
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInduction of potent apoptosis by an anti-CD20 aptamer via the crosslink of membrane CD20 on non-Hodgkin's lymphoma cells†
Cong Wu‡
a,
Wei Wan‡b,
Ji Zhua,
Hai Jina,
Tiejun Zhao*a and
Huafei Li *acd
*acd
aDepartment of Laboratory Diagnosis/Thoracic Surgery, Changhai Hospital Affiliated to the Second Military Medical University, 168 Changhai Road, Shanghai, 200433, P.R. China. E-mail: drzhaotiejun@126.com; huafey_lee@163.com; Fax: +86 21 81875336; Tel: +86 21 81875336
bDepartment of Orthopedic Oncology, Spine Tumor Center, Changzheng Hospital Affiliated to the Second Military Medical University, 415 Fengyang Road, Shanghai, 200433, P.R. China
cInternational Joint Cancer Institute, Translational Medicine Institute, The Second Military Medical University, 800 Xiangyin Road, Shanghai, 200433, P.R. China
dTumor Immunology and Gene Therapy Center, Eastern Hepatobiliary Surgery Hospital Affiliated to the Second Military Medical University, 225 Changhai Road, Shanghai, 200433, P.R. China
First published on 17th January 2017
Abstract
Targeted therapy opened a new era for the treatment of malignancies. Recently, there is growing interest in the development of novel targeted ligands except for monoclonal antibodies (mAbs). Aptamers are a class of therapeutic oligonucleotides form specific three dimensional structures dictated by their sequences. Aptamers can bind to a wide range of targets with high sensitivity and specificity, making it an ideal candidate for disease diagnosis and therapy. Herein, an anti-CD20 DNA aptamer (ACDA) was successfully screened from Harvard library by systematic evolution of ligands by exponential enrichment (SELEX). Experimental results demonstrated that ACDA can bind to surface CD20 than Rituximab Fab fragments with stronger binding affinity. With mass arming ACDA to a long chain polyethyleneimine (PEI) polymer, the resultant aptamer–polymer conjugates (P-ACDA) can induce potent caspase dependent apoptosis in targeting Non-Hodgkin's Lymphoma (NHL) cells via the crosslink of cellular CD20. All the results indicated that ACDA not only can be developed as targeting ligands for specifically delivering diagnostic or therapeutic drugs, but also can themselves be effective therapeutic candidates in treating NHL, which deserves for further investigation in the clinic.
Introduction
The introduction of mechanism-based targeted therapies to treat human malignancies has been heralded as one of the fruits of three decades of remarkable progress of research into the mechanisms of cancer pathogenesis.1 Until recently, most of the hallmark-targeting cancer drugs developed present inhibitory activity against a specific molecular target, while having relatively fewer off-target effects.1,2 Among the most prolific of targeted agents, monoclonal antibodies (mAbs) currently not only dominate in diagnostics, but also as highly successful therapeutics.3,4 Depending on their mechanism of action, mAbs can induce targeted cell-specific killing and/or enhance target cell susceptibility to chemo- or radiotherapy by modulating particular signal transduction pathways.5 Besides, toxic substances, such as radioisotopes or cytotoxic agents, can be accurately delivered to malignant cells with the guiding of certain mAbs by the construction of antibody–drug conjugates (ADCs).6,7CD20 is a B-cell differentiating antigen, which selectively expressed on the surface of mature and malignant B-cells.8 Currently, 5 anti-CD20 mAbs have been approved by the U.S. FDA (Food and Drug Administration): Rituximab (RTX, Rituxan), Ofatumumab (Arzerra), Obinutuzumab (Gazyva), the radio-conjugates Bexxar (Tositumomab-I131) and Zevalin (Ibritumomab tiuxetan).9,10 The first FDA-approved RTX leads to target cell depletion mainly through complement-dependent cytotoxicity (CDC) and antibody-dependent cellular cytotoxicity (ADCC), which is used in all phases of conventional treatment, including first-line therapy, maintenance and salvage therapy against CD20+ Non-Hodgkin's Lymphoma (NHL) in the clinic.11,12 Bexxar and Zevalin are two radio-conjugates, bound respectively, to radioactive Yttrium90 and Iodine131.13,14 Ofatumumab and Obinutuzumab were two most recently approved mAbs in treating CLL (chronic lymphocytic leukemia), both recognize a distinctly different sites on the CD20 molecule than Rituximab.15,16 Teeling JL revealed that Ofatumumab, which is a more potent inducer of CDC, specifically binds the smaller loop proximal to the cell membrane, while RTX's binding is restricted to the larger extracellular domain.15,17 Comparing with the type I anti-CD20 mAbs (including RTX and Ofatumumab), the type II mAb, Obinutuzumab, can directly induce programmed cell death (PCD) more efficiently.18,19 Our recent studies demonstrated that co-localization of CD20-TNFR1 after type II anti-CD20 mAb ligation can stimulate de novo ceramide synthesis and consequently induce remarkable lysosomal permeabilization, which is the main mechanism of lysosome-mediated cell death induced by type II mAbs.12 Recently, there is growing interest in the development of novel CD20 targeted therapeutics, highlighting the increased demand for novel targeted ligands, among which aptamers might be the most potential molecules.
