
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous synthesis of CuInS2 quantum dots†
T. Akdas‡
 *ac,
M. Haderleinac,
J. Walterac,
B. Apeleo Zubiribc,
E. Spieckerbc and
W. Peukertac
*ac,
M. Haderleinac,
J. Walterac,
B. Apeleo Zubiribc,
E. Spieckerbc and
W. Peukertac
aInstitute of Particle Technology (LFG), Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Cauerstr. 4, 91058 Erlangen, Germany. E-mail: tugce.akdas@cea.fr
bCenter for Nanoanalysis and Electron Microscopy (CENEM), Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Cauerstr. 6, 91058 Erlangen, Germany
cCluster of Excellence – Engineering of Advanced Materials (EAM), Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Nägelsbachstrasse 49b, 91052 Erlangen, Germany
First published on 6th February 2017
Abstract
In colloidal semiconductor nanocrystal synthesis, many activities have been focused on the control over the mean particle size of the product. However, for device applications additional requirements apply, e.g. the necessity for a narrow particle size distribution (PSD). In the present work, we investigate the impact of reactor characteristics on the synthesis of CuInS2 quantum dots (QDs). Therefore, the synthesis is carried out in three different reactors, namely in a fully scalable and continuous tubular reactor, in a commercial microreactor and in a batch three-neck round-bottom flask. All reaction products from the three reactor types have the same crystal structure and inorganic composition. Slight differences in optical properties are mainly ascribed to differences in the PSD, which is confirmed by sedimentation velocity experiments. Furthermore, the necessity for using different post-processing routines coupled with our findings by analytical ultracentrifugation (AUC) hint at changes in the organic matrix surrounding the CuInS2 QDs. Our study demonstrates the massive impact of heat transfer on the synthesis and the final PSD of CuInS2 QDs synthesized via heating-up and allows us to draw conclusions on their formation.
1 Introduction
CuInS2 QDs are attractive candidates for biomedical1,2 and electronic3–5 applications and a non-toxic alternative to Cd- and Pb-based QDs. Various batch methods exist for the synthesis of CuInS2 QDs,6–11 suitable for the synthesis and optimization of small amounts of material. Practical applications require the production of larger amounts of high quality QDs. For CdSe QDs, as the most widely studied system,12–18 optimization and scaling-up of the batch process19,20 have been reported. While these studies yielded well-defined samples with relatively narrow particle size distributions (PSD), the transfer of these empirical findings to other material systems remains a challenge. The factors determining the PSD of a QD sample are not yet fully understood.21 Actually, very little information on mixing, heat and mass transfer and the related build-up of the thermodynamic driving force of particle formation, i.e. the supersaturation, is available for most QD batch synthesis protocols.Continuous processes offer advantages such as high reproducibility, large production rate, in situ monitoring and simple automation coupled with a more effective heat transfer.22–26 Furthermore, micromixing devices are well-characterized in terms of critical steps of particle formation.27–31 Several groups have utilized microfluidic systems for continuous syntheses of nanoparticles of various types and shapes,32–38 QDs39–43 and core–shell systems.44–46 Various actions have resulted in narrow PSDs, such as the use of segmented flow,47–49 multistage48,50 and high-temperature high-pressure reactors,43,51,52 the latter enabling syntheses under supercritical conditions. While the use of multistage reactors allows for separation of nucleation and growth, the other two systems aim at minimization of the effects of solvent viscosity and axial dispersion. A recent study by Jiao et al.53 has evaluated the impact of residence time, fluid velocity and reactor dimensions on PSD of thermally decomposed Fe3O4 nanoparticles. The aforementioned studies are focused on mass transfer as a key to improve PSDs of binary systems, to the best of our knowledge detailed studies on the impact of heat transfer on syntheses outcome have not been reported so far.
Recently, continuous synthesis of CuInS2 QDs has been published50,54 and optimizations have been carried out focusing on the synthesis parameters and on the photoluminescence quantum yield (PL QY). Detailed studies, e.g. on their formation mechanism or on the impact of heat and mass transfer on the synthesis outcome are still missing. Especially the PSD of CuInS2 QDs which has been shown to be broad in the batch synthesis55 is a critical parameter which is worth further attention. In the present contribution, we describe a continuous route to produce CuInS2 QDs based on flow chemistry and compare this route to the batch synthesis of CuInS2 QDs. In-depth characterization of the products with regard to crystal structure, composition, optical properties and PSD hints on differences in particle formation in the two flow reactors (tubular reactor TR and microreactor MR) compared to synthesis in the batch reactor (three necked round bottom flask). The findings are compared to state-of-the-art nucleation and growth models and the dependency of particle formation on temperature evolution is discussed for the batch and the continuous flow syntheses. Our experiments reveal the importance of technical aspects in the heat-up synthesis of QDs and show that the challenging transfer from batch to continuous process not only opens promising options for economical production of larger quantities of multicomponent QDs, but also promises to yield products with narrower PSD and simplified post-processing.
