fluoroquinolones
Firstly, to confirm if both Spx and Cpx would dock at the constriction zone of the channel we started by using a docking box that included the entire OmpF channel (for both 4KRA and 2OMF models). The results (see ESI, Table S1†) showed that the zwitterionic Cpx is the only molecule that has the most negative binding free energy docking solution near the constriction zone of 2OMF model protein; while both FQs (and the respective zwitterionic, charged and neutral forms) dock near the constriction zone for the 4KRA model protein. These first results support the higher ability of Cpx to interact with the constriction zone residues of OmpF. Moreover, these results suggest a higher adaptation of the constriction zone of the 4KRA structure to bind the FQs.The size of the channel for both OmpF structures, calculated using BetaCavityWeb software, shows that the 4KRA protein has a narrower constriction zone (3.28 Å for 4KRA and 3.72 Å for 2OMF).46 Moreover, supplementary comparison of the constriction zone conformation of both OmpF structures shows that, even though almost all residues of the constriction zone preserve their position in both protein structures, E116 completely changes its position (see ESI, Fig. S1†).
It is also important to remember that a recent study, focused on the comparison of the full X-ray structure of both proteins, pointed out differences in the binding of antibiotics to OmpF from E. coli when compared to S. typhi, although the relevance of these differences for the binding process was not clarified.39 Our results reflect the differences between the OmpF channels from the two species, indicating that the preferred binding-site for molecules that are able to efficiently cross the porin channel (i.e. Cpx zwitterionic) is located near the constriction zone for both OmpF models. We thus advanced by testing all the molecules using a docking box adjusted to this region (defined with the help of the OmpF/Cpx co-crystallized structure – 4KRA).
First, to validate the docking protocol, we compared the most negative binding free energy result obtained for Cpx zwitterionic with the 4KRA model, to the Cpx crystallographic position. The result (see ESI, Fig. S2†) shows that the zwitterionic Cpx docks in a similar position than the crystallographic Cpx, but with a significant change on its rotational orientation (RMSD = 4.95 Å). A further analysis of the distances between both molecules and the 4KRA residues shows that the NH3+ group of the docking solution Cpx can interact by hydrogen-bonds with the carbonyl groups of D30 and Q34, while the same group in crystallographic Cpx interacts mainly with D108. Moreover, the carboxylic acid group of the Cpx docking result seems to interact slightly differently with the arginine's of the constriction zone (R60, R77 and R130), when compared with the crystallographic Cpx. Furthermore, the first docking solution that has a pose similar to the crystallographic Cpx has an energy gap of about 1.0 kcal mol−1 when compared to Cpx's most negative binding free energy result. Nevertheless, since the transition of a molecule through a protein channel is, as known, a process that involves successive changes in interactions between the molecules and the residues of the inner part of the protein channel, it is expectable that the ligand–channel interactions are less specific than in the more common cases of more static ligand–receptor binding. Considering thus that the docking protocol used is valid we advanced by testing all the molecules using a docking box adjusted to the constriction region and the ΔΔGbinding values obtained are summarized in Table 2.
The results show that the binding energies for the zwitterionic forms are more negative than the binding energies for the negative and neutral forms, for both Cpx and Spx. These results suggest that the constriction zone of both model proteins should be, as expected, more specific to bind FQs in the zwitterionic form.