Aptamers are a class of single-stranded DNA or RNA oligonucleotides (15–100 nt), which have firstly been described in 1990s as a new class of receptor molecules.20,21 In contrast to antisense oligonucleotides and small interfering RNAs (siRNAs) that inhibit translation of proteins by Watson–Crick base-pairing to their respective messenger RNAs, aptamers bind to existing individual target molecules with high affinity and specificity via a unique three dimensional interaction, analogous to mAbs.22 Aptamers possess a number of advantages over mAbs, which make them attractive pharmaceutical candidates. (1) Aptamers are less immunogenic than mAbs, making them better suited for systemic administration and long term therapy.23 (2) Most aptamers bind to target molecules with higher affinity, demonstrating typical dissociation constants in the pico- to nano-molar range.22 (3) Aptamers are structurally stable across a wide range of temperature and storage conditions, maintaining the ability to form their unique tertiary structures, while mAbs must be maintained in a temperature controlled environment to preserve structure and function.24 (4) Aptamers are artificially generated and do not require the labor intensive and considerably expensive methods of in vivo selection needed to produce mAbs.22 (5) Aptamers are synthesized consistently and with great accuracy every time, while antibodies can vary batch by batch, and require routine performance validation.25 (6) Unlike antibodies which can only be generated to antigens with immuno-stimulatory capacities, aptamers can be raised to virtually any targets including ions, small molecules, proteins, virus and even whole cells.26–28 However, no therapeutic aptamer or aptamer derivative targeting CD20 has been developed for treating B cell malignancies.
In this study, an anti-CD20 DNA aptamer (ACDA) was successfully fabricated by cell-SELEX (Systematic Evolution of Ligands by Exponential enrichment). Comparing with RTX, ACDA can bind to surface CD20 with higher affinity. More importantly, by mass arming of ACDAs to a polyethylenimine (PEI) polymer, the resulting aptamer nanoarray can elicit potent caspase dependent apoptosis in CD20 positive NHL cells.
Materials and methods
Cell lines and antibodies
Two NHL cell lines (Raji and Ramos) and wild type Chinese hamster ovary (CHO/WT) cells were obtained from the American Type Culture Collection (ATCC). The CD20+ CHO cell line (CHO/CD20P) was generated in our laboratory by expressing CD20 in a CHO/WT cells using the Lenti-X™ HTX Packaging System (TaKaRa, California, USA). NHL cells (Raji and Ramos) and CHO cells (CHO/WT and CHO/CD20P) were respectively maintained in RPMI-1640 and DMEM media (Gibco, California, USA) supplemented with 10% fetal calf serum (FCS, Gibco, California, USA) at 37 °C, 5% CO2. Rituximab was purchased from Roche Co. Ltd (Switzerland). Alexa Fluor 488 labeled Rabbit anti-Human IgG (H+L) secondary antibody was purchased from Invitrogen Co. Ltd (California, USA).Generation of Rituximab Fab fragments
