
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Complexes between cationic pyridylphenylene dendrimers and ovine prion protein: do hydrophobic interactions matter?†
S. Sorokinaa,
P. Semenyukb,
Yu. Stroylovab,
V. Muronetzb and
Z. Shifrina *a
*a
aA. N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, Moscow, Vavilova str., 28, 119991, Russian Federation. E-mail: shifrina@ineos.ac.ru; Tel: +7 4991359355
bBelozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow, Leninskye gory, 1/40, 119234, Russian Federation
First published on 15th March 2017
Abstract
Here, the interactions between cationic pyridylphenylene dendrimers of the second, third and fourth generations and full-length ovine prion protein (PrP) were studied using isothermal titration calorimetry (ITC), dynamic light scattering (DLS), and tryptophan fluorescence measurements. A molecular dynamic (MD) study was performed to predict the most possible binding sites for the dendrimer interactions with the protein. All the dendrimers used acted as effective quenchers of fluorescence of the tryptophan residues. The quenching constants calculated according to the Stern–Volmer equation allowed us to quantitatively estimate the efficiency of the dendrimer–protein interactions. ITC data revealed the driving force of the complexation: electrostatic interactions assisted by hydrophobic interactions. Due to the latter, the dendrimer and PrP form complexes which are stable towards the addition of a salt and of the oppositely charged polymer. These results allowed us to propose the mechanism and the model of the pyridylphenylene dendrimer interactions with full-length PrP.
Introduction
Applications of synthetic macromolecules in biomedicine have received considerable attention. Despite numerous reports on applications of synthetic polyelectrolytes for this purpose,1–5 polydispersity of synthetic polymers hampers studies of biomolecule–polymer complexes and makes the interpretation of the results difficult. The absence of full and unambiguous information on complexes does not allow one to predict their performance as diagnostic and therapeutic agents. Therefore, a number of publications in the field are dedicated to monodisperse synthetic polymers, namely dendrimers.6–9 Dendrimers are individual macromolecules of a well-defined structure and a globular shape. They can combine a high molecular weight with a large number of surface functional groups.10,11 These features of dendrimers explain their widespread use in biomedical applications.Dendrimers are actively used as drug delivery systems, vectors for gene therapy, anticancer and antiviral agents.6,9,12,13 The dendrimer application for treatment of neurodegenerative disorders is of particular interest. Prion protein (PrP) is amyloidogenic protein with molecular weight of 23 kDa, which causes several neurodegenerative diseases including scrapie in sheep, bovine spongiform encephalopathy, curu and Creutzfeldt–Jacob disease in humans.14–18 It was demonstrated that dendrimers prevent amyloid aggregation and inhibit fibril formation of PrP.19–23 Furthermore, PAMAM and PPI dendrimers were found to decrease the amyloid PrP content in vivo.24–26 The above applications imply the complex formation between a dendrimer and a corresponding biomolecule. Thus, the detailed analysis of the properties, compositions and characteristics of the complexes formed, the driving force and mechanism of the interactions are extremely important for understanding of the processes leading to the desired therapeutic effect. Such knowledge will allow one to vary the molecular characteristics of macromolecules and, as a result, their complexes in a designed manner to achieve the optimal effect and avoid undesirable side effects.
Among the different classes of dendrimers a special place belongs to cationic pyridyl containing aromatic dendrimers.27 The undeniable advantages of these dendrimers are the constancy of charge and shape as well as their independence of pH due to presence of quaternary nitrogen in the dendrimer pyridine moieties.27 This allows for predictability of the dendrimer interactions with other macromolecules, in particular, with proteins.
Recently we reported that cationic pyridylphenylene dendrimers are able to effectively disrupt amyloid aggregates of the ovine PrP.28 Therefore, the detailed clarification of the mechanism of the interactions of PrP with dendrimers may be an important step for further development of approaches to a disease treatment. The results obtained in our preceding work indicate the decrease of amyloidogenic capability of the PrP after treatment with the dendrimers.28 Nevertheless, the detailed mechanism of their interactions with the protein, analysis of the complexes formed as well as the protein structural changes upon the influence of dendrimers remained unexplored.
Here we report on the formation and structure of the complexes between full-length ovine PrP and cationic pyridylphenylene dendrimers of three generations. We elucidate the mechanism of their interactions using isothermal titration calorimetry (ITC), dynamic light scattering (DLS), and fluorescence quenching. To identify the dendrimer binding sites on the protein molecule, we used molecular dynamic (MD) simulations which were explored earlier to study the mechanism of amyloid aggregation of PrP and protein–polyelectrolyte interactions by different groups.29–32
Experimental section
Materials
Salts, buffers, dextran sulfate and starting materials for dendrimer syntheses were purchased from Sigma-Aldrich and used as received. The plasmid carrying a gene of ovine PrP-V136R154Q171 (25-233) variant was a gift from Prof. H. Rezaei (Institute National de la Recherche Agronomique, Virologie et Immunologie Moleculaires, Jouy-en-Josas, France).Dendrimers
The syntheses of cationic pyridylphenylene dendrimers were described in our preceding paper.27 The dendrimers of the second, third and fourth generations used in the study are depicted in Scheme 1. Numbers of the pyridynium cations and, therefore, the charges of the molecules are 15, 50 and 115 for the second (G2), third (G3) and fourth (G4) generations, respectively.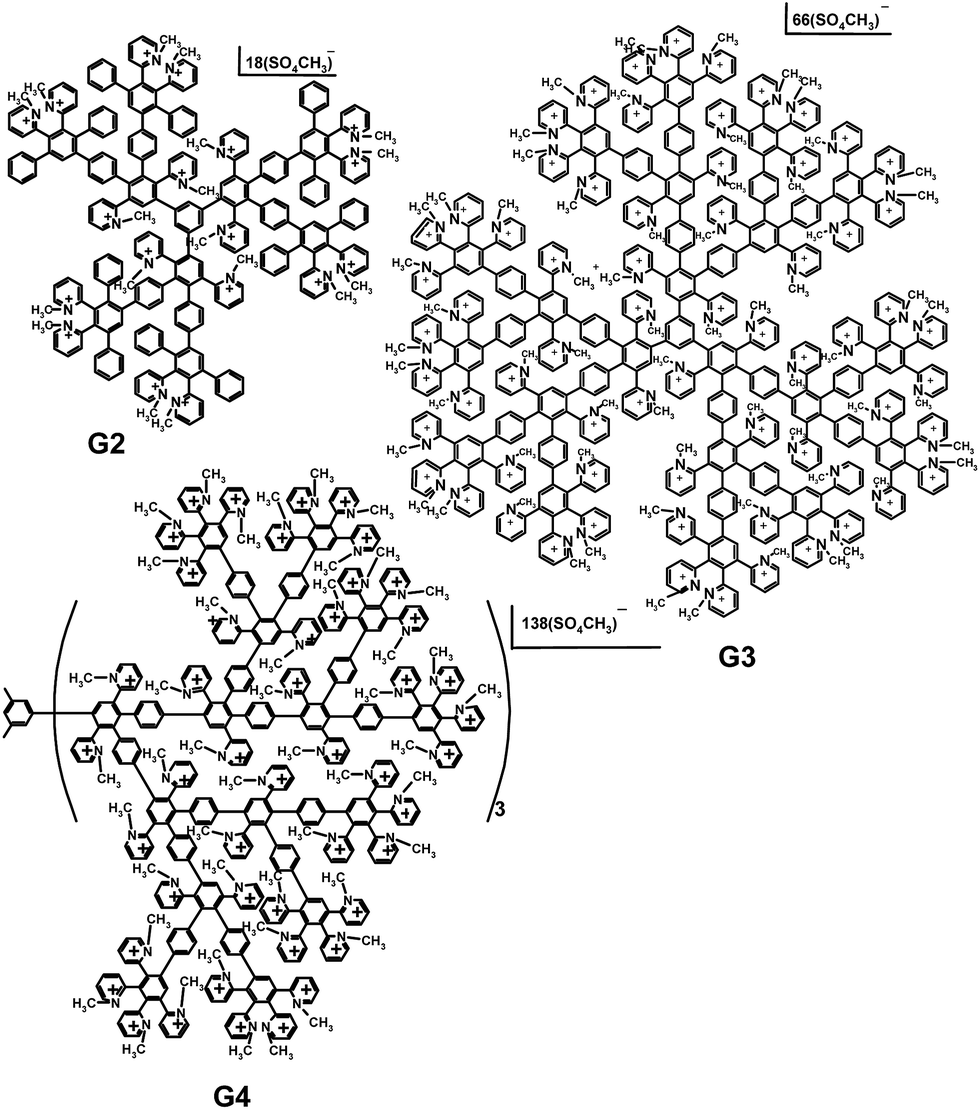 | ||
