
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation mechanism of hafnium oxide nanoparticles by a hydrothermal route
Yingying Wan and
Xingping Zhou *
*
College of Chemistry, Chemical Engineering and Biotechnology, Donghua University, Shanghai 201620, PR China. E-mail: xpzhou@dhu.edu.cn; Fax: +86-21-67792657; Tel: +86-21-67792657
First published on 23rd January 2017
Abstract
Hafnium oxide nanoparticles (NPs) were synthesized by a hydrothermal route, using hafnium tetrachloride (HfCl4) as the starting material and sodium hydroxide (NaOH) to adjust the pH. Through changing the aging temperature, concentration of NaOH and reaction time, both pure tetragonal hafnium oxide (t-HfO2) and pure monoclinic hafnium oxide (m-HfO2) were obtained. X-ray diffraction (XRD) spectra and transmission electron microscopy (TEM) images indicated that the shapes of t-HfO2 NPs and m-HfO2 NPs were near-spherical and spindle-like, respectively. The formation of t-HfO2 NPs or m-HfO2 NPs is probably related to their crystal cell structure, thermodynamic and kinetic stabilities. Tetragonal HfO2 is produced originally in the process of the formation of monoclinic HfO2. A higher temperature, lower concentration of NaOH, longer reaction time and addition of m-HfO2 seeds are beneficial for the formation of m-HfO2 NPs. By analysis and calculation of the equilibrium constants involving hydrolysis of hafnium ions, the changes in the mole fractions of hafnium hydro-complexes with pH were determined. The Hf(OH)62− ion is assigned to the precursory hydro-complex for the formation of HfO2 nanoparticles transformed from Hf(OH)4 gel according to a comparison between the influences of pH on the equilibrium and the formation of HfO2 particles. Moreover, the formation of HfO2 NPs was obviously promoted and the size was reduced by addition of seeds, suggesting that the formation of HfO2 NPs is controlled by the surface-deposition reaction. The above results are of great importance for studying nano-inorganic solution chemistry.
Introduction
Nanostructured materials have a lot of important applications in various fields because of their unique properties.1–3 Hafnium is known as the “little brother” of titanium and zirconium. Its dioxide (HfO2) is a material with a number of technologically attractive properties such as high melting point (2758 °C), high dielectric constant (≈30), high chemical stability, a wide band gap (>5.0 eV), and high neutron absorption cross section.4,5 It often plays an important role in the continuous down-scaling of integrated circuits since new insulating materials with a high dielectric constant are being researched to replace SiO2 as a gate dielectric.6,7 In terms of structural characteristics, hafnium oxide exists in three polymorphic structures, namely monoclinic (m-HfO2) at low temperature, tetragonal (t-HfO2) above 2050 K, and cubic (c-HfO2) at around 2803 K.8–10 Each structure has different applications.The optical, electrical and other properties of HfO2 nanoparticles (NPs) are strongly affected by their size, morphology, and surface characteristics.11,12 Therefore, it is necessary to consider the required specifications of HfO2 NPs before choosing the synthesis method. Up to now, researchers have developed some synthesis approaches such as gel–sol method,13 microemulsion processes14 and precipitation.10,15 However, these methods have some drawbacks, such as complicated operation, low yield and agglomerated products. In addition, all these ways only can obtain the m-HfO2 NPs. Preparation of t-HfO2 NPs has been rarely reported so far. More importantly, the formation mechanism and crystal form or morphology control of HfO2 NPs are less reported.
Recently, many efforts have been undertaken to improve the synthesis method. As a promising approach, hydrothermal synthesis16–18 is widely used to synthesize nanocrystalline oxide materials due to the mild reaction condition, simple operation and no roasting. A. Sahraneshin16 synthesized three differently shaped HfO2 NPs via a surfactant-assisted hydrothermal reaction in highly alkaline media. The products obtained were flower-like nanostructures (20.0 nm in diameter), polycrystalline nanoagglomerates (25.0 nm in diameter), and water-dispersible single nanoparticles (4.0 nm in diameter). In addition, S. A. Eliziario8 successfully obtained the HfO2 NPs by the microwave hydrothermal method. The temperature chosen was 140 °C and these nanostructures exhibited an average width in the range from 15.0 to 75.0 nm as well as an average height between 85.0 and 105.0 nm. The hydrothermal method allows to control size, morphology and the composition of the products, and to obtain homogeneous and dispersed nanoparticles, through varying parameters such as temperature, pressure, duration of the process, concentration and acidity (pH).19–21 The control of particle size and morphology is the result of processes of coarsening and redissolution–recrystallization which takes place under conditions of high pressure and temperature. The main advantages of hydrothermal method are simplicity and convenience.
In common, hydrothermal method still needs improvement in terms of yield, monodispersity, structural perfection, and size control. For synthesizing some nanoparticles, seeding is a very effective way to control particle size, to determine the crystal form22 and even to study the nucleation and growth mechanisms. Some researchers have made in-depth studies on this technology. Our previous studies reported that the addition of seeds has been able to systematically control the particle morphology and studied the nucleation and growth mechanisms of ZrO2 (ref. 22) and TiO2.23 The seeds inhibit aggregation and the formation of secondary particles, so as to obtain the product wanted. Generally, the surface-deposition of monomers is not often the rate-determining step for the formation of nanoparticles unless the formation is accelerated and the particle size is controlled by seeding. Also, in this case, the size-distribution of nanoparticles is commonly narrowed.
In this paper, we used a hydrothermal route to prepare hafnium oxide nanoparticles. Effects of temperature, NaOH concentration, duration and crystal form of seeds were investigated to study the formation mechanisms of HfO2 NPs from aspects of precursory complexes, thermodynamic and kinetic stabilities.
Experimental
Materials
HfCl4 (AR, 99.0%), sodium hydroxide (AR, 96.0%), sodium dodecyl-benzenesulfonate (AR, 88.0%), dodecylamine (CP) were all purchased from Sinopharm Chemical Reagent Co., Ltd.Synthesis
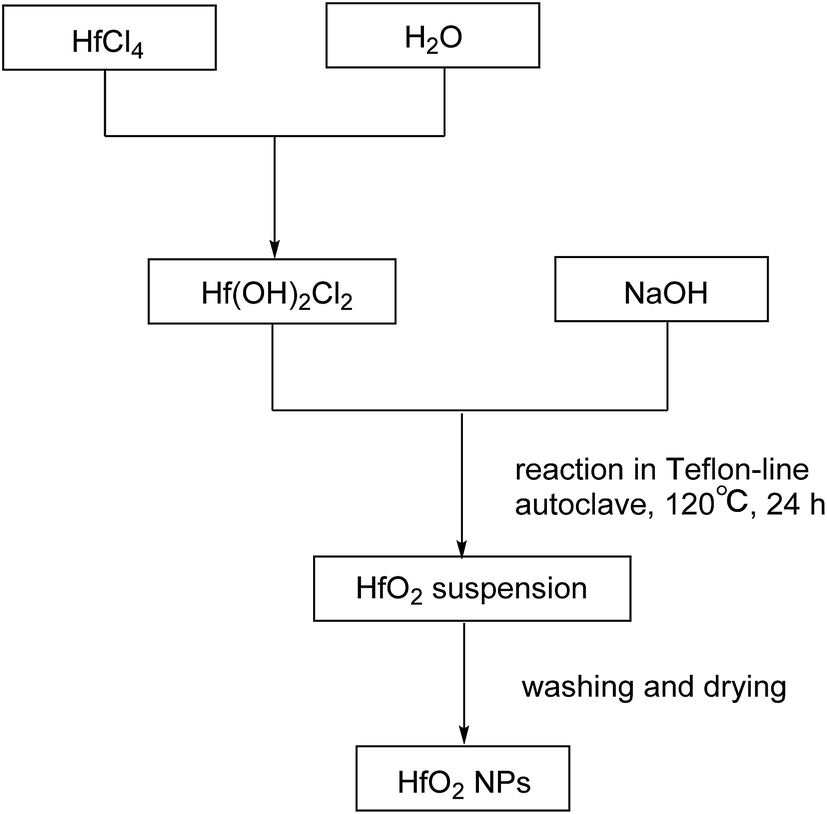 | ||
| Scheme 1 Preparation of HfO2 NPs. | ||
The products were purified by centrifugation for three cycles with alcohol and de-ionized water alternately after the autoclave was cooled down. Finally the precipitate was dried at 50 °C for 24 h. The procedures are summarized in Scheme 2.