The Fab fragments of Rituximab were generated by cleaving Rituximab using Papain (Sigma-Aldrich, St. Louis, USA) following previous studies with minor revisions.29,30 Briefly, 2 mg mL−1 Rituximab was incubated with 0.1 mg mL−1 papain in digestion buffer (20 mM cysteine monohydrochloride in phosphate buffer, pH 10) at 37 °C for 24 h. The Fc fragments were removed by affinity chromatography on Protein A-Sepharose (GE Healthcare).Selection of anti-CD20 aptamers
The anti-CD20 aptamers was selected by Cell-SELEX following previous studies.31,32 The initial population of DNA oligonucleotides (Harvard library) was purchased from Integrated DNA Technologies (Lowa, USA). The librarian oligonucleotide sequence is 5′CTCCTCTGACTGTAACCACG-N60-GCATAGGTAGTCCAGAAGCC3′. The cell SELEX process was shown in Fig. 1A. Prior to each round of selection the DNA sample is denatured at 95 °C × 10 min and snap cooled on ice for 5 min for preconditioning. Positive selection denotes selection performed using CHO/CD20P cells, following by negative selection in CHO/WT cells. For rounds 1–5, no negative selection was performed. For round 10–15, positive selection was performed in Rituximab bound CHO/CD20P cells. For each round, the aptamers were eluted from target cells by heating the cellular suspension to 95 °C with gently vortexing for 10 min. To prepare each pool for the next subsequent of selection, the DNA sample was purified using agarose gel extraction and the 100 bp band is purified with the GeneJET extraction kit (Thermo Fisher Scientific, California, USA). The collected DNA pool is PCR amplified and digested using Lambda exonuclease (Thermo Fisher Scientific, California, USA) to generate a single stranded aptamer. The selected aptamer (ACDA) was sequenced by Tianjin Dongfang Tongchuang Biological technology Co. Ltd (Tianjin, China) and its secondary structure was predicted by the mfold Web Server (http://www.unafold.rna.albany.edu/?q=mfold).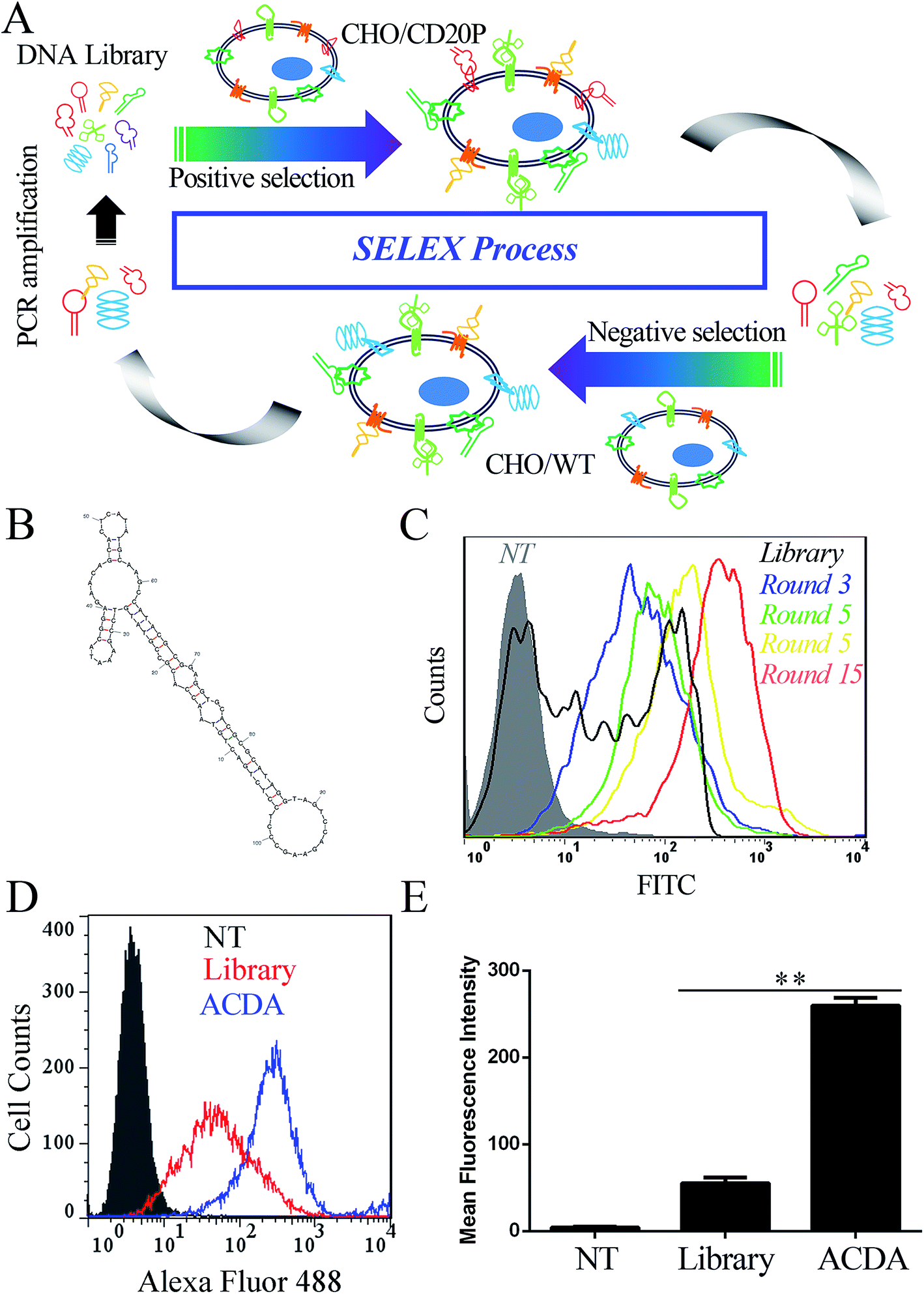 | ||
| Fig. 1 Generation and characterization of anti-CD20 DNA aptamers. (A) Generalized selection scheme of cell-SELEX using CD20 positive (CHO/CD20P) and negative (CHO/WT) cells. (B) Purported secondary structure of the selected anti-CD20 aptamer candidate. (C) Binding avidity of aptamer pools with CHO/CD20P cells after different round of cell-SELEX by FCM. (D and E) Binding affinity comparing DNA library and the selected anti-CD20 DNA aptamer (ACDA) with naturally CD20 positive Raji cells by FCM, data are shown as mean ± SD (n = 3), **p < 0.01. | ||
Fluorescence labeling of ACDAs and Rituximab Fab fragments
