
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hybrid luminescent porous silicon for efficient drug loading and release†
A. Ghafarinazari a,
M. Scarpab,
G. Zoccatellic,
M. Comes Franchinid,
E. Locatelli*d and
N. Daldosso*a
a,
M. Scarpab,
G. Zoccatellic,
M. Comes Franchinid,
E. Locatelli*d and
N. Daldosso*a
aDepartment of Computer Science, University of Verona, Strada le Grazie 15, 37134 Verona, Italy. E-mail: nicola.daldosso@univr.it
bDepartment of Physics, Istituto Nazionale Biostrutture Biosistemi, University of Trento, Via Sommarive 14, 38123 Trento, Italy
cDepartment of Biotechnology, University of Verona, Strada le Grazie 15, 37134 Verona, Italy
dDepartment of Industrial Chemistry, University of Bologna, Viale Risorgimento 4, 40136 Bologna, Italy. E-mail: erica.locatelli2@unibo.it
First published on 20th January 2017
Abstract
In recent decades, biocompatible and light emitting porous silicon (pSi) showed the possibility for use in biomedical applications. Unfortunately, until now the biological applications of light-emitting pSi have been severely limited due the immediate surface oxidation of this nanostructured material, followed by loss of optical properties and degradation in aqueous solutions. In view of drug delivery, the red–ox activity of pSi damages the carried drug. Herein, we have investigated the stabilization of pSi microparticles in aqueous solution by covalent attachment of polymers to a short-chain organic coating grafted onto inorganic silicon. The morphological, chemical and optical stability obtained using polyethylene glycol confirm this biocompatible polymer as a superior shell matrix nanostructure for use in biological environments.
1 Introduction
Space and time controlled delivery of drugs is a pharmacological challenge since it would reduce the waste and the side effects of drugs, thus improving their therapeutic properties.1,2 Drug delivery systems (DDS) should offer enough room for drug loading, provide an internal surface with an affinity for the cargo and penetrate homogeneously and efficiently the target tissue or barrier. Indeed, DDS control the release kinetics of the drug in the desired location. In this regard, porous materials are interesting because they offer the potential to reduce systemic toxicity and increase the amount of loaded drug.3 Among them, porous silicon (pSi) appears a suitable material for the realization of DDS because of: (i) its large pore volume,4 (ii) its “tuneable” surface chemistry,5 and (iii) its low toxicity associated with safe biodegradation pathways.6 Porous silicon has been investigated by many groups as DDS and still is a field of intense research.7–11 Moreover, well assessed fabrication protocols produce pSi microparticles with intense photoluminescence (PL) in the visible region, which can be tracked in cells by fluorescence imaging in real time.12,13 Another promising application of pSi for medicine is photo-thermal therapy, which is based on absorption of light and on its transformation to heat in order to kill the target (cell, tissue).14 These properties make pSi particles based systems particularly suitable for both therapy and diagnosis of diseases, allowing for real-time monitoring of the therapeutic efficacy and for a reduction of drugs administration, which are the basis of the novel and promising field of “theranostic”.15However, un-resolved questions still limit the effective utilization of the pSi as delivery system: (a) the fast degradation of pSi in in vitro conditions16 and in harsh biological environments,17 (b) the irreversible quenching of the pSi luminescence due to interaction with water or other biomolecules,18 (c) the decomposition of a carried drug by redox activity of pSi.19
One of the main applications of pSi is for bioimaging due to light emission in the biological window (BW). Absorption of pSi in BW is quite low, but this issue can be solved, for example, by using two photon absorption (TPA). Indeed, instead of using typical excitation at 350 or 400 nm, by means of TPA (with excitation wavelength of 700 or 800 nm, which perfectly matches BW) it is possible to get efficient light emission thus being used in bioimaging.20 The other issue is the optical stability of pSi in biological media, e.g. water based solutions. In fact, while pSi covered by a shell of acrylic groups (pSi–COOH functionalization) introduced by a hydrosilylation reaction shows efficient PL in ethanol for very long time,21 a stable light emission in biological media, such as phosphate-buffered saline (PBS) solution, still remains a big challenge.16 Some protocols have been proposed to postpone pSi degradation up to hours, for example: thermal oxidation resulted in 4 h stability12 while silica coating led to 8 h stability.22 Another example for PL stability is the reaction of pSi with borax, which increased the half-life from less than one day to 2 days.23 Previous published protocol with PEG obtained 100% dissolution in PBS just in 3 days.17
Moreover, recently rapid thermal oxidation succeeded in stabilization for about 5 days.6,24 Then, optical stability of pSi in biological media remains an issue for application in biomedicine. Oxidation mechanisms of freshly pSi in aqueous solutions have been already deeply investigated by Ogata et al.25 The problem of nanostructure stability in biological environment is however quite general. Nanomaterials, in fact, can often present chemical un-stability due to the large exposed surface, but also biological reactivity, since they undergo opsonisation and up-take by cells of the immune system.12 Opportune modification of the surface enables limiting these effects, meanwhile conferring desired properties to the final product.26
Among all the possibilities, the use of polyethylene glycol (PEG) has been shown appealing for applications in biomedical field. Indeed, PEG can ensure water stabilization to the nanosystems,27 meanwhile being biocompatible and able to reduce, once in vivo, opsonisation phenomena, which usually are the main factor affecting particles circulation and earlier elimination from the bloodstream.28 PEG is not the only polymer used for this purpose; recently, biopolymers derived from natural sources have showed promising and interesting capacity.29 Among others, chitosan is the product of the de-acetylation of chitin, the second most abundant biopolymer in the world generally found in crustacean shells.30 It is also used to form nano or micro water-soluble particles able to incorporate drugs or active agents.31 Finally, both PEG and chitosan, due to their carboxylic and amino groups respectively, offer the possibility of being linked to biomolecules (peptides, proteins, monoclonal antibodies and aptamers) thus providing to the final DDS potential targetable features.
On the other hand, the interaction between pSi and drugs must be carefully considered to design an effective delivery system without any side effect on the drug itself. As already reported, pSi in biological systems induces a strong redox activity.5 In fact, oxidation of pSi produces free electrons that can reduce drugs,32 proteins,33 and cells components.34 It is well known that the oxidation of pSi can reduce water to hydrogen according to reaction (1) with a large Gibbs energy, and in general, standard electrode potential of silicon is about 1.697 V.35 Thus, modification of pSi surface is needed to both limit the degradation of drugs due to their reduction and to increase the affinity of the drug for the pSi surface.
| Si + 2H2O → SiO2 + 2H2 (ΔG° = −382 kJ) | (1) |
In the literature, the most used way to modify pSi surface, in order to avoid drug reduction, is oxidation. Sailor et al.19,32,36 proposed complete oxidation at high temperature (800 °C) to reach this goal; in fact, the silicon skeleton was completely oxidized to ensure the drug not to be reduced or degraded by contact with elemental silicon during the particle dissolution-drug release phase. On the contrary, Barnes et al.33 proposed a mild oxidation at 400 °C to avoid reduction of proteins. Based on free kinetics model,37 we proposed that mild oxidation leads to remove surface moieties (such as hydroxyl agents) and the surface oxidation is obeying Avrami–Erofeev model. By increasing the oxidation conditions, i.e. longer time and temperature, oxygen diffuses through oxidized surface layer to oxidize the underlying silicon according to 3D diffusion model.
However, oxidation quenches the visible light emission of pSi,38 which is fundamental for bioimaging39 and photo-thermal therapy.14 Moreover, the oxidation induces a reduction of the pores volume because of the swelling of the pore walls thus significantly reducing the drug loading amount. As an example, oxidation at 400 °C and 800 °C led to 3-fold and 10-fold reduction of loading capacity of human serum albumin33 and cobinamide,36 respectively.
In the present study, we have addressed the possibility of using surface coating to control redox activity of pSi microparticles in biological systems thus maintaining the light emission properties for long time. Moreover, by using cobinamide, which is a vitamin 12 analogue and behaves as a probe of the redox activity of pSi,36 we investigated the effect of the surface chemistry of pSi on drug stability, loading amount and release profile.