2 Results and discussion
2.1 Transfer of batch synthesis to continuous flow reactors
In the batch synthesis of CuInS2 QDs according to Zhong et al.,8 the reactants copper acetate (CuAc), indium acetate (InAc3), 1-dodecanethiol (1-DDT) and 1-octadecene (1-ODE) are simply mixed and heated up to 240 °C under inert atmosphere. Prerequisite to transfer this synthesis to a flow reactor is a clear and homogeneous reaction solution which can be pumped through the reactor channels without clogging them. As the reactants do not dissolve in 1-ODE at room temperature, a suitable routine for the preparation of a homogeneous solution which can then be introduced into the flow reactors in the second step was evaluated. Similar to Tian et al.54 we found that a clear yellow precursor solution is obtained at 170 °C.In Scheme 1, a possible reaction path for CuInS2 QD synthesis is given which is based on the two-step nucleation and growth model established for CdSe QDs.15,17 First, the reagents dissolve and form a precursor solution upon heating to 170 °C. Further temperature increase leads to the decomposition of the precursors in the second step. This presumably results in “activated” monomers which eventually nucleate if their concentration is high enough and grow yielding CuInS2 QDs in the third step. Considering the synthesis of CuInS2 from single-source molecular precursors7,10,56,57 and the large variety of possible thiolate complexes,58,59 the in situ formation of a tetragonal precursor complex8 is very probable. We propose precursor decomposition at temperatures below 240 °C as our experiments revealed that particle formation is readily possible at ∼220 °C (see Fig. S3 in ESI†). Our previous investigations60 revealed that side-products such as dodecyloctadecylsulfide are formed during reaction.
 | ||
| Scheme 1 Illustration of the reaction path as a three step process, starting from the single reagents to the final product. | ||
2.2 Flow synthesis of CuInS2 QDs
All reactions discussed in the following were carried out by preparing a precursor solution under inert conditions at 170 °C first, then pumping that solution into the reactors preheated to 240 °C. First, the results obtained in the self-build TR will be discussed, followed by a comparison of the as-obtained products with the products of synthesis in MR and in batch.Synthesis in TR was performed with mean residence times ranging from ![[small tau, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d4.gif) = 5 min to = 20 min. Structural characterization of the products revealed CuInS2 QDs with tetragonal crystal structure (see Fig. S3 and S6 in ESI†) and a composition of Cu
= 5 min to = 20 min. Structural characterization of the products revealed CuInS2 QDs with tetragonal crystal structure (see Fig. S3 and S6 in ESI†) and a composition of Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :In:S = 1:0.9 (±0.08):1.8 (±0.16). TEM imaging provided additional information on the crystallinity and particle size of the CuInS2 QDs. BF-TEM and STEM images of the product with = 5 min are shown in Fig. 1. The QDs are crystalline with a size of about 2 nm and show a narrow PSD. The investigation of single particles reveals that the crystal lattice planes {112}CuInS2 can be observed with highest probability (lattice plane distance d112CuInS2 = 0.31973 nm [ICSD no. 186714]).
:In:S = 1:0.9 (±0.08):1.8 (±0.16). TEM imaging provided additional information on the crystallinity and particle size of the CuInS2 QDs. BF-TEM and STEM images of the product with = 5 min are shown in Fig. 1. The QDs are crystalline with a size of about 2 nm and show a narrow PSD. The investigation of single particles reveals that the crystal lattice planes {112}CuInS2 can be observed with highest probability (lattice plane distance d112CuInS2 = 0.31973 nm [ICSD no. 186714]).
 | ||
| Fig. 1 (a) BF-TEM image of the 5 min sample showing CuInS2 QDs with a size of around 1.5 nm to 2 nm – the insets show HRTEM images of two exemplary QDs with indicated {112}CuInS2 lattice planes (yellow lines) with a distance of d112CuInS2 = 0.31973 nm. (b) STEM image of the 5 min sample showing the CuInS2 QDs exhibiting a bright contrast. | ||
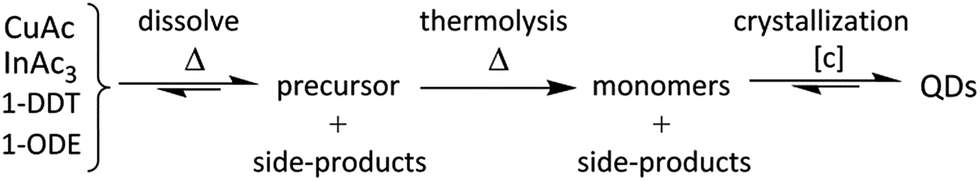
Optical characterization of products is shown in Fig. 2a and b. The QDs synthesized with = 5 min reveal an absorption spectrum with one single feature at ∼520 nm and an absorption onset at ∼650 nm. With increasing , both the feature and the absorption onset shift to longer wavelengths (∼560 nm and ∼700 nm), indicating growth of the QDs with increasing , and thus, increasing reaction times. A similar tendency is observed in the emission spectra (Fig. 2b) which show a slight shift of the maxima from ∼660 nm to ∼680 nm from = 5 min to = 20 min. Furthermore, the emission intensity increases and the relatively broad signals seem to consist of only one feature, although not being perfectly symmetrical. Interestingly, the differences between the optical properties of the two samples = 10 min and = 20 min are relatively small in terms of exact position and shape of the absorption spectrum, however, a clear intensity increase accompanied by just a small shift is discernible in the emission spectrum. Recent reports have shown that the PL QYs can be raised up to 57% (ref. 50 and 54) by coating the core particles with a ZnS shell. The focus of this work however was not to optimize the PL QY, but to gain understanding on the impact of various technical aspects on the CuInS2 core product. Various factors can explain an increase of the radiative recombination pathways relative to non-radiative ones, such as changes in (i) the PSD, (ii) the surface structure and/or (iii) crystal structure due to potential reorganization of the atoms and, finally, (iv) the nature and bonding of stabilizing ligands on the surface. Most probably, a combination of factors leads to the observed changes. In the following, solely the effect of the PSD will be investigated.