| Scheme 1 Cationic pyridylphenylene dendrimers of the second (G2), third (G3) and fourth (G4) generations. | ||
Production of the recombinant ovine prion protein
Expression of full-length recombinant ovine PrP was carried out in E. coli culture transformed with a plasmid carrying a gene of ovine PrP. There are several polymorphisms in the gene in sheep, coding PrP, which are associated with different susceptibility to disease. These polymorphisms include a dimorphism at codon 136, 154 and 171, which can respectively lead to amino acids, A/V, R/H and Q/R. We expressed a gene of PrP VRQ variant, coding full-length amino acid sequence without N-terminal signal peptide and C-terminal peptide, with one additional serine residue on the N-terminus. Purification procedure described in33 allows one to obtain a correctly folded monomeric PrP. PrP is produced as inclusion bodies. After the induction of the protein expression with 1 mM of IPTG, the E. coli cells were cultured overnight, centrifuged at 6000 rpm, 25 min and the pellet was thoroughly washed with 50 mM of Tris–HCl buffer, pH = 7. Bacterial pellets were lysed with a buffer, containing 0.5 mg mL−1 of lysozyme and 100 μL of the protease inhibitor cocktail, treated with ultrasound and centrifuged at 8000g, 15 min to obtain the insoluble inclusion bodies containing PrP. Then the inclusion bodies were solubilized with 6 M of guanidine–HCl and PrP was purified using affinity chromatography on the Ni-chelated sepharose due to the prion affinity to metals. SDS PAGE was used to confirm the protein purity. The protein was further lyophilized and stored at −20 °C.Dendrimer–PrP complex preparation
For the complex preparation, an appropriate buffer was added to the required amount of the dendrimer powder and stirred to obtain a homogeneous solution. The dendrimers demonstrated a good solubility in all the buffers used. PrP was dissolved in 100 mM of sodium acetate buffer, pH = 4.0 and transferred into a working buffer via desalting column filled with Sephadex G25. The protein concentration in a stock solution was determined using the Bradford method. In a typical procedure, suitable amounts of the dendrimers and protein stock solutions were mixed together with an extra amount of the buffer if needed, to reach the desired concentrations. Samples were equilibrated at least 2 hours prior any measurement.Isothermal titration calorimetry
ITC experiments were performed using a VP-ITC calorimeter (Microcal, USA) at 25 °C. A solution of PrP (10 μM) in the 20 mM of KH2PO4 buffer pH = 7.5 was titrated by successive 10 μL injections of the 100 μM dendrimer solution in the same buffer. The time intervals between the injections were 5 min. Binding isotherms were corrected by subtracting the dendrimer dilution isotherms determined by titrating the dendrimers solutions into buffer. Data were analyzed using MicroCal Origin 7.0 software with the “one set of sites” model. All samples were degassed before the experiment.Dynamic light scattering
Volume distributions of hydrodynamic diameters were obtained on Zetasizer Nano-ZS Instrument Malvern Instruments, Malvern. The data were recorded at the scattering angle 173° and temperature 25 °C.To analyze the complex stability upon the competitive action of an oppositely charged polymer, namely dextran sulfate (DS) of 15 kDa molecular weight, the solution containing 10 μM of PrP and 25 μM of a dendrimer in 10 mM of MOPS buffer, pH = 7.5 was titrated with DS. The DS concentrations were ranged from 12.5 to 175 μM. After the addition of DS, samples were allowed to equilibrate for 20 min before the measurement.
Fluorescence spectra of the tryptophan residues
Fluorescence spectra were recorded with FluoroMax-3 spectrofluorimeter (Jobin Yvon) at 25 °C. For the experiment, the 5 μM of PrP solution in the 10 mM of MOPS buffer, pH = 7.5 was titrated by a dendrimer. The final dendrimer concentrations in the range of 0.5 to 2 μM were achieved. Samples were stirred and tryptophan emission spectra were taken in 1 cm path length quartz cuvettes using the 295 nm excitation wavelength.To assess the complex stability under the influence of NaCl, a stock solution of the salt was added stepwise to the PrP–dendrimer complex in the 10 mM of MOPS buffer, pH = 7.5 to a final concentration of 2.5 M. The interval between titrations was 3 min. The titration was performed in 1 cm path length quartz cuvettes under continuous stirring at 25 °C. The fluorescence intensity was measured at 295 nm excitation and 352 nm emission wavelengths. The PrP concentration was 5 μM in a solution.
Molecular dynamics simulations
Molecular dynamics simulations were performed with the use of GROMACS 5.1 software.34 The GROMOS 54a7 force field was used. The structure of PrP was retrieved from PDB database, PDB ID 1tqb (chain A). The dendrimer topology was created using the PRODRG server.35 A set of simulations of the system consisting of the one protein and five dendrimer molecules in random orientations was performed. For each dendrimer, we performed five independent simulations with different start positions of the dendrimer molecules. The simulation box was orthogonal with the distance from the border to protein of 5 nm. The distance between the protein molecule and dendrimer molecules was 1–3 nm. In all cases, low molecular weight ions (normally Cl−) for the dendrimer charge compensation were added and short simulations for pre-equilibration were performed. The length of main simulations was 250 ns, the step was 2 fs. The duration was chosen according to the graphs of the protein RMSD and protein–dendrimer salt bridges that reached a plateau. Periodic boundary conditions and the particle mesh Ewald method for handling long-range electrostatic interaction were used. The simulations were performed using a NPT ensemble. Temperature in the simulation box was stabilized at 300 K using velocity rescale thermostat. Pressure coupling was performed with the Berendsen algorithm.36To analyze protein–dendrimer interactions, we calculated a number of bonds. Salt bridges were defined as a pair of negatively charged groups of PrP and positively charged groups of the dendrimer with a distance less than 0.35 nm. A number of nonpolar contacts was defined as a number of phenyl groups of dendrimer within 0.35 nm of the protein. The influence of the binding on the protein structure was also analyzed with calculation of a time dependence of RMSD of the protein molecule excluding protons. Besides, RMSD distribution along the PrP sequence averaged at the last 10 ns of simulations in comparison with the start structure was calculated to determine structure changes induced by interaction. Secondary structure analysis was performed using dssp.37