2 Experimental procedure
All chemicals were purchased from Sigma-Aldrich and used as received (all ACS grade). All aqueous solutions were prepared with ultrapure water obtained by using an ultrafiltration system (Milli-Q, Millipore) with resistivity above 20 MΩ cm−1.2.1 Preparation of pSi–COOH, pSi–NH2 and pSiO2 microparticles
Porous silicon layer was formed by electrochemical etching of p-type Si wafer, (100) oriented, with 10–20 Ω cm resistivity (University wafers, Boston MA). The etching was done at 80 mA cm−2 constant current for 5 min in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ethanol:HF (48%) solution. The porous silicon layer was sonicated for 15 min in anhydrous toluene (80 mL) under nitrogen atmosphere to get pSi microparticles. To introduce the carboxylic (COOH) or amine functionalities (NH2) on the pSi surface, the so prepared pSi was sonicated in toluene containing NHS ester of 5 mM acrylic acid (pSi–CONHS). Then, to allow the hydrosilylation reaction to occur, the so obtained pSi powder suspension was illuminated with white light (250 W) for 2 h, under argon atmosphere. After this step, the pSi–CONHS powder was centrifuged and rinsed with toluene. To remove the NHS group and obtain microparticles exposing the free carboxylic group (pSi–COOH), the NHS group was hydrolysed in ethanol and then the powder was rinsed several times with the same solvent.
:1 ethanol:HF (48%) solution. The porous silicon layer was sonicated for 15 min in anhydrous toluene (80 mL) under nitrogen atmosphere to get pSi microparticles. To introduce the carboxylic (COOH) or amine functionalities (NH2) on the pSi surface, the so prepared pSi was sonicated in toluene containing NHS ester of 5 mM acrylic acid (pSi–CONHS). Then, to allow the hydrosilylation reaction to occur, the so obtained pSi powder suspension was illuminated with white light (250 W) for 2 h, under argon atmosphere. After this step, the pSi–CONHS powder was centrifuged and rinsed with toluene. To remove the NHS group and obtain microparticles exposing the free carboxylic group (pSi–COOH), the NHS group was hydrolysed in ethanol and then the powder was rinsed several times with the same solvent.
To obtain microparticles exposing amino groups (pSi–NH2), the pSi–COOH powders were suspended in toluene and left to react with trioxa-1,13-tridecanediamine, under gentle shacking. Dicyclohexylcarbodiimide (200 μM) was added to increase the reaction yield. Morphological and optical properties of these samples were already studied in detail.21 When necessary, the pSi powders were dried removing the solvents by a gentle nitrogen flow in order to avoid air oxidation. The aging of pSi in air in our pSi samples and the related PL modifications have been already studied in the past.40
In this paper, the main goal is to compare pSi microparticles with the different surface coatings, as previously described, with completely oxidized pSi microparticles presented in literature. These oxidized porous silicon (pSiO2) microparticles were obtained by oxidation of pSi at 1000 °C for 10 min by heating rate of 3 min K−1.37
2.2 Coating of pSi–COOH microparticles with PEG or chitosan
As for pSi coated by PEG (pSi–PEG1), polyethylene glycol with amino and carboxylic acid end groups (HCl·H2N–PEG–COOH, MW ∼ 3 kDa) was purchased from Rapp Polymere GmbH (Tübingen, Germany). First, pSi–COOH (0.3 mg) were dispersed in anhydrous tetrahydrofuran (THF, 1 mL). Carbonyldiimidazole (CDI, 0.8 μmol) was added to the reaction mixture under nitrogen. After 90 min, HCl·H2N–PEG–COOH (50 mg, 1.6 μmol) dissolved in 1 mL of THF and diisopropylethylamine (DIPEA, 10 μL, 50 μmol) were added. The reaction was stirred in vortex for 24 hours at room temperature. After that, particles were collected in centrifuge (13100 rpm, 4 min), washed twice with 2 mL of a mixture 1:1 THF:water and centrifuged again before dispersion in ultrapure water (1 mL). Schematic passages of this reaction were shown in path A in Fig. S1a on the ESI.† In order to better understand the effect of PEG on pSi, the ratio of PEG/pSi was increased 2 and 3 times (samples pSi–PEG2 and pSi–PEG3, respectively). Moreover, native PEG (MW ∼ 3 kDa) was used as a reference.
In the case of chitosan functionalization (pSi–chitosan), to a solution of the pSi–COOH in ethanol (1 mL, 0.3 mg), chitosan dissolved in acetate buffer (pH = 4.3, 0.25 mg mL−1, 1.25 mg) was added drop wise. The system was left to incubate with mechanical stirring for 24 hours. After that, particles were collected in centrifuge (13100 rpm, 4 min), washed twice with 2 mL of water and centrifuged again before re-dispersion in ultrapure water (1 mL). Schematic passages of this reaction were shown in path B in Fig. S1b.† Table 1 summarizes the different samples investigated in this work.
| pSi | Native porous silicon |
| pSiO2 | Thermally oxidized pSi |
| pSi–COOH | pSi functionalized to expose carboxyl group |
| pSi–PEG1 | pSi–COOH functionalized with PEG |
| pSi–PEG2 | pSi–COOH functionalized with double amount of PEG |
| pSi–PEG3 | pSi–COOH functionalized with triple amount of PEG |
| PEG | Native PEG polymer |
| pSi–NH2 | pSi–COOH functionalized by diamine |
| pSi–chitosan | pSi–COOH functionalized with chitosan |
2.3 Synthesis of cobinamide
Cobinamide was synthesized from hydroxocobalamin (OH–Cbi, purchased from Sigma-Aldrich) by acid hydrolysis using HCl as proposed by Broderick et al.41 Purity of Cbi was confirmed both spectrophotometrically by comparison with published spectra42,43 and by high performance liquid chromatography (HPLC) by using a C18 reversed phase column eluted isocratically in 100 mM NaH2PO, pH 4.0, and 15% methanol (v/v).44 The column effluent was monitored at multiple wavelengths (168–351 nm) by a diode array detector. Fig. S2† shows HPLC results, which confirmed at 380 s that Cbi was obtained and had similar optical density with the references. When stored at −20 °C, Cbi was spectroscopically stable for at least one month.2.4 Structural investigation
ζ-Potential of the samples was measured by the Zetasizer Nano-S (Malvern) instrument, working with a 532 nm laser beam, and clear disposable Z cells. The results were expressed as an average of three measurements. The Phase Analysis Light Scattering principle was used to quantify the ζ-potential and electrophoretic mobility. The concentration of the samples was maintained at ∼0.1 wt% and the experiment was conducted at 25 °C. Diluted solutions of NaOH and HCl were utilized for making pH variation.The surface chemistry was investigated by Fourier Transform Infra-Red (FTIR) spectroscopy. The spectra were acquired by a micro-FTIR Nicolet iN10 instrument, in the spectral range of 700–4000 cm−1 with about 4 cm−1 resolution. Furthermore, scanning electron microscopy images were acquired with a SEM Zeiss instrument EVO 50′ EP.
2.5 Emission stability in biological media
In order to check workability of the samples as bioimaging agent, photoluminescence (PL) measurements were performed by Horiba Jobin-Yvon Nanolog instrument. The configuration setup was: excitation at 350 nm, 2 nm slit size, 1200 g mm−1 density grating (blazed at 500 nm), cut-off filtration at 370 nm, and 0.1 second integration time.To estimate the light efficiency of the samples, quantum yield (Φ) measurements have been carried out by the comparative method.45,46 In this regard, rhodamine 101 was selected as reference, having about 100% quantum yield.19 The quantum yield of a sample was calculated via Φs = Φr·Is/Ir·Ar/As·(ns/nr)2, where I is the integrated emission intensity, A is the absorbance, and n is the refractive index of the buffer solution. The subscript s and r refer to the sample and the reference solutions, respectively. The samples were further diluted to keep the absorbance less than 0.1 in order to minimize non-uniform irradiation.47
Phosphate-buffered saline (PBS, pH 7.4) was used as simulator of biological fluids. About 30 μg from each dried sample was suspended in 3 mL of PBS, at a concentration of about 10 μg mL−1. Then, PL spectrum of the samples was monitored as a function of time.