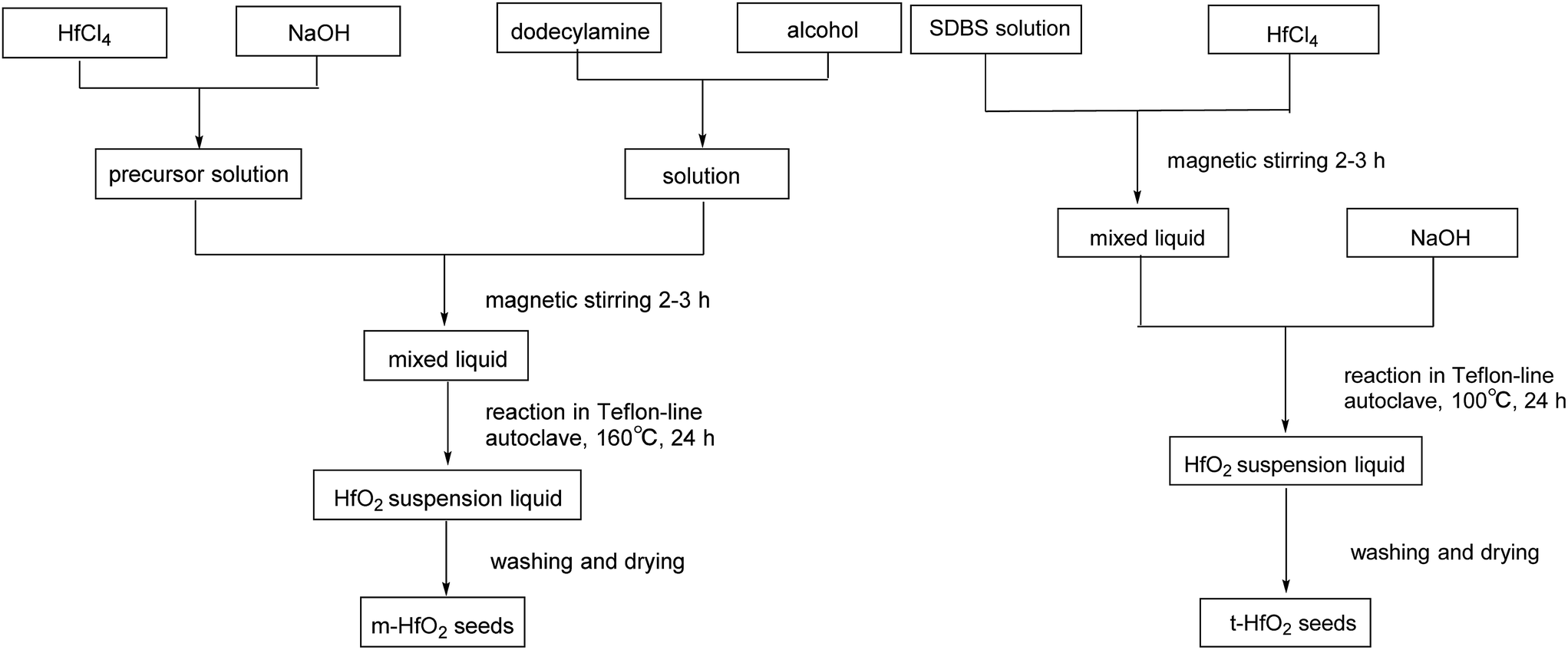 | ||
| Scheme 2 Preparation of HfO2 seeds. | ||
Characterization
The different morphologies and phases presenting in the solids were measured using X-ray powder diffraction (XRD, D/max, Rigaku, Tokyo, Japan) with Cu-Kα radiation (λ = 1.5418 Å) in a 2θ–θ setup, the 2θ angle was scanned from 0 to 90°. JADE software (MDI JADE 7 Materials Data XRD Pattern Processing, Identification, and Quantification) was used to evaluate/analyze the XRD patterns. The JCPDS card no. 06-0318 and the JCPDS card no. 53-0550 were used as the XRD standard files of m-HfO2 and t-HfO2, respectively. The pH was measured by Sartorius PB-10.Transmission electron microscopy (TEM) images were recorded with a JEOL JEM 2100F transmission electron-microscope, to analyze the product size and morphology. The samples used for TEM observations were prepared by dispersing the NPs in ethanol followed by ultrasonic vibration for 30 min, and then placing a drop of this dispersion onto a copper grid before loading into the instrument.
Results and discussion
Preparation of HfO2
Fig. 1 shows the XRD spectra of both tetragonal hafnium oxide nanoparticles (t-HfO2 NPs) and monoclinic hafnium oxide nanoparticles (m-HfO2 NPs) prepared. The tetragonal phase of HfO2 was obtained under otherwise the standard conditions at 100 °C, the monoclinic phase of HfO2 was obtained at 160 °C. In Fig. 1(a), the major characteristics peaks found at 30.48°, 35.30°, 50.68°, 60.26° on the 2θ scale correspond to the {111}, {200}, {220} and {311} planes respectively, in good agreements with the standard PDF card of t-HfO2 (JCPDS: no. 53-0550). The corresponding TEM image of near-spherical t-HfO2 particles (4.0 nm) is presented in Fig. 2(a). In Fig. 1(b), the major characteristics peaks found at 24.64°, 28.30°, 31.76°, 50.46° on the 2θ scale correspond to the {011}, {−111}, {111}, {220} planes respectively, in good agreements with the standard PDF card of m-HfO2 (JCPDS: no. 06-0318). The corresponding TEM images of spindle-like m-HfO2 particles (ca. 100 nm × 50 nm) with a high aspect ratio is presented in Fig. 2(b). These nanoparticles are uniform, but some particles gather together.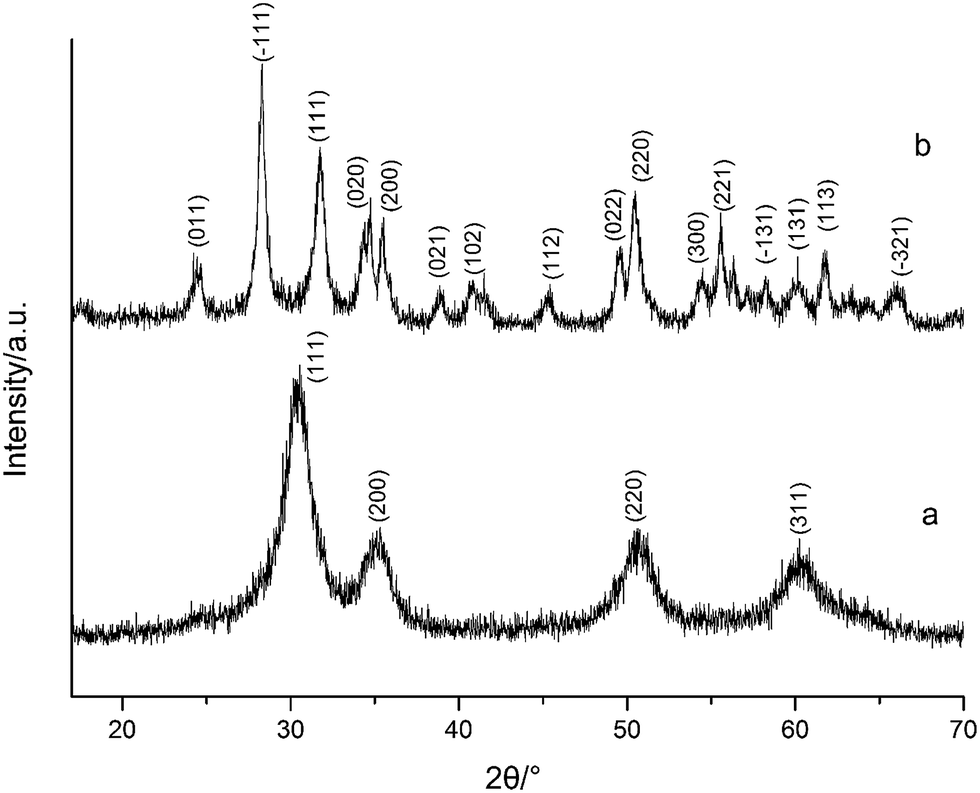 | ||
| Fig. 1 XRD spectra of HfO2 NPs prepared under otherwise the standard conditions (3.0 M NaOH and 24 h) at different temperatures: (a) 100 °C, (b) 160 °C. | ||
 | ||
| Fig. 2 HRTEM images (1) and TEM images (2) of HfO2 NPs prepared under otherwise the standard conditions (3.0 M NaOH and 24 h) at different temperatures: (a) 100 °C, (b) 160 °C. | ||
The high-resolution TEM images shown in Fig. 2(a1) indicates high crystallinity of the structure, the d spacing of lattice fringes of 0.25 nm and 0.29 nm are indexed to the (200) and (111) plane of t-HfO2, respectively. In Fig. 2(b1), the d spacing of lattice fringes of 0.28 nm and 0.32 nm are indexed to the (111) and (−111) plane of m-HfO2, respectively. All the d spacing values are in close agreement with those from t-HfO2 (JCPDS: no. 53-0550) and m-HfO2 (JCPDS: no. 06-0318).