Results
Isothermal titration calorimetry
ITC method was applied to study the interactions between PrP and dendrimers. Fig. 1 represents the binding isotherms obtained for the G2 and G3 dendrimers. The data clearly indicate the different behavior of G2 and G3 when interacted with PrP. The protein titration with the G2 dendrimer is accompanied with a significant heat release which is less pronounced for G3 (Fig. 1B). Nevertheless, both processes show an exothermic effect. The binding constant (K) for G3 is much higher than that for G2. The calculated thermodynamic parameters imply an entropy contribution in a high binding strength in case of G3, which might be attributed to more pronounced hydrophobic interactions. It should also be noted that a control titration of dendrimers (Fig. 1, top panels, curve 2) into buffer demonstrated a significant heat effect which is presumably caused by the dissociation of the dendrimer assemblies. Therefore, the binding constants as well as the complex stoichiometry are approximate. Regarding the sum of these processes as well as the possible protein conformational changes induced by the dendrimer binding, an accurate determination of a binding constant requires a very complex binding model. Here, we used a simple “one set of sites” model to estimate apparent values. For the G4, the dilution energy appeared to be comparable with the heat effect of the protein–dendrimer interaction that hampered the interpretation of the results and was not pursued.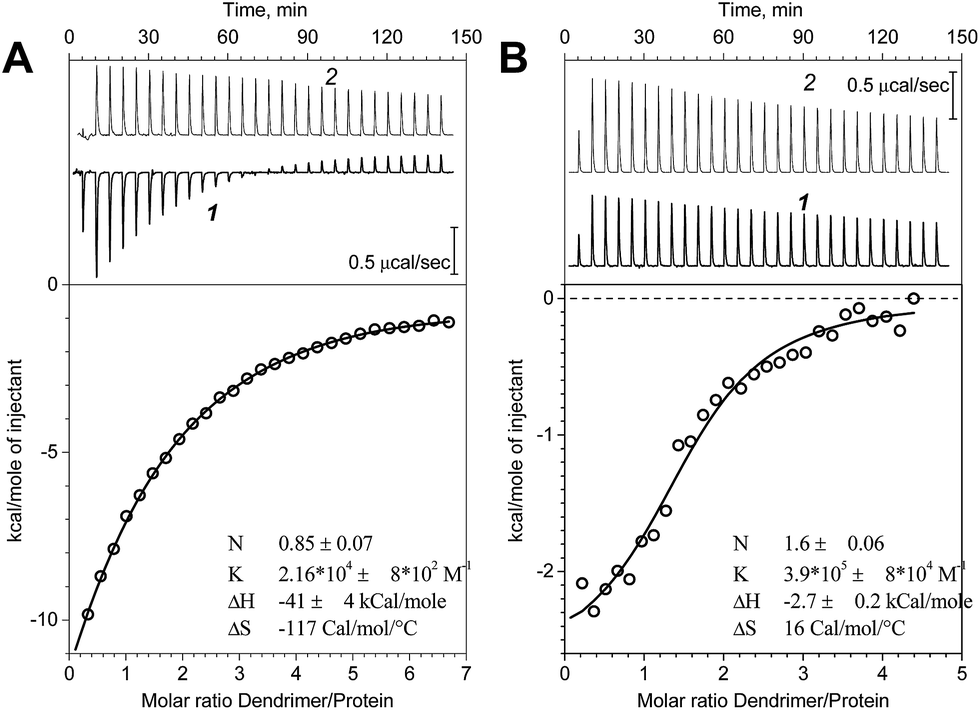 | ||
| Fig. 1 Calorimetric titration of PrP with the G2 (A) and G3 (B). Top panels represent raw data (1) and the control titration of dendrimers in buffer (2); bottom panels represent binding isotherms. The stoichiometry (N), association constant (K), enthalpy (ΔH) and entropy (ΔS) values obtained according to the “one site of sites” model are shown in the bottom panels. | ||
Molecular dynamics simulations
For the further analysis of the interaction mechanism, MD simulations of the systems containing PrP and G2 or G3 were performed. G4 was not studied with MD simulations because of its large size. The potential binding sites have been determined (Fig. 2). According to the results of simulations, PrP is capable of simultaneous binding up to 3–4 molecules of G2 and only two molecules of G3. The difference is likely caused by spatial limitations, because the binding sites are relatively small to bind more than one G3 molecule.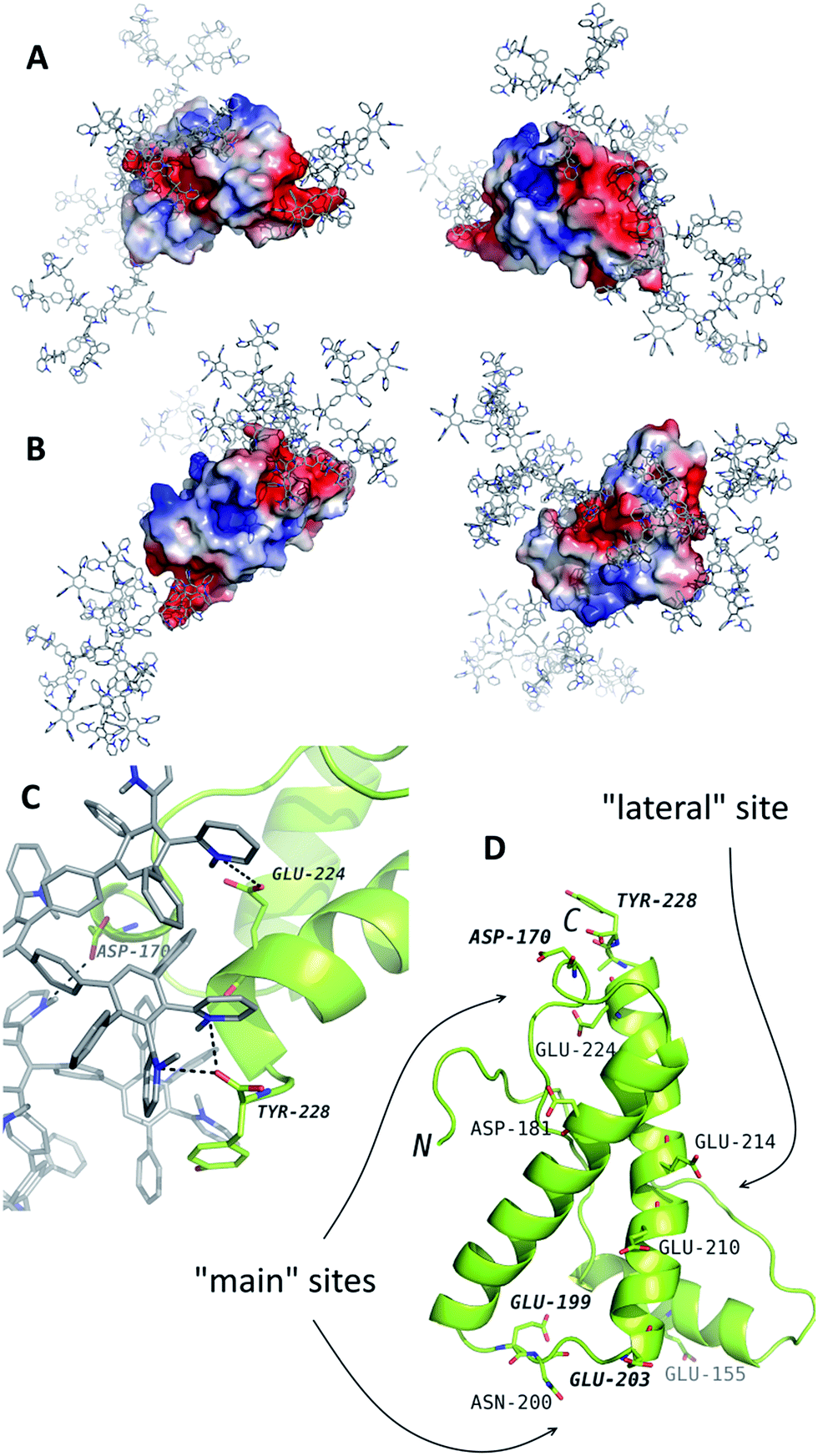 | ||
| Fig. 2 The results of molecular dynamics simulations of PrP (the structure from the PDB entry 1tqb) with G2 (A) and G3 (B). The typical binding positions are shown. Dendrimers are shown in gray with blue nitrogen, the protein is colored according to electrostatic potential of the surface, where positively charged regions are blue, and negatively charged regions are red. The panel (C) represents an example of interactions in details. The dendrimer-binding residues are shown in the panel (D). | ||
The binding sites determined by the MD simulations are shown in Fig. 2. There are two main sites enriched by acidic residues, and one more G2 molecule can bind to a “lateral” side of PrP. Binding of the G3 to the latter site blocks interaction with two main sites and vice versa: if two G3 molecules bind to the main site, no binding occurs at the “lateral” site. All predicted binding sites are negatively charged regions of the protein surface and include the Asp and Glu residues and C-terminal Tyr (Fig. 2D).
As is shown in Fig. 3A, the binding was accompanied by the ion pair formation. All dendrimer molecules, once bound to the protein, were bound up to the final step of the simulation, and the binding positions were almost invariable after 20–30 ns. On the other hand, nonpolar interactions were also involved in the binding, and a number of nonpolar carbon atoms within 0.35 nm of the protein increased slowly (Fig. 3B). It is noteworthy that a total number of ion pairs in case of G3 is the same or higher than that for G2 (Fig. 3A, S1A and B†); hence each G3 molecule forms more number of ion pairs. On the other hand, excepting the one simulation, a number of nonpolar contacts between the protein and G3 is much less as compared to G2 (Fig. 3B, S1C and D†), and there were simulations without any nonpolar contacts in case of G3 (Fig. S1D†). We believe this difference is due to the more prominent presence of phenylene groups in G2 dendrimer (Scheme 1).
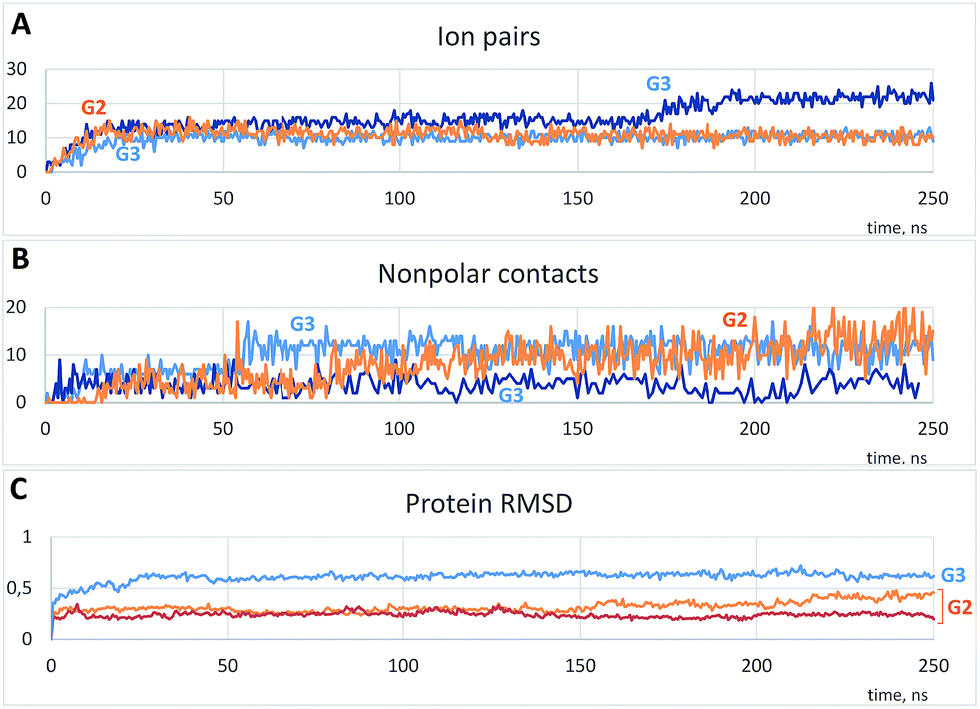 | ||
| Fig. 3 Typical time dependences of a number of ion pairs (A) and nonpolar contacts (B) between protein and dendrimer molecules and protein root-mean-square deviations (RMSD) (C). (A and B) Represent one example with G2 and two independent simulations with G3; (C) represents two curves for G2 and a curve for G3. See Fig. S1† for all simulations. | ||
Changes of the protein structure were also investigated after the binding. According to the RMSD time dependences, the influence of G3 on the protein structure was more pronounced than that of G2 (Fig. 3C and S1E and F†). However, in a few simulations of the protein with G2 RMSD increased after 120 ns of the simulation (two typical curves are shown in Fig. 3C).