Protocol 1 (rigid docking) – fluoroquinolones docking profile
The next step to further analyse and compare the interaction of Spx and Cpx with the two protein structures was to build up an affinity profile along the protein channel. This profile do not intend to sample the translocation coordinate (as umbrella sampling or steered molecular dynamics typically do get detailed information about the translocation43–45), but instead to individualize the affinity of the molecules to different specific regions of the channel of the two organisms.We have used six docking boxes with the exact same size but different positions throughout the channel. For this approach, only the zwitterionic form of the molecules was used, since it became clear from the previous experiments that this is the form that has a higher affinity for the OmpF channel. Fig. 4 depicts the results of this evaluation, considering 3 main regions along the protein channel (extracellular-facing region, constriction zone and intracellular-facing region) and the centre-of-mass location of the pose with the most negative free binding energy within the channel together with its relative ΔGbinding. The analysis of the results shows that, for both models, Cpx zwitterionic is the molecule with the highest binding affinity to the constriction zone confirming the previously obtained results. For the 2OMF structure, the ΔGbinding is slightly more negative for Cpx compared to Spx along the protein channel but the pattern of energy variation along the channel is very similar for both FQs. Though, for the 4KRA structure it is clear that the pattern of energy variation along the channel for Cpx and Spx is completely different. Overall, these results support the proposed observations that different pore properties could have an impact on the binding of molecules to these proteins. However, in this case, despite the docking profiles being different, the conclusions are not unrelated: Cpx still presents a higher affinity towards the pore when comparing with Spx, a result that is clearly exacerbated in the 4KRA structure.
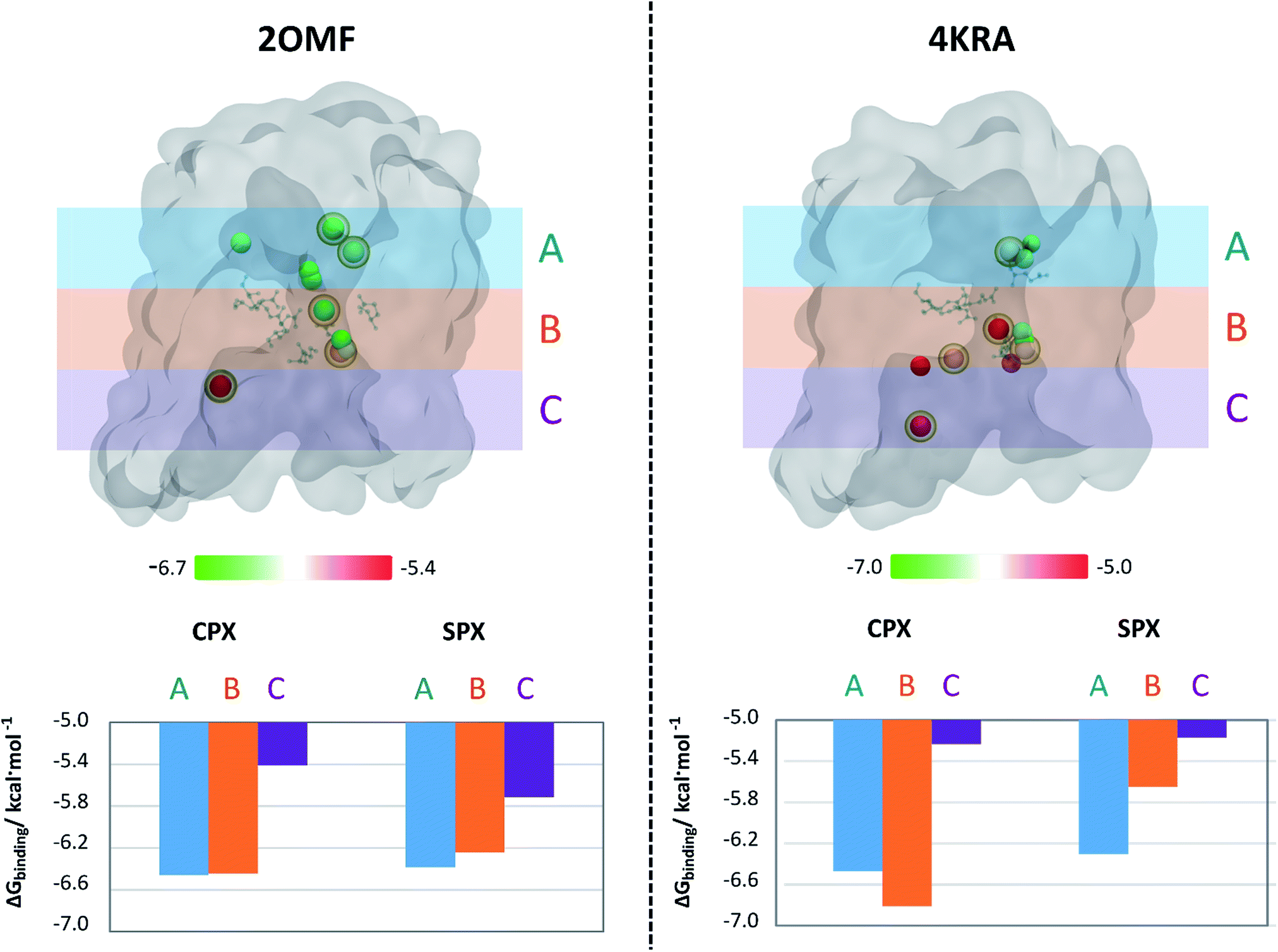 | ||
| Fig. 4 Position of the docking solutions along three different zones in the OmpF channel and the respective average ΔGbinding for each zone. The extracellular-facing region of the OmpF channel is represented by (A), the constriction zone by (B) and the intracellular-facing region by (C). The pose with the most negative binding energy for each position was considered. Proteins are represented in surface (silver); Cpx and Spx molecules' center-of-mass is represented in balls and coloured according to the ΔGbinding of each solution. Spx solutions are marked by a yellow hallow. Since the 6 defined docking boxes are superimposable in some regions, some docking solutions can also be superimposed. The constriction zone residues are represented by ball-and-stick, in blue. | ||