The Rituximab Fab fragments were fluorescence-labeled by using Alexa Fluor 488/555 labelling kit (Thermo Fisher Scientific, California, USA) following the manufacturer procedures. Briefly, 50 μL of 1 M bicarbonate were added to 0.5 mL 2 mg mL−1 purified Rituximab Fabs. Then the Fab solution was mixed with the vial of reactive dye provided by the labelling kit and reacted in room temperature with continuous stirring for 1 h protected from light. The fluorescence labeled Fab fragments were purified by the provided purification columns. The 3′-NH2 modified ACDAs was labeled in the same way.Binding avidity assessment
The binding avidity was assessed by flow cytometer (FCM) and confocal microscopy. For FCM, different concentration (800, 400, 200, 100, 50, 25 and 12.5 nM) of Alexa Fluor 488 labeled ACDA and RTX Fab was incubated with Raji and Ramos cells for 1 hour at 37 °C in the dark, respectively. After washing with PBS for twice, the mean fluorescence intensity (MFI) was determined by a FCM (FACS Calibur, BD Biosciences, USA). For confocal microscopy, cells were incubated with 400 nM ACDA (Alexa Fluor 555 labeled) and RTX Fab (Alexa Fluor 488 labeled) for 1 hour, respectively. After washing, the stained cells were observed using a Confocal Laser Scanning Microscope (CLSM, Zeiss, Germany).8Competitive binding assay
Raji cells (1 × 105) were incubated with 400 nM Alexa Fluor 488 labeled RTX Fab and increasing concentrations (25–800 nM) of competing agents (Tratuzumab Fab, Rituximab Fab or ACDA) for 1 h at 37 °C. Cells incubated with 400 nM Alexa Fluor 488 labeled RTX Fab only was used as a control group. After washing with PBS for twice, the MFI of each sample was evaluated by FCM. The ability of ACDA to compete with RTX Fab for binding to CD20 is evaluated by the decreased degree of MFI in targeting Raji cells compared with control group (MFIsample/MFIcontrol).33 The ability of RTX Fab to compete with ACDA was evaluated in the same way.In vitro crosslink of ACDA
In vitro crosslink of ACDA was realized by mass arming ACDAs to PEI polymer (molecular weight: 70 kDa, Sigma-Aldrich, St Louis, USA) as described in our previous studies (Fig. 3A).8,34 Briefly, the PEI polymer (1 mg mL−1) was incubated with MPEGS linker under stirring and N2 bubbling at room temperature for 4 h (PEI![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MPEGS = 1:15). Then, ∼SH modified ACDA were slowly dropped into the MPEGS–PEI suspension and the reaction was conducted in an N2 environment for 8 hours. Unconjugated aptamers were separated by dialysis, the resulted PEI–MPEGS–ACDA (P-ACDA) was preserved in 4 °C for future usage. The PEI–MPEGS–Fab (P-Fab) was generated in the same way. The size distribution of P-Fab and P-ACDA was determined with a dynamic laser light scattering (DLS) instrument (ALV/CGS3, Germany). The crosslinking of surface CD20 on Raji cells by P-ACDA and P-Fab was evaluated by confocal microscopy as mentioned in “Binding activity assessment”.