As it is seen from the RMSD values averaged per every residue (Fig. 4A and S2†), the major part of the protein structure is intact in both cases, but one of the “main” binding sites (residues 190–200, including Glu199 and Asn200) can be affected by G2. As for G3, all simulations fall into two groups according to the binding position. If G3 is bound to the “lateral” binding site, the major part of the protein structure is still intact, but if the dendrimer is bound to “main” sites, the protein structure changes are much more pronounced, and almost whole protein is affected (Fig. 4A). Nevertheless, the total RMSD values for all simulations with G3 are higher than those for the G2 simulations.
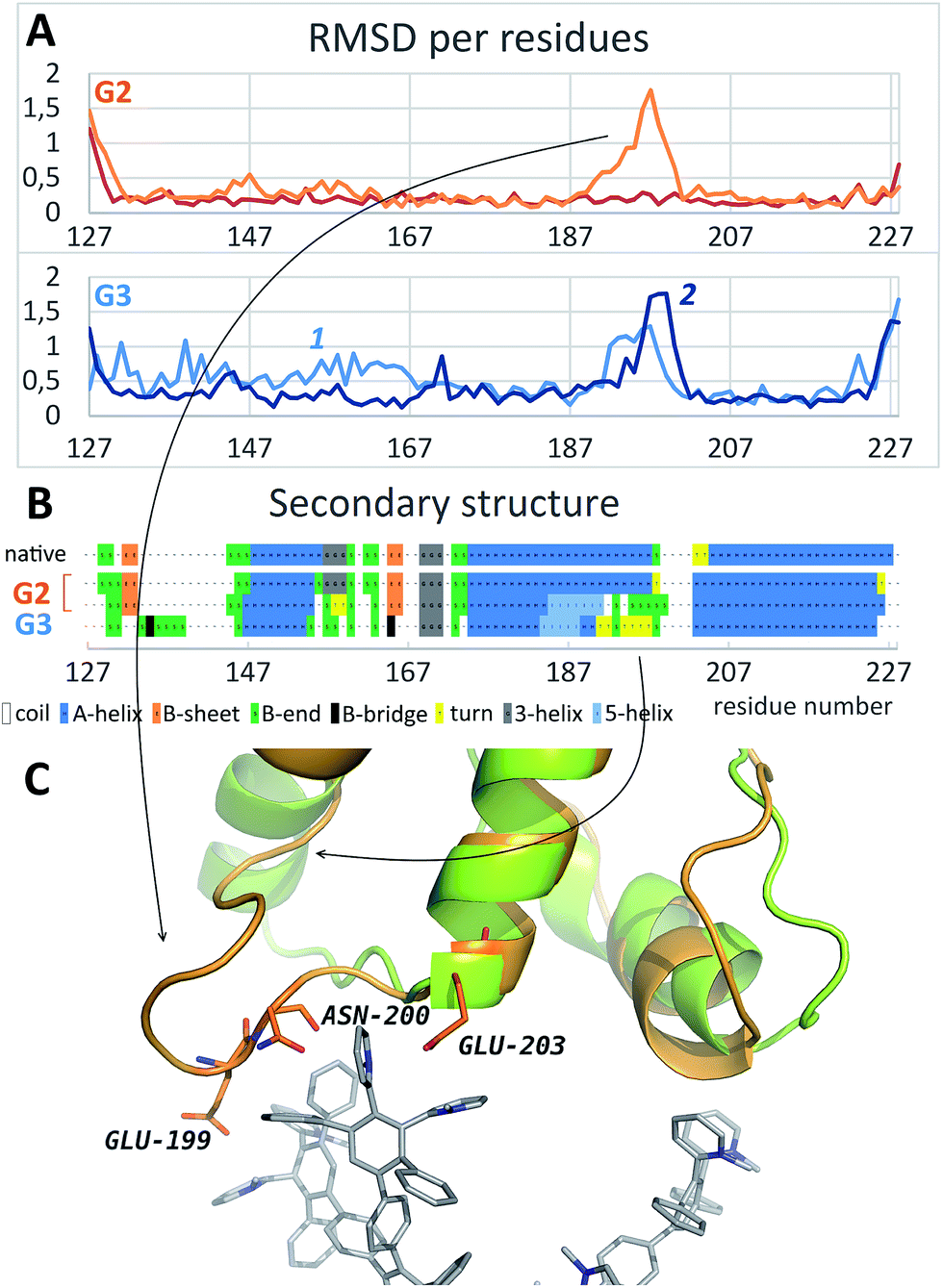 | ||
| Fig. 4 (A) Typical curves of the protein RMSD per residues averaged on the last 10 ns of simulation with G2 (top) and G3 (bottom); curves 1 and 2 represent binding of the dendrimer with lateral and main sites, respectively. (B) Secondary structure of the native PrP before and after the binding of G2 and G3. (C) Comparison of the PrP structure before (green) and after (orange) dendrimer binding (only one of the main binding sites is shown). See also Fig. S2† for RMSD data for all independent trajectories. | ||
Structural changes of the bound protein are shown in Fig. 4C. The binding of the dendrimer to the main sites mentioned above causes rearrangement of the N- and C-terminal regions as well as 190–200 region. Structural changes involve secondary structure changes: a 175–160 alpha-helix partly disappears (Fig. 4B). In the case of G3 binding, 130–170 region is also affected in contrast to G2 binding.
Dynamic light scattering
The hydrodynamic diameters (Dh) of the ovine PrP–cationic pyridylphenylene dendrimer complexes formed were estimated using DLS (Fig. 5). While Dh of PrP is 4 nm, an interaction with the dendrimers leads to the formation of well-defined particles with hydrodynamic diameters of 7, 7.5 and 18 nm for the G2, G3 and G4, respectively. Thus, there is an increase of the hydrodynamic diameter upon complexation.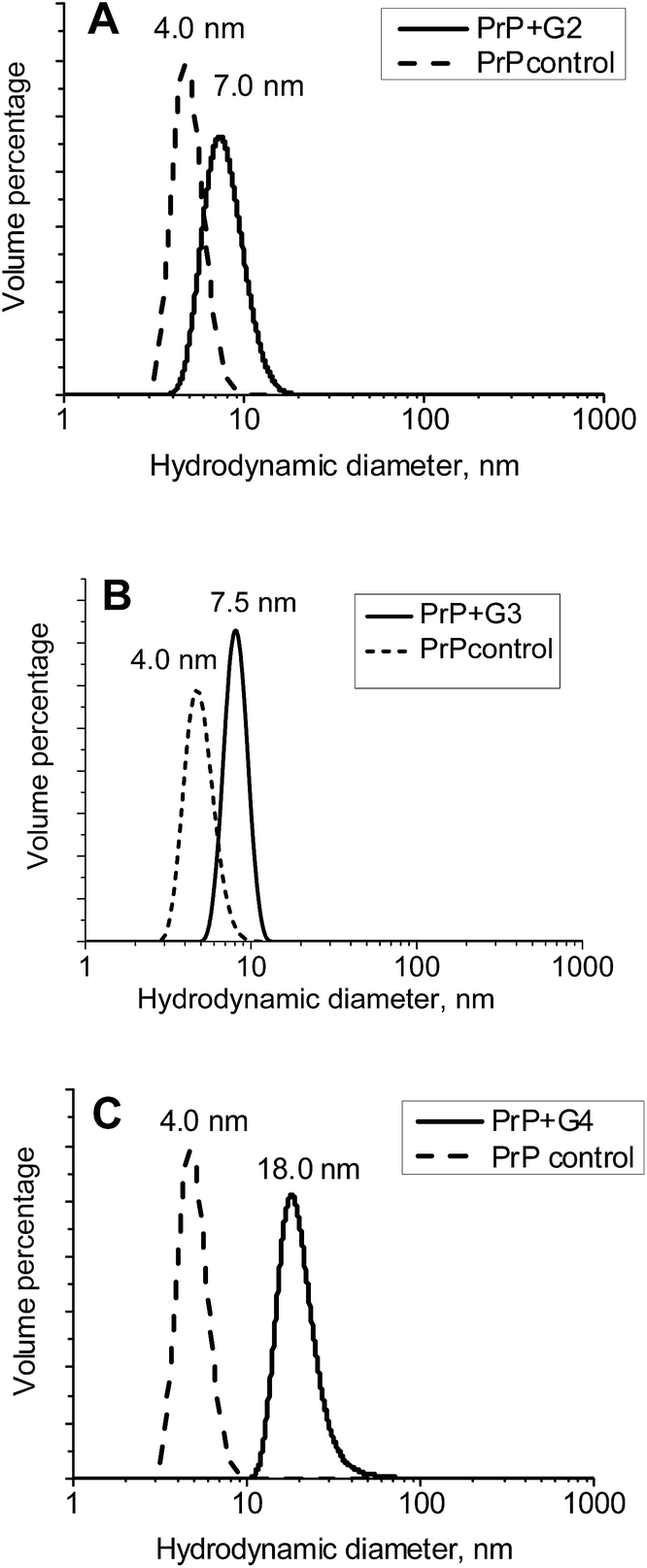 | ||
| Fig. 5 Volume distributions of the hydrodynamic diameters for complexes of PrP with the G2 (A), G3 (B) and G4 (C). | ||
Fluorescence study
Intrinsic fluorescence of proteins is widely used to study their structure as well as the association and denaturation processes. It is primarily caused by the presence of the tryptophan residues whose indole rings are shown to be uniquely sensitive fluorophores. Moreover, a tryptophan molecule is extremely sensitive to the fluorescence quenching by different compounds including N-alkylpyridinium and picolinium salts.38,39The presence of the seven tryptophan residues in the ovine PrP molecule allowed us to apply the fluorescence quenching method for studying the complexation process with the dendrimers because of their quaternary pyridinium groups that interact with tryptophan, resulting in protein fluorescence quenching. It is also noteworthy that the dendrimer absorption spectra do not overlap with the tryptophan emission.