Protocol 2 (flexible docking) – fluoroquinolones
To account for the dynamics of the channel residues that could have an impact in the binding affinity, we have resorted to a second protocol that considered key residues to be flexible within the constriction zone (see methods section for details). We have performed molecular docking with a docking box that covered the entire constriction zone (similar to the one used in rigid docking). Up to this point, we have shown that (1) 4KRA is the model with an apparently higher adaptation to bind FQs at the constriction zone; and (2) Cpx in the zwitterionic form is the FQ with the highest affinity to the constriction zone. We expect that, for the 2OMF structure the flexibilization of the constriction zone residues should influence the docking results. For the 4KRA structure, since the protein was co-crystalized with Cpx, the protein should, theoretically, already be in the best conformation for FQs association and thus no improvement in the binding would be introduced by the flexibilization of the constriction zone residues. However, a comparison of the both rigid and flexible docking solutions for zwitterionic Cpx with 4KRA, shows that there is a variation in the position of the arginine residues in the most energetically favourable docking solutions (see ESI, Fig. S3†). This results in a more negative ΔGbinding for the flexible docking pose when compared with the rigid docking. It is also important to refer that, unlike the Cpx rigid docking solution, the Cpx flexible docking solution interacts very closely with the R130, R77, R60, and E116 residues of the constriction zone, being those interactions more similar to the ones observed for the Cpx X-ray (RMSD = 4.12 Å, see ESI, Fig. S3B†). Thus, these results allow us to infer that, due to the high mobility and numerous interactions that a molecule can make inside the protein channel, the flexibilization of the constriction zone residues is important to get the more stable conformation of the FQ. Moreover, it is important to point that the low resolution of the 4KRA protein model (3.32 Å) provides a level of uncertainty for the position of the lateral chains of the amino acids in the X-ray model. The relative binding free energies for the FQs and the flexibilized OmpF structures are listed in Table 3.The results clearly show that the flexibilization of the protein residues leads to an increase in the variation between the binding energies of the zwitterionic forms when compared to the negative or neutral forms. Once more, these results show that the affinity of the Cpx zwitterionic form is greater, compared to the other Cpx species and to Spx. In Fig. 5, some of the main interactions observed are presented, considering the most negative free binding energy pose of zwitterionic Cpx and Spx. Many ionic interactions and hydrogen bonds are established between the constriction zone residues and FQs, which are essential for the channel's selectivity.9–12