:MPEGS = 1:15). Then, ∼SH modified ACDA were slowly dropped into the MPEGS–PEI suspension and the reaction was conducted in an N2 environment for 8 hours. Unconjugated aptamers were separated by dialysis, the resulted PEI–MPEGS–ACDA (P-ACDA) was preserved in 4 °C for future usage. The PEI–MPEGS–Fab (P-Fab) was generated in the same way. The size distribution of P-Fab and P-ACDA was determined with a dynamic laser light scattering (DLS) instrument (ALV/CGS3, Germany). The crosslinking of surface CD20 on Raji cells by P-ACDA and P-Fab was evaluated by confocal microscopy as mentioned in “Binding activity assessment”.
Weight-average molar mass analysis by static light scattering
The static light scattering (SLS) measurements were carried out by the DLS instrument as described in our previous studies.35 The weight-average molar mass (Mm) was calculated by the following equation:36| KCp/R(q) = 1/Mm + 2A2C |
Annexin V & PI staining
After a 16 hour treatment with Fab, P-Fab, ACDA and P-ACDA (with equal amount of 400 nM Fab or ACDA), NHL cells were stained with Alexa Fluor-488 labeled Annexin V antibody & propidium iodide (PI) and analyzed by two-channel FCM of FL-1 (annexin-V) and FL-2 (PI). For apoptosis inhibition assays, a cell-permeable pan-caspase inhibitor, Z-VAD-FMK (Promega Corporation, Fitchburg, USA), was added 1 h prior to the addition of therapeutic agents.Mitochondrial membrane potentials (MMP) and caspase activation
Raji cells were incubated with Fab, P-Fab, ACDA and P-ACDA (with equal amount of 400 nM Fab or ACDA) for 16 h. After washing, the JC-1 probe (Beyotime Biotechnology, Shanghai China) and Vybrant® FAM Poly Caspase Assay Kit (Invitrogen, California, USA) was respectively employed to measure mitochondrial depolarization and caspase activation in Raji cells by FCM following manufacturer's protocol.Western blotting
After washing, cells were lysed in cell lysis buffer for western (Beyotime Biotechnology, Shanghai China). The cell lysates were subjected to SDS-PAGE and immunoblotted with cleaved caspase-3/9 antibodies (Cell Signaling Technology, USA).Statistical analysis
Error bars represent the standard deviation (SD) of 3 independent samples unless otherwise stated. Statistical analysis was performed by Student's unpaired t test or ANOVA to identify significant differences unless otherwise indicated. The number of asterisks displayed on the figures represents the degree of statistical significance as determined by p values as follows: *p < 0.05, **p < 0.01. All data were processed with SPSS 10.0 software.Results
Successful isolation of anti-CD20 DNA aptamers (ACDA)