Effects of reaction conditions on the formation of HfO2 nanoparticles
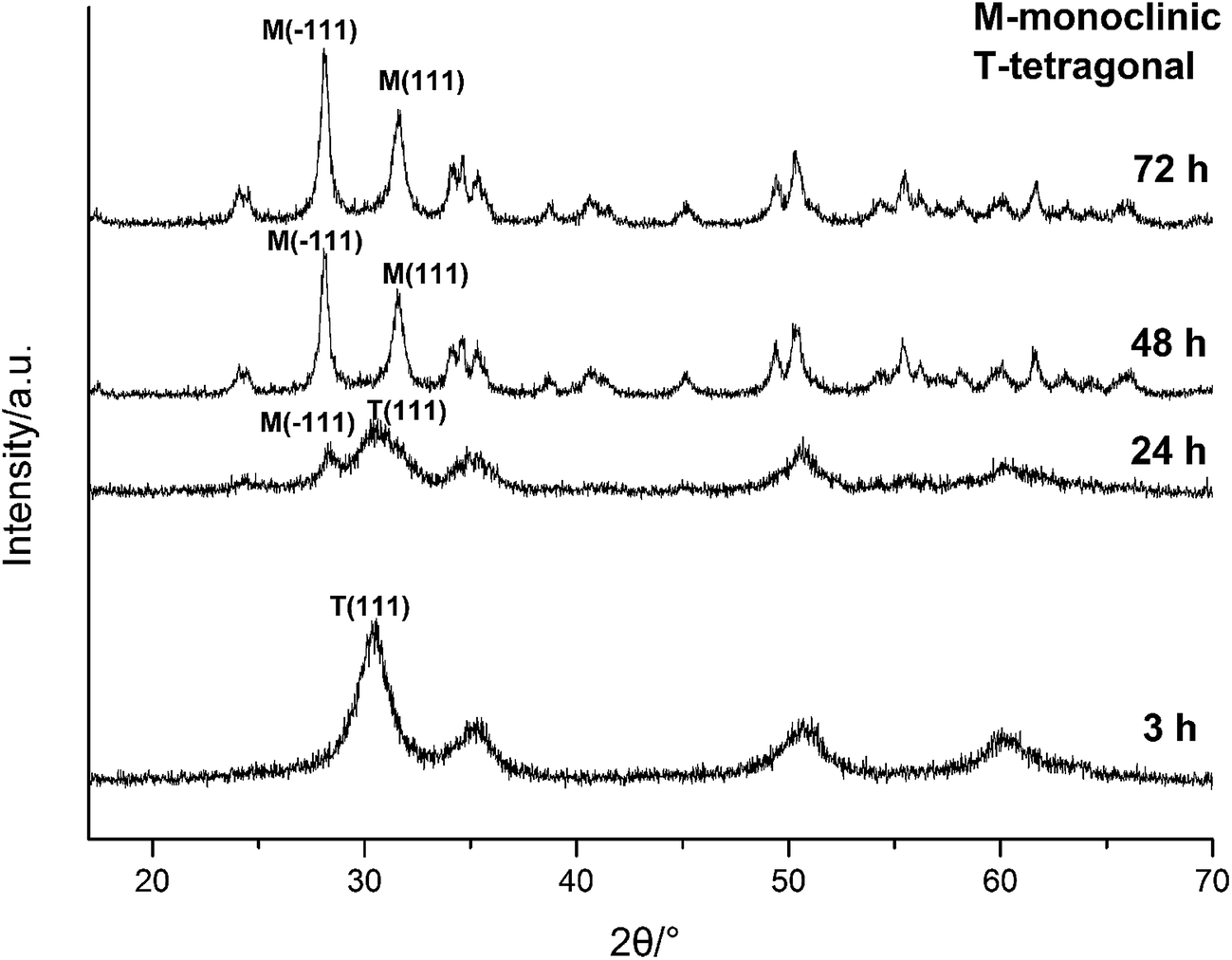 | ||
| Fig. 3 XRD spectra of HfO2 NPs prepared under otherwise the standard conditions (3.0 M NaOH and 120 °C) with different reaction time. | ||
 | ||
| Fig. 4 TEM images of HfO2 NPs prepared under otherwise the standard conditions (3.0 M NaOH and 120 °C) with different reaction time: (a) 3 h, (b) 24 h, (c) 72 h. | ||
The above analysis reflected a phenomenon that the m-HfO2 was not formed at beginning, but transformed from the t-HfO2. In the early stage of reaction, the t-HfO2 NPs were firstly generated and then aggregated to form large secondary particles, which subsequently re-crystallized to produce m-HfO2 NPs. This fact indicates that m-HfO2 NPs probably have lower kinetic stability and higher thermodynamic stability in comparison to t-HfO2 NPs.
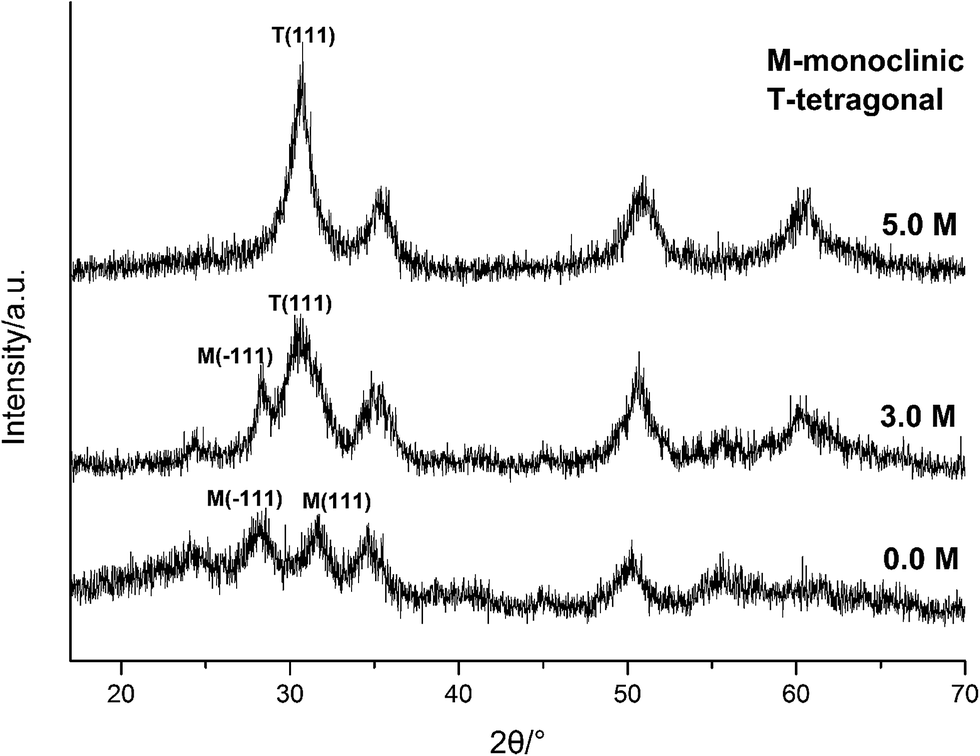 | ||
| Fig. 5 XRD spectra of HfO2 NPs prepared under otherwise the standard conditions (120 °C and 24 h) with different concentrations of NaOH. | ||
 | ||
| Fig. 6 TEM images of HfO2 NPs prepared under otherwise the standard conditions (120 °C and 24 h) with different concentrations of NaOH: (a) 5.0 M, (b) 3.0 M, (c) 0 M. | ||
Carefully considering the influence of pH or alkaline concentration on the formation of metal oxide from metal hydroxide, some metal hydro-complexes usually play a key role to the formation of metal oxide like TiO2 (ref. 24 and 25) and ZrO2.22,26 Jia et al.27 suggested that the pH of the reaction mixture can control the dissociation rate of the precursor, which significantly affects the supply of metal ions or metal hydro-complexes in the reaction environment and thus the growth rate of the metal-oxide nanocrystals. For HfO2 particles, precursory complex Hf(OH)x4−x (x ≥ 5), has been suggested28 from its influence on the particle size. On the other hand, A. Sahraneshin16 proposed that the concentration of alkali can affect the growth of (111) plane of HfO2 which was unstable with higher surface energy. For the current synthesis of HfO2, with no doubt, the high concentration of NaOH is of benefit to the formation of primary t-HfO2 NPs, this may be related to the effects of NaOH concentration on the precursor complex, which will be discussed in details in the latter section.
