DOI:
10.1039/C6RA26511A
(Paper)
RSC Adv., 2017,
7, 5046-5054
PAMAM/5-fluorouracil drug conjugate for targeting E6 and E7 oncoproteins in cervical cancer: a combined experimental/in silico approach†
Received
8th November 2016
, Accepted 27th December 2016
First published on 17th January 2017
Abstract
In the present study, poly(amidoamine)/5-fluorouracil (PAMAM/5-FU) was prepared and used as a conjugate system for delivering drugs to target E6 and E7 oncoproteins, which are predominant in cervical cancers. Specifically, molecular docking analysis was used to investigate the interaction between the PAMAM/5-FU and E6/E7 oncoproteins, which showed that the PAMAM/5-FU conjugate had a higher affinity for the oncoprotein than for 5-FU. Different generations of PAMAM dendrimers (0.5G, 1.0G, 1.5G, 2.0G, and 2.5G) were synthesized, characterized and tested as drug carriers for 5-FU. The PAMAM and PAMAM/5-FU drug conjugate showed less toxicity over COS-7 and HeLa cell lines. Laser confocal imaging and western blotting for tumor suppressor proteins pRb and p53 were used to confirm the interaction of PAMAM/5-FU with E6/E7 oncoproteins. Hematological analysis of PAMAM/5-FU using BALB/c female mice with cervical cancer confirmed the less toxic nature of this material. Based on these results, the developed PAMAM/5-FU conjugate is a potential candidate for the treatment of cervical cancer.
1. Introduction
Cancer is one of the most important diseases worldwide, with 12.7 million new cases and 7.6 million fatalities annually.1 Cervical cancer is a problematic and life-threatening health condition and has been prominently discussed in United Nations high-level meetings on non-communicable diseases.2 Human papillomaviruses (HPVs), which account for approximately 40–50% of all cervical cancers, infect epithelial tissues through micro-abrasions or other epithelial trauma.3 Accumulating evidence has clearly demonstrated that the expression of two high-risk viral oncogenes, E6 and E7, is essential for malignant transformation and maintenance of the malignant phenotype of cervical cancer cells, which occurs via interaction and elimination of key tumor-suppressive proteins.4 Various drugs and their combinations are currently used to treat cancers; however, repeated exposure to these drugs creates multi-drug resistance in cancer patients.5
5-Fluorouracil (5-FU) is a drug widely applied to treat cancers because its metabolites can intercalate with DNA, whereas in cervical cancer it inhibits the progression of cancer through effective interaction with E6 and E7 oncoproteins.6 Hsu et al. demonstrated that the 5-FU has effectively inhibited the activity of E6 and E7 oncoproteins, resulting in an increased p53 protein expression and decreased pRB expression. However, application of this drug has been hindered because there is a little difference between the minimum effective dose, maximum tolerated dose and development of drug resistance after prolonged treatment.7,8 Accordingly, the drug must be incorporated into nanocarriers to provide optimal and sustained drug release at the cancer site.
Different types of nanocarriers including liposomes, polymers, poly-ion complex micelles (PIC), cell-penetrating peptides (CPPs), polycholesterol substituted cyclen and dendrimers have been utilized in anticancer drug delivery systems.9–11 Among these, dendrimers are polymers with highly branched architectures of nanometer dimensions are showed the potential applications in bio-imaging, drug delivery, and gene transfer.12,13 Dendrimer is a well-known polymer, which is capable of binding with drugs and proteins through covalent, noncovalent, hydrophobic and hydrophilic interactions.14 As compared to other polymers, dendrimers showed less toxicity in animals and now they are under human trials for commercial usage in cancer treatment.15,16 A wide variety of molecules have been successfully encapsulated inside the dendrimers including methotrexate,17 5-FU,18 paclitaxel,19 flurbiprofen,20 cisplatin,21 and doxorubicin.22 Myc et al.17 used PAMAM–folic acid–methotrexate drug-conjugate for treating the tumor and they observed nonimmunogenic nature of this polymer. Tripathi et al.18 synthesized fatty acid grafted PAMAM dendrimers and they demonstrated 53% of 5-FU loading. Gupta et al.19 demonstrated dendrimer mediated drug solubility using less-soluble paclitaxel in PAMAM dendrimer. Asthana et al.20 loaded antiinflammatory drug flurbiprofen in PAMAM dendrimers and they found 5 fold drug distribution in animal model. Kulhari et al.21 prepared carboxylate-terminated PAMAM–cisplatin conjugate to treat testicular cancer and they found sustainable drug release in in vitro condition. Choi et al.22 prepared light sensitive PAMAM–doxorubicin conjugate to treat HeLa cells in in vivo condition. Recently, Giri et al.23 studied the interaction of PAMAM with human serum albumin protein using molecular dynamics at different pH and the dendrimer showed strong interaction at lower pH. Based on these findings, dendrimers are exceptionally advanced polymers for drug delivery as well as ability over regulating specific protein at low pH, which is highly beneficial to cancer therapy.
Molecular docking is used to understand the interaction between proteins and ligands. Recently, PAMAM, chitosan, and metal organic framework were studied using a molecular docking analysis.24–26 Luis et al.27 employed a molecular modeling approach to study the interaction between PAMAM and 5-FU. Kumar et al.28 conducted a docking analysis of several phytochemicals with E6 oncoprotein and showed effective inhibition of the protein. However, to the best of our knowledge, no molecular docking analysis for the interaction between PAMAM/5-FU and E6/E7 oncoproteins has been conducted to date. Application of nanomaterials for drug delivery has positive and negative aspects.29 Drug delivery based on nanomaterials can accumulate inside the body after the drug is delivered, which may cause a severe immunological response. Dobrovolskaia et al.30 considered blood platelets to be essential to understanding the toxicological effects of nanomaterials. Because nanomaterials in animals can activate platelets and show apparent changes in their count and platelet aggregation, they can be used as markers for the drug carrier thrombogenic properties and hemocompatibility. In this study, PAMAM/5-FU conjugate was subjected to in vitro drug release of 5-FU for targeting the E6/E7 proteins of cervical cancer cells. The molecular docking analysis was carried out to determine the interaction between PAMAM/5-FU conjugate and E6/E7 oncoproteins in cervical cancer. In order to confirm the safety of PAMAM/5-FU conjugate, hematological analyses were conducted in the BALB/c cervical cancer mouse model.
2. Experimental section
2.1. Materials
Ethylenediamine and methyl acrylate were purchased from TCL (Korea). Analytical grade methanol, DMF, N,N′-dicyclohexylurea, and other chemicals of reagent grade were purchased from Samchun Pure Chemicals (Korea). Distilled water was used throughout the experiments unless or otherwise indicated. 5-FU, 4′,6-diamidino-2-phenylindole (DAPI), and fluorescein isothiocyanate (FITC) were purchased from Sigma-Aldrich.
2.2. Protein, ligand retrieval, preparation, and molecular docking
Human papillomavirus E6 (1VZN) and E7 (2B9D) oncoproteins were retrieved from PDB data resources. The proton nuclear magnetic resonance (1HNMR) structure of 5-FU (CID 3385) and PAMAM (CID 4140276) dendrimer were retrieved from PubChem. To provide atomic-scale insights of molecular interaction with PAMAM, up to 2.5G of the dendrimers were considered for molecular docking. The crystal structures of E6 and E7 were prepared and the missing residues were modeled using Schrödinger Suite (Protein Preparation Wizard). The pre and post-translational effects of amino acids were taken into consideration; therefore, ionization and tautomerization were checked using the Epik module for a pH range of 7 to 9. An optimized potentials for liquid simulations (OPLS) force field was used for charges, bond angle, and torsion parameters. The structures of the E6 and E7 oncoproteins were pre-processed prior to docking and molecular simulation. The protocol used for protein preparation is described in detail in the ESI† (protocol 1). After preparation of proteins and ligands, docking analysis was conducted using a macromodel described in the ESI† (protocol 2).