2.6 Interaction of pSi with Cbi
To load Cbi into pSi, we used the procedures already proposed in the literature.36,48 A small amount of dried pSi (0.5–1 mg) admixed with the same amount of Cbi, was incubated for 2 h in 1.5 mL of deionized water at pH 6 (adjusted by HCl), under weak shaking. Cbi loaded particles were then washed three times with water, recollected by centrifugation and dried in freeze-dryer. Then, the powder was dispersed in 1 mL of aqueous PBS.As for the loading capacity and release rate studies, pSi microparticles loaded with Cbi were dispersed in 1 mL of PBS at a particle concentration of 100 μg mL−1 at room temperature for 2 h under mild shaking. The supernatant containing released Cbi was collected by 5 seconds centrifugation, at set times and replaced with fresh water or buffer. Concentrations of released Cbi were determined by adding excess potassium cyanide to the solution to convert Cbi to the dicyano form and then by measuring the optical absorbance at 370 nm (ε = 30000 M−1 cm−1) according to ref. 36.
Optical density (absorbance) spectra were recorded on a Thermo Scientific Evolution 201 UV-Visible Double Beam spectrophotometer (Thermo Scientific Inc.) with 1 cm matched Quartz cells, in a range of wavelength between 250 and 800 nm. Optical density of samples was analysed for one week in order to check stability of the loaded drug. This monitoring period is more than enough for DDS applications.49
3 Results and discussion
3.1 Structural investigation
To investigate surface electric charge, ζ-potential of samples was monitored at different pH for all the samples in PBS (Fig. 1). The results show pSi–COOH particles having a negative ζ-potential, with a value of −26.2 mV, at physiological pH (i.e. 7), due to the presence of carboxyl groups onto the surface. Addition of diamine and obtainment of pSi–NH2, leads to have positive surface charge at physiological pH without significant effect on the morphology of the samples.12 Moreover, ζ-potential variation of free Cbi in PBS as a function of pH is similar to the one of pSi–NH2, which could be related to the amine functions of both samples.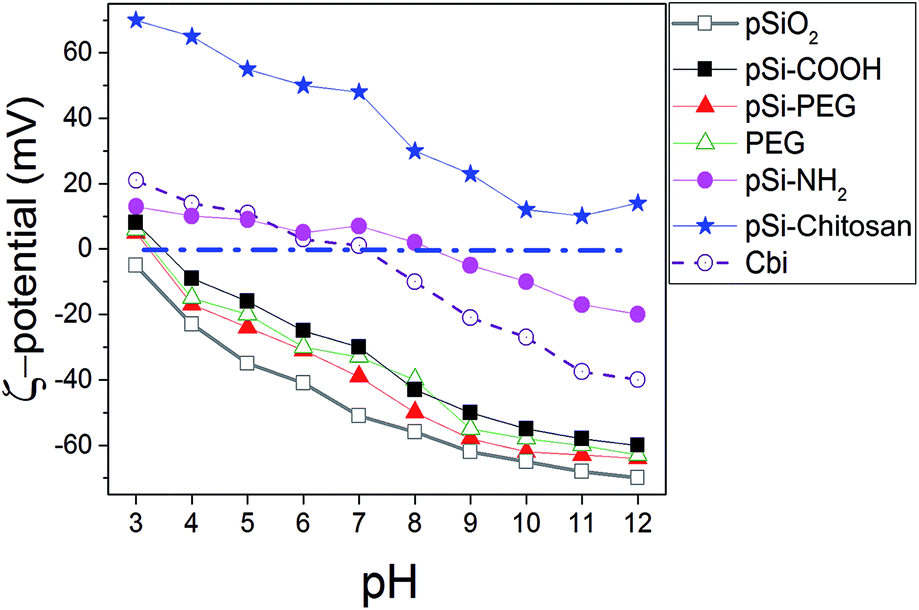 | ||
| Fig. 1 Variation of ζ-potential as a function of the pH for Cbi and different surface coverage of pSi in PBS. | ||
In order to allow structural stability and optical properties preservation in water and/or biological liquids, various surface coatings were considered and studied. Firstly, the surface modification with PEG through covalent attachment was attempted. We selected a PEG functionalized at the two ends with an amino and a carboxyl group (H2N–PEG–COOH) respectively, in order to create an amide bond between the amino group of PEG and the carboxyl group present on the pSi–COOH surface, thus leaving the same surface termination (–COOH) at the end of PEG chain for further possible conjugations (i.e. DDS targeting). This is reflected on the fact that the highly negative ζ-potential (−37.9 mV in pH of 7) is maintained. In case of coating with a double amount of PEG (pSi–PEG2), the results in terms of ζ-potential values were similar to those found for pSi–PEG1 and pure PEG, as previously reported.17 In fact, variation of ζ-potential for samples with similar surface functionality (–COOH), which are pSi–COOH, pSi–PEG, and PEG, is in the order of the experimental error. Moreover, according to ref. 50, the pSiO2 has ζ value slightly lower than other samples which is consistent with the ζ-potential of silica.51
Finally, the surface modification with chitosan biopolymer, through physical adsorption and electrostatic interaction, was also investigated (pSi–chitosan). Indeed chitosan, thanks to its abundance of positively charged amino groups, can electrostatically interact with the negative charged pSi–COOH, thus keeping protected the particles between its chains, meanwhile leaving free amino groups for further conjugation and modification.52 Indeed, ζ-potential analysis showed a highly positive value +48.3 mV, due to the amino groups of chitosan, meaning that an efficient coating of the native particles has been obtained. ζ-Potential changes of chitosan functionalized particles are in line with recent published data.53
SEM analysis was performed in order to check particle sizes and to assess their morphology, in particular after the organic coating. Native pSi micro-particles are typically elongated (i.e. rod like shape) with few micron size (Fig. 2a). This is due to the fragmentation of the porous silicon layer, which occurs along the vertically oriented pores. Micro-particles size distribution obtained from optical image analysis is large (few microns for the major axis) as expected on the basis of the fabrication approach.12 By surface functionalization to obtain pSi–COOH (Fig. 2b) roughness of surface increased based on corrosive chemical reactions. By increasing the magnification of SEM, the diameter of the pores has been observed to be about 30 nm (Fig. 2c). As it was mentioned before,12 there is not significant variation on morphology by addition of diamine to get pSi–NH2 (Fig. 2d). SEM analysis of pSi–PEG1 showed a homogenous sample with rods like particles clearly visible (Fig. 2e). As for pSi–chitosan sample, SEM analysis revealed, indeed, the presence of microparticles, spheres-like shaped, homogeneously dispersed in the sample (Fig. 2f) with a dimensions of about 2–5 μm. In fact, based on the chemical reaction during the functionalization, homogeneity of pSi–COOH sample morphology was increased.
 | ||
| Fig. 2 SEM images of native pSi (a), pSi–COOH sample in one-micron scale to show particle rod-like shape (b) and in nanometre scale to better point out surface porosity (c), pSi–NH2 sample (d), pSi–PEG1 sample in 10-micron scale (e) with a higher magnification in the inset, and pSi–chitosan sample in 20-micron scale (f). | ||
To validate the organic functionalization procedure, FTIR analysis was performed, which showed that the expected functional groups were correctly grafted (Fig. 3). For the sake of clarity, all spectra have been normalized and shifted along the y axis. The native pSi particles present the characteristic vibration mode at 2100–2300 cm−1, and also the peak at 790–800 cm−1, which are due to SiH.54 On the contrary, pSi–COOH sample did not present any characteristic SiH vibration mode at 2100–2300 cm−1, but a strong peak at 1060–1110 cm−1 due to the presence of asymmetric stretching of Si–O–Si bond.55 Interestingly, also a signal at 1715 cm−1 appears, which is the typical stretching vibration of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O containing compounds thus confirming the presence of COOH groups onto the surface.57 The other peak at 873 cm−1 is related to Si–O bending.
O containing compounds thus confirming the presence of COOH groups onto the surface.57 The other peak at 873 cm−1 is related to Si–O bending.