The wavelength of the fluorescence maximum for PrP was observed at 352 nm (Fig. 6, black line). The dendrimers also emitted when excited at the same wavelength as PrP (see Fig. 7 for G2), however, showing the emission peak at 465 nm. Therefore, this emission does not hamper the assessment of the changes in the tryptophan spectrum upon the dendrimer addition. Fig. 6 shows an anticipated decrease of the tryptophan fluorescence intensity upon the interaction with the dendrimers. Indeed, upon the addition of 0.5 μM of G2 (Fig. 6A), the emission spectrum is characterized by minor suppression of the fluorescence intensity of the tryptophan residues and emergence of the shoulder at 465 nm, while the addition of 2 μM of the dendrimer results in the more pronounced decrease accompanied by the growth of the dendrimer fluorescence. Moreover, the effect observed depends on the dendrimer generation.
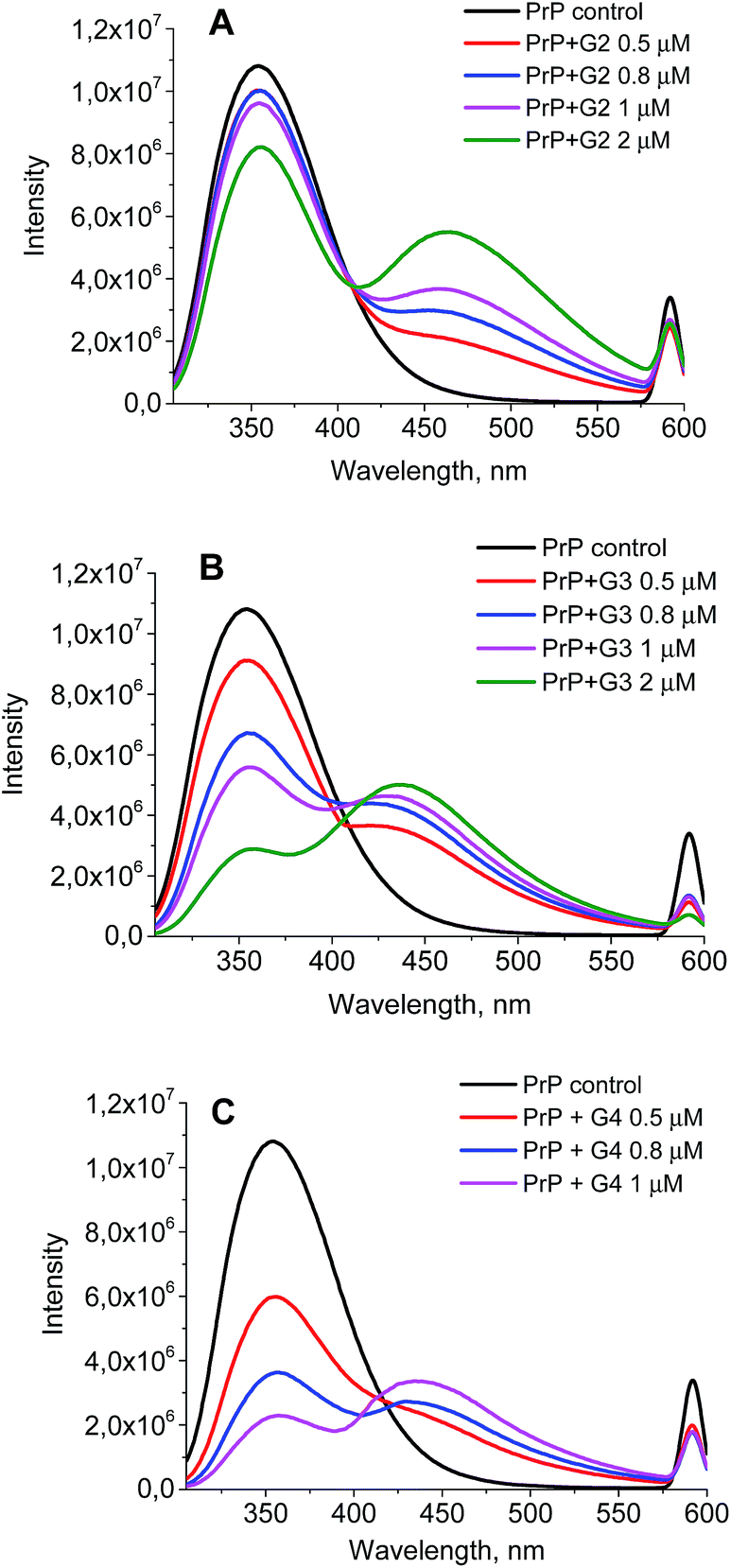 | ||
| Fig. 6 The quenching curves of the intrinsic PrP fluorescence upon the interaction with G2 (A), G3 (B), and G4 (C). | ||
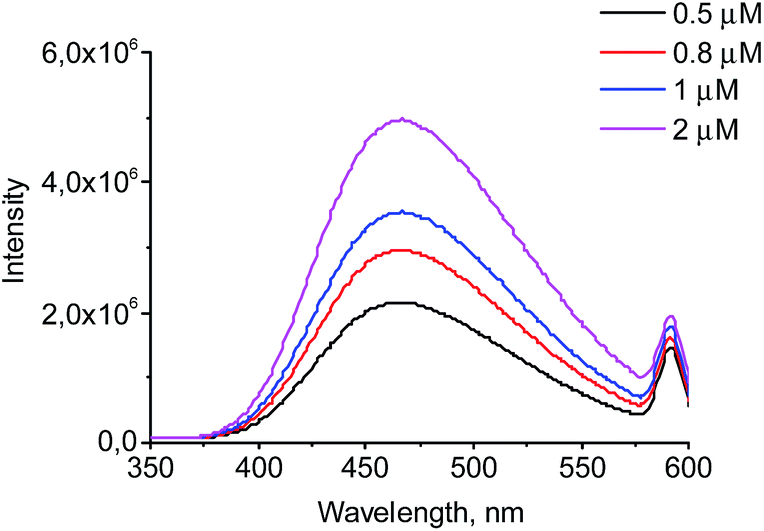 | ||
| Fig. 7 Fluorescence emission spectra for G2. | ||
A quenching efficiency increases with an increase of the dendrimer generation. The most pronounced changes in the PrP fluorescence spectrum occurred upon the addition of G4, while the lowest impact was observed for G2 (Fig. 8).