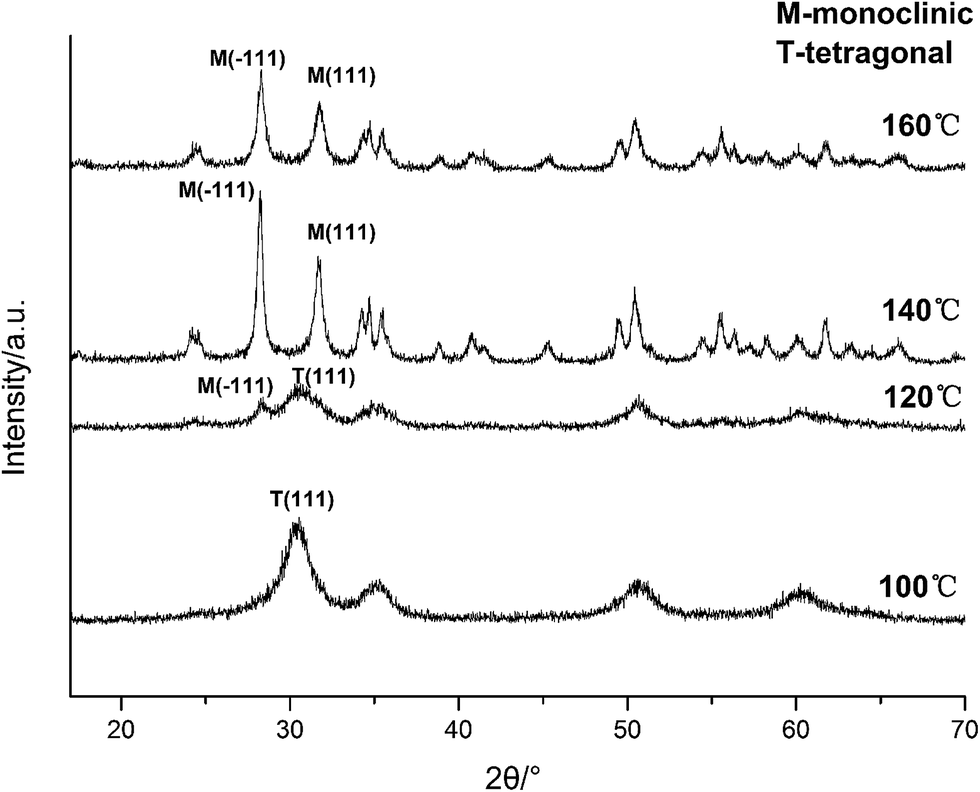 | ||
| Fig. 7 XRD spectra of HfO2 NPs prepared under otherwise the standard conditions (3.0 M NaOH and 24 h) at different temperatures. | ||
 | ||
| Fig. 8 TEM images of HfO2 NPs prepared under otherwise the standard conditions (3.0 M NaOH and 24 h) at different temperatures: (a) 100 °C, (b) 120 °C, (c) 160 °C. | ||
Based on the XRD and TEM observation in Fig. 7 and 8, the reaction temperature is also the key factor that influences particle morphology under the fixed alkaline concentration and reaction time. It has been reported that with the increase of temperature, the particles are more likely to collide to form a large particle even with different form at enough high temperature.23–25 In this work, t-HfO2 NPs may collide mutually and transform to m-HfO2 NPs with the increase of temperature. Therefore, higher temperature, lower concentration of NaOH, and longer reaction time are beneficial for the formation of monoclinic hafnium oxide nanoparticles.
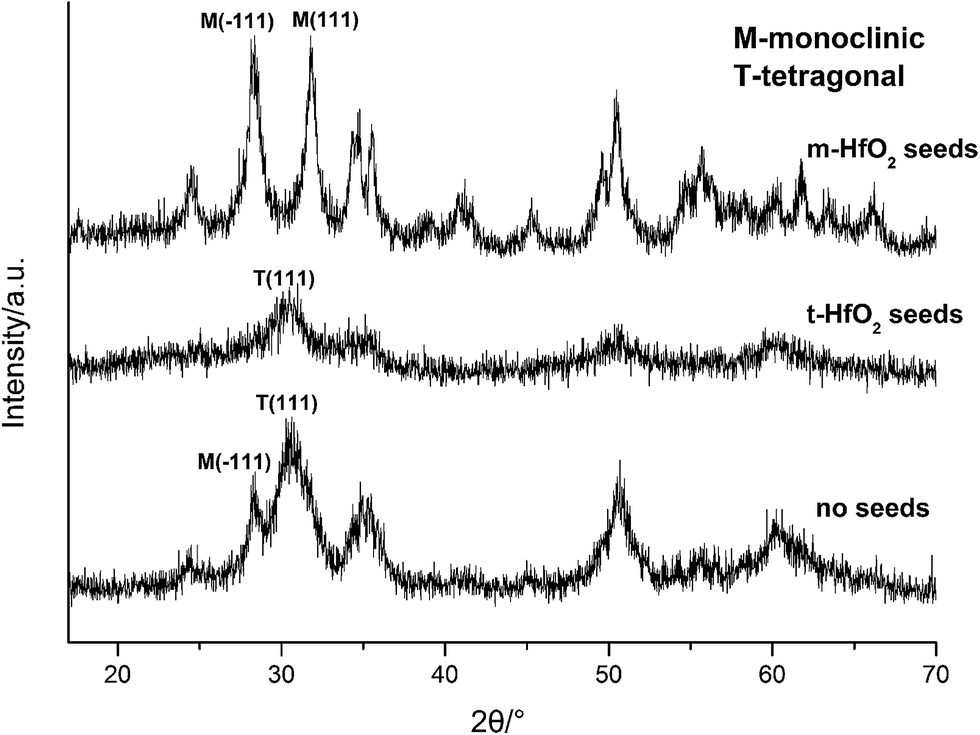 | ||
| Fig. 9 XRD spectra of HfO2 NPs prepared with different seeds. | ||
Formation mechanisms of HfO2 by a hydrothermal route
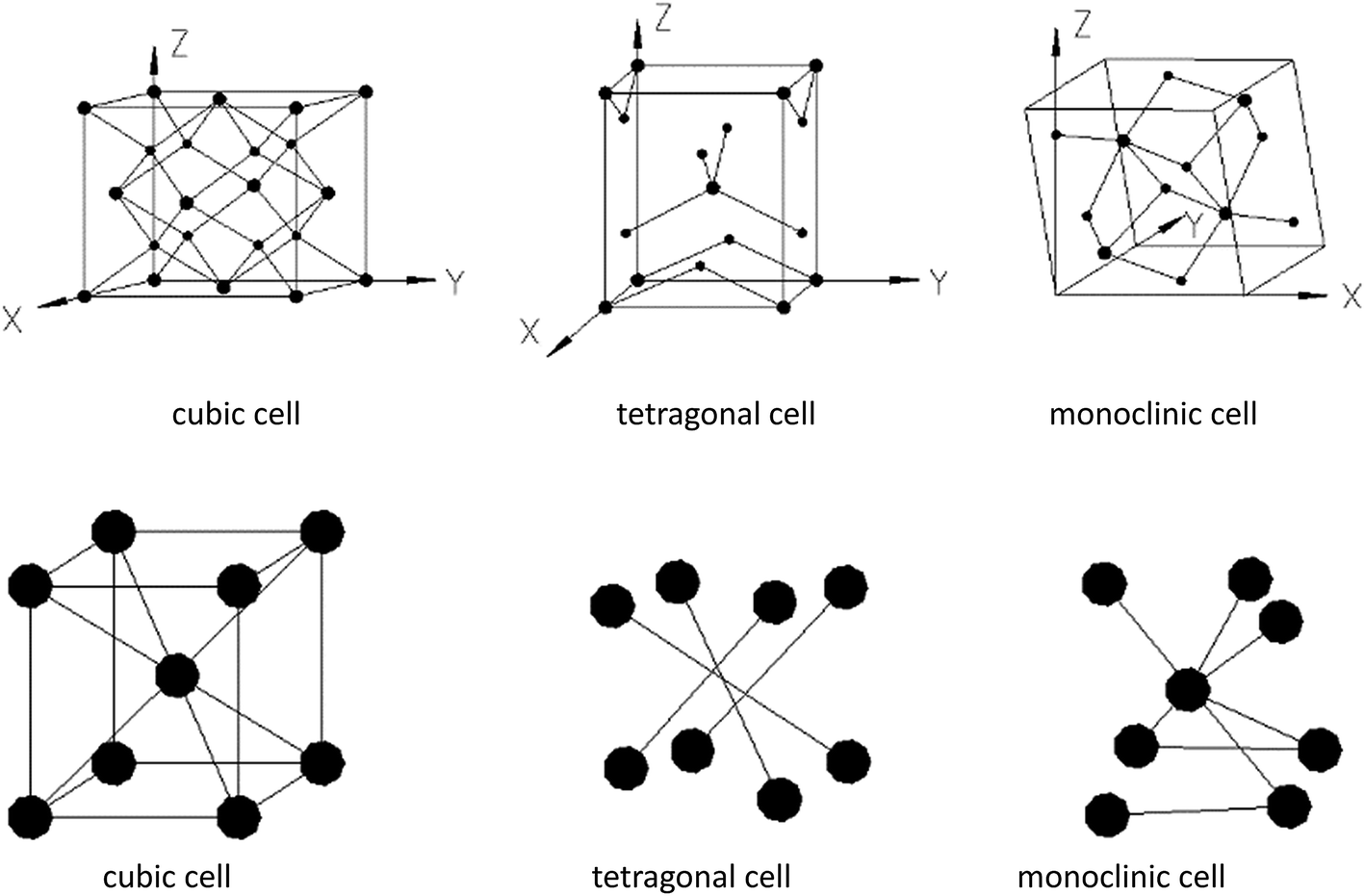 | ||
| Fig. 10 Structures of different HfO2 crystal cells. | ||
In terms of structures of t-HfO2 and m-HfO2, t-HfO2 has better geometric symmetry compared with m-HfO2, and a rule of thumb is that better symmetry products usually have good kinetic stability and are easy to be produced preferentially. Generally, the occurrence of any reaction or variance is comprehensively selected or controlled by both factors of thermodynamics and kinetics, and the reaction in the initial stage is controlled mainly by factor of kinetics.
In our experiment, t-HfO2 NPs are easier to form compared with m-HfO2 NPs and then the t-HfO2 gradually transforms to m-HfO2 with temperature rising or time going. In our previous work,28 the m-HfO2 NPs were obtained at 160 °C with 3.0 M NaOH for 24 h, but in the early stage of this reaction (1 h and 3 h), the products were t-HfO2 NPs. It can be deduced that generally t-HfO2 is produced originally and m-HfO2 will be obtained by transformation from t-HfO2. We have mentioned that