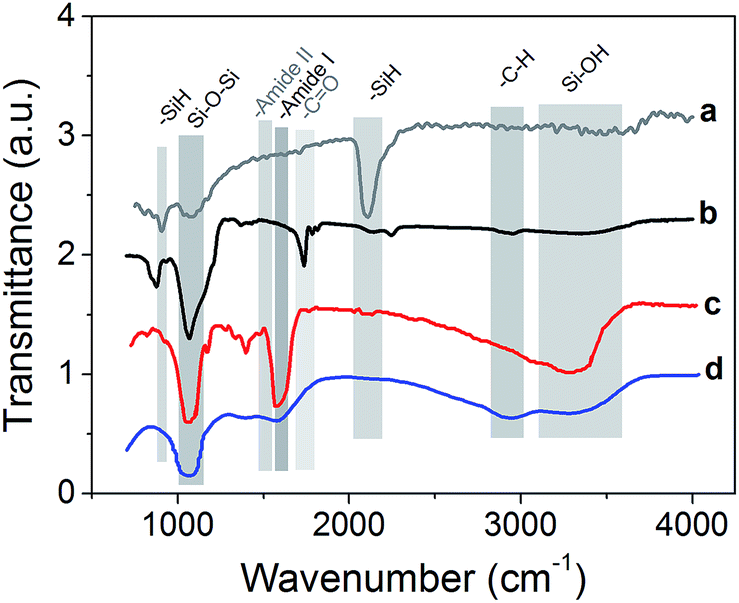 | ||
| Fig. 3 FTIR measurements of the pSi microparticles before and after surface functionalization and successive coating: native psi (a), pSi–COOH functionalized (b) and surface modified by PEG (c) and chitosan (d). | ||
In the case of PEG functionalization, it is possible to observe a strong peak at 1046 cm−1 as typical stretching vibration in ethers, the presence of the amide bond vibrations of –CO at 1644 cm−1 (amide I band) and of –NH stretching at 3200–3400 cm−1 which overlaps with Si–OH peak.56 After the coating of pSi–COOH samples with chitosan, the peak around 1070 cm−1 was retained and a new band is found at 1550–1600 cm−1 due to the presence of primary amides, which are abundant in chitosan. Also Si–OH vibrations appeared at 3200–3600 cm−1 as a broad band, which is typical of –OH modes, while alkyl –CH stretching modes appeared at 2850–2950 cm−1.57
3.2 Emission stability in biological media
Fig. 4 reports PL spectra of all the samples excited at 350 nm. Based on pSi–COOH and pSi–NH2 have more compatibility in ethanol, PL of these samples was acquired in ethanol where functionalized pSi has been shown to be stable for years.21 Conversely, for the other samples PL spectra were obtained in PBS, because PEG and chitosan have high affinity for aqueous environments. PL of all samples was normalized by comparison with pSi–COOH, as reference (see Table 2). As it can be seen, pSi–COOH has a peak in the red region (about 665 nm).58 It is worth noting that the sharp peak at about 390 nm is due to the Raman effect of water.59 After functionalization and further suspension in PBS, the pSi–PEG and the pSi–chitosan PL bands are very similar in shape, presenting just a blue-shift with respect to pSi–COOH sample. The blue-shift of the red band is consistent with quantum effects.60 Chitosan, alone, has a low emission and, as it can be seen from Fig. 4, there is no correlation between PL of chitosan and pSi–chitosan spectra.61 It must be noticed that pSiO2 has no optical emission band in the range of interest.37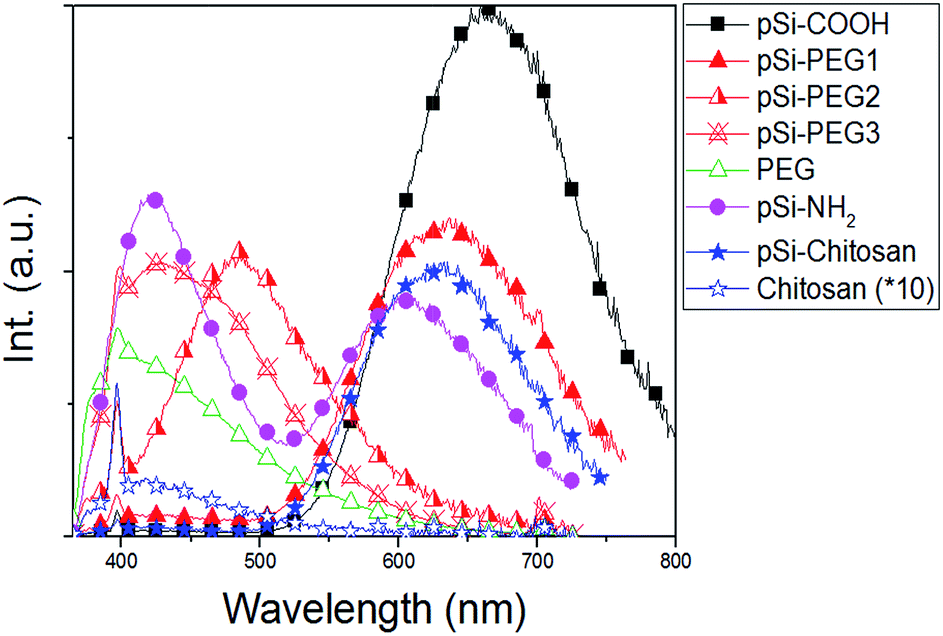 | ||
| Fig. 4 PL spectra of samples in Table 1 (excitation at 350 nm). PL curves normalized based on Φ value from Table 2. Chitosan (×10) signal is reported for comparison with pSi–chitosan. | ||
| Sample | Φ (%) | λ(max) (nm) | FWHM (nm) |
|---|---|---|---|
| pSi–COOH | 1.7 | 660 | 154 |
| pSi–NH2 red band | 0.7 | 603 | 140 |
| pSi–NH2 blue band | 0.6 | 422 | 88 |
| pSi–PEG1 | 1.1 | 636 | 153 |
| pSi–PEG2 | 0.9 | 485 | 115 |
| pSi–PEG3 | 0.8 | 430 | 131 |
| PEG | 0.5 | 410 | 110 |
| pSi–chitosan | 0.8 | 635 | 139 |
| Chitosan | 0.02 | 419 | 105 |
PL of pSi–PEG2 and pSi–PEG3 has been shifted more, about 180 nm and 235 nm, respectively. It must be considered that even though the microparticles functionalized with both PEG concentrations present similar molecular structure and size, the larger amount of PEG (i.e.: HCl·H2N–PEG–COOH) used during functionalization probably leads to a stronger surface modification. The correlation between the spectral shift and the concentration of the reactant (NH–PEG–COOH) suggests that some modification of the light emitting silicon structures occurs during the functionalization, probably due to silicon oxidation catalysed by the amino group, as already reported for other amines.40 For the sake of clarity, PL of pure PEG has been measured: as it can be seen, PEG, itself, has a significant emission band in the blue region (400–450 nm).62
The emission quantum yield (Φ) of all samples dispersed in PBS has been estimated by comparative method (Table 2). The obtained values for pSi samples (between 0.8% and 1.1%) are not significantly reduced if compared to that of the pSi–COOH that is about 1.7%. On the other hand, pure chitosan has negligible light emission efficiency.
Unlike the blue shift, the Φ reduction is not the same after PEG and chitosan functionalization. As for the pSi–PEG1 sample, Φ is reduced of about 33%, as previously reported.18 A similar decrease of the quantum yield is observed for pSi–NH2. However, quantum yield is further reduced by increasing PEG amount (down to 0.9 and 0.8 for the pSi–PEG2 and pSi–PEG3 samples, respectively). As for pSi–chitosan sample, the quantum yield is about 0.8, which is roughly half of pSi–COOH sample and the same as for pSi–PEG3. The reduction with respect to pSi–PEG1 sample can be attributed to the fact that the chitosan matrix acts as higher optical absorber especially in the blue range.63 In fact, it appears more opaque due to the larger passivation upon the pSi–COOH structures. These quantum yield reductions by surface functionalization are in line with recent experimental data of pSi by others.64
Until now, we obtained a long-lasting PL stabilization in organic solvents (toluene and ethanol) for years,12 but the PL half-life in aqueous environment remained of the order of few hours. The change over time of the PL spectral features of pSi samples in PBS have been studied and compared in Fig. 5. This figure shows variation of the red emission band for all the tested samples (solid lines), and variation of the second band in the blue region only for pSi–chitosan and pSi–NH2 samples (dash lines). The intensities have been normalized by eqn (2), in order to let them be appreciable in the same figure.