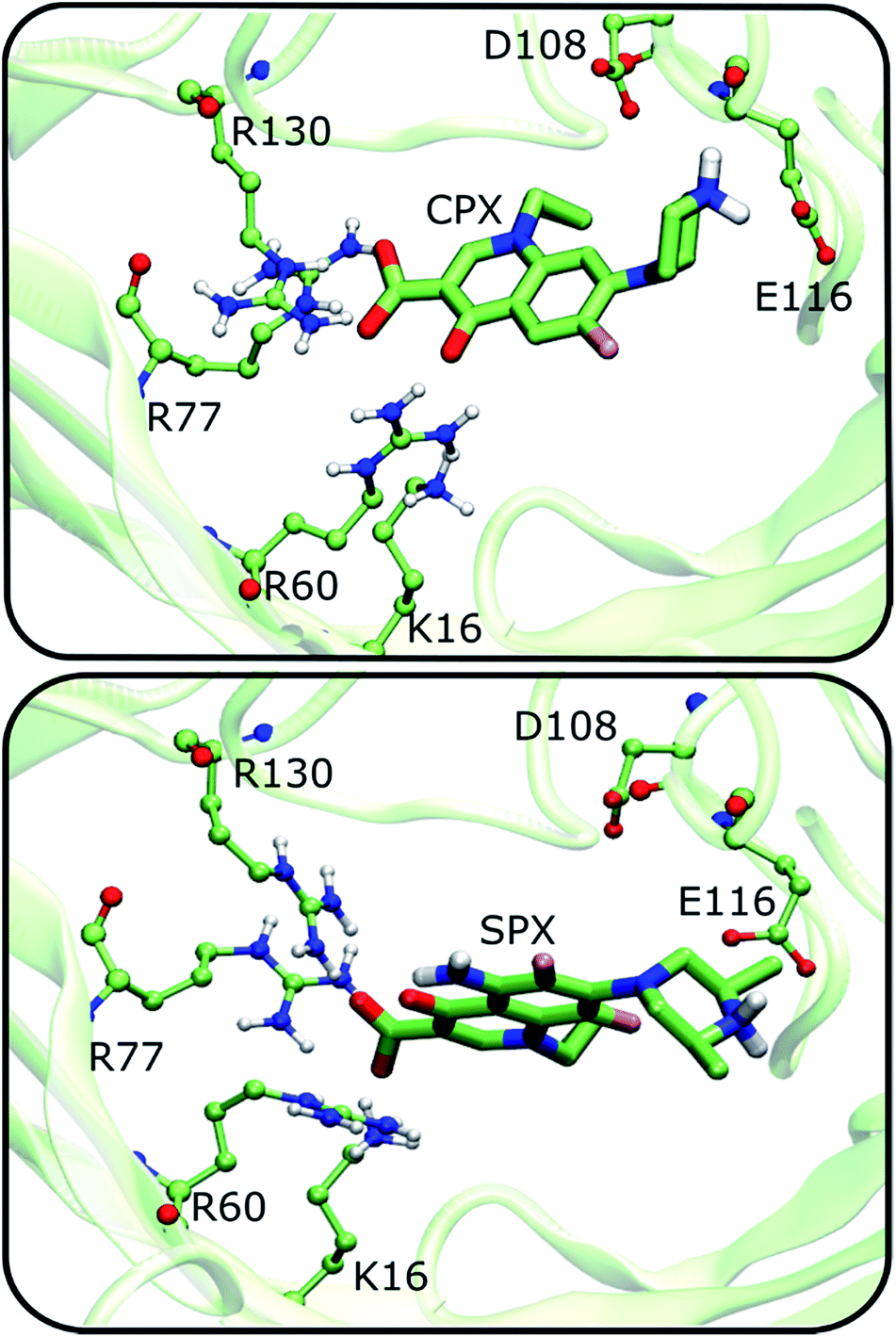 | ||
| Fig. 5 Interaction of the Cpx and Spx most negative energy docking solutions with the main residues constituting the constriction zone of the OmpF protein. Flexible docking procedure was considered and 4KRA used as OmpF structure model. | ||
Copper-complexed fluoroquinolones – protocol 1 and 2 (rigid and flexible docking) at the constriction zone
The complexation of FQs with copper is an approach studied to counteract the bacterial resistance to these antibiotics. The hypothesis is that the copper complexes of FQs might have a different route of entry in the bacterial cell, being their influx porin-independent and presumably promoted by a favourable lipid–antibiotic interaction.17–21,25,26Both rigid and flexible docking experiments were designed to evaluate the interaction of the copper complexes with the main residues of the constriction zone and compare with the results obtained for the free-drugs. A similar approach to the one took for free-FQ was performed using a docking box that only considers the constriction zone and using the same flexibilized residues. Results are listed on Table 4.
| Docking | ΔΔGbinding/kcal mol−1 | |||
|---|---|---|---|---|
| Rigid | Flexible | |||
| Ligand | 4KRA | 2OMF | 4KRA | 2OMF |
| a All the listed ΔΔGbinding values represent the most negative energy docking solution except the values marked with *a, b, or c, which represent the first docking solution that was localized near the constriction zone, since the most negative energy docking solution was outside this zone. a 28th, b 31st and c 2nd docking solutions. | ||||
| Cpx zwitterionic | 0.0 | 0.0 | 0.0 | 0.0 |
| Spx zwitterionic | 1.6 | 1.6 | 1.2 | 1.4 |
| CuCpxPhen | 1.2*a | −0.1 | 2.6*c | 2.6 |
| CuSpxPhen | 1.0*b | 0.3 | 2.9 | 3.1 |
It is first important to point out that the use of Mulliken charges, leaded to similar results, since the binding energy of the zwitterionic Cpx is more negative than the one of the zwitterionic Spx in both docking protocols. This result allows the validation of the Mulliken charge model. Overall, both rigid and flexible docking results show similar binding energies between the copper complexes. Previous microbiological results pointed for a decreased CuCpxPhen antibacterial activity when porin is not present. It is, however, possible that the conformational changes in the bacterial membrane caused by porin deletion would affect the CuCpxPhen entry in the cell even if it does not specifically cross the porin channel.