under adequate temperature and concentration of NaOH, the products were a mixture of t-HfO2 and m-HfO2. Moreover, based on the influence of temperature and concentration of NaOH on products, it is confirmed that t-HfO2 is produced originally and m-HfO2 is obtained by transformation from t-HfO2. Therefore, it can be concluded that t-HfO2 is stable kinetically and m-HfO2 is stable thermodynamically.
To further confirm it above, the aging time of the reaction at 100 °C with 3.0 M NaOH was extended from 24 h to 168 h, the XRD spectra are shown in Fig. 11. Obviously, the products were mainly t-HfO2 NPs after 24 h, but after 168 h, the products were mainly m-HfO2 NPs. Passage of time led to the weakening of peaks of t-HfO2 and the appearance of the peaks of m-HfO2 in Fig. 11. After 168 h, it was surprising to see that all of t-HfO2 NPs was transformed to m-HfO2 NPs. The result offers a clear proof that the m-HfO2 is stable thermodynamically and the m-HfO2 is commonly transformed from t-HfO2.
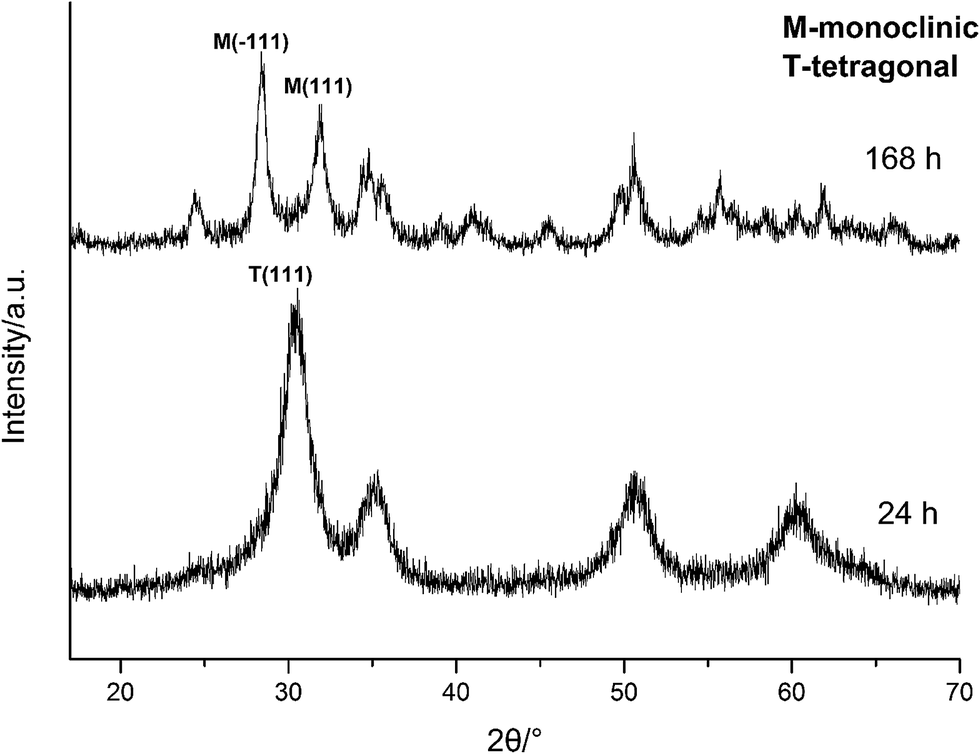 | ||
| Fig. 11 XRD spectra of HfO2 NPs prepared under otherwise the standard conditions (3.0 M NaOH and 100 °C) with different reaction time. | ||
The formation of t-HfO2 with high geometric symmetry becomes more possible in alkaline circumstance. Contrarily, the formation of m-HfO2 is probably inhibited when the concentration of NaOH in solution is enough high. It is reasonable that high concentration of NaOH can inhibit transformation from t-HfO2 to m-HfO2, because high concentration of NaOH can offer more coordinating oxygen ions and then more oxygen ions are beneficial for formation of oxides with high coordination number. These oxygen ions increase the possibility of the occurrence of the structure with high coordination number and high symmetry, and reduce that of the occurrence of distortion, leading to the formation of t-HfO2 NPs. In a word, high concentration of NaOH is beneficial for formation of t-HfO2 and inhibits the formation of m-HfO2, this has been confirmed by our previous report.28 More interestingly, it is also found that with the increased concentration of NaOH, the size of the obtained t-HfO2 decreased in the concentrated NaOH aqueous solution.