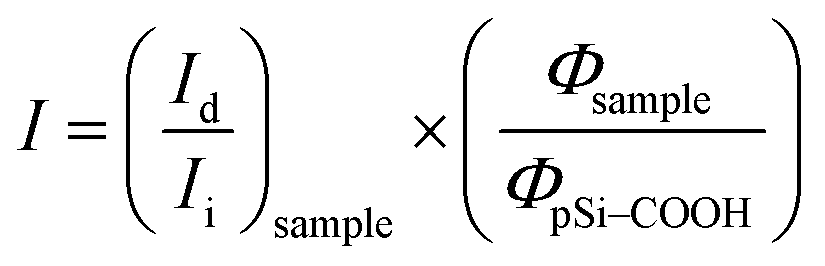 | (2) |
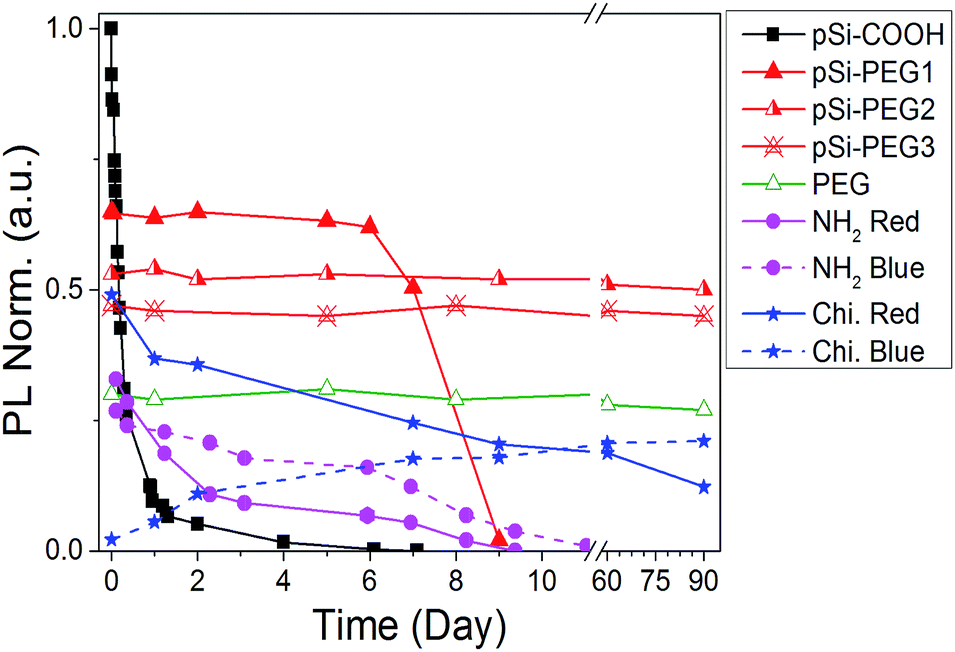 | ||
| Fig. 5 PL intensity vs. time (day) in PBS buffer. Blue band of pSi–NH2 and pSi–chitosan are indicated by dash lines. | ||
The half-life of the pSi–COOH in PBS is about 5 h, which is longer than what reported by other authors (about 3 h).65 The half-life increases to more than 7 days after suspension in PBS for the pSi–PEG1, without any blue shift. It is worth noting that one-week optical stability is more than what is generally required for bioimaging applications and it is also promising for photo-thermal therapy.66
By increasing the amount of PEG (pSi–PEG2 and pSi–PEG3), Φ reduces of about 30% and PL peak shifts up to about 200 nm from red to blue spectral region (Table 2). After this initial variation, the PL band is stable up to three months (90 days). To the best of our knowledge, this stability of the visible light emission (in both intensity and shape) in PBS for Si-based nano or micro-structures has not been reported till now. Moreover, these data may indicate that the PL band could be further tuned by varying the PEG amount thus having an additional post-processing tool for wavelength shifting without influencing the intensity stability over the time.
Similar behaviour was observed for pSi–chitosan showing a long-term optical stability in PBS (Fig. S3†): the red band (at about 635 nm) decreases with time (days), while the blue one (at about 400 nm) appears and increases with time (days). The decrease of the red emission (about 30% of the initial value after 15 days) can be due to the slow but progressive oxidation of luminescent silicon microparticles67 that reduces the dimensions of the nanocrystallites and introduces oxygen defects.68 For this reason, the red emission (related to quantum confinement) progressively decreases in intensity and slightly shifts to shorter wavelength (from about 635 nm to about 600 nm after 90 days). Conversely, the optical emission at 400 nm (blue band) has significantly more emission thus suggesting that it might be attributed to the surface defects at the nanocrystallites induced by nitrogen containing compounds ascribed to the amino groups,69 which are progressively released because of the dissolution of chitosan and interaction with pSi.70,71
According to the above results, we were able to stabilize the PL intensity and feature it up to months in a high ionic strength aqueous buffer such as PBS. To the best of our knowledge, this is the first functionalization protocol that stabilizes visible PL of functionalized pSi for such a long period: 90 days.
3.3 Interaction of pSi with Cbi
As previously said, the surface chemistry of pSi plays a fundamental role in the adsorption of drugs within the porous matrix. In order to understand the mechanisms regulating such an interaction, we first exploit optical (UV-Visible) absorption to monitor the absorbance spectrum of Cbi. The spectra of the cobinamide solution after interaction with pSi for 3 h revealed significant changes compared to that of the reference cobinamide solution (Fig. 6).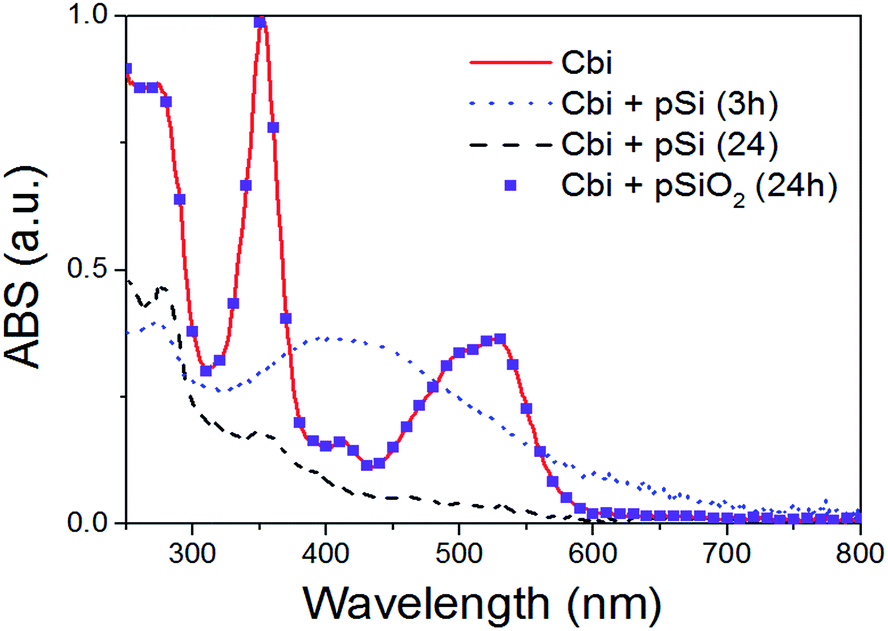 | ||
| Fig. 6 Optical absorption of Cbi after the interaction with pSi, or pSiO2, in PBS as a function of time. | ||
The characteristic optical absorption peaks of cobinamide are at about 500 and 525 nm (cobalt is in Co3+ state).45 After 3 h (big dot line of Fig. 6), the peaks at about 500 and 525 nm disappeared, while a large absorption peak centred at 430 nm raises according to the reduction of Co3+ centre to Co2+ by interaction with the Si surface. After longer interaction (24 h) it occurs also a complete loss of the cobalt–corrin ring coordination as clearly shown by the disappearance of this peak, that means that Cbi is completely decomposed.46 On the other hand, our results showed that Cbi is stable when in contact with pSiO2 in PBS solution (see dots in Fig. 6) in agreement with previous findings.36
Therefore, we experimentally investigated which different surface functionalization is efficient to avoid alteration (e.g. reducing process) of the loaded Cbi. Table 3 shows the reaction times of Cbi reduction and its decomposition as a function of the different functionalized pSi samples. In this table, the column “step 1” shows the time measured to get the maximum intensity of the 430 nm peak (i.e. Co3+ → Co2+ transformation) while “step 2” the time of complete Cbi decomposition associated to the disappearing of it.