However, the comparison between the free energies of binding of the copper complexes and its respective free-FQ depends on the type of docking employed. For rigid docking, the results show that Cpx zwitterionic only has a more negative free energy of binding compared to its copper complex for the 4KRA structure and Spx zwitterionic has a less negative free energy of binding compared to CuSpxPhen in both protein structures. These results, can be though partially explained by a higher capacity of the copper complex to make interactions with different residues outside the constriction zone, especially in the upper part of the OmpF protein channel (which was not flexibilized), since it is a bigger molecule when compared to the free drug. These interactions may however not carry an increased ability for the copper complexes to cross the OmpF channel: previous studies on antibiotic permeation have also shown that the interaction of drugs with residues outside the constriction zone has the opposite effect, hampering the transport through the channel.7 In fact, when the flexibilization of the constriction zone is considered, the results clearly show a smaller binding affinity for the copper complexes, when compared to the zwitterionic free drugs. Moreover, the affinity of the copper complexes to the OmpF channel was not relevantly enhanced by the flexibilization of the constriction zone residues: there is not a big change between their binding affinities to OmpF in the rigid and the flexible docking approaches. This suggests that the copper-complexes of FQs do not have a strong affinity to interact with the constriction zone residues and their favourable binding energies in the rigid docking protocol are mainly related with interaction with residues outside the constriction area. Therefore, considering the importance of the interaction with the constriction residues for drug permeation across the porins and the dynamic nature of the transport through a protein channel, the transport of the free-zwitterionic FQs should be kinetically more favourable than the transport of the copper complexes. The unlikeliness of the copper complexes permeation through the OmpF channel is also supported by the fact that the FQs copper complexes have around 700 to 800 Da and only molecules with a maximum size of ∼600 Da are expected to be able to cross the protein channel.47,48
Taken together, these results support our assumption that the copper complexes of FQs should cross the bacterial cell by a mechanism independent on porins. These results, combined with preliminary studies from our group that point for a cytotoxicity against human cells over than 10× higher than their MIC values, support these complexes as putatively suitable to be used against resistant bacteria.