2.3. Synthesis of PAMAM dendrimer
The PAMAM dendrimers were synthesized based on a divergent method described by Tomalia and Frechet.31 The first step in the synthesis of PAMAM dendrimer was a Michael addition reaction of the primary amine, ethylenediamine (EDA) to the double bond of an acrylic ester (methyl acrylate). The reaction continued for up to 5 h while stirring under room temperature. After 5 h, the reaction mixture was kept undisturbed for 2 days. This reaction results in a tetraester (Scheme S1†). The resulting tetraester represents the half generation (G-0.5) of PAMAM dendrimer. The second stage of the synthesis was an amidation reaction. The ester group of methyl acrylate reacted with the amino group of EDA. As a result, the full generation (G0) of PAMAM dendrimer was formed. Now the outer layer of the dendrimer consists of free amino groups. In the amidation reaction, the EDA was taken in a large excess to suppress the formation of intramolecular cycles and clusters.31 Both steps were carried out in methanol. By repeating these two steps, the following higher generations up to the G2.5 were synthesized. According to the divergent method for the synthesis of PAMAM dendrimers, no purification steps were required. After completion of the reaction, the reagents present in excess were removed to prevent the formation of side-products. Table S1† shows the amount of EDA and methyl acrylate used for the preparation of different generations of PAMAM.
2.4. Cytotoxicity studies (MTT assay)
To measure cell viability, we conducted an MTT assay using HeLa cells isolated from cervical cancer. Cells were seeded in a 96-well plate at 1.5 × 104 cells per well, then incubated in 100 μL of medium for 24 h. Following incubation, cells were treated with different generations of dendrimers (PAMAM 1G, 1.5G, 2G, 2.5G), after which the MTT reagent (2 mg mL−1) dissolved in DMSO was added to each well, and the cells were incubated for 4 h. The absorbance of each well was measured at 570 nm.
2.5. Incorporation of drug into dendrimer
A total of 3 mg of 5-FU was dissolved in 10 mL of double distilled water, after which 100 μL of dendrimer was added to a 50 mL of double distilled water. Next, 4 mL of dendrimer solution was mixed with 1 mL of 5-FU solution and stirred for 24 h. The sample was then centrifuged and the supernatant was removed, and lyophilized to obtain freeze-dried drug loaded dendrimer.
2.6. In vitro drug release and stability
Drug encapsulated dendrimer solution was packed into the dialysis bag (Dialysis Membrane-50, Hi-Media) and kept in double distilled water as a dissolution medium (pH 7.0) for the dialysis process. Samples were sonicated for 1 min and later agitated at 200 rpm to liberate the drug. Next, 4 mL of the dissolution medium was recovered every 30, 60, 90, 120, 150, and 180 minutes while replacing the same volume of the dissolution medium, after which the drug content was determined by UV-visible analysis (λmax 266 nm). The stability of the formulation was evaluated at three different temperatures (4°, 24°, and 50 °C).
2.7. Western blot analysis
Floating and adherent HeLa cells were harvested after treatment with the 5-FU and PAMAM/5-FU for 24 h, then lysed in RIPA buffer supplemented with protease inhibitor with brief sonication.32 Next, 50 μg of the protein was separated on a 4–15% SDS–PAGE gradient gel and transferred to nitrocellulose membrane. Western analysis was performed using the primary antibodies such as p53 (Santa Cruz-126), pRB (Santa Cruz-169028), and β-Actin (Santa Cruz-47778).
2.8. DAPI staining, synthesis of drug–dendrimer–FITC conjugates for cellular internalization
To localize the cellular nucleus, the cells were first stained with DAPI. The dendrimer and drug were conjugated with FITC to track them in the DAPI stained cell line. Briefly, 0.02 g of (FITC) was conjugated with 0.067 g of glutaric acid to form carboxyl-terminated FITC using 0.09 mg of dicyclohexylcarbodiimide (DCC) as a coupling agent. Further, the drug–dendrimer conjugates and FITC–glutaric acid moiety were dissolved in anhydrous DMSO and another 0.04 g of DCC were added. The reaction mixture was kept in the stirrer environment for 5 days at room temperature, after which it was filtered to remove N,N′-dicyclohexylurea. The solution was subsequently filtered and dialyzed against DMSO for 24 h. Finally, the FITC conjugated drug–dendrimer complex was studied for molecular cell internalization against cervical cancer cell lines.
2.9. In vivo cancer study and hematological studies
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) in Inha University and conducted in accordance with the guidance from the Experimental Animal Research Committee of Inha University. For the experiment, 6 to 8 week old nude BALB/c female mice cervical cancer model with uniform weight were selected and divided into three groups of six animals each. The first group was kept as a control, the second was treated with the 5-FU and the third was treated with the drug–dendrimer, PAMAM/5-FU complex. All groups were maintained on the same regular controlled minimal diet and PAMAM/5-FU conjugate in saline was injected into the tail. After 14 days, blood was collected and analyzed for hemoglobin level, red blood cells (RBC), white blood cells (WBC), differential monocytes, lymphocytes, and neutrophil counts.
2.10. Characterization
TEM analysis was conducted at an acceleration voltage of 200 kV (JEOL, JEM-2010F). 5 μL of dendrimer was dropped over the carbon-coated copper nets and left for 1 min. The excess PAMAM was removed and washed with 2% sodium phosphotungstate (pH 6.5) and dried in the room temperature. This negative-stained sample was utilized for TEM analysis. Fourier transform infrared (FTIR) spectroscopy was recorded on a VERTEX 80 V spectrometer (Bruker) under ambient condition. The viscosity of the dendrimer was analyzed using a Brookfield viscometer and the chemical composition was analyzed with a Bruker 1HNMR spectroscopy. Zeta potential and dynamic light scattering (DLS) measurements were conducted on a Malvern Zetasizer Nano instrument (laser source of 633 nm at room temperature).