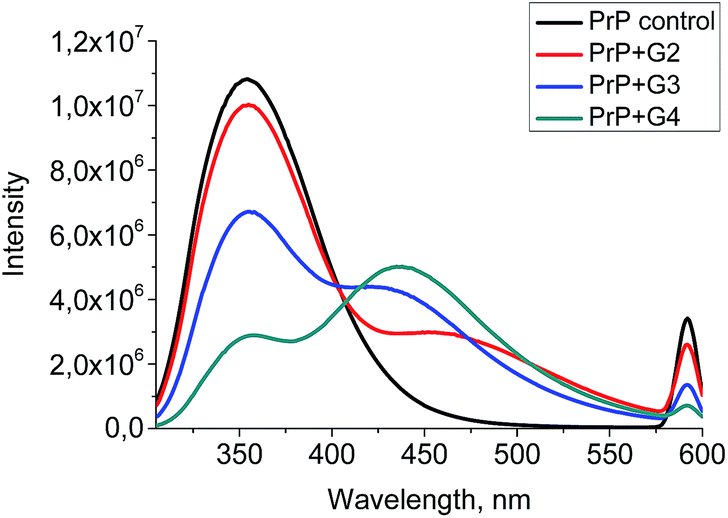 | ||
| Fig. 8 The quenching curves of the intrinsic PrP fluorescence upon the addition of 0.5 μM of the cationic pyridylphenylene dendrimers of different generations. | ||
The fluorescence quenching in proteins is described by the Stern–Volmer equation:40,41
| F0/F = 1 + KSV[Q], |
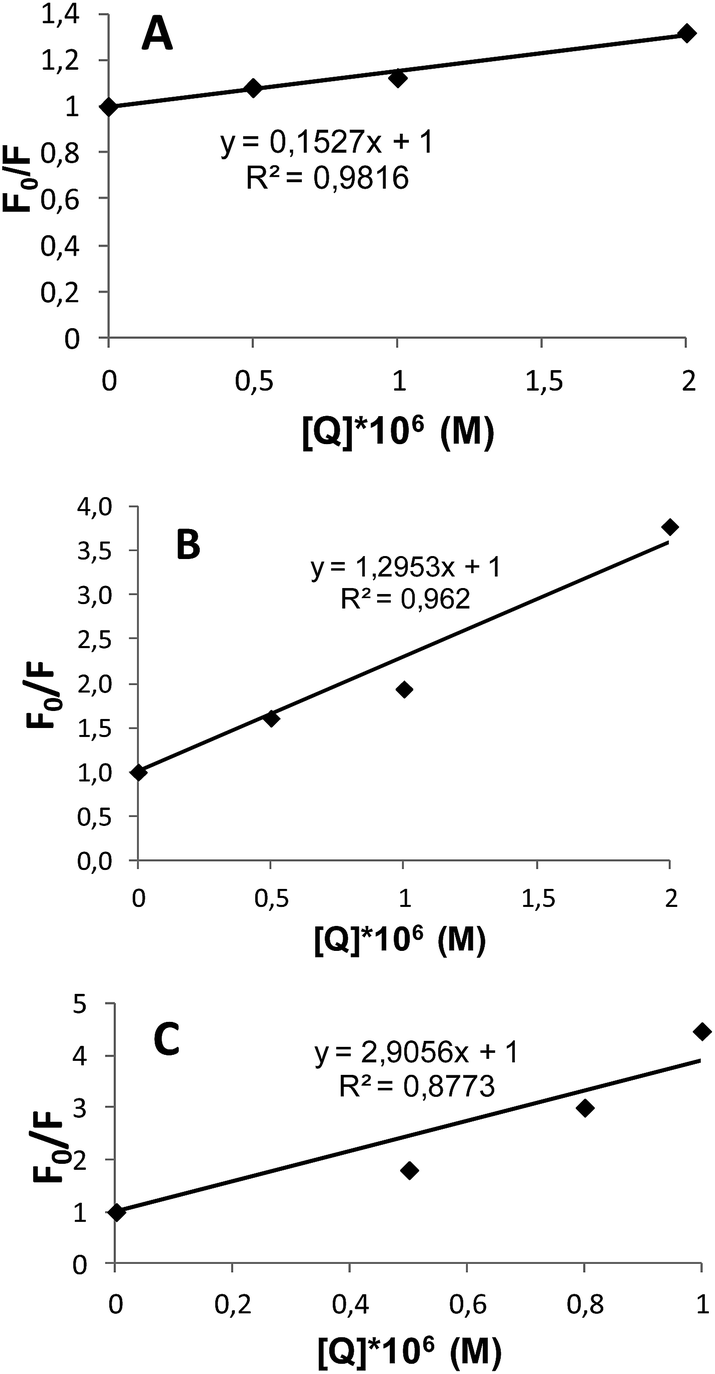 | ||
| Fig. 9 The Stern–Volmer plots for G2 (A), G3 (B) and G4 (C). | ||
| Dendrimer | KSV (M−1) |
|---|---|
| G2 | 1.5 × 105 |
| G3 | 1.35 × 106 |
| G4 | 2.95 × 106 |
Moreover, the fluorescence of the tryptophan residues is known to be extremely sensitive to the changes in their microenvironment, hence the fluorescence is used to assess the protein conformational changes upon the interaction with a ligand. Generally, a red shift of the fluorescence maximum indicates that the tryptophan residues are exposed to the solvent, while a blue shift is a consequence of the screening of tryptophan by a protein matrix.42 The addition of the dendrimers results in a peak broadening and its slight red shift. The effect increases from G2 to G4.
Stability of the complexes
To assess the complex stability, the titration of the PrP–dendrimer complexes with a NaCl solution was performed.The complex dissociation is expected to be accompanied with an increase of the tryptophan fluorescence. However, the fluorimetric titration of the PrP–dendrimer complexes with the salt solution shows no any significant changes in the fluorescence intensity for all the dendrimers studied (Fig. 10). The strongest changes appeared for the PrP–G2 complex although the fluorescence intensity increase did not exceed 10% of the initial value. For G3 and G4, the increase of the fluorescence intensity did not exceed 5% of the initial value and further increase of the salt concentration did not affect the titration curve shape.
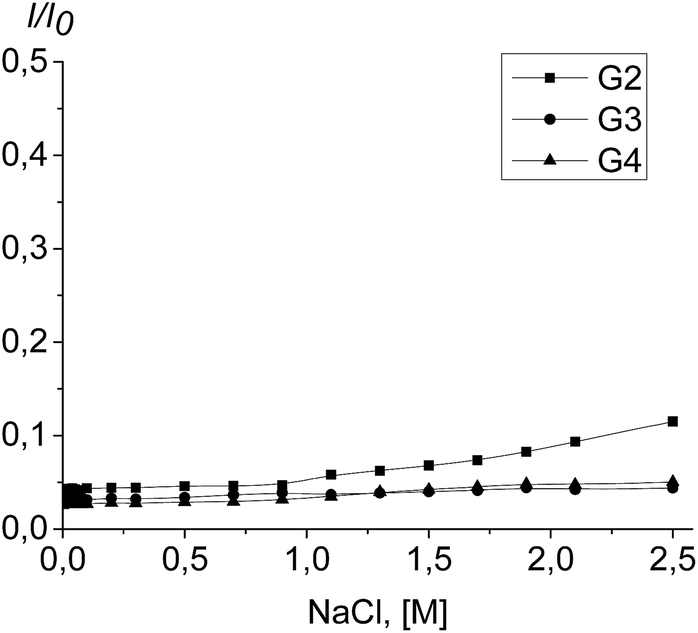 | ||
| Fig. 10 Fluorimetric titration of the PrP–dendrimer complexes with the NaCl solution. | ||
Additionally, the complex stability was studied via the competitive interaction with the oppositely charged dextran sulfate (DS). Ideally, electrostatic interactions of the polycationic dendrimer with polyanion should result in a displacement of the dendrimer from the protein complex and the formation of new DS–dendrimer and PrP–DS complexes due to a higher number of charges in DS, leading to entropically favorable conditions.43 As the DS Dh is approximately equal to that of G2 or G3, the hydrodynamic diameter of the DS–PrP complexes should be similar to that of PrP–dendrimer, i.e., we expected to observe the hydrodynamic diameter of new complexes at 7.5 nm for G3 (Fig. 11). However, to our surprise a stepwise addition of DS to the PrP–G3 complexes did not lead to the release of the individual components or the formation of new complexes with the Dh close to 7.5 nm. Instead, the formation of large aggregates with hydrodynamic diameters increasing with every portion of DS was observed. Finally, the samples demonstrated a bimodal distribution of the particle sizes, revealing the formation of triple DS–dendrimer–PrP complexes due to the DS sorption on the surface of the existing conjugates. The similar results were obtained for the G2 and G4 dendrimers.
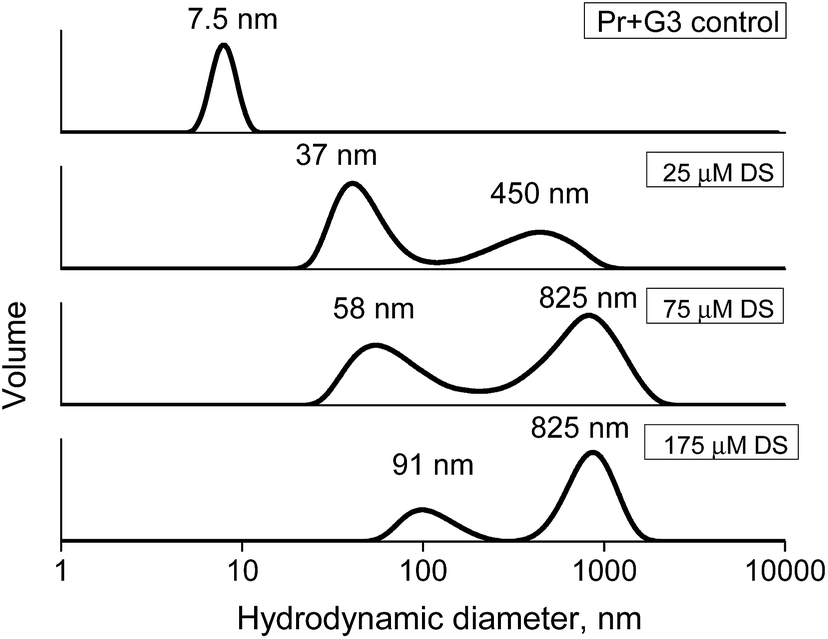 | ||
| Fig. 11 Hydrodynamic diameters of the PrP–G3 complexes upon the addition of dextran sulfate. | ||
Discussion
In this paper a full-length ovine PrP was chosen to study the interactions with the cationic pyridylphenylene dendrimers of three generations: G2, G3, and G4. Both ITC and DLS results clearly demonstrated an effective complexation of these dendrimers with PrP. However, an isoelectric point of PrP is reported to be 9.6. That means the dendrimer and protein both possess a net positive charge under experimental conditions (pH = 7.5). Nevertheless, the observed heat effect for the dendrimer binding to PrP proves the involvement of electrostatic forces which may be attributed to the heterogeneity of the charge localization within the protein molecule. Having the net positive charge, the protein still preserves the sites with negative charges, which open the opportunities for binding of cationic dendrimers. MD simulations also proved this hypothesis. The predicted binding sites for the G2 and G3 dendrimers are located at the protein surface areas containing no positively charged amino acid residues (Fig. 2). The similar behavior was reported for polycation interactions with the protein under its isoelectric point as well as for other similarly charged polymers.44–46 These observations indicate the necessity of the local oppositely charged sites for the effective interactions of polyelectrolytes.On the other hand, the positive value of entropy for the dendrimer–protein interactions (the ITC data) confirms the contribution of hydrophobic interactions. Thus, the dendrimer–protein complex formation might be explained by joint hydrophobic and electrostatic interactions between dendrimers and proteins. At the same time, the heat effect for the G2 binding with the protein is higher than that for G3, which is consistent with the MD results where the total number of bound dendrimer molecules is higher for the G2.