Conclusion
Despite porins being general diffusion protein channels, it has become clear that the transport of antibiotics across these channels does not occur by a simple passive diffusion event. The interaction of antibiotics with the residues inside the porin channel have been proved to play an important role in the transport of the molecules. Furthermore, specific interaction with the constriction zone residues appear to be essential to facilitate the transport of drugs through the channel.9–12 In this work the interaction of two FQs (Cpx and Spx) and their copper complexes (CuCpxPhen and CuSpxPhen) with OmpF protein was studied to explore the channel selectivity for each of these molecules.Experimental results, obtained by SPR, show a smaller binding affinity of Spx to the OmpF porin, when compared to Cpx, being this result in agreement with the assumption that Cpx has a higher affinity than Spx to cross the OmpF channel. SPR results show, however, similar binding affinities for the free-drugs when compared to their respective copper(II) complexes. Nevertheless, it is important to take into account that the equilibrium steady-state binding affinity calculated by SPR does not necessarily express the binding to the constriction zone residues. In fact, molecular docking results clearly show a smaller affinity of the copper(II) ternary complexes to the constriction zone residues when compared to free-FQs. These results, combined with the size of these complexes (700–800 Da) support the idea that the large binding affinities between the copper complexes and the protein found in the experimental studies are mainly related to interactions in the upper part of OmpF protein and not to the penetration of these molecules through the channel protein.
Furthermore, molecular docking results evidenced that the OmpF channel is especially selective for the zwitterionic form of FQs since these present a higher interaction with the constriction zone residues. In addition, the docking results support a higher selectivity of Cpx relative to Spx.
To our knowledge, molecular docking has never been applied extensively to protein channels, such as OmpF. Nevertheless, our results seem to be sufficient to validate this technique as a tool to compare the affinity of different molecules to the walls of the channel protein, with obvious implications to permeation, when there is a specific group of residues that are very relevant for the crossing process (in this case, the constriction zone residues).
Overall, the combination of experimental and theoretical studies presented in this work support the fact that the FQs studied in the free form have a higher affinity for the OmpF protein than in the copper-complexed form. These findings provide insights that will be helpful to proceed with the study of metal-complexes as alternatives to free fluoroquinolones in resistant infections, avoiding the high-cost and time-consuming processes of discovering and designing new antibiotics.
Acknowledgements
This work was funded through projects UID/MULTI/04378/2013-POCI/01/0145/FERDER/007728 with financial support from FCT/MEC through national funds and co-financed by FEDER, under the Partnership Agreement PT2020 and FCT (EXCL-II/QEQ-COM/0394/2012). MF and CFS thanks The Medical Biochemistry and Biophysics Doctoral Program (M2B-PhD) and FCT, Portugal for a PhD fellowship (SFRH/BD/52382/2013, PD/BD/114178/2016). JTSC thanks FCT for a PhD grant (SFRH/BD/87434/2012). SL and IG thanks FCT for a post-doc fellowship, (SFRH/BPD/110140/2015, SFRH/BPD/63850/2009). Authors thank to Dr Frederico Silva, IBMC/I3S, for assistance with SPR experiments.References
- V. T. Andriole, Clin. Infect. Dis., 2005, 41(suppl. 2), S113–S119 CrossRef CAS PubMed.