On the other hand, temperature is often an important factor to determine the reaction direction and even to determine the crystal form of products. In this work, it has been found that the high temperature is beneficial for the formation of m-HfO2. It could be caused by particles annealing and the difference in the structures of the two forms of HfO2. Generally, large crystals can be formed by annealing of small crystals, along with the change in the crystal forms in some cases. Therefore, with the rise in temperature, the transformation of t-HfO2 to m-HfO2 can be explained in terms of the collision and the distortion in the process of the annealing. It has been reported that with the increase of temperature, the particles are more likely to collide, resulting in the reduction of coordination number, thus forming a large particle.23–25 As a result, these small t-HfO2 crystals were transformed to large m-HfO2 crystals due to the reduction of coordination number.
In Fig. 12, after accomplishment of reaction between hafnium hydroxide chlorides and OH− in the water, the precursory complex Hf(OH)x4−x (x ≥ 5) gather and react in the water. Through nucleation of solute monomers without seeds, the meta-stable t-HfO2 NPs appear. Under appropriate conditions, the product is a mixture of t-HfO2 NPs and m-HfO2 NPs because some t-HfO2 NPs transform to m-HfO2 NPs. However, the main product is m-HfO2 NPs under higher temperature and lower alkali, whereas the product under the opposite conditions is t-HfO2 NPs. In other words, higher temperature and lower alkali concentration are beneficial for the formation of m-HfO2. For better control of crystal forms, the seeds are added. When the adequate t-HfO2 seeds exist, the solute monomers will deposit on t-HfO2 seeds and the size of formed t-HfO2 NPs become larger and larger, inhibiting aggregation and the formation of secondary particles, and then preventing the production of m-HfO2 NPs. Obviously, the t-HfO2 seeds promote the growth of t-HfO2 and inhibit the formation of m-HfO2. In the other hand, when the m-HfO2 seeds are added, the solute monomers will deposit on the pre-existing m-HfO2 seeds, promoting the formation of m-HfO2 and inhibiting the formation of t-HfO2. In a word, the product is t-HfO2 in the presence of t-HfO2 seeds, and that is m-HfO2 in the presence of m-HfO2 seeds.
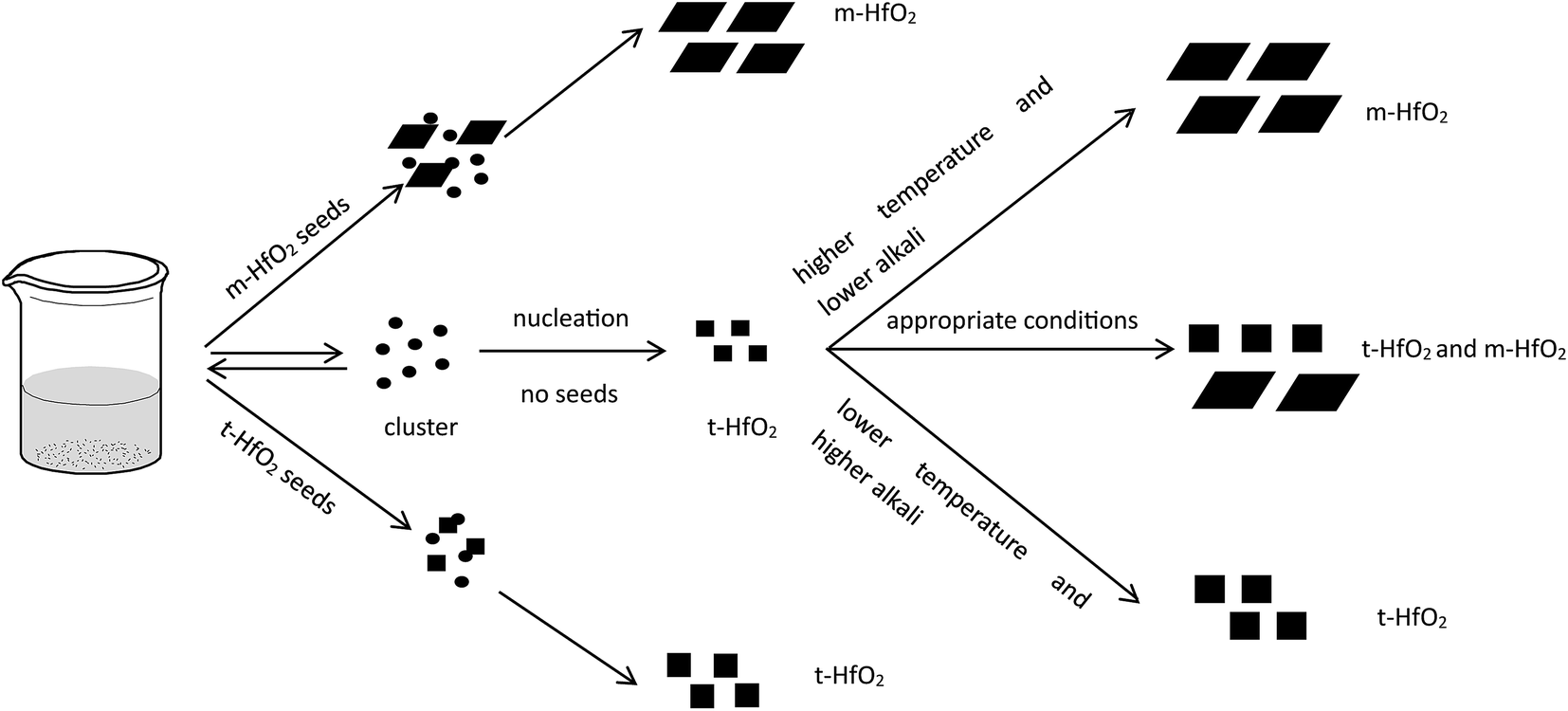 | ||
| Fig. 12 Formation and transformation processes of HfO2 NPs with and without seeds. | ||
For nucleation, the overall Gibbs free energy change,30 ΔG, should be considered. ΔG, is the sum of the free energy due to the formation of the cluster and that due to the new surface created. In Fig. 13(b), ΔG has a positive maximum at a critical size, r*. This maximum free energy change is the activation energy for nucleation. Nuclei or clusters larger than the critical size will further decrease their free energy for growth and become stable ones that grow to form particles.