Another evidence of the effective interactions is obtained from the fluorescence quenching. An intrinsic fluorescence of the tryptophan residues was chosen as a sensitive tool to assess the protein conformational changes. The excitation wavelength was set to be 295 nm to avoid the contribution of tyrosine residues. The tyrosine absorption is known to be the smallest and only the tryptophan emission is observed at this wavelength. All the dendrimers studied acted as effective quenchers probably due to the presence of the pyridinium moieties. Therefore, the changes observed in the tryptophan emission spectrum upon interaction with dendrimers may serve as a reliable evidence of an effective complexation.
The most noticeable changes in fluorescence occurred upon the addition of the G4. The calculation of the Stern–Volmer constants allowed one to estimate the efficiency of the interaction. The data in Table 1 show an increase of the interaction strength with an increase of the generation number. Such a behavior differs from that of the linear systems, where the decrease of the polymerization degree leads to the fluorescence quenching enhancement, in particular in case, of oligomeric quenchers.47 Apparently, despite the tendency of the dendrimer hydrophobic parts to be surrounded by a hydrophobic protein matrix, the dendrimer is unable to penetrate the inner parts of the protein molecule due to rigidity of the dendrimer structure. Therefore, the size decrease with the generation number decrease did not lead to the enhancement of the fluorescence quenching.
The tryptophan residues in proteins are reported to exist in five discrete states according to the position of the fluorescence maximum which is dependent on the tryptophan localization and microenvironment.48 Therefore, the changes in the protein emission spectrum reflect its structural changes. The fluorescence maximum of PrP at 352 nm indicates that fluorophore molecules are exposed to the solvent and surrounded by highly mobile water.42 The fluorescence quenching by dendrimers also proved a high availability of the tryptophan residues to dendrimer molecules. The observed peak broadening accompanied by the red shift shows that tryptophans become more exposed to the solvent after the dendrimer addition. KSV values also confirmed high availability of the tryptophan residues and are in agreement with an emission maximum wavelength.
The fluorescence quenching effect of the tryptophan residues was also used to study the stability of the protein–dendrimer complexes. It is known that electrostatic interactions become weaker with the ionic strength increase. Therefore a subsequent addition of sodium chloride to the prion–dendrimer complex should result in a complex dissociation which leads to the increase of the tryptophan fluorescence intensity. Nevertheless, the titration of the complexes with NaCl did not show any noticeable changes in the fluorescence intensity for all the dendrimers studied (Fig. 8). Thus, the PrP–dendrimer complexes are stable in a wide range of ionic strengths and therefore, resistant to the salt addition.
Competitive interactions of the complexes with an oppositely charged polymer also did not lead to dissociation. Indeed, an addition of DS results in a gradual increase of the hydrodynamic diameters of particles and formation of large aggregates instead of dissociation. Together with the fluorimetric titration experiments, these results indicate the crucial role of hydrophobic interactions due to the presence of the phenylene groups in the inner parts of the dendrimer molecule.
The impressive stability of the complexes obtained may be of high importance for the treatment of neurodegenerative disorders with the dendrimers. The disruption of amyloid fibrils is reported to be one of the possible therapies for these diseases.49,50 However, the protein released from the aggregates should not serve as a nucleation center for a new aggregation or re-aggregate once more. This may be realized only in case of strong association of the protein with ligand. Another strategy is the stabilization of the native protein state and the prevention of its further aggregation.51–53 Again, the complex formed has to possess a high stability. It is known that sulfated polymers tightly bound to the protein protect the latter from aggregation more efficiently than polyphosphate.54 Furthermore, polymers that are capable of hydrophobic interactions with the protein are the most efficient in the aggregation suppression55 and the disruption of pre-formed aggregates.5 On the other hand, an importance of hydrophobic interactions for amyloid transformation is stated in some works.56,57 From this point of view, the hydrophobic interactions play a crucial role and might be a significant advantage of the proposed dendrimers. It is noteworthy that one of three binding sites predicted using MD simulations included the 190–200 loop, which can be involved in the amyloid transformation of PrP.29 Furthermore, the dendrimer interaction with this site caused structural rearrangement of the above loop and a partial loss of the secondary structure in the 180–190 region (Fig. 4). Therefore, binding of dendrimers may alter the amyloid transformation of PrP and further amyloid aggregation. It is also noteworthy that the effect observed is exceptionally stable. The properties of the complexes formed do not change under environmental conditions as the cationic pyridylphenylene dendrimers have a rigid structure with the constant spatial architecture and charge. They also disrupt the PrP amyloid aggregates.28 This is in contrast to interactions of flexible PAMAM or PPI dendrimers commonly studied as anti-amyloid agents because these interactions are pH-dependent,19,58,59 i.e., their complex formation with PrP and the complex dissociation depend on pH and ionic strength changes.59,60
To summarize, the pyridylphenylene dendrimer interactions with ovine PrP are driven by the hydrophobic and electrostatic interactions. According to intrinsic fluorescence measurements, the binding of the dendrimer with the protein leads to its minor misfolding. The impact of the dendrimers on the secondary structure of proteins increased from low to high generation dendrimers as is confirmed by the KSV values. The ITC results show the smaller heat effect along with the larger binding constant for G3 than those for G2, indicating an increase of the contribution of hydrophobic interactions with the increase of the generation number. Despite the smaller number of binding sites available for G3 compared with that for G2 (according to MD simulations), it has a more pronounced impact on the protein molecule. The G3–PrP complex is more stable upon the salt addition that is in a good agreement with thermodynamic parameters obtained from the ITC data.
Conclusions
The cationic pyridylphenylene dendrimers were found to form stable complexes with ovine PrP. The PrP molecule has two main sites possessing negative charge for the dendrimer binding and one more in the lateral side allowing the electrostatic interactions. Due to the presence of the phenylene groups in the dendrimer molecule, the hydrophobic interactions are also involved in the binding. The contribution of the hydrophobic interactions increased from low to high generation dendrimers. The more pronounced hydrophobic interactions resulted in more efficient binding and higher dendrimer impact on the protein molecule. Moreover, increase of the hydrophobic interactions lead to increase of the complex stability. Thus the hydrophobic interactions play a crucial role in outstanding stability of the complexes and high affinity and strength of the dendrimer binding with PrP. These results may be of interest for more deep insight to interaction process of dendrimers and PrP and promising further for treatment of neurodegenerative disorders.Acknowledgements
The work was supported by Russian Foundation for Basic Research, projects No. 16-33-00593, 16-34-60089, 15-38-70034.Notes and references
- B. Greenwald, Y. H. Choe, J. McGuire and C. D. Conover, Adv. Drug Delivery Rev., 2003, 55, 217 CrossRef PubMed.
- B. S. Lele, H. Murata, K. Matyjaszewski and A. J. Russell, Biomacromolecules, 2005, 6, 3380 CrossRef CAS PubMed.
- F. Wurm, C. Dingels, H. Frey and H. A. Klok, Biomacromolecules, 2012, 13, 1161 CrossRef CAS PubMed.
- C. M. Niemeyer, Angew. Chem., Int. Ed., 2010, 49, 1200 CrossRef CAS PubMed.
- P. I. Semenyuk, E. V. Moiseeva, Y. Y. Stroylova, M. Lotti, V. A. Izumrudov and V. I. Muronetz, Arch. Biochem. Biophys., 2015, 567, 22 CrossRef CAS PubMed.
- S. Svenson and D. A. Tomalia, Adv. Drug Delivery Rev., 2005, 57, 2106 CrossRef CAS PubMed.