 | ||
| Fig. 2 Temporal evolution of (a) absorption and (b) emission spectra of CuInS2 QDs synthesized in a tubular reactor (TR) with residence times of 5 min, 10 min and 20 min. | ||
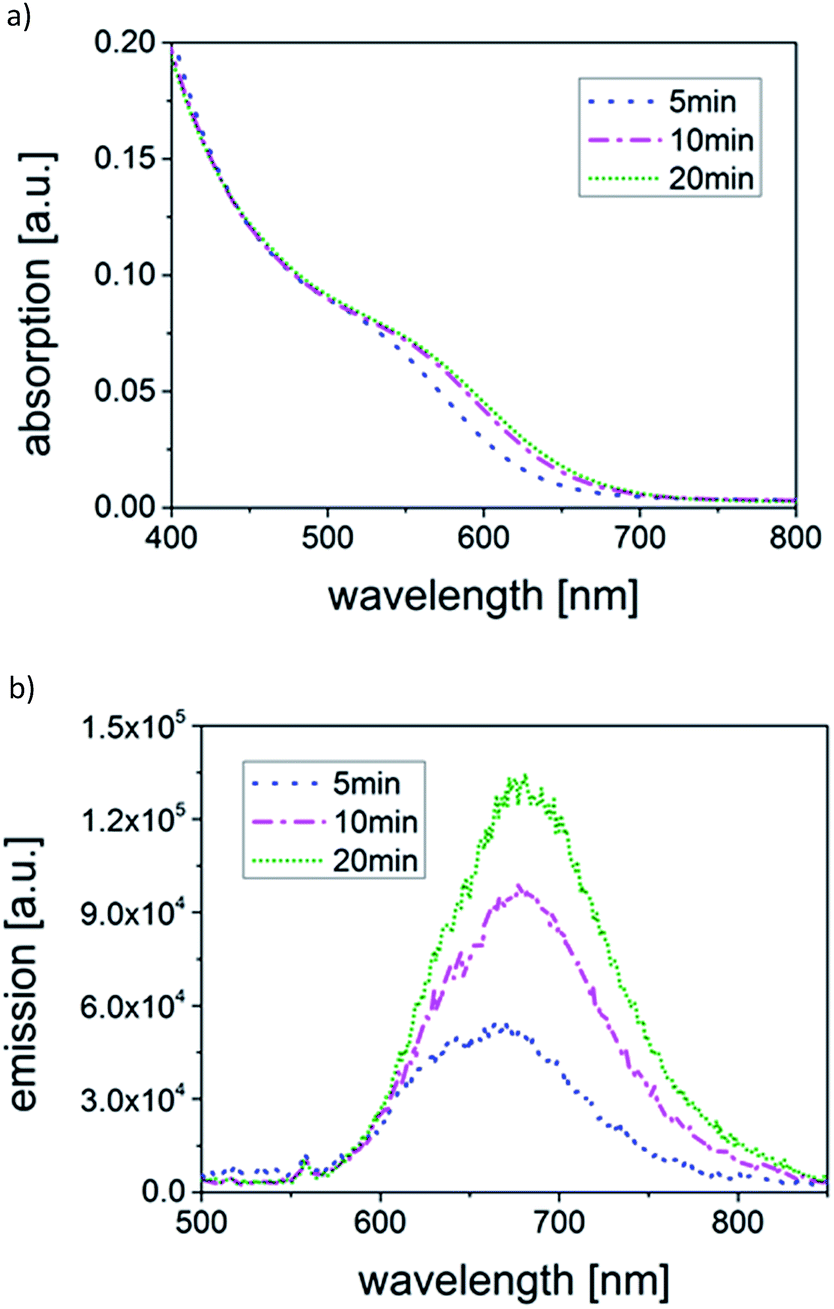
To precisely determine the changes in the PSD during growth, sedimentation velocity experiments were carried out via AUC. The AUC experiments reveal a very narrow PSD ( = 5 min) which then shifts to larger sedimentation coefficients with increasing as shown in Fig. 3. A very slight broadening is recognized (the normalized results are compared in Fig. S7 in ESI†). Calculations of the mean core diameter xc using a core–shell model and the multidimensional approach resulted in mean xc of 2.41 nm ( = 5 min), 2.61 nm ( = 10 min) and 2.68 nm ( = 20 min), respectively. The growth rate based on mean diameters can be estimated to initially 2.4 nm h−1 (between = 5 min and 10 min) slowing down to only 0.42 nm h−1 (between 10 and 20 min). More information on AUC results can be found in Table S2.† The AUC results corroborate the growth of the particles as suggested by the redshift of absorption and emission spectra and, thus, demonstrate the connection of the particle size with the optical properties.