The time courses of Cbi reduction experimentally recorded can be attributed to the surface charge: samples with negative charge (pSi–COOH, pSi–PEGs) have low or no effect on Cbi modification. In fact, carboxyl functionalization significantly reduces the reaction rates of about a factor 3 with respect to native pSi for both steps: Co3+ reduction in 10 h instead of 3 h and Cbi complete decomposition 3 days instead of 1 day. More attractively, we report here that the PEG functionalization has no effect on the structure of Cbi, similarly to what already reported for completely oxidized pSi. It is worth noting that no optical absorption modification was observed after one week for both pSiO2 and PEG functionalized pSi samples.
On the other hand, samples with positive surface charge (pSi–NH2 and pSi–chitosan) showed very fast degradation (in the order of few seconds for step 1 and about 1 hour for step 2) of the Cbi. The fast reaction (few seconds) of Cbi with samples containing amine functional groups could be related to the reduction of Cbi by nitrogen impurities as previously reported.44 In fact, based on this report, kinetic rate of this reduction is in the order of seconds.
After the chemical stability assessment of the Cbi drug once incorporated into the silicon porous matrix, the other fundamental features to be tested for a reliable DDS are (i) the loading capacity and efficiency and (ii) the release time profile. Cbi loading efficiency was measured by extracting the loaded cobinamide from the pSi microparticle matrix by a treatment with a sodium hydroxide solution (pH 9), that dissolved the Si and SiO2 components thus releasing the drug payload into the solution. We quantified the Cbi amount by converting it into the dicyano form (by adding excess potassium cyanide) and by measuring the optical absorbance at 370 nm.36
The results are reported in Table 4. The cobinamide loadings were 110 and 32 μg of Cbi per mg of microparticle, in average, for the pSi and pSiO2, respectively. The loading capacity dramatically reduced by silicon oxidation because of the reduction of available surface area. This fact strongly limits the use of oxidized pSi for DDS. Loading capacity for pSi–PEG and pSi–COOH is about 225 and 380 μg of Cbi per mg of pSi, respectively. Indeed, the loading capacity increased significantly with respect to oxidized pSi (2 and 5 fold, respectively). The loading capacity for pSi–PEG2 is in the same range as that of native pSi. Functionalized PEG samples show less capacity with respect to carboxyl ones probably due to the longer chains of PEG, which lead to a reduction of the pores volume available for the Cbi loading. This is confirmed by the fact that with increasing PEG amount (see pSi–PEG2) the pores volume is less and less available to carry the drug. This effect can be however mitigated by an increase of the pores size in the native pSi by playing with the electrochemical etching parameters in the fabrication of the porous silicon.72
| Sample | Loading capacity (μg of Cbi per mg of pSi) | ζ-Potentiala (mV) |
|---|---|---|
| a Measured at pH 6, which is used for loading Cbi. | ||
| Native pSi | 110 ± 15 | −14 |
| pSiO2 | 32 ± 2 | −56 |
| pSi–COOH | 380 ± 32 | −25 |
| pSi–PEG1 | 225 ± 18 | −31 |
| pSi–PEG2 | 120 ± 10 | −33 |
| pSi–PEG3 | 176 ± 14 | −32 |
| PEG | 52 ± 9 | −30 |
| pSi–NH2 | 12 ± 2 | 2 |
| pSi–chitosan | 18 ± 1 | 30 |
It is worth noting that the large loading capacity (attributed to electrostatic interaction) here measured in pSi–COOH sample (about 40%), has not ever seen even by the oxidation trapping36 and other methodologies.50 However, as shown above, pSi–COOH has not good optical stability in PBS in comparison with samples functionalized with PEG. Therefore, pSi–PEG with a loading capacity up to 225 μg of Cbi per mg of microparticle is the best candidate for a reliable DDS.
On the other hand, positive surface charge leads to a poor loading capacity with respect to native pSi, and even less than the oxidized one (pSiO2): about 12 and 18 μg Cbi per mg of microparticles for pSi–NH2 and pSi–chitosan samples, respectively.
As a further and last step of this experimental study, we verified the release of Cbi from pSi. Results are summarized in Fig. 7, where the Cbi release is monitored as a function of time (days). As it is obvious, when there is not attraction between pSi and Cbi, Cbi will release from pSi easily. According to this fact and Table 4, we can categorize the Cbi release into two groups, respectively faster and slower if compared to a medium release rate represented by native pSi and pSiO2 samples. As it can be clearly observed in Fig. 7, we found faster release rate for the positively charged samples and slower rate for the negatively charged ones (refer to the ζ-potential values in the Table 4).
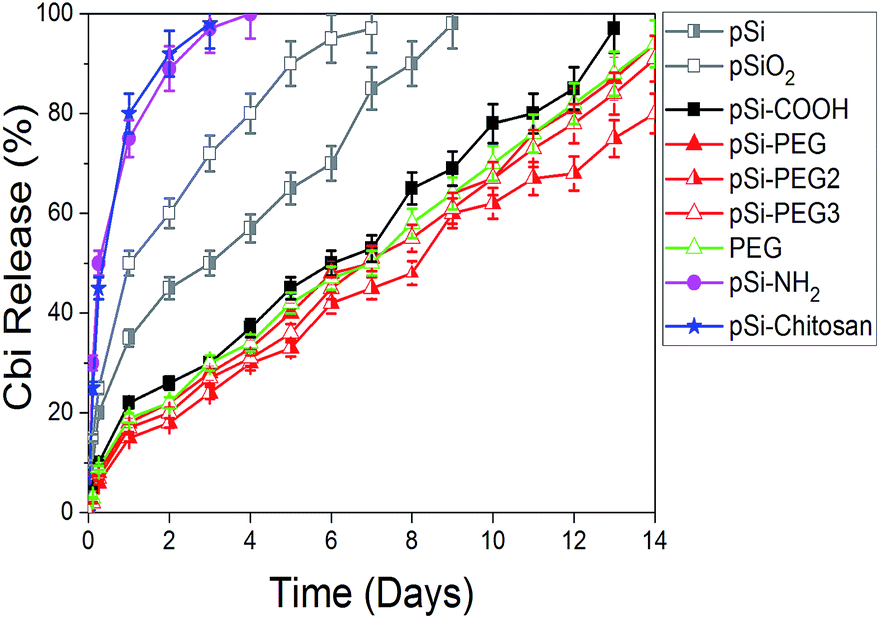 | ||
| Fig. 7 First two-weeks release profile of Cbi into PBS from pSi samples as a function of the surface coating. | ||
In the first case, pSi–NH2 and pSi–chitosan samples show about 50% of the drug released within few hours and more than 95% within 3 days. This fast release might be attributed to the electrostatic repulsion of similar surface charge. Release rate from chitosan and amine functionalized samples are quite the same, in opposite of having different ζ-potentials. The expected faster release rate of Cbi in the pSi–chitosan sample is probably contrasted by the fact that the long polymeric chains of chitosan can act as traps for the Cbi molecules.
In the second group associated to the negatively charged samples, the Cbi release is quite linear with time with a slope of about 6% release per day. Rate of pSi–PEG and pSi–PEG2 is slightly lower than that of pSi–COOH, which could be related to the trapping of Cbi in the longer chains of PEG samples.
Moreover, the release rate by native pSi is lower based on trapping of Cbi during oxidation of pSi. Indeed, surface oxidation of pSi in PBS leads to closing of porosity due to oxygen absorption.37 This phenomenon cases trapping of Cbi.19 Then release of Cbi needs first this oxidized layer to dissolve in PBS, which is time consuming.