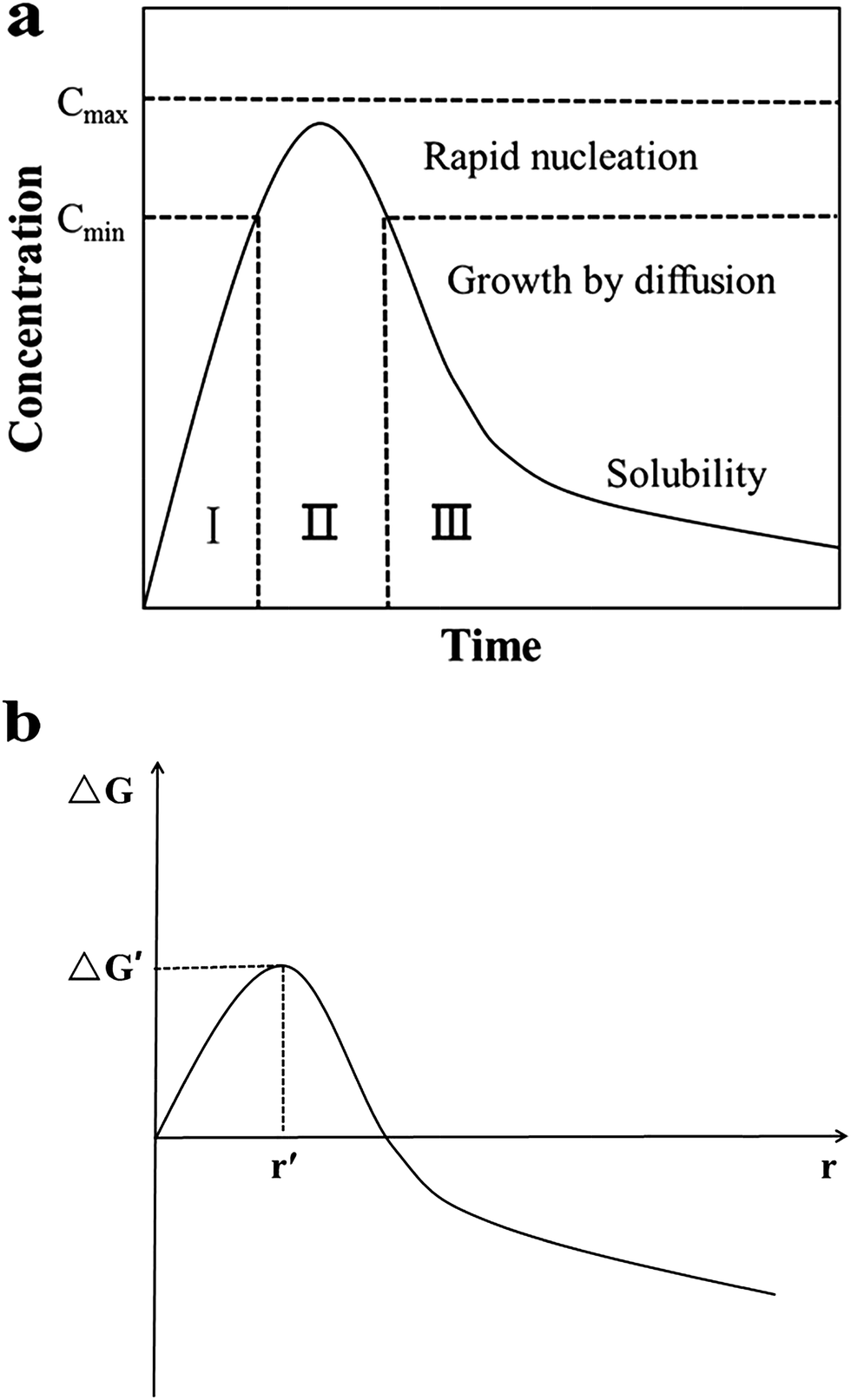 | ||
| Fig. 13 (a) LaMer model schematic diagram32 and (b) illustration of the overall free energy ΔG as a function of the growth particle size r.30 | ||
Usually, it is somewhat hard to make ΔG < 0 in the absence of seeds. However, once the seeds preformed stably are added, spontaneous nucleation is inhibited, and then they grow by deposition of monomers on their surfaces to become large particles. At this time, the overall free energy change ΔG, could be much lowered even to less than 0. That means, the formation process of stable solute clusters or particles is skipped (Fig. 13(a)) and the reaction is accelerated. Therefore, the resulting final nanoparticles are smaller in size and more evenly dispersed. Undoubtedly, the seeding technique is useful for the systematic control of crystal form and for the study of the growth and nucleation mechanisms.31
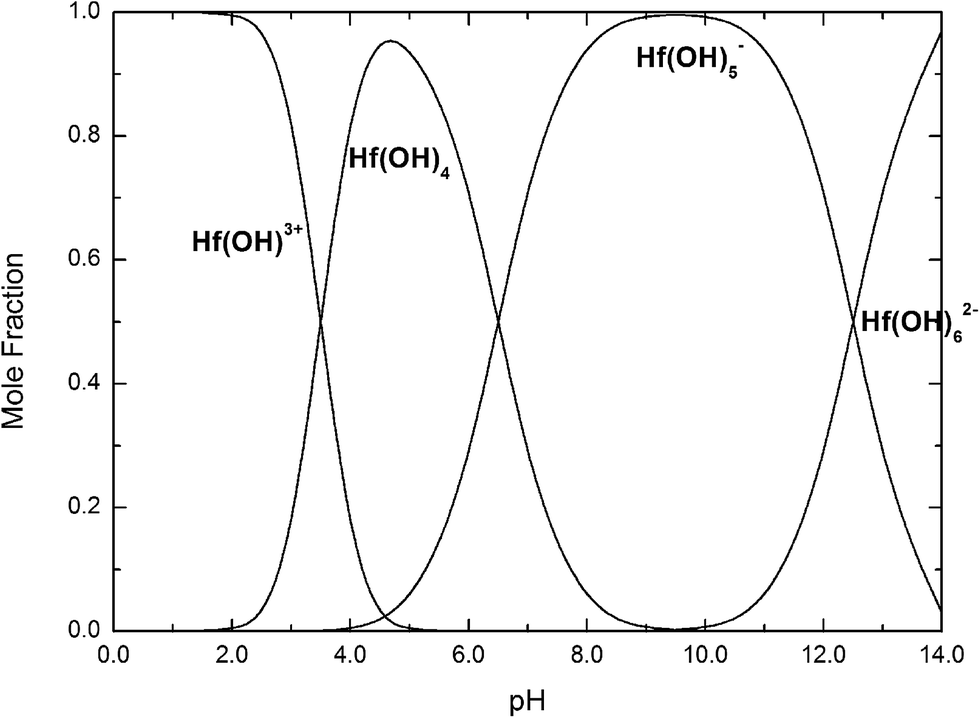 | ||
| Fig. 14 Mole fractions of Hf(OH)3+, Hf(OH)4, Hf(OH)5−, Hf(OH)62− complexes as a function of pH at 25 °C. | ||
If we take into account that the overall reaction for the transformation from Hf(OH)4 gel to HfO2 sol is Hf(OH)4 → HfO2 + 2H2O, the precursory complex must only be a catalyst or an intermediate, and thus Hf(OH)4 complex, constantly furnished from the Hf(OH)4 gel with the progress of the reaction, may also contribute to the poly-condensation reaction in the formation of HfO2 particles. The transformation is similar to production of TiO2 from Ti(OH)4,24 CeO2 from Ce(OH)4 (ref. 34) and ZrO2 from Zr(OH)4.22,35 The following elementary processes may be involved in the overall reaction shown in Fig. 15(a and b), deduced from those of TiO2 (ref. 24) and CeO2.34
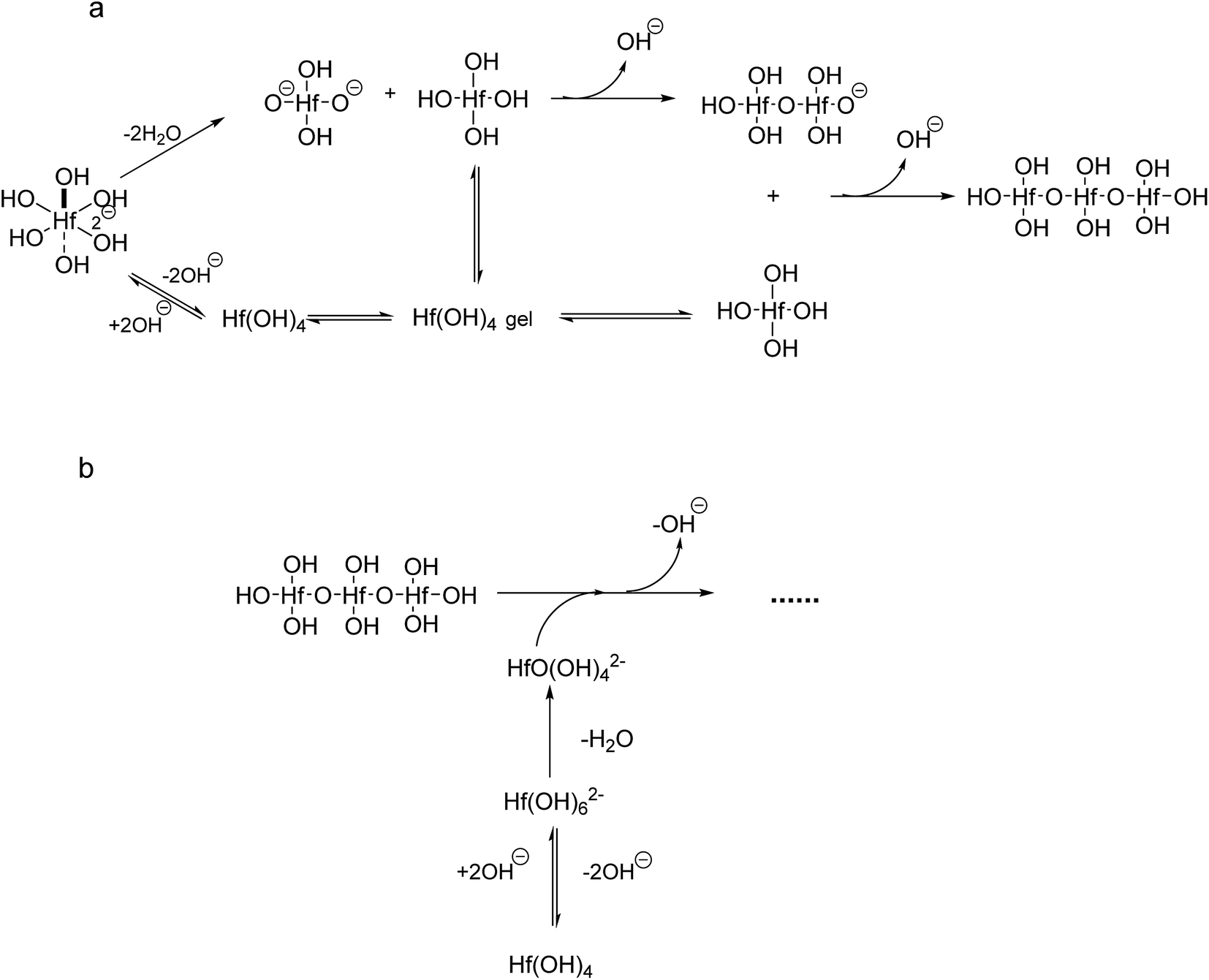 | ||
| Fig. 15 Formation of HfO2 from Hf(OH)62− and Hf(OH)4. | ||
Conclusions
The HfO2 nanoparticles (NPs) were prepared successfully by a hydrothermal route. The phase transformation of HfO2 occurs through changing the reaction temperature, the concentration of NaOH, aging time and crystal form of seeds. XRD spectra and TEM images revealed the near-spherical particles belonged to tetragonal HfO2 (t-HfO2), and the spindle-like particles belonged to monoclinic HfO2 (m-HfO2). Based on the kinetic and thermodynamic analyses, it can be induced that higher temperature, lower concentration of NaOH and longer reaction time are beneficial for the formation of m-HfO2 nanoparticles. Moreover, it is confirmed that t-HfO2 generally produces originally and m-HfO2 is obtained by transformation from t-HfO2.In the standard conditions at 120 °C with 3.0 M NaOH for 24 h, the product is a mixture of t-HfO2 and m-HfO2 NPs in the absence of seeds. When m-HfO2 seeds are added, the resultant is pure m-HfO2 NPs, while t-HfO2 seeds are added, under the same conditions the particles are pure t-HfO2 NPs, proving the non-aggregation growth of nanoparticles. Not only the crystal form of HfO2 can be well controlled, but also the particles size can be altered. The addition of HfO2 seeds decreases the size of nanoparticles. In the presence of seeds, the declined activated energy for the reaction is due to the decrease of ΔG, resulting from the skipping of spontaneous nucleation stage, and then causing separation of nucleation from growth.