 | ||
| Fig. 3 Temporal evolution of the SV-AUC of CuInS2 QDs synthesized in a TR with mean residence times of 5 min, 10 min and 20 min. | ||
2.3 Comparison of batch and flow product
In the following, the products from synthesis in different reactors will be investigated and compared in terms of crystal structure, composition, optical properties and PSD.XRD analyses revealed tetragonal crystal structures with three main diffractions at exactly the same diffraction angles in all three cases (see Fig. S6a in ESI†). Additional TEM investigations of the TR (Fig. 1) corroborated these results. The inorganic composition of the samples was found to be almost identical, i.e., Cu1.0In0.9S1.8 for the TR product, Cu1.0In1.0S1.9 for the MR product and Cu1.0In0.8S2.0 for the batch product (slight deviations are within the experimental error, i.e. ±10% for both indium and sulfur). However, the organic content of the samples differed, making an adjustment of the post-processing procedure necessary. While many centrifugation cycles were necessary for purification of the batch product,60 just 2–3 cycles of centrifugation using a combination of methanol with acetone or only acetone as poor solvent were sufficient for purification of the continuously synthesized products. The TGA curves given in Fig. S6b (in ESI†) reveal that in all cases a final organic content of ∼45 wt% was reached. The large number of centrifugation cycles necessary to purify the batch product indicates once more that these nanocrystals are surrounded by a huge and rigid organic matrix which seems not to be the case for the products from flow synthesis (see discussion at the end of this section). From an economical point of view, continuous flow synthesis results in products which allow for an easier post-processing saving time and money.
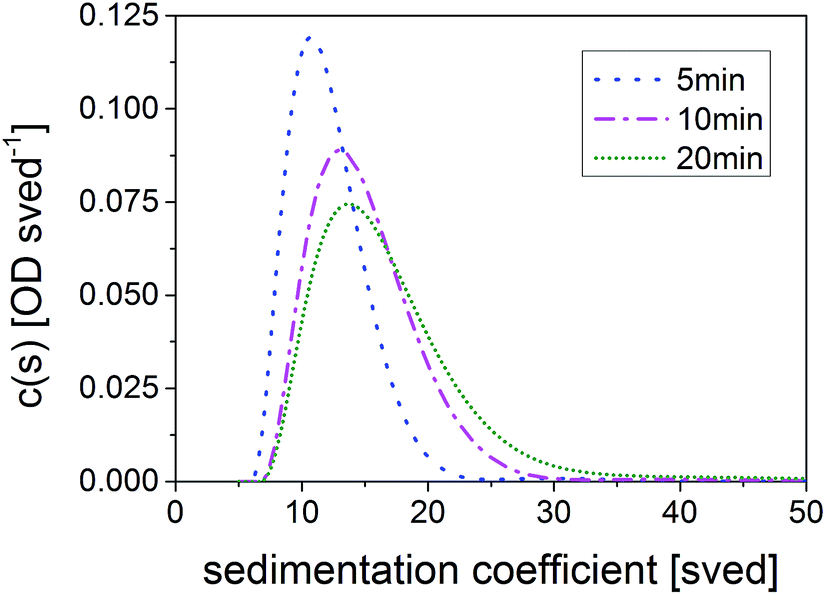
Turning to optical properties, it should be mentioned first that the time-dependent growth of the QDs and the corresponding shifts in the absorption spectra were observed for products synthesized in all three reactors. Comparing the results in more detail, it becomes obvious that samples synthesized with = 5 min in TR and = 3 min in MR demonstrate almost identical absorption (Fig. 4) and emission features (Fig. S6c in ESI†). The closest respective batch sample showing an absorption onset at the same wavelength range was synthesized after 45 min and reveals a broader absorption spectrum with one or two less distinct absorption features. The reason for the significant differences in reaction times will be discussed in Section 3.5.
 | ||
| Fig. 4 Comparison of the absorption of CuInS2 QDs synthesized in batch and in continuous flow, respectively. The reaction temperature was 240 °C in all reactions; corresponding were 5 min in the TR, 3.0 min in the microreactor (MR) and 45 min in the batch reactor. | ||
To examine the PSD of the samples, PSDs derived by sedimentation coefficient distributions were compared. Comparing the QDs synthesized in TR and in batch (see Fig. 5 and additionally Fig. S7 (ESI†)), it becomes obvious that the PSD of the samples synthesized in the TR is much narrower than the PSD of the ones synthesized in the batch reactor. The particle sizes range from 1.9 nm to 3.3 nm for synthesis in the TR, while sizes ranging from 1.6 nm to 3.7 nm are obtained in batch. Thus, the overall width of the PSD could be reduced from 2.1 nm in batch to 1.4 nm in TR (reduction by ∼30%).
 | ||
| Fig. 5 Comparison of the PSDs obtained from SV-AUC experiments of CuInS2 QDs synthesized in the TR and in the batch reactor. | ||
On closer inspection, similarities but also a deviation between the AUC results and the absorption spectra become apparent. First, smaller particles are present in the batch product, which are detected by AUC and are also found in the absorption spectra (less distinct second feature at ∼480 nm, Fig. 4). Second, the AUC identifies larger particles in the batch product as the upper ends of the PSD deviate for the two products. This finding, however, is not supported by the absorption spectra as the absorption onsets of the TR and the batch samples are located at nearly equal wavelengths. Since we can exclude the existence of products with different crystal structure and varying inorganic composition from our TEM, XRD and ICP-OES studies, it is very likely that aggregation occurred during the batch synthesis - a finding that was also proposed in our previous study.55 The organic molecules seem to form ligand-mediated bridges between the nanocrystals and thereby link them to each other, resulting in the detection of larger particle sizes in AUC. As the aggregates are not visible in the absorption spectrum, it is likely to conclude that electronic communication between single nanocrystals does not exist. Presumably, the aggregation of the nanocrystals in the batch synthesis and their strong encapsulation in an organic matrix resulting in tedious purification procedures are linked to each other. Furthermore, these findings might serve to explain the moderate separation efficiencies in case of size-selective precipitation.