Cbi has no PL properties in the visible range. Then, PL of loaded samples is just based on pSi. Additionally, native pSi and pSiO2 samples should be avoided since pSiO2 does not have PL properties, and native pSi quenched during 3 h after the loading of Cbi. Also, PL properties of Cbi loaded pSi–NH2 and pSi–chitosan samples were similar with unloaded ones, which it could be related to negligible amount of loading (less than 2%).
As previously showed in the Fig. 5, pSi–COOH sample presents a dramatic quenching in PBS. However, the loading of Cbi leads to significantly mitigate the optical quenching rate: Fig. S4a† shows about a factor 2 of improvement in time (days). This improvement of the light emission stability is attributed to the facts that Cbi works as a barrier for surface oxidation of silicon. On the other hand, Fig. S4b† demonstrates that Cbi does not have any effects, as expected, on PL band of the pSi–COOH sample thus preserving the optical properties (in peak maximum wavelength and shape).
As for PEG functionalized samples, we did not observe any variation of the optical properties as a function of Cbi loading. The reason is attributed to the electrostatically attraction of Cbi with PEG, without any interaction with the silicon core structure. In fact, the pSi–PEG was already stable from the optical point of view right in reason of a better passivation of the silicon surface.
All these results are in favour of the idea that a reliable delivery drug system should consist of a carrier that does not damage either the drug but maintain or even improve the optical properties of the carrier itself thus paving the way to an intrinsic (without the need of additional fluorophores) combined imaging.
4 Conclusions
In this experimental study, luminescent porous silicon (pSi) microparticles have been obtained based on anodization method. In order to reach structural and optical stabilization in biological media (such as water or PBS), different surface functionalization procedures were investigated. Attachment of the different functional groups onto the pSi surface were confirmed by FTIR analysis. Among them, PEG and chitosan were the more effective polymer for pSi stabilization, but with opposite electric surface charge as determined by ζ-potential measurements.Visible light emission of pSi stabilized for over three months has been reported for the first time for both PEG and chitosan coating of pSi microparticles. In fact, it is very suitable for biomedical applications to offer orthogonal potential functionalization since they bear in the outer shell carboxylic and amino functional groups, negatively and positively charged, respectively.
In fact, in addition to the poor stability in biological media, the concern about pSi in biomedicine is the reducing activity of pSi itself that degrades the drug. To test the improved performances of polymer coated pSi as DDS, Cbi was selected as a reference drug based on its high sensitivity to reduction. Experimental data demonstrated that PEG functionalization leads to avoid reduction and decomposition of Cbi; in addition, it leads to improve the loading amount and the release profile. Moreover, Cbi improves PL stability.
In conclusion, this experimental study shows that pSi microparticles, opportunely functionalized, are a reliable and promising candidate for theranostics due to the high drug loading capacity, the possibility of controlling fast or long release rate accordingly, no drug deterioration, and to the long-term optical stability in biological liquids.
Acknowledgements
This work has been done within the Nanomedicine Initiative funded by Fondazione Cariverona (Verona). University of Bologna is also gratefully acknowledged. We thank Prof. Antonella Motta and Lorenzo Moschini, Department of Material Engineering, University of Trento for acquisition of SEM images. We are grateful to Dr Martino Bianconi for his technical assistance in HPLC measurements.References
- L. T. Canham, Adv. Mater., 1995, 7, 1033 CrossRef CAS.
- J. Salonen, A. M. Kaukonen, J. Hirvonen and P. V. Lehto, J. Pharm. Sci., 2008, 97, 632 CrossRef CAS PubMed.
- Q. Fang, J. Wang, S. Gu and R. B. Kaspar, J. Am. Chem. Soc., 2015, 137(26), 8352 CrossRef CAS PubMed.
- O. Bisi, S. Ossicini and L. Pavesi, Surf. Sci. Rep., 2000, 38, 1 CrossRef CAS.
- M. L. Sailor, Chemical reactivity and surface chemistry of porous silicon, in Handbook of Porous silicon, Springer International Publishing, Switzerland, 2014, p. 335 Search PubMed.
- S. P. Low and N. H. Voelcker, Biodegradability of porous silicon, in Handbook of Porous silicon, Springer International Publishing, Switzerland, 2014, p. 381 Search PubMed.
- C. F. Wang, E. M. Mäkilä, M. H. Kaasalainen and D. Liu, Biomaterials, 2014, 35(4), 1257 CrossRef CAS PubMed.
- C. F. Wang, M. P. Sarparanta, E. M. Mäkilä and M. L. K. Hyvönen, Biomaterials, 2015, 48, 108 CrossRef CAS PubMed.
- H. Hou, A. Nieto, F. Ma, W. R. Freeman and M. J. Sailor, J. Controlled Release, 2014, 178, 46 CrossRef CAS PubMed.
- E. J. Anglin, L. Cheng, W. R. Freeman and M. J. Sailor, Adv. Drug Delivery, 2008, 60, 1266 CrossRef CAS PubMed.
- R. J. Martín-Palma, J. Hernández-Montelongo, V. Torres-Costa, M. Manso-Silván and Á. Muñoz-Noval, Expert Opin. Drug Delivery, 2014, 11, 1273 CrossRef PubMed.
- N. Daldosso, A. Ghafarinazari, P. Cortelletti, L. Marongiu, M. Donini, V. Paterlini, P. Bettotti, R. Guider, E. Froner, S. Dusi and M. Scarpa, J. Mater. Chem. B, 2014, 2, 6345 RSC.
- J. H. Park, L. Gu, G. von Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Nat. Mater., 2009, 8(4), 331 CrossRef CAS PubMed.
- M. A. Avidad, A. Nieto, A. Salinas-Castillo, L. Capitan-Vallvey, G. Miskelly and M. J. Sailor, Nano Res. Lett., 2014, 9(1), 1 CrossRef PubMed.
- G. M. Lanza, Nat. Nanotechnol., 2015, 10, 301 CrossRef CAS PubMed.
- S. Näkki, J. Rytkönen, T. Nissinen, C. Florea and J. Riikonen, Acta Biomater., 2015, 13, 207 CrossRef PubMed.
- B. Godin, J. Gu, R. E. Serda, R. Bhavane, E. Tasciotti, C. Chiapinni, X. Lu, T. Tanaka, P. Decuzzi and M. Ferrari, J. Biomed. Mater. Res., Part A, 2010, 94(4), 1236 Search PubMed.
- R. R. Chandler-Henderson, B. Sweryda-Krawiec and J. L. Coffer, J. Phys. Chem., 1995, 99, 8851 CrossRef CAS.
- E. C. Wu, J. S. Andrew, A. Buyanin, J. M. Kinsella and M. J. Sailor, Biomaterials, 2011, 32, 1957 CrossRef CAS PubMed.
- E. Secret, M. Maynadier, A. Gallud and A. Chaix, Adv. Mater., 2014, 26(45), 7643 CrossRef CAS PubMed.
- A. Ghafarinazari, V. Paterlini, P. Cortelletti, P. Bettotti, M. Scarpa and N. Daldosso, J. Nanosci. Nanotechnol., 2017, 17(2), 1240 Search PubMed.
- N. K. Hon, Z. Shaposhnik, E. D. Diebold, F. Tamanoi and B. Jalali, ECS Trans., 2013, 45, 7 CrossRef.
- J. Joo, J. F. Cruz, S. Vijayakumar, J. Grondek and M. J. Sailor, Adv. Funct. Mater., 2014, 24, 5688 CrossRef CAS.
- K. H. Wu, C. W. Li and J. H. Liu, Microelectron. Eng., 2015, 148, 70 CrossRef CAS.
- Y. H. Ogata, T. Tsuboi, T. Sakka and S. Naito, J. Porous Mater., 2000, 7, 63 CrossRef CAS.
- E. Locatelli, I. Monaco and M. Comes Franchini, RSC Adv., 2015, 5, 21681 RSC.
- B. C. Mei, K. Susumu, I. L. Medintz and H. Mattoussi, Natur. Prot., 2009, 4(3), 412 CrossRef CAS PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288 CrossRef CAS PubMed.
- S. K. Nitta and K. Numata, Int. J. Mole. Scien., 2013, 14, 1629 CrossRef CAS PubMed.
- M. N. V. R. Kumar, Reac. Func. Poly., 2000, 46, 1 CrossRef CAS.