- L. P. Wu, M. Ficker, J. B. Christensen, P. N. Trohopoulos and S. M. Moghimi, Bioconjugate Chem., 2015, 26, 1198 CrossRef CAS PubMed.
- W. D. Jang, K. M. K. Selim, C. H. Lee and I. K. Kang, Prog. Polym. Sci., 2009, 34, 1 CrossRef CAS.
- Y. Y. Cheng, Z. H. Xu, M. L. Ma and T. W. Xu, J. Pharmaceut. Sci., 2008, 97, 123 CrossRef CAS PubMed.
- D. A. Tomalia, Prog. Polym. Sci., 2005, 30, 294 CrossRef CAS.
- J. Fréchet and D. Tomalia, in Dendrimers and other dendritic polymers, ed. D. Tomalia, 2001 Search PubMed.
- C. Dufes, I. F. Uchegbu and A. G. Schatzlein, Adv. Drug Delivery Rev., 2005, 57, 2177 CrossRef CAS PubMed.
- J. Lazniewska, K. Milowska and T. Gabryelak, WIREs Nanomedicine and Nanobiotechnology, 2012, 4, 469 CrossRef CAS PubMed.
- C. M. Dobson, Nature, 2003, 426, 884 CrossRef CAS PubMed.
- S. B. Prusiner, Sci. Am., 1995, 272, 48 CrossRef CAS PubMed.
- P. Gambetti, Z. Dong, J. Yuan, X. Xiao, M. Zheng, A. Alshekhlee, R. Castellani, M. Cohen, M. A. Barria, D. Gonzalez-Romero, E. D. Belay, L. B. Schonberger, K. Marder, C. Harris, J. R. Burke, T. Montine, T. Wisniewski, D. W. Dickson, C. Soto, C. M. Hulette, J. A. Mastrianni, Q. Z. Kong and W. Q. Zou, Ann. Neurol., 2008, 63, 697 CrossRef CAS PubMed.
- S. B. Prusiner and S. J. Dearmond, Amyloid, 1995, 2, 39 CrossRef CAS.
- M. A. Baldwin, F. E. Cohen and S. B. Prusiner, J. Biol. Chem., 1995, 270, 19197 CrossRef CAS PubMed.
- B. Klajnert, M. Cortijo-Arellano, J. Cladera and M. Bryszewska, Biochem. Biophys. Res. Commun., 2006, 345, 21 CrossRef CAS PubMed.
- Y. B. Lim, C. E. Mays, Y. Kim, W. B. Titlow and C. Ryou, Biomaterials, 2010, 31, 2025 CrossRef CAS PubMed.
- H. Cordes, U. Boas, P. Olsen and P. M. H. Heegaard, Biomacromolecules, 2007, 8, 3578 CrossRef CAS PubMed.
- J. M. McCarthy, M. Franke, U. K. Resenberger, S. Waldron, J. C. Simpson, J. Tatzelt, D. Appelhans and M. S. Rogers, PLoS One, 2013, 8, e55282 CAS.
- P. M. H. Heegaard, H. G. Pedersen, J. Flink and U. Boas, FEBS Lett., 2004, 577, 127 CrossRef CAS PubMed.
- J. M. McCarthy, B. R. Moreno, D. Filippini, H. Komber, M. Maly, M. Cernescu, B. Brutschy, D. Appelhans and M. S. Rogers, Biomacromolecules, 2013, 14, 27 CrossRef CAS PubMed.
- S. Supattapone, H. O. B. Nguyen, F. E. Cohen, S. B. Prusiner and M. R. Scott, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 14529 CrossRef CAS.
- S. Supattapone, H. Wille, L. Uyechi, J. Safar, P. Tremblay, F. C. Szoka, F. E. Cohen, S. B. Prusiner and M. R. Scott, J. Virol., 2001, 75, 3453 CrossRef CAS PubMed.
- Z. B. Shifrina, N. V. Kuchkina, P. N. Rutkevich, T. N. Vlasik, A. D. Sushko and V. A. Izumrudov, Macromolecules, 2009, 42, 9548 CrossRef CAS.
- S. A. Sorokina, Y. Y. Stroylova, Z. B. Shifrina and V. I. Muronetz, Macromol. Biosci., 2016, 16, 266 CrossRef CAS PubMed.
- N. Chakroun, A. Fornili, S. Prigent, J. Kleinjung, C. A. Dreiss, H. Rezaei and F. Fraternali, J. Chem. Theory Comput., 2013, 9, 2455 CrossRef CAS PubMed.
- S. Menon and N. Sengupta, Mol. BioSyst., 2015, 11, 1443 RSC.
- S. Zhou, D. Shi, X. Liu, H. Liu and X. Yao, Sci. Rep., 2016, 6, 21804 CrossRef CAS PubMed.
- A. A. Sofronova, D. B. Evstafyeva, V. A. Izumrudov, V. I. Muronetz and P. I. Semenyuk, Polymer, 2017, 113, 39–45 CrossRef CAS.
- H. Rezaei, D. Marc, Y. Choiset, M. Takahashi, G. H. B. Hoa, T. Haertle, J. Grosclaude and P. Debey, Eur. J. Biochem., 2000, 267, 2833 CrossRef CAS PubMed.
- S. Pronk, S. Páll, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. van der Spoel, B. Hess and E. Lindahl, Bioinformatics, 2013, 29, 845 CrossRef CAS PubMed.
- A. W. Schüttelkopf and D. M. F. van Aalten, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1355 CrossRef PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684 CrossRef CAS.
- R. P. Joosten, T. A. te Beek, E. Krieger, M. L. Hekkelman, R. W. Hooft, R. Schneider, C. Sander and G. Vriend, Nucleic Acids Res., 2011, 39, D411 CrossRef CAS PubMed.
- G. A. Davis, J. Chem. Soc., Chem. Commun., 1973, 728 RSC.
- M. Shinitzsky and B. Rivnay, Biochemistry, 1977, 16, 982 CrossRef.
- M. R. Eftink, Methods Biochem. Anal., 1991, 35, 127 CAS.
- B. Klajnert and M. Bryszewska, Bioelectrochemistry, 2002, 55, 33 CrossRef CAS PubMed.
- Y. K. Reshetnyak and E. A. Burstein, Biophys. J., 2001, 81, 1710 CrossRef CAS PubMed.
- I. N. Shalova, I. N. Naletova, L. Saso, V. I. Muronetz and V. A. Izumrudov, Macromol. Biosci., 2007, 7, 929 CrossRef CAS PubMed.
- A. N. Gupta, H. B. Bohidar and V. K. Aswal, J. Phys. Chem. B, 2007, 111, 10137 CrossRef CAS PubMed.
- S. Wang, K. Chen, L. Li and X. Guo, Biomacromolecules, 2013, 14, 818 CrossRef CAS PubMed.
- A. A. Sofronova, V. A. Izumrudov, V. I. Muronetz and P. I. Semenyuk, Polymer, 2017, 108, 281 CrossRef CAS.
- V. A. Izumrudov, N. V. Kuchkina, A. L. Rusanov and Z. B. Shifrina, Polym. Sci., Ser. A, 2009, 51, 229 CrossRef.
- E. A. Burstein, S. M. Abornev and Y. K. Reshetnyak, Biophys. J., 2001, 81, 1699 CrossRef CAS PubMed.
- X. Y. Meng, L. A. Munishkina, A. L. Fink and V. N. Uversky, Biochemistry, 2009, 48, 8206 CrossRef CAS PubMed.
- T. Hard and C. Lendel, J. Mol. Biol., 2012, 421, 441 CrossRef PubMed.
- M. Citron, Nat. Rev. Drug Discovery, 2010, 9, 387 CrossRef CAS PubMed.
- W. Hoyer, C. Gronwall, A. Jonsson, S. Stahl and T. Hard, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5099 CrossRef CAS PubMed.
- C. M. Wischik, P. C. Edwards, R. Y. K. Lai, M. Roth and C. R. Harrington, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 11213 CrossRef CAS.
- P. I. Semenyuk, V. I. Muronetz, T. Haertle and V. A. Izumrudov, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 4800 CrossRef CAS PubMed.
- N. Martin, D. Ma, A. Herbet, D. Boquet, F. M. Winnik and C. Tribet, Biomacromolecules, 2014, 15, 2952 CrossRef CAS PubMed.
- A. K. Jana, J. C. Jose and N. Sengupta, Phys. Chem. Chem. Phys., 2012, 15, 837 RSC.
- S. G. Kang, T. Huynh, Z. Xia, Y. Zhang, H. Fang, G. Wei and R. Zhou, J. Am. Chem. Soc., 2013, 135, 3150 CrossRef CAS PubMed.
- B. Klajnert, M. Cortijo-Arellano and J. Cladera, FEBS J., 2006, 273, 239 Search PubMed.
- L. Giehm, C. Christensen, U. Boas, P. A. H. Heegaard and D. E. Otzen, Biopolymers, 2008, 89, 522 CrossRef CAS PubMed.
- B. Klajnert, J. Cladera and M. Bryszewska, Biomacromolecules, 2006, 7, 2186 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26563d |
| This journal is © The Royal Society of Chemistry 2017 |