Summarizing, a relatively narrow PSD for the TR product ( = 5 min) is confirmed by AUC, absorption spectroscopy and TEM. For the batch product, in turn, the existence of smaller species was proven by AUC, absorption and TEM55 confirming it's broader PSD. Additionally, aggregation was determined in the batch product by AUC.
2.4 Influence of reactor characteristics on CuInS2 synthesis
Our results demonstrate the successful synthesis of CuInS2 QDs with tetragonal crystal structure in batch and continuous operation mode. As shown above, the main difference between batch and flow products is the width of their PSD which is narrower for products synthesized in flow reactors. As the reagents and the preparation procedures were identical in all three reactors, it is very likely that the main reasons for product variations originate from the impact of different reactor characteristics on the reaction kinetics.To investigate the impact of the reactors, all three of them were characterized with regard to their flow profile and heat transfer (see Section 1 in ESI†). While turbulent mixing was assured in the flask by stirring the reaction solution vigorously, the flow was laminar in both flow reactors. Comparing the products obtained in the three reactors, the impact of mixing on the synthesis product seems to be marginal in terms of impact on crystal structure and inorganic composition. Therefore, we conclude that intense mixing is not necessary to ensure a successful synthesis of CuInS2 QDs, provided that a stable precursor solution has been previously prepared in a separate process step.
When turning to heat transfer, temperature evolution to the reaction temperature (240 °C) is most efficient in the MR and least efficient in the flask, see Fig. S2 and corresponding calculations and discussions in ESI.† The more efficient heat transfer in flow reactors has serious consequences for both, the duration of the heating period (few seconds in MR and ∼3 min in TR, opposed to ∼13 min in flask) and the duration of the reaction itself which is shortened down to a few minutes in the flow reactors. The improved heat transfer has a huge impact on reaction kinetics of QD formation and side-reactions, the latter resulting in changes in the organic matrix around the nanocrystals (as proven by TGA and AUC results). In the following, the impact of heat transfer will be discussed in detail for both, the continuous and the batch syntheses.
Based on LaMer-type nucleation and growth and the strong dependency of reaction kinetics on temperature, two scenarios are depicted in Scheme 2 illustrating the formation of CuInS2 QDs in different reactors. In the flow reactors used in this study, the heating period is relatively short and 240 °C is reached quickly. This results in quasi simultaneous decomposition of the precursors followed by a sudden nucleation event, similar to the theoretical ‘burst’ nucleation known from the hot injection method (illustrated by the fast heating curve, blue line, in Scheme 2). As the wall temperature in flow reactors is constant, (i.e. no temperature fluctuations occur), and as there is ideally no mixing between earlier and later entering fluid (i.e. no backmixing), a defined stream of reaction solution is flowing along the tube. The nuclei formed in the beginning grow and have all a similar growth history, resulting in a relatively narrow PSD of the product, as confirmed by AUC analysis. The fact that the reaction proceeds relatively fast in the flow reactors (a few minutes opposed to 45 min in batch) might originate from two effects. First, the effective and homogeneous heat transfer in the tube enhances the decomposition of the precursor and, thus, the formation of monomer units. This does not only influence the formation of nuclei but also their growth. Second, also the kinetics of the side-reactions are varied, affecting the organic matrix and, eventually, colloidal stability of the particles. As the organic matrix in the TR product seems to be less rigid, growth of the particles might be accelerated.