In this study, 15 rounds of cell-SELEX were carried out to screen out aptamers specifically binding to CD20 in CHO/CD20P cells, with wild type CHO cells as counter selection. It should be mentioned that the CD20 expression of CHO/CD20P cells were verified by FCM (Fig. S1†) before the SELEX process. To evaluate selection process, CHO/CD20P cells were incubated with Alexa Fluor-488 labelled aptamer pools derived from DNA library and the products of rounds 3, 5, 10 and 15. FCM results (Fig. 1C) showed that cells incubated with DNA library (black curve) exhibit a moderate shift in fluorescence. With the rounds increased, incubated cells exhibit increasing rightward shifts in fluorescence, with round 15 possessing the greatest movement. This means that after 15 rounds of selection, we have achieved the aptamer pool which contains significantly enriched ssDNA sequences selectively recognizing CHO/CD20P cells rather than CHO/WT cells. Besides, the resultant ACDA can efficiently bind to naturally CD20 expressed Raji cells than DNA library with stronger affinity (Fig. 1D and E). DNA sequencing results of the ACDA are as following: 5′-CTCCTCTGAC TGTAACCACG CCGTATGTCC GAAATACGGA GAACAGCACT CATATGCAAG CCATACGCGG AGGTGCACGC GCATAGGTAG TCCAGAAGCC-3′, the secondary structure of which was predicted by the mfold Web Server with the results showing in Fig. 1B.ACDA can bind to CD20+ NHL cells than RTX Fab with better affinity
The FCM results (Fig. 2A and B) revealed that the ACDA aptamer exhibited relatively appreciable binding affinity to cellular CD20 on both NHL cells, with the dissociation constant (Kd) both less than 80 nM (KdRaji: 69.1 ± 7.58 nM, KdRamos: 71.0 ± 7.3 nM), comparing with the Kds of Rituximab Fab fragments more than 100 nM (KdRaji: 107.1 ± 11.0 nM, KdRamos: 128.1 ± 13.3 nM). The excellent binding avidity of ACDA was further evaluated by CLSM (Fig. 2C). The green fluorescence on cellular surface was from the Fab fragments of Rituximab, and the red fluorescence from ACDAs. CLSM results demonstrated that the exposure of NHL cells to Rituximab Fab or ACDA led to decoration of cellular surface with regular green or red fluorescence, respectively, which indicates excellent binding avidity between ACDA and CD20 molecules (Fig. 2C). Competitive binding assay results showed that the binding of Rituximab Fab to cellular CD20 was obviously affected by ACDA, and vice versa. However, the influence of ACDA on Rituximab Fab's binding to CD20 was much greater than that of Rituximab Fab on ACDA as indicated by the competitive binding curve evaluated in Raji cells (Fig. 2D and E). All the above results indicated that the binding activity with surface CD20 on NHL cells of ACDA was stronger than that of Rituximab Fab fragments.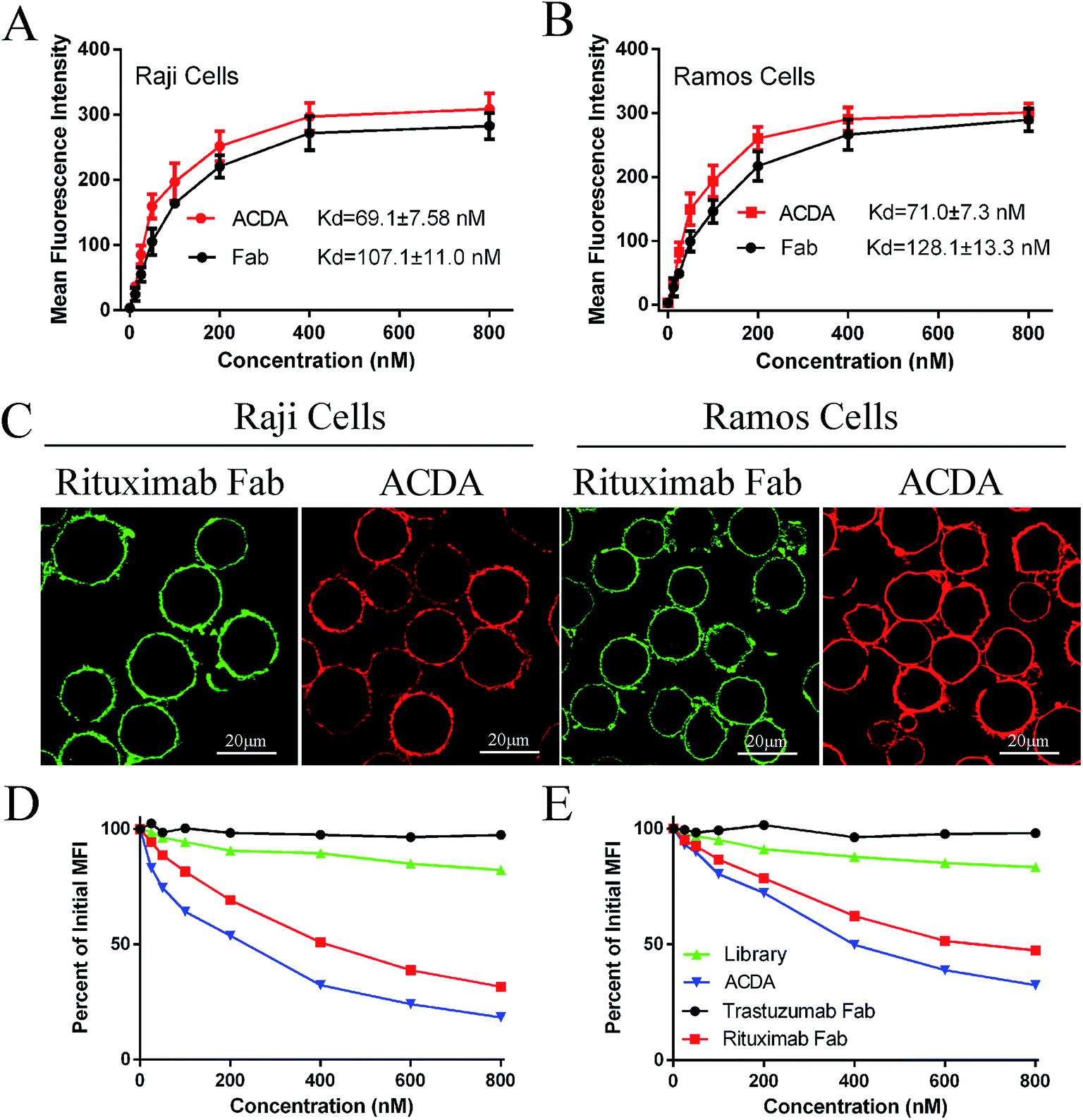 | ||