- A. M. Piras, S. Sandreschi, G. Maisetta and S. Esin, Pharma. Res., 2015, 32(7), 2259 CrossRef CAS PubMed.
- E. C. Wu, J. S. Andrew, A. Buyanin, J. M. Kinsella and M. J. Sailor, Chem. Commun., 2011, 47(20), 5699 RSC.
- K. L. Jarvis, T. J. Barnes and C. A. Prestidge, Langmuir, 2010, 26, 14316 CrossRef CAS PubMed.
- S. Low, L. Canham and N. Voelcker, Bio Mater., 2006, 27(26), 4538 CrossRef CAS PubMed.
- CRC Handbook of Chemistry and Physics, ed. R. David, CRC Press, Boca Raton, FL, 87th edn, 2006, ISBN 0-8493-0487-3 Search PubMed.
- N. L. Fry, G. R. Boss and M. J. Sailor, Chem. Mater., 2014, 26, 2758 CrossRef CAS PubMed.
- A. Ghafarinazari, E. Zera, A. Lion, M. Scarpa, G. D. Soraru and N. Daldosso, Thermo. Acta, 2016, 623, 6 Search PubMed.
- G. Aggarwal, P. Mishra, B. Joshi and S. S. Islam, J. Porous Mater., 2014, 21, 23 CrossRef CAS.
- F. Peng, Y. Su, Y. Zhong, C. Fan and S. T. Lee, Acc. Chem. Res., 2014, 47(2), 612 CrossRef CAS PubMed.
- E. Froner, R. Adamo, Z. Gaburro, B. Margesin, L. Pavesi, E. Rigo and M. Scarpa, J. Nanopart. Res., 2006, 8(6), 1071 CrossRef CAS.
- K. E. Broderick, V. Singh, S. Zhuang, A. Kambo, J. C. Chen, V. S. Sharma, R. B. Pilz and G. R. Boss, J. Biol. Chem., 2005, 208(10), 8678 CrossRef PubMed.
- K. Proinsias, M. Giedyk and D. Gryko, Chem. Soc. Rev., 2013, 42, 6605 RSC.
- V. S. Sharma, R. B. Pilz, G. R. Boss and D. Magde, Biochemistry, 2003, 42(29), 8900 CrossRef CAS PubMed.
- S. H. Ford, A. Nichols and M. Shambee, J. Inorg. Biochem., 1991, 41, 235 CrossRef CAS PubMed.
- D. Timmerman, J. Valenta, K. Dohnalová, W. De Boer and T. Gregorkiewicz, Nat. Nanotechnol., 2011, 6(11), 710 CrossRef CAS PubMed.
- A. Ogunsipe, J.-Y. Chen and T. Nyokong, New J. Chem., 2004, 28(7), 822 RSC.
- J. H. Ahire, Q. Wang, P. R. Coxon, G. Malhotra, R. Brydson, R. Chen and Y. Chao, ACS Appl. Mater. Interfaces, 2012, 4, 3285 CAS.
- T. Tanaka, L. S. Mangala, P. E. Vivas-Mejia, R. Nieves-Alicea, A. P. Mann, E. Mora, H. D. Han, M. K. Shahzad, X. Liu and R. Bhavane, Cancer Res., 2010, 70(9), 3687 CrossRef CAS PubMed.
- J. Salonen, Drug delivery with porous silicon, in Handbook of Porous silicon, Springer International Publishing, Switzerland, 2014, p. 909 Search PubMed.
- M. Kaasalainen, E. Mäkilä, J. Riikonen, M. Kovalainen, K. Järvinen, K. H. Herzig, V. P. Lehto and J. Salonen, Int. J. Pharm., 2012, 431, 230 CrossRef CAS PubMed.
- J. M. Rosenholm, T. Czuryszkiewicz, F. Kleitz, J. B. Rosenholm and M. Lindén, Langmuir, 2007, 23, 4315 CrossRef CAS PubMed.
- N. Shrestha, M. A. Shahbazi, F. Araújo, H. Zhang, E. M. Mäkilä, J. Kauppila, B. Sarmento, J. J. Salonen, J. T. Hirvonen and H. A. Santos, Biomaterials, 2014, 35(25), 7172 CrossRef CAS PubMed.
- S. H. Chang, H. T. Victor Lin, G. J. Wu and G. Jane Tsai, Carbohydr. Polym., 2015, 134, 74 CrossRef CAS PubMed.
- Y. Ogata, H. Niki, T. Sakka and M. Iwasaki, J. Electrochem. Soc., 1995, 142, 195 CrossRef CAS.
- G. Korotcenkov and B. K. Cho, Crit. Rev. Solid State Mater. Sci., 2010, 35(3), 153 CrossRef CAS.
- C. T. Johnston and Y. O. Aochi, Methods of soil analysis. Part 3-chemical methods, 1996, p. 269 Search PubMed.
- J. Coates, Interpretation of infrared spectra, a practical approach, in Encyclopedia of analytical chemistry, 2000 Search PubMed.
- G. B. Li, L. S. Liao, X. B. Liu, X.-Y. Hou and X. Wang, Appl. Phys. Lett., 1997, 70, 1284 CrossRef CAS.
- I. R. Rao, Nature, 1930, 125, 600 Search PubMed.
- J. Rytkönen, R. Miettinen, M. Kaasalainen, V.-P. Lehto, J. Salonen and A. Närvänen, J. Nanomater., 2012, 896562 Search PubMed.
- H. Huang, F. Liu, S. Chen, Q. Zhao, B. Liao, Y. Long, Y. Zeng and X. Xia, Biosens. Bioelectron., 2013, 42, 539 CrossRef CAS PubMed.
- H. Xu, T. Zhu and R. P. Yu, Study on the absorption and fluorescence spectra of ethylene glycol and glycerol, Guangpuxue Yu Guangpu Fenxi, 2007, 27(7), 1381 CAS.
- D. E. Azofeifa, H. J. Arguedas and W. E. Vargas, Opt. Mater., 2012, 35(2), 175 CrossRef CAS.
- K. Dohnalová, A. N. Poddubny and A. A. Prokofiev, Light: Sci. Appl., 2013, 2, 47 CrossRef.
- N. K. Hon, Z. Shaposhnik, E. D. Diebold, F. Tamanoi and B. Jalali, J. Biomed. Mater. Res., 2012, 100(12), 3416 CrossRef PubMed.
- M. A. Avidad, A. Nieto, A. Salinas-Castillo, L. Capitan-Vallvey, G. Miskelly and M. J. Sailor, Nano Res. Lett., 2014, 9(1), 1 CrossRef PubMed.
- A. G. Cullis, L. T. Canham and P. D. J. Calcott, J. Appl. Phys., 1997, 82, 909 CrossRef CAS.
- B. Gelloz, Biodegradability of porous silicon, in Handbook of Porous silicon, Springer International Publishing, Switzerland, 2014, p. 307 Search PubMed.
- W. De Benedetti, S. Chiu, R. Radlinger, J. Ellison, B. A. Manhat, J. Z. Zhang and J. Shi, J. Phys. Chem. C, 2015, 119(17), 9595 CAS.
- G. Ledoux, J. Gong, F. Huisken and O. Guillois, Appl. Phys. Lett., 2002, 80(25), 4834 CrossRef CAS.
- Y. Cai and Y. Lapitsky, Colloids Surf., B, 2014, 115, 100 CrossRef CAS PubMed.
- A. Loni, Porous silicon formation by anodization, in Handbook of Porous silicon, Springer International Publishing, Switzerland, 2014, p. 11 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1. Schematic representation of the pSi surface functionalization procedure. Fig. S2. HPLC trace of compounds released from hydroxocobalamin (OH–Cbi) by UV detection at 168–351 nm (a). Optical density at times of 289 s (b) and 380 s (c). Fig. S3. Optical emission spectra of pSi–chitosan sample after suspension in PBS as a function of time. Fig. S4. (a) Influence of Cbi loading on reduction of pSi–COOH PL intensity into PBS. (b) PL spectrum of pSi–COOH initially and after 3 h of incubation in PBS, and effect of Cbi loading. See DOI: 10.1039/c6ra27102b |
| This journal is © The Royal Society of Chemistry 2017 |