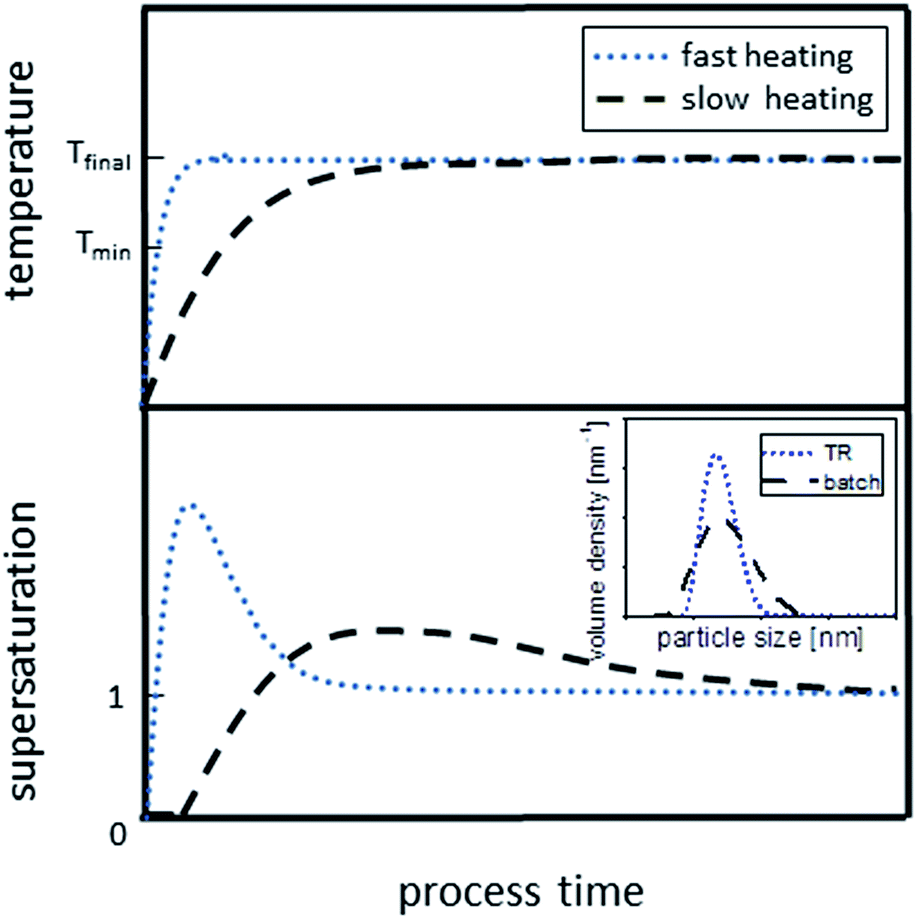 | ||
| Scheme 2 Effect of the heating rate on the build-up and decline of supersaturation. The fast heating curve (blue dotted line) depicts the situation in the flow reactors, while the slow heating curve illustrates the reaction progress in the batch reactor (black dashed line). Inset shows the effect on PSDs (compare Fig. 5). | ||
In the batch reactor used here, the heating period is comparably long, resulting in a slow reaction during heat-up as depicted in Scheme 2 (black dashed line). Considering that the precursor decomposes at temperatures ∼220 °C or even below (see Section 2.1), activated monomers can already form and accumulate in the solution over a long period of time before the desired reaction temperature is reached. Thus, a longer heating period as in the case of batch reaction increases the probability for nucleation to start prior reaching final reaction temperature. This results in a wide overlap of (i) formation of activated monomers, (ii) nucleation and (iii) growth and, thus, yields a wider PSD. The broad PSD becomes even more evident when considering the temperature fluctuations during the batch synthesis in the flask (electrical heating: 239 °C ± 5 °C). These fluctuations will definitely influence the monomer supply kinetics and might even result in spontaneous nucleation events during synthesis. Additionally, there is enough time for side-reactions to occur, leading to the consequences discussed in Section 2.3. This serves to explain broad and potentially even bimodal PSDs obtained from batch synthesis.
3 Conclusions
The potential of flow synthesis to narrow the PSD of nanoscale products had been proposed more than a decade ago, however, evidence was missing so far as most studies were performed on CdSe QDs which already have a narrow PSD in batch. The PSD of multicomponent QDs from batch synthesis, in contrast, is usually quite broad. In the present work, we successfully transferred the one-pot synthesis of CuInS2 QDs to two different flow reactors and investigated the impact of reactor characteristics on QD synthesis and products' PSD. Remarkably, continuous synthesis in flow reactors offered several advantages:First, the reaction time was dramatically reduced down to a few minutes in the flow reactors. Second, the PSD is significantly narrower in the flow syntheses compared to batch synthesis. Third, purification of the continuously synthesized product was faster and economically more feasible as the number of centrifugation cycles was reduced.
These improvements were mainly ascribed to a very efficient heat transfer in flow reactors impacting the reaction kinetics of both particle formation and side-reactions. Based on the LaMer model, a possible reaction scheme was developed for both continuous flow and batch syntheses. We are confident that an improved heat transfer in general has a positive impact on products' PSD for various material systems, especially when the synthesis procedure involves thermal decomposition of precursors. Minimization of solvent viscosity and dispersion effects is considered to further narrow the PSD for such multicomponent material systems. For future research, purification techniques compatible with continuous processing need to be evaluated with regard to both applicability and final product properties. A recent review by Shen et al.61 highlights both established and emerging (continuous) purification approaches and discusses them in the framework of surface chemistry and optical properties. Our finding of a less rigid organic surrounding for the flow product holds promise to facilitate the continuous purification. Furthermore, negligible differences between the products synthesized in MR and TR indicate that these processes were conducted in a regime where heating is not a limiting factor. Therefore, the applied TR is well-suited for upscaling.
Acknowledgements
The authors would like to thank the German Research Council (DFG) for financial support within the framework of its Excellence Initiative the Cluster of Excellence “Engineering of Advanced Materials” (DFG EXC 315), within the Priority Program SPP 1679 “Dynamische Simulation vernetzter Feststoffprozesse” and within project PE 427/28-1. Additionally, we would like to thank Bayer Technology Services (BTS) for financial support. T. A. is deeply grateful to P. Hoppe for ICP-OES measurements at the Institute of Particle Technology LFG, Friedrich-Alexander-Universität Erlangen-Nürnberg.References
- M. Bruchez, M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013–2016 CrossRef CAS PubMed.
- W. C. W. Chan and S. M. Nie, Science, 1998, 281, 2016–2018 CrossRef CAS PubMed.
- V. I. Klimov, A. A. Mikhailovsky, S. Xu, A. Malko, J. A. Hollingsworth, C. A. Leatherdale, H. J. Eisler and M. G. Bawendi, Science, 2000, 290, 314–317 CrossRef CAS PubMed.