- A. M. Emmerson and A. M. Jones, J. Antimicrob. Chemother., 2003, 51(suppl 1), 13–20 CrossRef CAS PubMed.
- E. M. Scholar, Am. J. Pharm. Educ., 2002, 66, 164–172 Search PubMed.
- H. Nikaido, Microbiol. Mol. Biol. Rev., 2003, 67, 593–656 CrossRef CAS PubMed.
- P. Neves, E. Berkane, P. Gameiro, M. Winterhalter and B. de Castro, Biophys. chem., 2005, 113, 123–128 CrossRef CAS PubMed.
- J. L. Vazquez, S. Merino, O. Domenech, M. Berlanga, M. Vinas, M. T. Montero and J. Hernandez-Borrell, Int. J. Pharm., 2001, 220, 53–62 CrossRef CAS PubMed.
- K. R. Mahendran, et al., J. Phys. Chem. B, 2010, 114, 5170–5179 CrossRef CAS PubMed.
- O. D. Novikova and T. F. Solovyeva, Biol. Membr., 2009, 26, 6–20 CAS.
- E. M. Nestorovich, C. Danelon, M. Winterhalter and S. M. Bezrukov, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9789–9794 CrossRef CAS PubMed.
- C. Danelon, E. M. Nestorovich, M. Winterhalter, M. Ceccarelli and S. M. Bezrukov, Biophys. J., 2006, 90, 1617–1627 CrossRef CAS PubMed.
- C. E. James, K. R. Mahendran, A. Molitor, J. M. Bolla, A. N. Bessonov, M. Winterhalter and J. M. Page, Plos One, 2009, 4, e5453 Search PubMed.
- J. M. Pages, C. E. James and M. Winterhalter, Nat. Rev. Microbiol., 2008, 6, 893–903 CrossRef CAS PubMed.
- P. Manoharan and K. M. Saravanan, J. Biomol. Struct. Dyn., 2016, 1–10 CAS.
- A. Serafin and A. Stanczak, Russ. J. Coord. Chem., 2009, 35, 81–95 CrossRef CAS.
- D. C. Hooper, Emerging Infect. Dis., 2001, 7, 337–341 CrossRef CAS PubMed.
- G. A. Jacoby, Clin. Infect. Dis., 2005, 41, S120–S126 CrossRef CAS PubMed.
- M. J. Feio, et al., J. Inorg. Biochem., 2014, 138, 129–143 CrossRef CAS PubMed.
- R. Saraiva, S. Lopes, M. Ferreira, F. Novais, E. Pereira, M. J. Feio and P. Gameiro, J. Inorg. Biochem., 2010, 104, 843–850 CrossRef CAS PubMed.
- S. C. Lopes, C. Ribeiro and P. Gameiro, Chem. Biol. Drug Des., 2013, 81, 265–274 CAS.
- C. Ribeiro, S. C. Lopes and P. Gameiro, J. Membr. Biol., 2011, 241, 117–125 CrossRef CAS PubMed.
- P. Gameiro, C. Rodrigues, T. Baptista, I. Sousa and B. de Castro, Int. J. Pharm., 2007, 334, 129–136 CrossRef CAS PubMed.
- S. Dorotikova, et al., J. Inorg. Biochem., 2015, 150, 160–173 CrossRef CAS PubMed.
- D. Inci, R. Aydin, O. Vatan, D. Yilmaz, H. M. Genckal, Y. Zorlu and T. Cavas, Spectrochim. Acta, Part A, 2015, 145, 313–324 CrossRef CAS PubMed.
- R. Krikavova, J. Vanco, Z. Travnicek, J. Hutyra and Z. Dvorak, J. Inorg. Biochem., 2016, 163, 8–17 CrossRef CAS PubMed.