| Fig. 2 Biorecognition of ACDA on surface CD20 of NHL cells. (A and B) CD20+ Raji (A) and Ramos (B) cells were respectively incubated with Alexa Fluor 488 labeled ACDA and Rituximab Fab at indicated concentration (12.5 nM, 25 nM, 50 nM, 100 nM, 200 nM, 400 nM and 800 nM) and the MFI (mean fluorescence intensity) was measured by FCM. The dissociation constant (Kd) was calculated by GraphPad software. (C) Cells incubated with RTX-Fab-488/ACDA-555 were observed with a confocal microscope. Scale bar: 20 μm. (D) Competitive binding assays. ACDAs, Trastuzumab Fab and Rituximab Fab fragments were evaluated for their abilities to compete with indicated Rituximab Fab-488 (D) or ACDA-555 for binding to cellular CD20. | ||
Fabrication and characterization of P-ACDA
It is well established that, with the cross-linking of secondary antibodies, RTX can induce caspase-dependent apoptosis in malignant B-cells.34,37,38 However, this cross-linking can hardly be realized in vivo. In our previous studies, a novel anti-CD20 mAb's nanoarray was finely prepared by in vitro mass arming anti-CD20 mAbs to a PEI polymer by nanotechnology, which can successfully elicit caspase dependent apoptosis in CD20+ NHL cells.34,39 In order to evaluate whether the crosslink of ACDA–CD20 complexes can induce target cell apoptosis, an anti-CD20 aptamer's nanoarray (P-ACDA) was constructed in the same way, with the schematic diagram shown in Fig. 3A. Besides, the antibodies' nanoarray (P-Fab) was fabricated as a parallel control. DLS results demonstrated that the hydrodynamic radius of P-ACDA and P-Fab was 91.1 ± 23.3 nm and 171.2 ± 41.5 nm, respectively (Fig. 3C). The valency of P-Fab/P-ACDA (the number of Fab/ACDA molecules per nanoarray) was calculated according to the following equation:35here, Mm(P-ACDA/P-Fab) is the weight-average molar mass of P-ACDA or P-Fab, which was estimated to be 1.86 × 106 and 1.21 × 106 g mol−1 by SLS analysis, respectively. The MW(PEI), MW(Fab) and MW(MPEGS) are respectively 70 kDa, 45 kDa and 1 kDa according to the product descriptions. The MW(ACDA) was estimated to be approximately 33 kDa (100 nt × 330 Da nt−1). Therefore, we can estimate that there are on average ∼33.5 ACDA and 38.9 Fab molecules per nanoarray (Table 1).
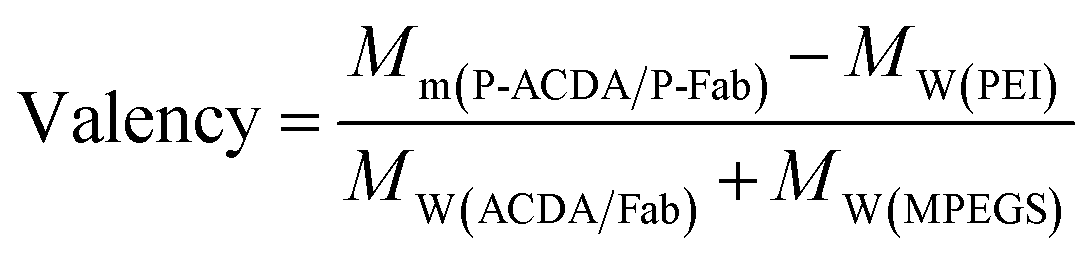
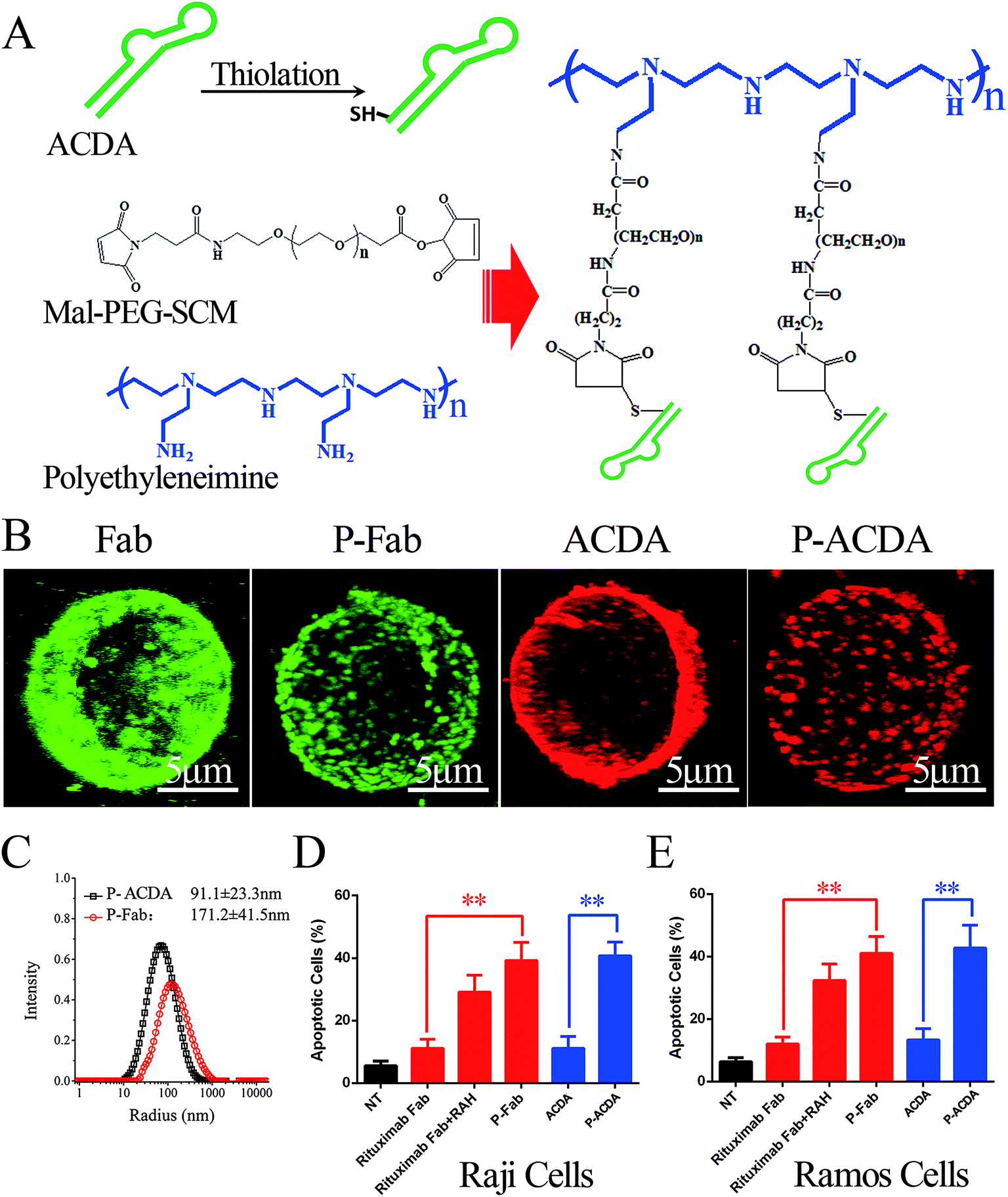 | ||
| Fig. 3 ACDA can evoke potent apoptosis by crosslinking surface CD20 on NHL cells. (A) Schematic plot of the fabrication of an anti-CD20 aptamer nanoarray (P-ACDA). (B) CLSM images of the distribution of CD20 molecule on surface of Raji cells after incubating with Rituximab Fabs, ACDA, P-Fab or P-ACDA for 1 h. 3D reconstruction is based on a confocal z-stack. Scale Bar: 5 μm. (C) Size distribution of P-Fab and P-ACDA nanoarray. (D and E) Apoptosis inducing ability comparing free and crosslinked anti-CD20 mAbs and aptamers against Raji (D) and Ramos (E) cells. Data are mean ± SD (n = 3), **p < 0.01. | ||
| Rh (nm) | Mm (g mol−1) | Valence | |
|---|---|---|---|
| a Rh: averaged hydrodynamic radius. Mm: weight-average molar mass. Valence: number of Fab or ACDA per polymer chain. | |||