The effect of solution pH on the formation of HfO2 NPs was learned by the analysis above. Meantime, the high concentration of NaOH promoted the formation of t-HfO2 NPs. Therefore, the hydro-complex Hf(OH)62− has been confirmed as the precursor complex to form HfO2 particles.
Acknowledgements
The authors gratefully acknowledge the Foundation of Shanghai Municipal Commission of Economy and Informatization (15XI-1-28).Notes and references
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS PubMed.
- D. L. Feldheim and C. D. Keating, Chem. Soc. Rev., 1998, 27, 1–12 RSC.
- S. Saito, Science, 1997, 278, 77–78 CrossRef CAS.
- J. Robertson, Rep. Prog. Phys., 2006, 69, 327–396 CrossRef CAS.
- J. De Roo, K. De Keukeleere, J. Feys, P. Lommens, Z. Hens and I. Van Driessche, J. Nanopart. Res., 2013, 15, 1778 CrossRef.
- G. D. Wilk, R. M. Wallace and J. M. Anthony, J. Appl. Phys., 2001, 89, 5243–5275 CrossRef CAS.
- J. Molina, R. Ortega, W. Calleja, P. Rosales, C. Zuniga and A. Torres, Mater. Sci. Eng., B, 2012, 177, 1501–1508 CrossRef CAS.
- S. A. Eliziario, L. S. Cavalcante, J. C. Sczancoski, P. S. Pizani, J. A. Varela, J. W. M. Espinosa and E. Longo, Nanoscale Res. Lett., 2009, 4, 1371–1379 CrossRef CAS PubMed.
- R. Terki, G. Bertrand, H. Aourag and C. Coddet, Mater. Lett., 2008, 62, 1484–1486 CrossRef CAS.
- V. Jayaraman, G. Bhavesh, S. Chinnathambi, S. Ganesan and P. Aruna, Mater. Express, 2014, 4, 375–383 CrossRef CAS.
- W. E. Buhro and V. L. Colvin, Nat. Mater., 2003, 2, 138–139 CrossRef CAS PubMed.
- H. B. Yao, M. R. Gao and S. H. Yu, Nanoscale, 2010, 2, 323–334 RSC.
- E. Tirosh and G. Markovich, Adv. Mater., 2007, 19, 2608–2612 CrossRef CAS.
- J. S. Quintero-Garcia, B. A. Puente-Urbina, L. A. Garcia-Cerda, O. S. Rodriguez-Fernandez and E. Mendoza-Mendoza, Mater. Lett., 2015, 159, 520–524 CrossRef CAS.
- A. Ramadoss, K. Krishnamoorthy and S. J. Kim, Mater. Lett., 2012, 75, 215–217 CrossRef CAS.
- A. Sahraneshin, S. Asahina, T. Togashi, V. Singh, S. Takami, D. Hojo, T. Arita, K. Minami and T. Adschiri, Cryst. Growth Des., 2012, 12, 5219–5226 CAS.
- P. E. Meskin, F. Y. Sharikov, V. K. Ivanov, B. R. Churagulov and Y. D. Tretyakov, Mater. Chem. Phys., 2007, 104, 439–443 CrossRef CAS.
- E. Montes, P. Ceron, T. R. Montalvo, J. Guzman, M. Garcia-Hipolito, A. B. Soto-Guzman, R. Garcia-Salcedo and C. Falcony, Appl. Radiat. Isot., 2014, 83, 196–199 CrossRef CAS PubMed.
- L. Xiang, Y. P. Yin and Y. Jin, J. Mater. Sci., 2002, 37, 349–352 CrossRef CAS.
- H. B. Yin, Y. Wada, T. Kitamura, S. Kambe, S. Murasawa, H. Mori, T. Sakata and S. Yanagida, J. Mater. Chem., 2001, 11, 1694–1703 RSC.
- Y. Q. Zheng, E. R. Shi, Z. Z. Chen, W. J. Li and X. F. Hu, J. Mater. Chem., 2001, 11, 1547–1551 RSC.
- T. T. Shi, Y. T. Cai, L. Liu and X. P. Zhou, Colloids Surf., A, 2015, 469, 83–92 CrossRef CAS.
- T. Sugimoto, X. P. Zhou and A. Muramatsu, J. Colloid Interface Sci., 2003, 259, 43–52 CrossRef CAS PubMed.
- T. Sugimoto, X. P. Zhou and A. Muramatsu, J. Colloid Interface Sci., 2002, 252, 339–346 CrossRef CAS PubMed.
- T. Sugimoto and X. P. Zhou, J. Colloid Interface Sci., 2002, 252, 347–353 CrossRef CAS PubMed.
- L. Liu, J. C. Xue and X. P. Zhou, Nanosci. Nanotechnol. Lett., 2014, 6, 346–352 CrossRef CAS.
- C. Jia, Y. Cheng, F. Bao, D. Chen and Y. Wang, J. Cryst. Growth, 2006, 294, 353–357 CrossRef CAS.
- J. J. Qi and X. P. Zhou, Colloids Surf., A, 2015, 487, 26–34 CrossRef CAS.
- X. P. Zhou, S. S. Li, X. L. Chen, Q. Zhu, Z. Q. Wang and J. Zhang, J. Nanosci. Nanotechnol., 2008, 8, 1392–1397 CAS.
- I. V. Markov, Crystal growth for beginners: fundamentals of nucleation, crystal growth and epitaxy, World Scientific, 2003 Search PubMed.
- S. Taotao, C. Yutian, L. Li and Z. Xingping, Colloids Surf., A, 2015, 469, 83–92 CrossRef.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS PubMed.
- D. Rai, Y. X. Xia, N. J. Hess, D. M. Strachan and B. P. McGrail, J. Solution Chem., 2001, 30, 949–967 CrossRef CAS.
- X. S. Zheng, L. Liu and X. P. Zhou, Colloid J., 2014, 76, 558–563 CrossRef CAS.
- R. R. Piticescu, C. Monty, D. Taloi, A. Motoc and S. Axinte, J. Eur. Ceram. Soc., 2001, 21, 2057–2060 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |