
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConformational stabilization of isatin Schiff bases – biologically active chemical probes†
Andrey S. Smirnovab,
Dmitriy N. Nikolaev c,
Vlad V. Gurzhiyb,
Sergey N. Smirnovb,
Vitaliy S. Suslonovb,
Alexander V. Garabadzhiua and
Pavel B. Davidovich*a
c,
Vlad V. Gurzhiyb,
Sergey N. Smirnovb,
Vitaliy S. Suslonovb,
Alexander V. Garabadzhiua and
Pavel B. Davidovich*a
aSaint-Petersburg Technological Institute, Moskovskii av. 26, St. Petersburg, 190013, Russia. E-mail: davidovich.pavel@technolog.edu.ru
bSaint-Petersburg State University, University emb. 7/9, St. Petersburg, 199034, Russia
cResearch Institute of Experimental Medicine, Akad. Pavlova str. 12, St. Petersburg, 197376, Russia
First published on 6th February 2017
Abstract
Isatin Schiff base derivatives have a wide range of biological effects. Unfortunately, these compounds possess a serious topological shortcoming: the conformational E ⇆ Z interconversion. Two ways of conformation stabilization are reported here: complexation with metals that stabilize the E-conformer and substitution in the 4th position of the isatin core stabilizing the Z-form.
Isatin1 is a naturally occurring alkaloid first discovered in plant species and later found as a blood circulation molecule that can easily cross the blood–brain barrier acting as an endogenous neurotransmitter activator.3 To date it is known that isatin and its analogues have a plethora of biological effects4 including antitumor activities.5 Modified by hyrdoxamic acids, isatin-3-oxymes are potent for T-cell lymphoma killing, acting as a micromolar histone deacetylase inhibitors.6 It was shown on a cellular leukemia model that N-substituted acrylate isatin derivatives promote cell cycle arrest and apoptosis induction.7 Isatin-β-thiosemicarbazones show selective proliferation inhibition towards multidrug-resistant cell lines.8 5-Sulfonamide and -aryl substituted isatins act as inhibitors of cancer associated matrix metalloproteinases.9,10 Isatin Schiff bases with benzenesulfonamide moiety have been recently identified as tumor associated carbonic anhydrase IX and XII inhibitors.11 Our previous work12 showed that isatin Schiff base derivatives (ISBDs) activate p53 transcription factor – the major oncosuppressor protein. Unfortunately, ISBDs poses a serious drawback – intramolecular isomeric interconversion. Since E and Z isomers significantly differ topologically, one becomes almost an impurity that may cause undesirable side effects. E ⇆ Z equilibrium occurs in most of the solvents (the ratio is slightly dependent on the solvent14) with preferable E-conformation, which makes circa 80–90%.13 It is recognized that indole like scaffolds are privileged in medicinal chemistry molecule space,15 thus making ISBDs more conformationally rigid is an important and actual objective.
This report is a proof-of-principle that shows how conformational interconversion may be stopped by applying two different approaches: substitution in the isatin aromatic moiety (ring A on Fig. 1) and complexation with d-metals. These data are supported by the structural X-ray solid state and NMR solution studies that are accompanied by DFT calculations. It is worth noting, that the structural findings in coordination chemistry of isatin–aromatic amines Schiff bases up-to-date remain scarce and are limited to one work describing homoleptic bischelate copper(II) complex with isatin-4-hexylaniline.16
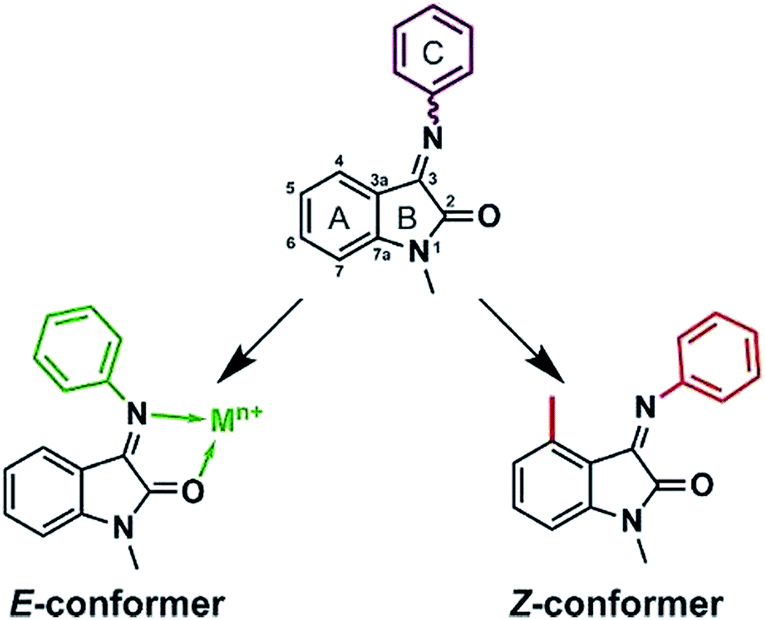 | ||
| Fig. 1 Two principal ways to fix phenyl ring orientation: by complexation with metals and substitution in isatin core. | ||
To study the metal complexation processes we have prepared the model compound – 1-methyl-3(phenylimino)indolinone-2-one (I) by the method described in our previous paper.14 As non-protected amide nitrogen (NH) is acidic enough to form contacts with d-metals, this potential reactive center was blocked by N-methylation.
XRD study showed that ISBD I adopts only E-conformation outlined in Fig. 2‡:
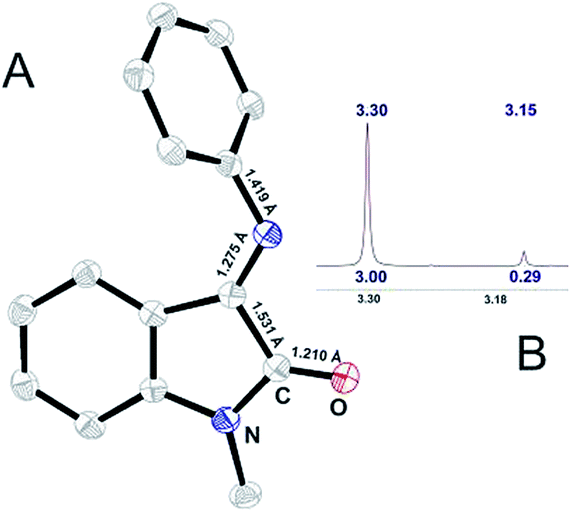 | ||
| Fig. 2 (A) ORTEP representation of X-ray determined molecular structure of I and (B) 1H NMR signal for N-methyl group of I. | ||
In solution interconversion results in methyl proton peak signal splitting and the E/Z ratio can be estimated as a peak surface relation. Solution phase NMR data (Fig. 2B) shows that 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 E- to Z-conformer ratio is settled in CDCl3 for ISBD I.
:1 E- to Z-conformer ratio is settled in CDCl3 for ISBD I.
We have expected that introduction of substituent in the 4th position (Fig. 1) can force the molecule to adopt Z-conformation, as a reason of steric repulsion between substituent and aromatic ring C. Using CAM-B3LYP functional that includes long-range corrections, total energy differences (ΔEtot = EZ − EE, kJ mol−1, for R4 = H, ΔEtot = 0) between two conformers with various substituents in ring A were estimated. For the most common isatin derivatives ΔEtot changes in the following order: H (0), Me (−23.5), CF3 (−30.0), Et (−28.3), OMe (−6.2), F (−5.4), Cl (−19.4), Br (−23.5), CN (−24.5). Hence using methyl substituent should be enough to make the molecule adopt Z-conformation. Staring from these assumptions, we have prepared 1,4,7-methylated ISBD with ring C represented by 2,6-diisopropylaniline (III, Fig. S2†) and aniline (II, Fig. 3):
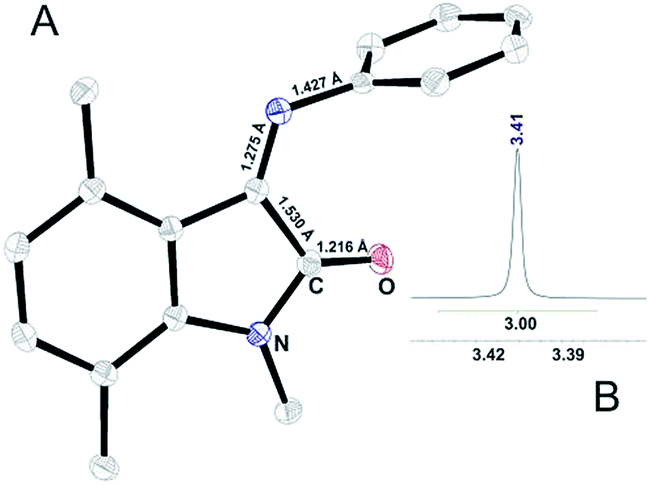 | ||
| Fig. 3 (A) ORTEP representation of X-ray determined molecular structure of II and (B) 1H NMR signal for N-methyl group of II. | ||
As depicted in Fig. 3, introduction of methyl group in the 4th position results in the crystallization of compound II Z-conformer. Up to date it is the first crystallized ISBD in Z-conformation. The absence of methyl signal splitting in 1H NMR spectrum (Fig. 3B) confirms that no interconversion occurs in solution. Thus, it can be stated that substitution in the isatin core can be used as a strategy to stabilize Z-form of ISBD and may be further applied for medicinal chemistry purposes.
Ligand flexibility reduction by metal complexation has long been known, but practical aspects of this effect remain limited. To validate hypothesis of ISBDs E-conformation stabilization by complexation with metals we have performed ligand I reactions with diamagnetic metal ions. Attempts to isolate complexes with RedOx stable biogenic diamagnetic earth metals and zinc salts were in vain. This could be the reason of non-complementarity between a soft base (ligand) and hard M(II)17 acids. Attempt to get the complex with Mg(II) ions resulted in formation of low melting point (110–112 °C) crystals (IV, Fig. 1S†) consisting of an equimolar ratio of both Schiff base and its hydrolyzed product – N-methyl-isatin. Therefore, it might be expected that coordination with some metals can induce nucleophilic attack of hydroxide anion (−OH) on C3 atom attenuating labile azomethine C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond promoting its hydrolytic cleavage. This assumption can be supported by Ghosh works,18,19 where the authors reported CN bond cleavage of benzoquinone ligand promoted by coordination to Rh(III) ions.
N bond promoting its hydrolytic cleavage. This assumption can be supported by Ghosh works,18,19 where the authors reported CN bond cleavage of benzoquinone ligand promoted by coordination to Rh(III) ions.
We have managed to isolate and structurally characterize two novel complexes with softer Cd(II) and Hg(II) acids. The complexes were prepared by the reaction of ligand double excess with HgBr2/CdBr2 salts dissolved in ethanol. The reaction resulted in isolation of crystalline coordination products as well as unreacted ligand. Upon coordination to Hg(II) ion carbonyl frequency shifts from 1734 cm−1 lower to 1708 cm−1, while the CN vibration slightly moves from 1604 cm−1 to 1606 cm−1. The Cd(II) complex carbonyl vibration exhibits shift towards 1707 cm−1, while the CN vibration remains almost unaffected 1608 cm−1 (Fig. 4):
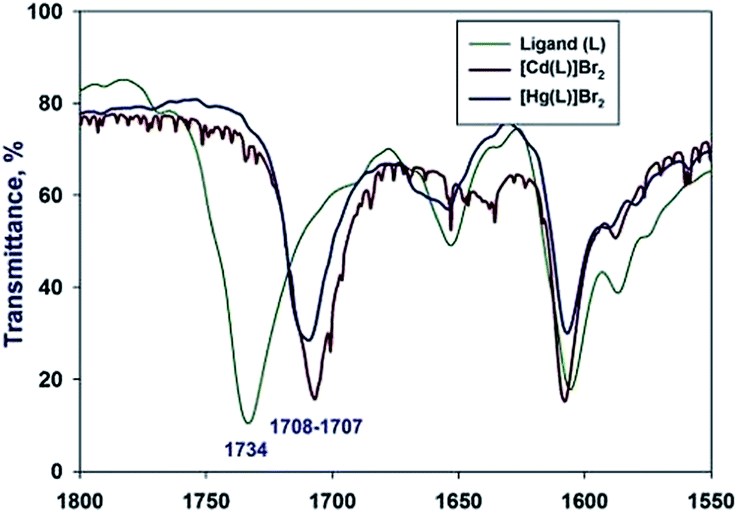 | ||
| Fig. 4 Superimposition of IR spectra for I, V and VI. | ||
Obtained Cd(II) complex was insoluble in almost all widely used solvents, what resulted in NMR experiment failure. Nonetheless, the MS analysis of Cd(II) complex in DMSO/MeOH mixture showed the predominant peak attributed to monochelate [Cd(L)Br]+ (M/Z = 428.92) complex with the minor peak corresponding to bischelate [Cd(L)2Br]+ (Fig. S18,† M/Z = 665.02) species.
The negligible solubility turned to be the issue of the polymeric crystal structure.§ It is seen in the figure below that Cd(II) ions form monochelate complex with ISBD I and are bridged by two bromide anions forming infinite chain with mean Cd to Cd distance of ∼3.95 Å (Fig. 5).
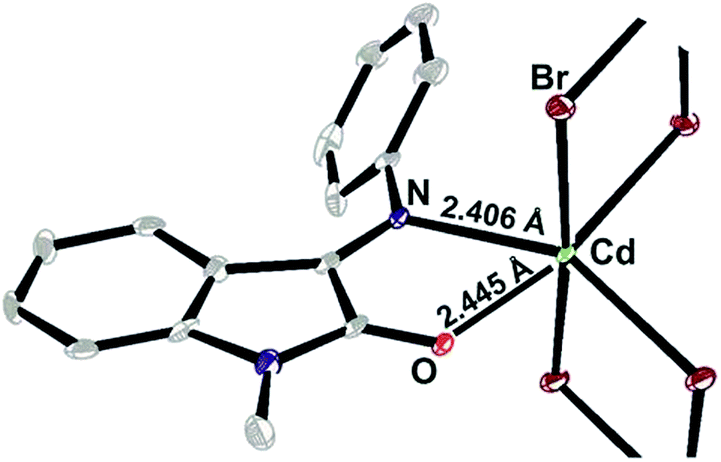 | ||
| Fig. 5 ORTEP plot of X-ray determined structure of V (coordination polymer). | ||
By the analogy with Cd(II), coordination of I to Hg(II) ions resulted in the N,O-monochelate complex formation [M(L)]Br2 (Fig. S19,† M/Z = 516.98). The bischelate complex [Hg(L)2]Br2 was also detected by MS method, but in a less extent (Fig. S19,† M/Z = 735.07). We have isolated a monocrystalline sample and characterized the molecular structure of monochelate complex [Hg(L)]Br2 (VI)¶ that is outlined in Fig. 6A:
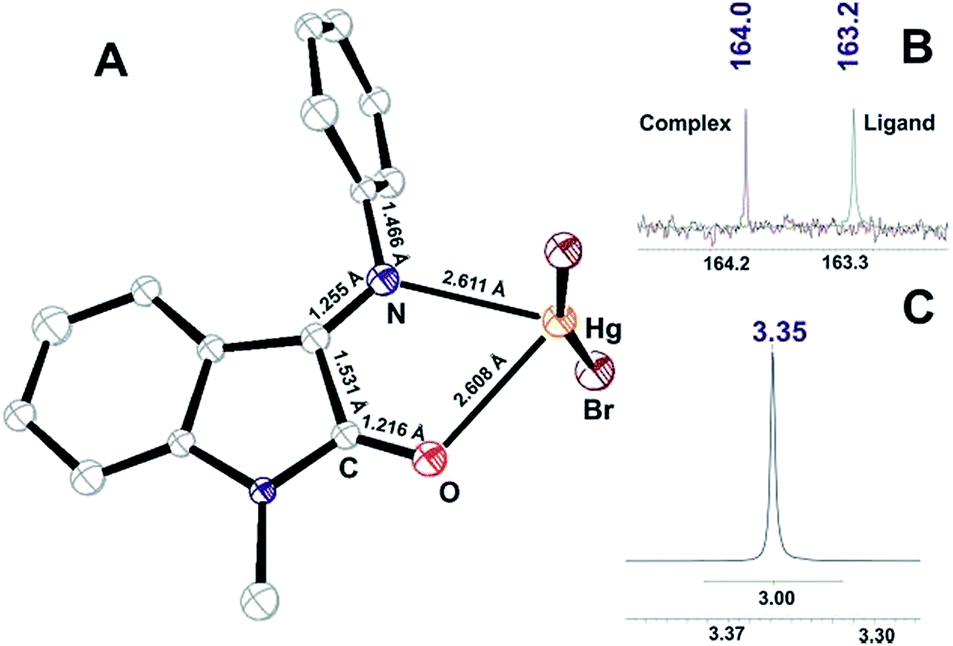 | ||
| Fig. 6 (A) ORTEP plot of X-ray determined molecular structure of VI; (B) 13C NMR signal for carbonyl of I and VI; (C) 1H NMR signal for methyl group of VI. | ||
The cell unit of VI is formed by the several types of slightly different elemental fragments of the monochelate mercury(II) complex (unequal Hg–N and Hg–O bond lengths). The weak interactions between Hg2+ cation and Br− anions of the neighboring molecules might be the reason of such inequalities (Fig. 7):
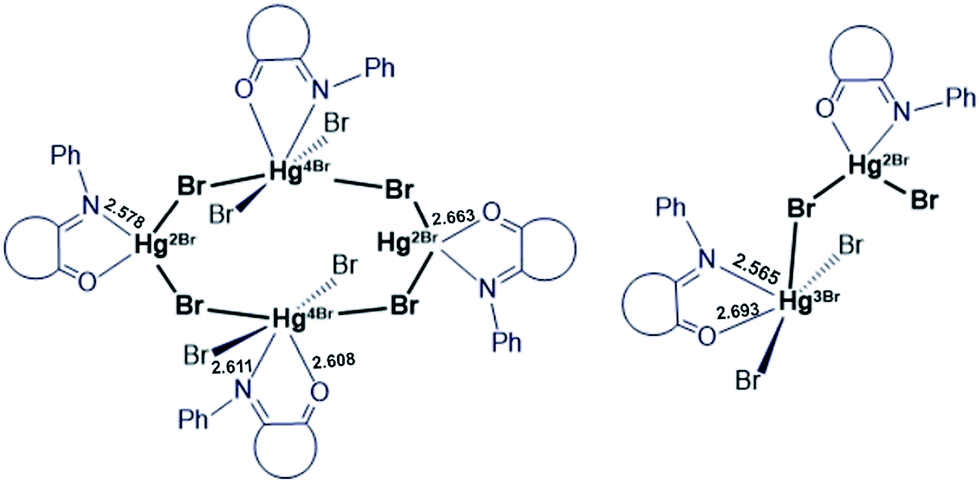 | ||
| Fig. 7 Schematic representation of Hg(II) – ISBD I supramolecular structures. | ||
The solution study of [Hg(L)2]Br2 complex showed that coordination resulted in 13C carbonyl signal shift to the lower field and total reduction of the Z-component according to 1H NMR N-methyl signal (Fig. 6B and C).
Thus, the current report elucidates two approaches of freezing E/Z isomeric interconversion within ISBDs that may further find implementation in the medicinal chemistry field. Substitution in the 4th position of the isatin core results in total reduction of E-conformation existence, whereas complexation of the isatin Schiff base with Hg(II) ions stabilizes the ligand E-conformation. X-ray powder patterns confirmed the isomeric purity of samples.
It's worthwhile to say that, our ongoing studies show that biogenic 3d-metal ions (Cu2+, Co2+, Ni2+, Mn2+) form stable complexes with ISBDs that are more likely to be studied in biochemical assays. Moreover, by the analogy with the homologue iminoquinone complexes catalyst,20 the combination of non-innocent ISBDs ligands with RedOx active metals can result in unexpected electrochemical properties of their complexes.
Acknowledgements
The work was supported by the Ministry of Education and Science of the Russian Federation (14.B25.31013) and by the Russian Foundation for Basic Research (16-34-50250). Physicochemical studies were performed at the Saint-Petersburg State University Centers for Magnetic Resonance, X-ray Diffraction Studies, and Chemical Analysis and Materials Research.Notes and references
- E. G. Gutierrez and A. K. Franz, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2001 Search PubMed.
- N. A. Aslam, S. A. Babu, S. Rani, S. Mahajan, J. Solanki, M. Yasuda and A. Baba, Eur. J. Org. Chem., 2015, 2015, 4168 CrossRef CAS.
- T. Sommer, K. Bjerregaard-Andersen, S. M. Simensen, J. K. Jensen, B. Jochimsen, P. J. Riss, M. Etzerodt and J. P. Morth, ACS Chem. Neurosci., 2015, 6, 1353 CrossRef CAS PubMed.
- A. Medvedev, O. Buneeva and V. Glover, Biologics, 2007, 1, 151 CAS.
- L. M. Kara, L. Vine, J. M. Locke and D. Skropeta, Adv. Anticancer Agents Med. Chem., 2013, 2, 254 Search PubMed.
- N. H. Nam, T. L. Huong, D. T. Mai Dung, P. T. Phuong Dung, D. T. Kim Oanh, D. Quyen, L. T. Thao, S. H. Park, K. R. Kim, B. W. Han, J. Yun, J. S. Kang, Y. Kim and S. B. Han, Eur. J. Med. Chem., 2013, 70, 477 CrossRef CAS PubMed.
- Y. Zhou, H.-Y. Zhao, K.-L. Han, Y. Yang, B.-B. Song, Q.-N. Guo, Z.-C. Fan, Y.-M. Zhang, Y.-O. Teng and P. Yu, Biochem. Biophys. Res. Commun., 2014, 450, 1650 CrossRef CAS PubMed.
- H. S. Ibrahim, S. M. Abou-Seri and H. A. Abdel-Aziz, Eur. J. Med. Chem., 2016, 122, 366 CrossRef CAS PubMed.
- S. Sjoli, A. I. Solli, O. Akselsen, Y. Jiang, E. Berg, T. V. Hansen, I. Sylte and J. O. Winberg, Biochim. Biophys. Acta, 2014, 1840, 3162 CrossRef PubMed.
- M. Agamennone, D. S. Belov, A. Laghezza, V. N. Ivanov, A. M. Novoselov, I. A. Andreev, N. K. Ratmanova, A. Altieri, P. Tortorella and A. V. Kurkin, ChemMedChem, 2016, 11, 1892 CrossRef CAS PubMed.
- O. Guzel-Akdemir, A. Akdemir, N. Karal and C. T. Supuran, Org. Biomol. Chem., 2015, 13, 6493 CAS.
- P. Davidovich, V. Aksenova, V. Petrova, D. Tentler, D. Orlova, S. Smirnov, V. Gurzhiy, A. L. Okorokov, A. Garabadzhiu, G. Melino, N. Barlev and V. Tribulovich, ACS Med. Chem. Lett., 2015, 6, 856 CrossRef CAS PubMed.
- A. González, J. Quirante, J. Nieto, M. R. Almeida, M. J. Saraiva, A. Planas, G. Arsequell and G. Valencia, Bioorg. Med. Chem. Lett., 2009, 19, 5270 CrossRef PubMed.
- P. Davidovich, D. Novikova, V. Tribulovich, S. Smirnov, V. Gurzhiy, G. Melino and A. Garabadzhiu, J. Mol. Struct., 2014, 1075, 450 CrossRef CAS.
- E. J. Barreiro, in Privileged Scaffolds in Medicinal Chemistry: Design, Synthesis, Evaluation, RCS, 2016, pp. 1–15 Search PubMed.
- A. Ercag, S. O. Yildirim, M. Akkurt, M. U. Ozgur and F. W. Heinemann, Chin. Chem. Lett., 2006, 17, 243 CAS.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533 CrossRef CAS.
- P. Saha, A. Saha Roy, T. Weyhermuller and P. Ghosh, Chem. Commun., 2014, 50, 13073 RSC.
- S. Maity, S. Kundu, S. Bera, T. Weyhermüller and P. Ghosh, Eur. J. Inorg. Chem., 2016, 2016, 3691 CrossRef CAS.
- D. L. J. Broere, R. Plessius and J. I. van der Vlugt, Chem. Soc. Rev., 2015, 44, 6886 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental, crystallographic details, NMR and MS details. CCDC 1505866–1505870 and 1506944. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra26779c |
| ‡ Here and further atoms are drawn in thermal ellipsoids at 50% probability level and hydrogens are omitted for clarity. Of note, compound I polymorph was earlier described.2 |
§ Crystal structures of I–VI were determined by the means of single crystal X-ray diffraction analysis using a Rigaku Oxford Diffraction Supernova Atlas (I, IV and VI) and an Excalibur Eos (II, III and V) diffractometers at a temperature of 100 K. I: (C15H12N2O), P21/c, a = 9.4554(3), b = 11.8767(3), c = 10.4401(2) Å, β = 91.739(2)°, V = 1171.88(5) Å3, Z = 4, R1 = 0.037, CCDC 1505866. II: (C17H16N2O), P21/c, a = 10.2483(3), b = 8.1749(2), c = 16.2529(6) Å, β = 102.182(3)°, V = 1330.99(8) Å3, Z = 4, R1 = 0.038, CCDC 1505868. III: (C23H28N2O), P21/c, a = 8.5182(6), b = 29.4795(14), c = 8.4782(5) Å, β = 115.150(8)°, V = 1927.1(2) Å3, Z = 4, R1 = 0.047, CCDC 1505867. IV: (C15H12N2O)·(C9H7NO2), P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 7.8377(2), b = 8.33995(19), c = 31.7116(6) Å, α = 85.8492(17)°, β = 85.7112(18)°, γ = 69.989(2)°, V = 1939.83(8) Å3, Z = 4, R1 = 0.061, CCDC 1505870. V: 0.5(C30H24Br4Cd2N4O2), P, a = 7.2618(3), b = 11.2036(10), c = 20.2141(11) Å, α = 93.876(6)°, β = 98.487(4)°, γ = 107.037(6)°, V = 1544.4(8) Å3, Z = 4, R1 = 0.069, CCDC 1506944. VI: (C30H24Br4Hg2N4O2), P21/c, a = 23.6723(11), b = 19.9737(5), c = 14.0418(4) Å, β = 101.024(3)°, V = 6516.8(4) Å3, Z = 8, R1 = 0.068, CCDC 1505869. , a = 7.8377(2), b = 8.33995(19), c = 31.7116(6) Å, α = 85.8492(17)°, β = 85.7112(18)°, γ = 69.989(2)°, V = 1939.83(8) Å3, Z = 4, R1 = 0.061, CCDC 1505870. V: 0.5(C30H24Br4Cd2N4O2), P, a = 7.2618(3), b = 11.2036(10), c = 20.2141(11) Å, α = 93.876(6)°, β = 98.487(4)°, γ = 107.037(6)°, V = 1544.4(8) Å3, Z = 4, R1 = 0.069, CCDC 1506944. VI: (C30H24Br4Hg2N4O2), P21/c, a = 23.6723(11), b = 19.9737(5), c = 14.0418(4) Å, β = 101.024(3)°, V = 6516.8(4) Å3, Z = 8, R1 = 0.068, CCDC 1505869. |
| ¶ Attempts to isolate Hg(II) complex with ligands II or III didn't succeed. |
| This journal is © The Royal Society of Chemistry 2017 |