- S. Coe, W. K. Woo, M. Bawendi and V. Bulovic, Nature, 2002, 420, 800–803 CrossRef CAS PubMed.
- V. I. Klimov, Semiconductor and metal nanocrystals: synthesis and electronic and optical properties, CRC Press, 2003 Search PubMed.
- P. M. Allen and M. G. Bawendi, J. Am. Chem. Soc., 2008, 130, 9240–9241 CrossRef CAS PubMed.
- S. L. Castro, S. G. Bailey, R. P. Raffaelle, K. K. Banger and A. F. Hepp, J. Phys. Chem. B, 2004, 108, 12429–12435 CrossRef CAS.
- H. Zhong, Y. Zhou, M. Ye, Y. He, J. Ye, C. He, C. Yang and Y. Li, Chem. Mater., 2008, 20, 6434–6443 CrossRef CAS.
- K. Yu, P. Ng, J. Ouyang, M. B. Zaman, A. Abulrob, T. N. Baral, D. Fatehi, Z. J. Jakubek, D. Kingston, X. Wu, X. Liu, C. Hebert, D. M. Leek and D. M. Whitfield, ACS Appl. Mater. Interfaces, 2013, 5, 2870–2880 CAS.
- S. L. Castro, S. G. Bailey, R. P. Raffaelle, K. K. Banger and A. F. Hepp, Chem. Mater., 2003, 15, 3142–3147 CrossRef CAS.
- J. J. Nairn, P. J. Shapiro, B. Twamley, T. Pounds, R. von Wandruszka, T. R. Fletcher, M. Williams, C. M. Wang and M. G. Norton, Nano Lett., 2006, 6, 1218–1223 CrossRef CAS PubMed.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- C. A. Leatherdale, W. K. Woo, F. V. Mikulec and M. G. Bawendi, J. Phys. Chem. B, 2002, 106, 7619–7622 CrossRef CAS.
- W. W. Yu, L. H. Qu, W. Z. Guo and X. G. Peng, Chem. Mater., 2003, 15, 2854–2860 CrossRef CAS.
- J. S. Owen, E. M. Chan, H. Liu and A. P. Alivisatos, J. Am. Chem. Soc., 2010, 132, 18206–18213 CrossRef CAS PubMed.
- S. Abe, R. K. Capek, B. De Geyter and Z. Hens, ACS Nano, 2012, 6, 42–53 CrossRef CAS PubMed.
- Z. Hens and R. K. Capek, Coord. Chem. Rev., 2014, 263, 217–228 CrossRef.
- B. Abecassis, C. Bouet, C. Garnero, D. Constantin, N. Lequeux, S. Ithurria, B. Dubertret, B. R. Pauw and D. Pontoni, Nano Lett., 2015, 15, 2620–2626 CrossRef CAS PubMed.
- E. M. Chan, C. Xu, A. W. Mao, G. Han, J. S. Owen, B. E. Cohen and D. J. Milliron, Nano Lett., 2010, 10, 1874–1885 CrossRef CAS PubMed.
- M. Protiere, N. Nerambourg, O. Renard and P. Reiss, Nanoscale Res. Lett., 2011, 6(1), 472 CrossRef PubMed.
- Z. Hens, Science, 2015, 348, 1211–1212 CrossRef CAS PubMed.
- J. P. McMullen and K. F. Jensen, in Annual Review of Analytical Chemistry, ed. E. S. Yeung and R. N. Zare, 2010, vol. 3, pp. 19–42 Search PubMed.
- A. J. deMello, Nature, 2006, 442, 394–402 CrossRef CAS PubMed.
- A. Abou-Hassan, O. Sandre and V. Cabuil, Angew. Chem., Int. Ed., 2010, 49, 6268–6286 CrossRef CAS PubMed.
- W. Ehrfeld, V. Hessel and H. Löwe, Microreactors, Wiley-VCH, Weinheim, 2000 Search PubMed.
- W. Ehrfeld, V. Hessel and V. Haverkamp, Microreactors, Ullmann's Encyclopedia of Industrial Chemistry, 2000 Search PubMed.
- M. A. J. Hartig, N. Jacobsen and W. Peukert, Chem. Eng. Sci., 2014, 109, 158–170 CrossRef CAS.
- J. Gradl and W. Peukert, Chem. Eng. Sci., 2009, 64, 709–720 CrossRef CAS.
- F. Schwertfirm, J. Gradl, H. C. Schwarzer, W. Peukert and M. Manhart, Int. J. Heat Fluid Flow, 2007, 28, 1429–1442 CrossRef CAS.
- J. Gradl, H. C. Schwarzer, F. Schwertfirm, M. Manhart and W. Peukert, Chem. Eng. Process., 2006, 45, 908–916 CrossRef CAS.
- H. C. Schwarzer and W. Peukert, Chem. Eng. Technol., 2002, 25, 657–661 CrossRef CAS.
- J. Boleininger, A. Kurz, V. Reuss and C. Soennichsen, Phys. Chem. Chem. Phys., 2006, 8, 3824–3827 RSC.
- C.-H. Weng, C.-C. Huang, C.-S. Yeh, H.-Y. Lei and G.-B. Lee, J. Micromech. Microeng., 2008, 18(3), 035019 CrossRef.