- M. Ferreira and P. Gameiro, J. Membr. Biol., 2015, 248, 125–136 CrossRef CAS PubMed.
- C. F. Sousa, M. Ferreira, B. Abreu, C. J. Medforth and P. Gameiro, Int. J. Pharm., 2015, 495, 761–770 CrossRef CAS PubMed.
- S. Heyse, T. Stora, E. Schmid, J. H. Lakey and H. Vogel, Biochim. Biophys. Acta, Rev. Biomembr., 1998, 1376, 319–338 CrossRef CAS.
- I. Xenarios and D. Eisenberg, Curr. Opin. Biotechnol., 2001, 12, 334–339 CrossRef CAS PubMed.
- S. G. Patching, Biochim. Biophys. Acta, 2014, 1838, 43–55 CrossRef CAS PubMed.
- M. Besenicar, P. Macek, J. H. Lakey and G. Anderluh, Chem. Phys. Lipids, 2006, 141, 169–178 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- A. Prilipov, P. S. Phale, P. Van Gelder, J. P. Rosenbusch and R. Koebnik, FEMS Microbiol. Lett., 1998, 163, 65–72 CrossRef CAS PubMed.
- J. L. Rigaud, Braz. J. Med. Biol. Res., 2002, 35, 753–766 CrossRef CAS PubMed.
- J. L. Rigaud, M. T. Paternostre and A. Bluzat, Biochemistry, 1988, 27, 2677–2688 CrossRef CAS PubMed.
- J. Knol, K. Sjollema and B. Poolman, Biochemistry, 1998, 37, 16410–16415 CrossRef CAS PubMed.
- M. T. Montero, D. Saiz, R. Sitges, J. L. Vazquez and J. H. Borrell, Int. J. Pharm., 1996, 138, 113–120 CrossRef CAS.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 3, 33 Search PubMed.
- M. J. Frisch, et al., Gaussian 09, 2009 Search PubMed.
- D. Balasubramaniam, A. Arockiasamy, P. D. Kumar, A. Sharma and S. Krishnaswamy, J. Struct. Biol., 2012, 178, 233–244 CrossRef CAS PubMed.
- S. F. Altschul, T. L. Madden, A. A. Schaffer, J. H. Zhang, Z. Zhang, W. Miller and D. J. Lipman, Nucleic Acids Res., 1997, 25, 3389–3402 CrossRef CAS PubMed.
- S. F. Altschul, J. C. Wootton, E. M. Gertz, R. Agarwala, A. Morgulis, A. A. Schaffer and Y. K. Yu, FEBS J., 2005, 272, 5101–5109 CrossRef CAS PubMed.
- R. Huey, G. M. Morris, A. J. Olson and D. S. Goodsell, J. Comput. Chem., 2007, 28, 1145–1152 CrossRef CAS PubMed.
- Y. B. Wang, X. Zhao, B. L. Sun, H. Yu and X. R. Huang, Arch. Biochem. Biophys., 2012, 524, 132–139 CrossRef CAS PubMed.
- B. K. Ziervogel and B. Roux, Structure, 2013, 21, 76–87 CrossRef CAS PubMed.
- A. Kumar, E. Hajjar, P. Ruggerone and M. Ceccarelli, J. Phys. Chem. B, 2010, 114, 9608–9616 CrossRef CAS PubMed.
- J. K. Kim, Y. S. Cho, M. Lee, R. A. Laskowski, S. E. Ryu, K. Sugihara and D. S. Kim, Nucleic Acids Res., 2015, 43, W413–W418 CrossRef PubMed.
- T. Nakae and J. Ishii, J. Bacteriol., 1978, 133, 1412–1418 CAS.
- A. H. Delcour, Biochim. Biophys. Acta, Proteins Proteomics, 2009, 1794, 808–816 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26466b |
| This journal is © The Royal Society of Chemistry 2017 |