3. Results and discussion
3.1. Characterization of PAMAM
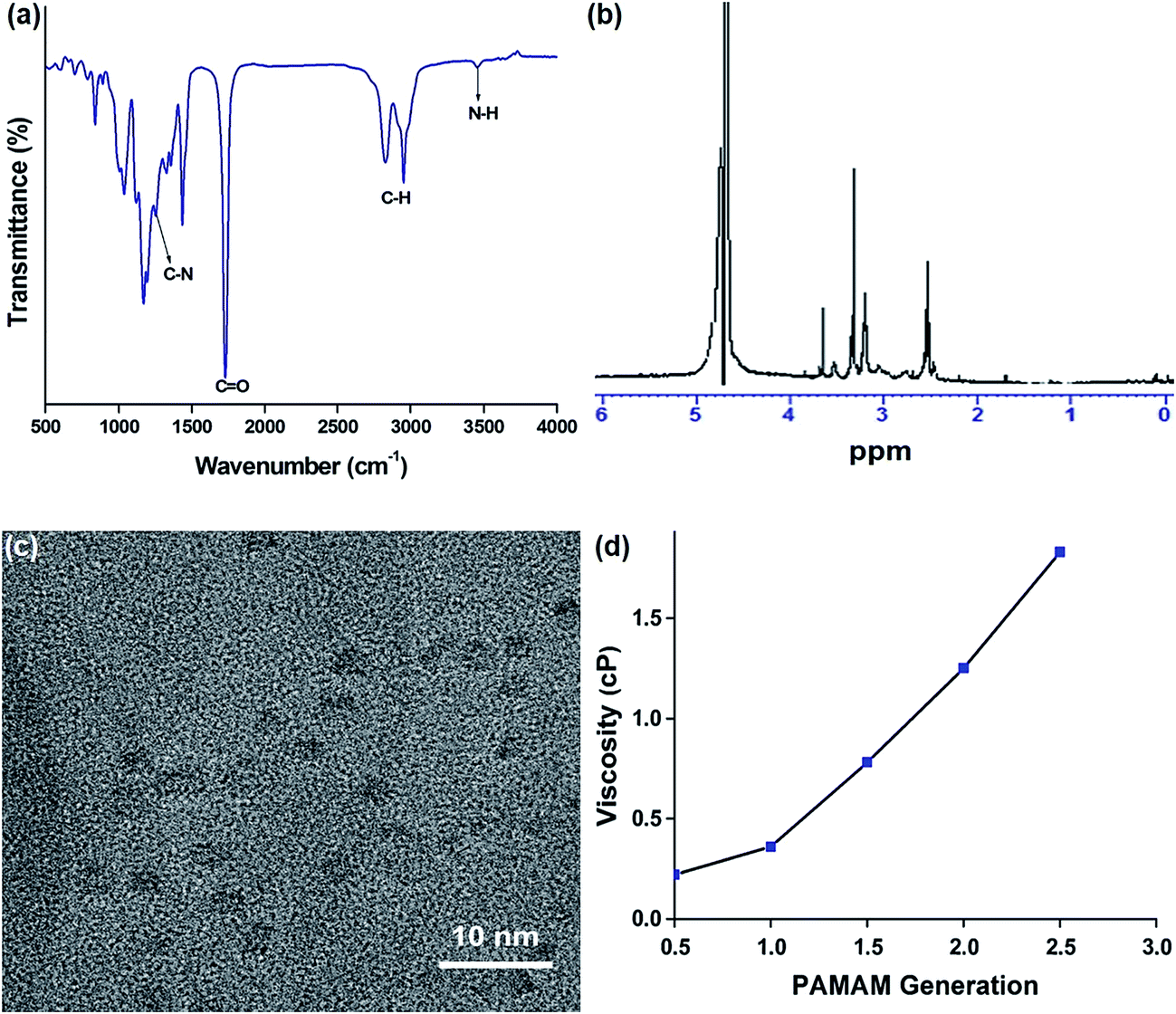
We successfully synthesized different generations of PAMAM dendrimers for drug delivery. In the present study, we utilized PAMAM (1.5G) dendrimer for drug delivery application because of its low toxicity, high drug loading and sustained drug release. Fig. 1a shows the FTIR spectrum of PAMAM (1.5G) dendrimer. The spectrum showed characteristic absorption bands: 3451 cm−1 (N–H stretching of primary amine), 2800–3000 cm−1 (C–H stretching), 1645 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of carbonyl group), 1257 cm−1 (C–N stretching), and 1437 cm−1 (C–H bending), which validated the successful synthesis of PAMAM (1.5G) dendrimer. Fig. 1b presents the NMR spectrum of PAMAM (1.5G) dendrimer to determine its structural integrity and purity. In the spectrum, a CO group attached methylene proton (–CH2CO) was observed at 2.58 ppm and an amide methylene proton (–CONHCH2CH2N–) was observed at 3.40 ppm.33 The addition of identical branches to the initial core and the fast increase of the molecular weight of the dendrimer results in the overlapping of signals of intensities strongly different from non-equivalent positions. Therefore, it is very difficult to resolve all the peaks for the PAMAM (1.5G) dendrimer. The TEM image of the as-synthesized PAMAM (1.5G) is shown in Fig. 1c. The polymer particles were spherical in shape with an average particle size of 2 nm. The DLS analysis also showed the average particle size of 2.3 nm which is consistent with the TEM result. Fig. 1d shows the viscosity measurements of different generations of PAMAM. The viscosity of PAMAM dendrimers increased with increasing generations, the similar results were reported previously.34
O stretching of carbonyl group), 1257 cm−1 (C–N stretching), and 1437 cm−1 (C–H bending), which validated the successful synthesis of PAMAM (1.5G) dendrimer. Fig. 1b presents the NMR spectrum of PAMAM (1.5G) dendrimer to determine its structural integrity and purity. In the spectrum, a CO group attached methylene proton (–CH2CO) was observed at 2.58 ppm and an amide methylene proton (–CONHCH2CH2N–) was observed at 3.40 ppm.33 The addition of identical branches to the initial core and the fast increase of the molecular weight of the dendrimer results in the overlapping of signals of intensities strongly different from non-equivalent positions. Therefore, it is very difficult to resolve all the peaks for the PAMAM (1.5G) dendrimer. The TEM image of the as-synthesized PAMAM (1.5G) is shown in Fig. 1c. The polymer particles were spherical in shape with an average particle size of 2 nm. The DLS analysis also showed the average particle size of 2.3 nm which is consistent with the TEM result. Fig. 1d shows the viscosity measurements of different generations of PAMAM. The viscosity of PAMAM dendrimers increased with increasing generations, the similar results were reported previously.34
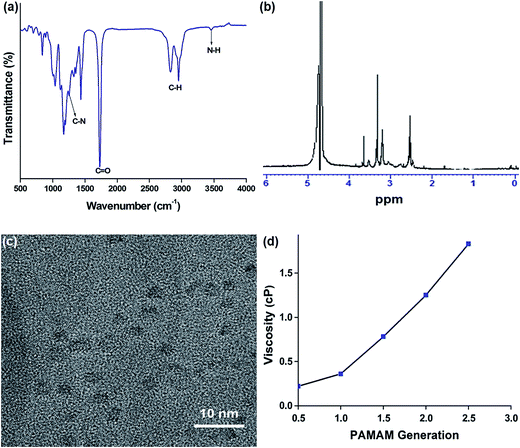 |
| | Fig. 1 (a) FTIR spectrum, (b) 1HNMR spectrum, (c) TEM image of 1.5G PAMAM, and (d) viscosity of different generations of PAMAM. | |
3.2. Molecular docking analysis of the interaction of E6 and E7 oncoproteins with 5-FU and PAMAM/5-FU conjugate
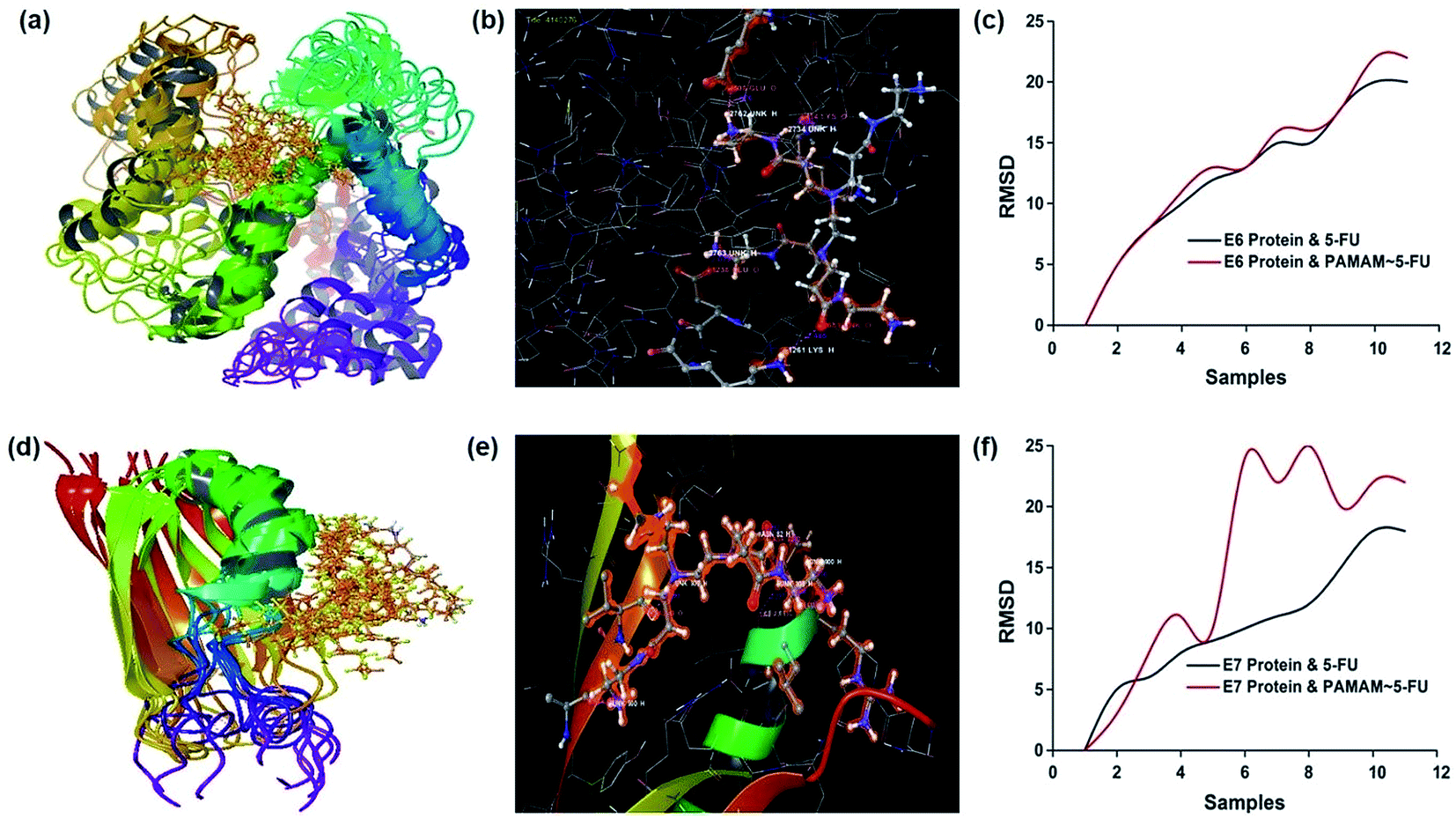
The conformational structure of E6 and E7 proteins and their interaction with 5-FU is shown in Fig. S1.† The docking studies showed that 5-FU interacted at two sites of the E6 proteins, through ALA 47 (O) and VAL 49 (H) of the receptors via H and O atoms with bond distances of 1.886 and 1.982 Å, respectively. The E7 oncoprotein with 5-FU exhibited three sites of interaction in which GLU 48 (O), LEU 22 (H) and SER 89 (H) of protein formed hydrogen bonds via H and O. The interaction of 5-FU with E6 and E7 established a Glide score value of −2.925 and −0.502, respectively. PAMAM/5-FU conjugate was docked with E6 and E7 oncoproteins and the observed interaction energy was −3.309 and −5.329, respectively. The conformational structures of the PAMAM/5-FU conjugate and oncoproteins along with the RMSD curve is shown in Fig. 2. These results confirmed that the PAMAM/5-FU conjugate exhibited a strong binding energy for its interaction with oncoproteins (E6 and E7) than the oncoprotein–5FU binding energy. To check the possible orientation of these molecules during the interaction, RMSD values were calculated for 10 samples with 1.0 picosecond equilibration and a trajectory of 100 picoseconds. The RMSD values showed more fluctuation because of the formation of a complex between the oncoprotein and the dendrimer in solution. The 5-FU and PAMAM/5-FU possess an equal number of binding orientations with E6, while E7 protein and PAMAM/5-FU exhibited more possible orientations. These results clearly showed that the E7 oncoprotein is easily down-regulated by the PAMAM/5-FU conjugate.
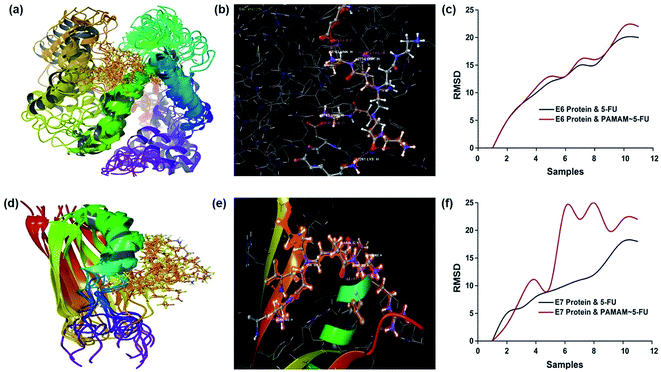 |
| | Fig. 2 Molecular docking analysis. (a and d) Conformational structure (b and e) interaction profile, and (c and f) RMSD plot of E6 and E7 protein over PAMAM/5-FU conjugate. | |
3.3. Cytotoxicity of dendrimer series
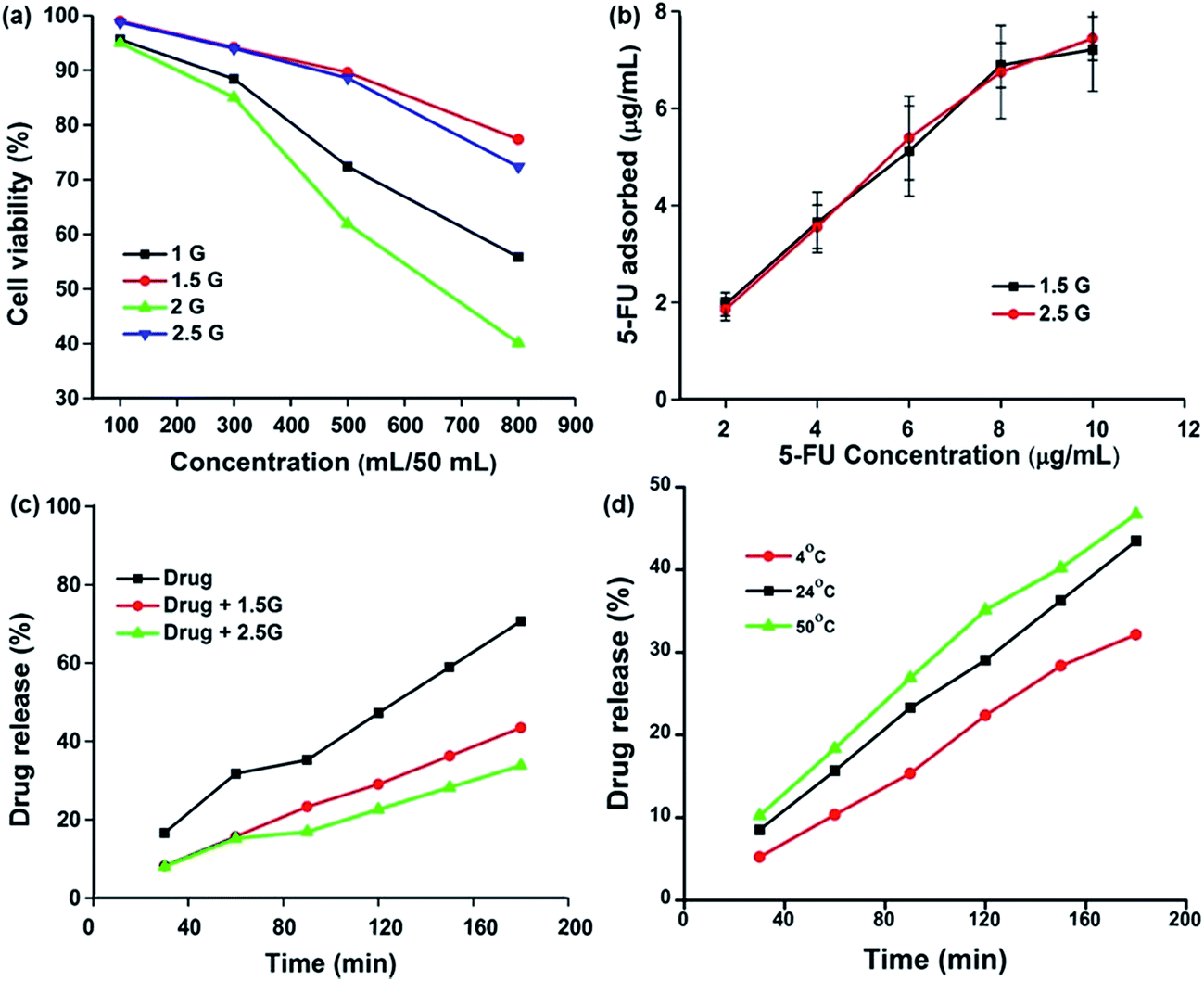
The 0.5G, 1.0G, 1.5G, 2.0G, and 2.5G dendrimers were tested by MTT assay (Fig. 3a). Briefly, various concentrations (100, 300, 500, and 800 μL) of dendrimers were added to 50 mL of double distilled water, after which 100 μL of the prepared solution was added to cell culture medium and incubated for further analysis. In 1.0G and 2.0G, cell clumping was observed along with 40 and 60% cell death, respectively. In contrast, 1.5G and 2.5G showed minimal cytotoxic effects of 20 and 25%, respectively. The effect of cell death over 1.0G and 2.0G was observed because of the amine groups present on their surface, while the presence of amide and carboxylic groups on the surface of 1.5G and 2.5G tended to have lower toxicity than that of the other co-generations. It has been reported that the cationic porphyrin dendrimer showed increased toxicity because of its electrostatic association with cell membranes.35 However, anionic PAMAM dendrimers were administered to mice bearing B16F10 tumors and showed no toxicity throughout the experimental period.36
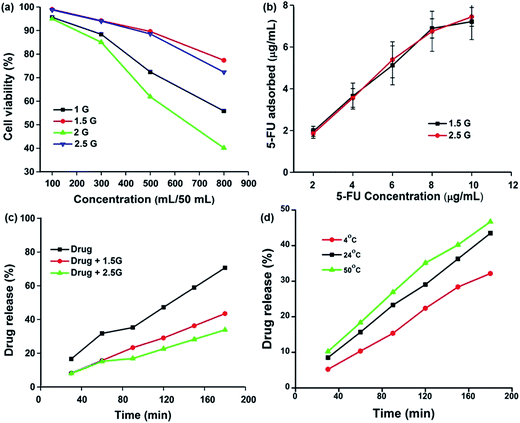 |
| | Fig. 3 (a) Cytotoxicity of different generations of PAMAM nanoparticles, (b) drug adsorption study, (c) drug release behavior of 1.5G and 2.5G PAMAM nanoparticles, and (d) stability of PAMAM/5-FU formulation at a different temperature. | |
3.4. Drug adsorption and release profile
The MTT assay results indicate that the PAMAM dendrimers of generations 1.0G and 2.0G were toxic; therefore, drug adsorption studies were only performed with 1.5G and 2.5G. To identify the loading and release kinetics of 5-FU on the PAMAM dendrimers (1.5G and 2.5G), the standard calibration curve was plotted against various concentrations of 5-FU (2, 4, 6, 8, and 10 μg mL−1). The loading and releasing kinetics were performed using PAMAM (1.5G and 2.5G) dendrimer for 24 h. As shown in Fig. 3b, 1.5G and 2.5G exhibited a higher capability of adsorbing 5-FU (∼70%), which was attributed to the (i) host (PAMAM)–guest (5-FU) interaction and (ii) the increased solubility of 5-FU because of the hydrophilic nature of PAMAM.31 The interaction of drug loading on PAMAM was confirmed by FT-IR, TEM, and DLS. Dendrimer conjugants were subjected to FTIR study (Fig. S2a†) to ensure the accumulation of drug into the dendrimers and there is a peak shift absorbed in most all the peaks NH stretching moved from 3451 cm−1 to 3366.14 cm−1. CH2 Asymmetric stretching moved from 2954 cm−1, 2827 cm−1 to 2972 cm−1, 2891 cm−1. C–N stretching moved from 1257 cm−1 to 1275 cm−1 and C–F stretching from 1034 cm−1 to 1088 cm−1. The detailed wavenumber shift is presented in Table S2.† TEM analysis showed that the size of the PAMAM (1.5G) dendrimer increased from 2 nm to 4 nm after drug encapsulation (Fig. S2b†), and this result was further validated by the DLS. The DLS analysis clearly showed an almost two-fold increase in size after drug adsorption (Table S3†). These findings confirmed that the 5-FU was encapsulated within the PAMAM dendrimer.
The results of drug release profile for the free 5-FU and PAMAM/5-FU of different generations (1.5G and 2.5G) in PBS (pH 7.4) are shown in Fig. 3c. At 30 min, 1.5G PAMAM dendrimer released 8.21% of the loaded drug into the buffer, whereas 7.98% of the drug was released from 2.5G PAMAM dendrimer. Both values were much lower than those of the pure drug release profile (16.64% at 30 min), After 180 min, 43.47 and 33.84% of the drug was released from the 1.5G and 2.5G PAMAM dendrimer, respectively. This drug release profile clearly follows zero-order kinetics and this may be attributed to the interaction between drug and dendrimer. The FTIR result shows that 5-FU covalently conjugated with the PAMAM. Patri et al.37 utilized surface modified PAMAM to switch the amount of methotrexate (MTX) release from PAMAM. Bhadra et al.38 also demonstrated zero-order kinetics using PAMAM/5-FU conjugate. The rate of drug release can be measured using the following eqn (1).
Fig. S2c† shows the plot between t (min) and C (concentration at particular time) and the slope gives the value of rate constant (K0). The slope values (K0) for drug, drug–1.5G PAMAM, drug–2.5G PAMAM are found to be 0.031, 0.019, 0.014 μg min−1, respectively. This result clearly indicates that there is a two-fold reduction in drug release, when the drug (5-FU) is incorporated into the polymer (PAMAM). The half-life period (t1/2) which gives the drug retention time in blood plasma will be calculated using the following eqn (2);
where
C0 is the initial concentration of drug in the carrier. The calculated
t1/2 values for drug, drug–1.5G PAMAM, drug–2.5G PAMAM are 129, 210, 285.71 min, respectively. This result shows that when 5-FU is incorporated into PAMAM, the half-life period for drug release is increased to two-fold. Even though, the 2.5G PAMAM dendrimer exhibited controlled drug release, it showed higher toxicity than 1.5G PAMAM dendrimers. Therefore, we further studied the effects of temperature on the drug release profile of 5-FU from the 1.5G PAMAM dendrimer. As shown in
Fig. 3d, the rate of drug release also depend on temperature, when the temperature increased the release rate also increased. Here in all three temperatures (4 °C, 24 °C, and 50 °C), there was no substantial difference in the percentage of drug release indicating that the formulated drug conjugate was highly stable due to the strong host–guest interaction between PAMAM and 5-FU.