- J. Wagner and J. M. Kohler, Nano Lett., 2005, 5, 685–691 CrossRef CAS PubMed.
- H. D. Jin and C.-H. Chang, J. Mater. Chem., 2011, 21, 12218–12220 RSC.
- K.-J. Kim, R. P. Oleksak, C. Pan, M. W. Knapp, P. B. Kreider, G. S. Herman and C.-H. Chang, RSC Adv., 2014, 4, 16418–16424 RSC.
- A. Shavel, D. Cadavid, M. Ibanez, A. Carrete and A. Cabot, J. Am. Chem. Soc., 2012, 134, 1438–1441 CrossRef CAS PubMed.
- C.-X. Zhao, L. He, S. Z. Qiao and A. P. J. Middelberg, Chem. Eng. Sci., 2011, 66, 1463–1479 CrossRef CAS.
- F. G. Bessoth, A. J. deMello and A. Manz, Anal. Commun., 1999, 36, 213–215 RSC.
- H. Nakamura, Y. Yamaguchi, M. Miyazaki, M. Uehara, H. Maeda and P. Mulvaney, Chem. Lett., 2002, 1072–1073, DOI:10.1246/cl.2002.1072.
- H. Nakamura, Y. Yamaguchi, M. Miyazaki, H. Maeda, M. Uehara and P. Mulvaney, Chem. Commun., 2002, 2844–2845, 10.1039/b208992k.
- E. M. Chan, A. P. Alivisatos and R. A. Mathies, J. Am. Chem. Soc., 2005, 127, 13854–13861 CrossRef CAS PubMed.
- S. Marre, J. Park, J. Rempel, J. Guan, M. G. Bawendi and K. F. Jensen, Adv. Mater., 2008, 20, 4830–4834 CrossRef CAS.
- H. Z. Wang, H. Nakamura, M. Uehara, Y. Yamaguchi, M. Miyazaki and H. Maeda, Adv. Funct. Mater., 2005, 15, 603–608 CrossRef CAS.
- W. Luan, H. Yang, N. Fan and S.-T. Tu, Nanoscale Res. Lett., 2008, 3, 134–139 CrossRef.
- H. Z. Wang, X. Y. Li, M. Uehara, Y. Yamaguchi, H. Nakamura, M. P. Miyazaki, H. Shimizu and H. Maeda, Chem. Commun., 2004, 48–49, 10.1039/b310644f.
- S. A. Khan, A. Günther, M. A. Schmidt and K. F. Jensen, Langmuir, 2004, 20, 8604–8611 CrossRef CAS PubMed.
- V. Misuk, M. Schmidt, S. Braukmann, K. Giannopoulos, D. Karl and H. Loewe, Chem. Eng. Technol., 2015, 38, 1150–1153 CrossRef CAS.
- A. M. Nightingale and J. C. de Mello, J. Mater. Chem., 2010, 20, 8454–8463 RSC.
- A. Yashina, I. Lignos, S. Stavrakis, J. Choo and A. J. deMello, J. Mater. Chem. C, 2016, 4, 6401–6408 RSC.
- J. Baek, P. M. Allen, M. G. Bawendi and K. F. Jensen, Angew. Chem., 2011, 123, 653–656 CrossRef.
- A. Chakrabarty, S. Marre, R. F. Landis, V. M. Rotello, U. Maitra, A. D. Guerzo and C. Aymonier, J. Mater. Chem. C, 2015, 3, 7561–7566 RSC.
- M. Jiao, J. Zeng, L. Jing, C. Liu and M. Gao, Chem. Mater., 2015, 27, 1299–1305 CrossRef CAS.
- S. Tian, M. Fu, W. Hoheisel and L. Mleczko, Chem. Eng. J., 2016, 289, 365–373 CrossRef CAS.
- T. Akdas, J. Walter, D. Segets, M. Distaso, B. Winter, B. Birajdar, E. Spiecker and W. Peukert, Nanoscale, 2015, 7(43), 18105–18118 RSC.
- W. Hirpo, S. Dhingra, A. C. Sutorik and M. G. Kanatzidis, J. Am. Chem. Soc., 1993, 115, 1597–1599 CrossRef CAS.
- K. K. Banger, J. Cowen and A. F. Hepp, Chem. Mater., 2001, 13, 3827–3729 CrossRef CAS.
- I. G. Dance, Polyhedron, 1986, 5, 1037–1104 CrossRef CAS.
- B. Krebs and G. Henkel, Angew. Chem., Int. Ed., 1991, 30, 769–788 CrossRef.
- T. Akdas, M. Distaso, S. Kuhri, B. Winter, B. Birajdar, E. Spiecker, D. M. Guldi and W. Peukert, J. Colloid Interface Sci., 2015, 445, 337–347 CrossRef CAS PubMed.
- Y. Shen, M. Y. Gee and A. B. Greytak, Chem. Commun., 2017, 53(5), 827–841 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Details on mixing and heat transfer; summary of AUC results; together with further syntheses results in various reactors and according comparisons. See DOI: 10.1039/c6ra27052b |
| ‡ Present address: CEA Grenoble, INAC, SyMMES, STEP, 17 rue des Martyrs, 38054 Grenoble, France. |
| This journal is © The Royal Society of Chemistry 2017 |