3.5. Microscopic evaluation of PAMAM/5FU conjugate in HeLa cell
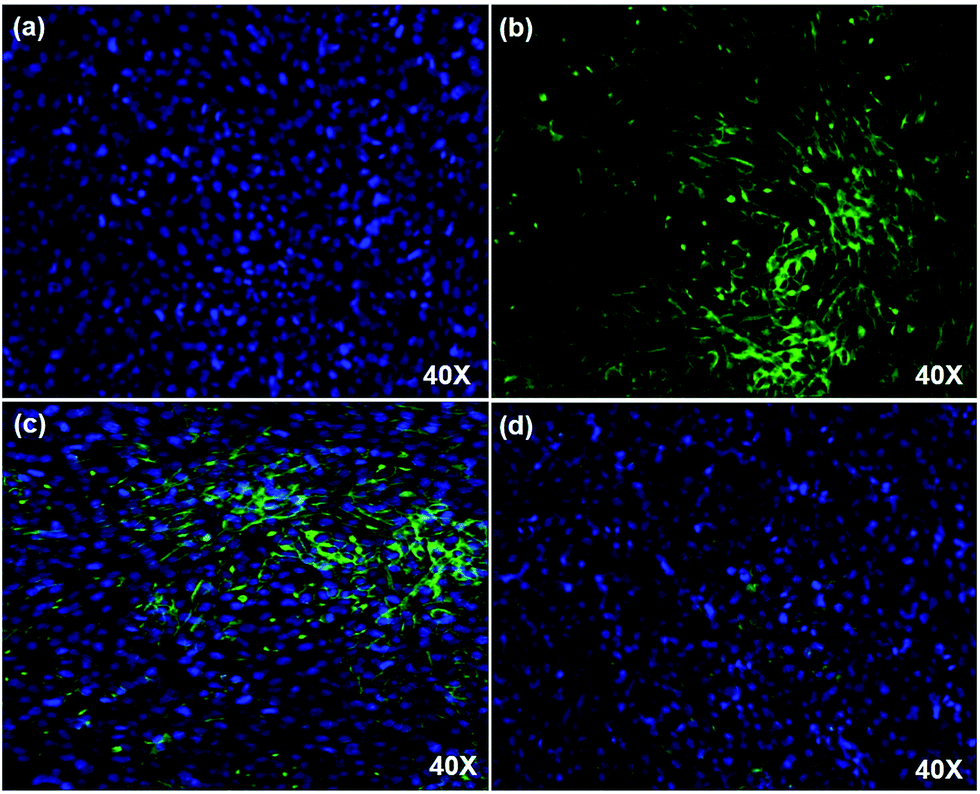
Fig. 4 confocal imaging of DAPI stained HeLa cells with FITC conjugated PAMAM/5-FU. The DAPI stained cells emit a blue color (Fig. 4a) due to the affinity of DAPI with double-stranded DNA present in the nucleus of the cell. The FITC was conjugated with PAMAM/5-FU to track the pathway of the delivery system inside the cell. The fluorescent intensity observed for the FITC–PAMAM/5-FU (Fig. 4b) was high when compared to that of FITC–5-FU (Fig. S4, ESI†), due to the interaction between PAMAM and FITC. Fig. 4c and d shows the confocal microscopic images of the FITC–PAMAM/5-FU and FITC–5FU conjugates with the HeLa cell. The images revealed that more FITC–PAMAM/5-FU conjugate was approached the cancer cells than that of FITC–5FU. The interaction between cancer cells and 5-FU tends to be lower than that with PAMAM/5-FU, resulting in the need for high levels of drug administration over a long period.39 Thus, this study revealed the necessity for a dendrimeric delivery vehicle to deliver the anti-cancer drug efficiently.
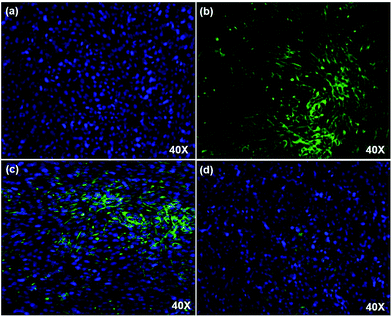 |
| | Fig. 4 Confocal imaging of (a) DAPI stained HeLa cell, (b) FITC–PAMAM/5-FU (c) FITC–PAMAM/5-FU in HeLa cell, and (d) FITC–5-FU in HeLa cell. | |
3.6. Downstream regulation of tumor suppressor proteins p53 and pRb upon inhibition on E6 and E7 oncoprotein with PAMAM/5-FU conjugate
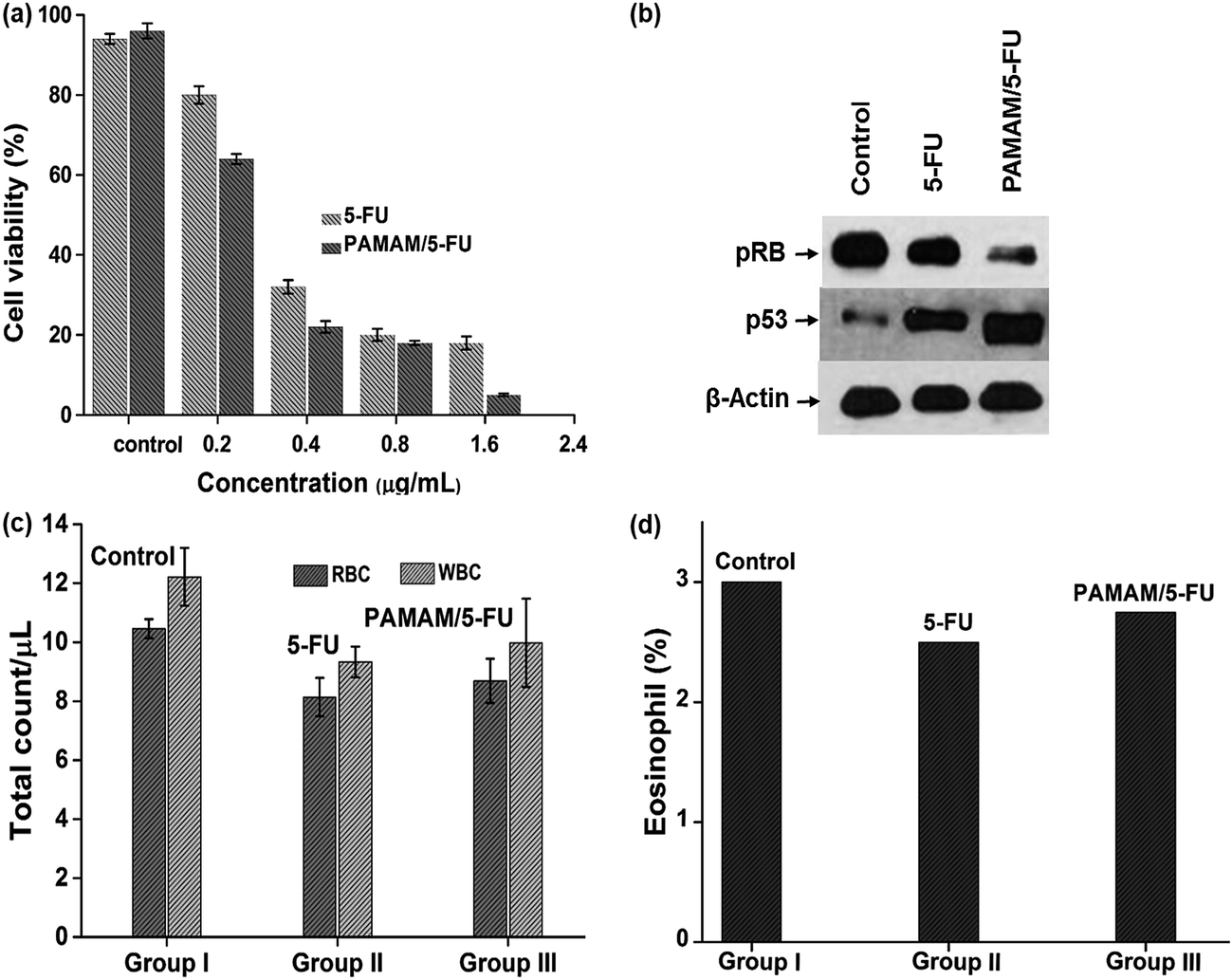
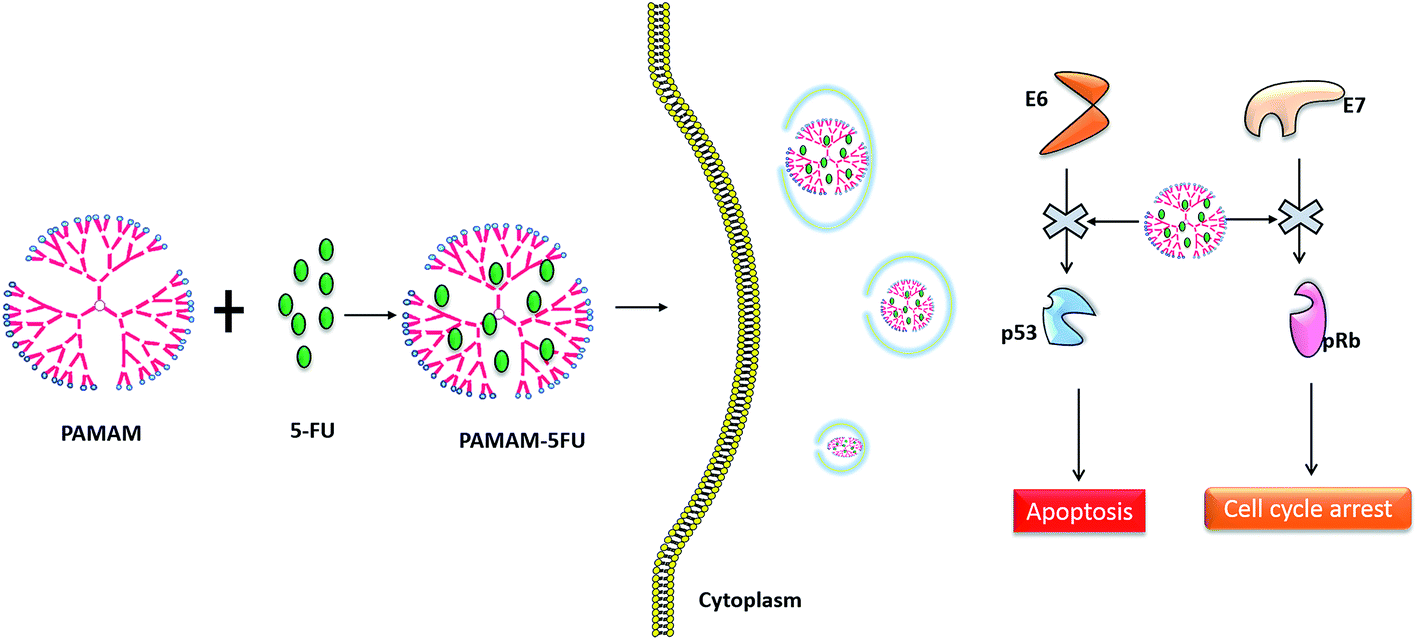
The effect of the PAMAM/5-FU conjugate on cell viability was compared with those of the free drug in the HeLa cell line (Fig. 5a). At 0.2 μg mL−1 of 5-FU and PAMAM/5-FU, cell viability decreased by ∼20% and 40%, respectively. When we increased the concentration of PAMAM/5-FU to 0.4 μg mL−1, 80% decrease in cell viability was observed, which is consistent with the results of previous studies.27 The increased cell toxicity may be due to the interaction PAMAM/5-FU with oncoproteins E6 and E7. To confirm the toxic effect of E6 and E7 proteins in the HeLa cell, we evaluated the interaction of two down-regulated tumor suppressor proteins p53 and pRB over the cancer cells (Fig. 5b) and their mechanism was presented in Scheme 1. In cervical cancer, the amount of pRB is always high due to the E7 protein binds with native pRB protein and inhibits the dephosphorylation process. In our experiment, we also observed a higher concentration of pRB in cancer cells. When we treated cancer cells with 5-FU alone, the amount of pRB was reduced slightly, while it was greatly reduced in response to treatment with PAMAM/5-FU. The p53 is an important tumor suppressor protein that induces cancer cells to undergo apoptosis and senescence. In cervical cancer, E6 protein degrades p53 protein so that the cells cannot enter into apoptosis and continuously grow. When we added the 5-FU to cancer cell the amount of p53 was increased, while PAMAM/5-FU lead to a greater increase in p53 expression. Similar results were observed in the literature.40 These results clearly indicate that PAMAM/5-FU has more potential to arrest the cancer cell cycle than pure 5-FU by inhibiting the E6 and E7 oncoproteins.
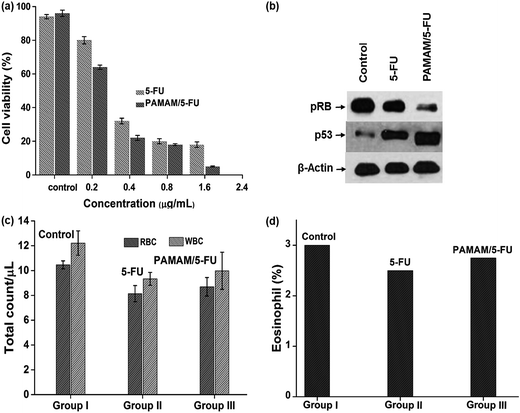 |
| | Fig. 5 (a) Cytotoxicity analysis of 5-FU and PAMAM/5-FU conjugate over HeLa cells, (b) Western blotting to evaluate the effects of 5-FU and PAMAM/5-FU conjugate over tumor suppressor proteins, (c) RBC and WBC count, and (d) eosinophil count. | |
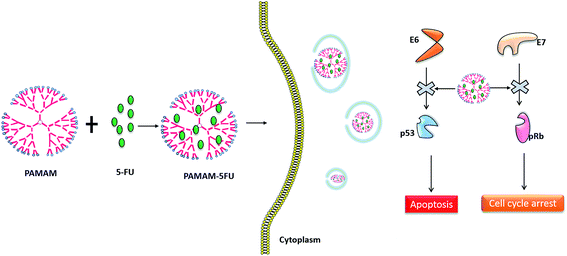 |
| | Scheme 1 PAMAM/5-FU conjugate binding with E6 and E7 oncoproteins in cervical cancer and preventing their interactions with the tumor suppressor protein p53 and pRb. | |
3.7. Hematological analysis of PAMAM/5-FU over BALB/c female mice with cervical cancer
The hematological analysis was conducted to confirm the safety concern over the as-synthesized PAMAM/5-FU conjugate. Animals were divided into three groups, control, 5-FU treatment group and PAMAM/5-FU conjugate treatment group. After 14 days of treatment, blood samples were collected from all groups for the analysis of total WBC and RBC counts, as shown in Fig. 5c. In the control, the WBC count was found to be 12.20 ± 1.08, while in the case of 5-FU and PAMAM/5-FU these values were 9.33 ± 0.84 and 9.98 ± 0.96, respectively. The total RBC count for the control animal was 10.46 ± 1.01, while those for the 5-FU and PAMAM/5-FU treated groups were 8.14 ± 0.48 and 8.69 ± 0.85, respectively, The eosinophil results (Fig. 5d) were 3.0 ± 0.32, 2.75 ± 0.29 and 2.5 ± 0.38 for the control, PAMAM/5-FU and 5-FU groups, respectively.41 In the above results, the amount of WBC, RBC, and eosinophil for 5-FU and PAMAM/5-FU was less than that of the control animal, this is due to the effect of 5-FU over immune system.42 When we compare the 5-FU and PAMAM/5-FU, the PAMAM/5-FU has fewer side effects than 5-FU alone, and similar results were reported in the literature.43 Thus, our results indicate that the 2.5G PAMAM (0.01 M) dendrimer was an effective delivery vehicle to release the anti-cancer drug 5-FU in a controlled manner, and the PAMAM/5-FU complex acted as a targeting moiety towards E6 and E7 oncoproteins.
4. Conclusions
In this study, molecular docking analysis of PAMMA–5FU with the oncoproteins (E6 and E7) revealed excellent downregulation activities of the oncoproteins relative to the pure 5-FU. Different generations of PAMAM (0.5G, 1.0G, 1.5G, 2.0G, and 2.5G) were synthesized and the low cytotoxicity was observed for 1.5G and 2.5G PAMAM. We utilized 1.5G and 2.5G PAMAM for the encapsulation of 5-FU for controlled drug delivery. Physio-chemical analyses revealed that ∼70% of the 5-FU was loaded onto the PAMAM (1.5G and 2.5G) and that the 2.5G PAMAM loaded 5-FU exhibited better-controlled drug release. Confocal imaging showed the interaction of PAMAM/5-FU with the nucleus of the HeLa cancer cells. The effectiveness of PAMAM/5-FU towards cancer killing ability was confirmed by the down-regulation of tumor oncogenic proteins p53 and pRB. The hematological analysis of the animals confirmed that the PAMAM/5-FU exhibited fewer side effects than 5-FU. The above results indicate that the PAMAM/5-FU is an effective and reliable drug conjugate for targeting E6 and E7 oncoproteins in HPV-mediated cervical cancer treatment.
Acknowledgements
This work was supported by the Basic Science Research Program (No. 2014R1A5A1009799 and NRF-2015R1A2A2A09001059) and the Framework of International Cooperation Program (NRF-2016K2A9A2A10005545) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning. This work was partly supported by Institute for Information & communications Technology Promotion (IITP) Grant (B0132-16-1001) funded by the Korea government (MSIP).
References
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2015, 65, 5–29 CrossRef PubMed.
- M. H. Forouzanfar, K. J. Foreman, A. M. Delossantos, R. Lozano, A. D. Lopez, C. J. Murray and M. Naghavi, Lancet, 2011, 378, 1461–1484 CrossRef.
- T. Rampias, E. Boutati, E. Pectasides, C. Sasaki, P. Kountourakis, P. Weinberger and A. Psyrri, Mol. Cancer Res., 2010, 8, 433–443 CrossRef CAS PubMed.
- L.-C. Hsu, R. M. Lee and R. L. White, Biochem. Biophys. Res. Commun., 2004, 320, 249–255 CrossRef CAS PubMed.
- C. Mohanty, M. Das, J. R Kanwar and S. K Sahoo, Curr. Drug Delivery, 2011, 8, 45–58 CrossRef CAS.
- M. Scheffner and N. J. Whitaker, Semin. Cancer Biol., 2003, 13, 59–67 CrossRef CAS PubMed.
- E. Gamelin and M. Boisdron-Celle, Crit Rev Oncol Hematol, 1999, 30, 71–79 CrossRef CAS PubMed.
- C. N. Arnold, A. Goel and C. R. Boland, Int. J. Cancer, 2003, 106, 66–73 CrossRef CAS PubMed.
- L. Nair, S. Jagadeeshan, S. A. Nair and G. V. Kumar, Int. J. Nanomedicine, 2011, 6, 1685 Search PubMed.
- A. Rengaraj, P. Puthiaraj, Y. Haldorai, N. S. Heo, S.-K. Hwang, Y.-K. Han, S. Kwon, W.-S. Ahn and Y. S. Huh, ACS Appl. Mater. Interfaces, 2016, 8, 8947–8955 CAS.
- K. Ariga, K. Kawakami, M. Ebara, Y. Kotsuchibashi, Q. Ji and J. P. Hill, New J. Chem., 2014, 38(11), 5149–5163 RSC.
- U. Boas and P. M. Heegaard, Chem. Soc. Rev., 2004, 33, 43–63 RSC.
- C. Dufès, I. F. Uchegbu and A. G. Schätzlein, Adv. Drug Delivery Rev., 2005, 57, 2177–2202 CrossRef PubMed.
- Y. Cheng, Z. Xu, M. Ma and T. Xu, J. Pharm. Sci., 2008, 97, 123–143 CrossRef CAS PubMed.
- C. Pan, C. Kumar, S. Bohl, U. Klingmueller and M. Mann, Mol. Cell. Proteomics, 2009, 8, 443–450 CAS.
- S. Sadekar and H. Ghandehari, Adv. Drug Delivery Rev., 2012, 64, 571–588 CrossRef CAS PubMed.
- A. Myc, T. B. Douce, N. Ahuja, A. Kotlyar, J. Kukowska-Latallo, T. P. Thomas and J. R. Baker Jr, Anticancer Drugs, 2008, 19, 143–149 CrossRef CAS PubMed.
- P. Tripathi, A. Khopade, S. Nagaich, S. Shrivastava, S. Jain and N. Jain, Die Pharmazie, 2002, 57, 261–264 CAS.
- U. Gupta, H. B. Agashe, A. Asthana and N. Jain, Biomacromolecules, 2006, 7, 649–658 CrossRef CAS PubMed.
- A. Asthana, A. S. Chauhan, P. V. Diwan and N. K. Jain, AAPS PharmSciTech, 2005, 6, E536–E542 CrossRef PubMed.
- H. Kulhari, D. Pooja, M. K. Singh and A. S. Chauhan, Drug Dev. Ind. Pharm., 2015, 41, 232–238 CrossRef CAS PubMed.
- S. K. Choi, T. Thomas, M.-H. Li, A. Kotlyar, A. Desai and J. R. Baker Jr, Chem. Commun., 2010, 46, 2632–2634 RSC.
- J. Giri, M. S. Diallo, A. J. Simpson, Y. Liu, W. A. Goddard III, R. Kumar and G. C. Woods, ACS Nano, 2011, 5, 3456–3468 CrossRef CAS PubMed.
- C. Anju, N. Anusooya, M. Deeasree, O. Deepak and P. Namboori, Int. J. Pharm. Sci. Health care., 2012, 2, 59–66 Search PubMed.
- S. Sanyakamdhorn, D. Agudelo and H.-A. Tajmir-Riahi, Biomacromolecules, 2013, 14, 557–563 CrossRef CAS PubMed.
- M. O. Rodrigues, M. V. de Paula, K. A. Wanderley, I. B. Vasconcelos, S. Alves and T. A. Soares, Int. J. Quantum Chem., 2012, 112, 3346–3355 CrossRef CAS.
- L. F. Barraza, V. A. Jiménez and J. B. Alderete, Macromol. Chem. Phys., 2015, 216, 1689–1701 CrossRef CAS.
- S. Kumar, L. Jena, S. Galande, S. Daf, K. Mohod and A. K. Varma, Genomics Inform., 2014, 12, 64–70 CrossRef PubMed.
- S. Sharifi, S. Behzadi, S. Laurent, M. L. Forrest, P. Stroeve and M. Mahmoudi, Chem. Soc. Rev., 2012, 41, 2323–2343 RSC.
- M. A. Dobrovolskaia, A. K. Patri, T. M. Potter, J. C. Rodriguez, J. B. Hall and S. E. McNeil, Nanomedicine, 2012, 17, 245–256 CrossRef PubMed.
- D. A. Tomalia and J. M. Frechet, Introduction to the dendritic state, Wiley Online Library, 2002 Search PubMed.
- L.-C. Hsu and R. L. White, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12983–12988 CrossRef CAS.
- M. H. Chiu and E. J. Prenner, J. Pharm. Bioall. Sci., 2011, 3, 39 CrossRef CAS PubMed.
- T. H. Mourey, S. Turner, M. Rubinstein, J. Fréchet, C. Hawker and K. Wooley, Macromolecules, 1992, 25, 2401–2406 CrossRef CAS.
- J. m. Solassol, C. Crozet, V. Perrier, J. Leclaire, F. Beranger, A.-M. Caminade, B. Meunier, D. Dormont, J.-P. Majoral and S. Lehmann, J. Gen. Virol., 2004, 85, 1791–1799 CrossRef CAS PubMed.
- A. Malik, S. Chaudhary, G. Garg and A. Tomar, Adv. Biol. Res., 2012, 6, 165–169 CAS.
- A. K. Patri, J. F. Kukowska-Latallo and J. R. Baker, Adv. Drug Delivery Rev., 2005, 57, 2203–2214 CrossRef CAS PubMed.
- D. Bhadra, S. Bhadra, S. Jain and N. K. Jain, Int. J. Pharm., 2003, 257, 111–124 CrossRef CAS PubMed.
- H. D. Williams, N. L. Trevaskis, S. A. Charman, R. M. Shanker, W. N. Charman, C. W. Pouton and C. J. Porter, Pharmacol. Rev., 2013, 65, 315–499 CrossRef PubMed.
- I. Matai, A. Sachdev and P. Gopinath, Biomater. Sci., 2015, 3, 457–468 RSC.
- R. A. Larson, R. K. Dodge, C. P. Burns, E. J. Lee, R. M. Stone, P. Schulman, D. Duggan, F. Davey, R. Sobol and S. Frankel, Blood, 1995, 85, 2025–2037 CAS.
- L. Zitvogel, L. Apetoh, F. Ghiringhelli, F. André, A. Tesniere and G. Kroemer, J. Clin. Invest., 2008, 118, 1991–2001 CrossRef CAS PubMed.
- D. Bhadra, S. Bhadra, S. Jain and N. Jain, Int. J. Pharm., 2003, 257, 111–124 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26511a |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a