| P-ACDA | 91.1 ± 23.3 | 1.21 × 106 | 33.5 |
| P-Fab | 171.2 ± 41.5 | 1.86 × 106 | 38.9 |
In vitro anti-NHL ability of ACDA
In order to determine the influence of P-ACDA on CD20 crosslinking, confocal microscopy was employed to probe the distribution of CD20 on Raji cell membranes after aptamer ligation. As shown in Fig. 3B, CD20 microcluster formation can be observed after treatment with P-ACDA or P-Fab, while neither free aptamers nor Fab fragments can have the ability to induce micro-cluster formation of CD20 molecules. Fig. 3C and D demonstrates that although free Rituximab Fab fragments and ACDA can hardly induce apparent apoptosis, the crosslink of cellular CD20 via P-ACDA or P-Fab can elicit a significantly higher level of apoptosis as indicated by Annexin V positive subsets in both NHL cells (Raji and Ramos).Involvement of caspase in P-ACDA induced apoptosis
It is widely accepted that the caspases, a family of cysteine proteases, function as central regulators of apoptosis.40 Caspase activation can be regulated mainly through two signaling pathways: the extrinsic pathway, which involves Fas and TNFR stimulation, and the intrinsic pathway, which may be the primary means of activating apoptotic caspase in lymphoma cells treated by crosslinked anti-CD20 mAbs.41,42 The intrinsic pathway triggers the mitochondrial depolarization, which can oligomerize with apoptotic protease activating facter-1 (Apaf-1) and activate pro-caspase-9/3 and eventually leads to cell apoptosis.41,42 For the determination of caspase involvement in P-ACDA induced cell death, the mitochondrial depolarization was firstly assessed by FCM post JC-1 staining, cells undergone mitochondrial depolarization was indicated by a decrease in FL-2 fluorescence intensity (JC-1 red). As shown in Fig. 4A and B, neither free Rituximab Fab nor ACDA can cause significant changes in mitochondrial membrane potential (MMP), while both P-ACDA and P-Fab treated Raji cells experienced a remarkable mitochondrial depolarization. The direct detection of caspase activation was detected by Western blotting, with the results shown in Fig. 4C. Apoptosis inhibition assays (Fig. 4D) revealed that Z-VAD-FMK (a cell-permeable pan-caspase inhibitor) over a range of concentrations from 10 to 30 μM can significantly prevent P-ACDA and P-Fab induced apoptosis in a dose dependent way.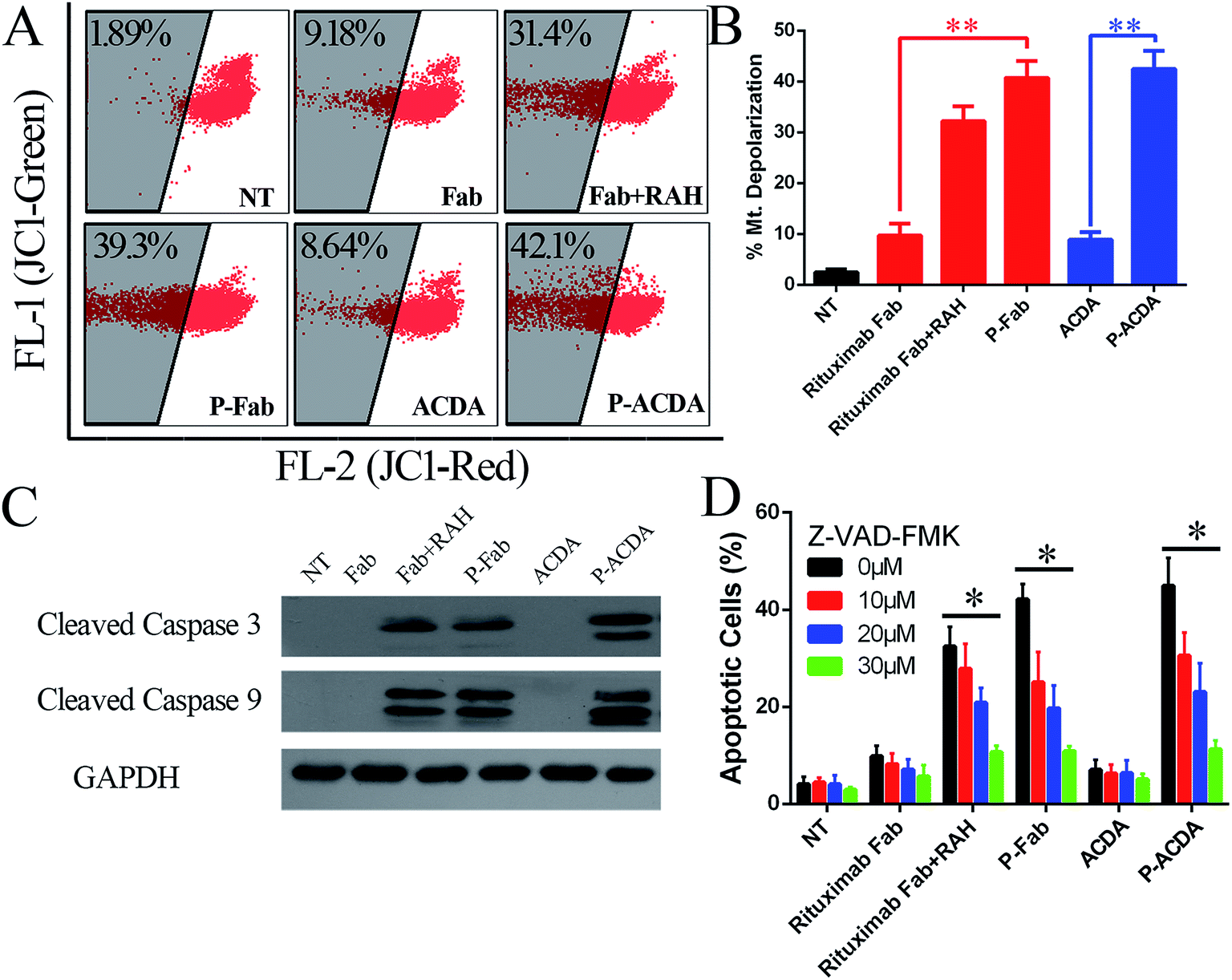 | ||
| Fig. 4 Involvement of caspase in P-ACDA induced cell death. (A and B) Detection of the mitochondrial membrane potentials of anti-CD20 mAb/aptamer treated Raji cells. Data are mean ± SD (n = 3) (**P < 0.01). (C) Western blotting analysis for cleaved caspase 3 and 9 in anti-CD20 mAb/aptamer treated Raji cells. (D) Crosslinked anti-CD20 mAb/aptamer induced apoptosis can completely be prevented by a pan-caspase inhibitor (Z-VAD-FMK). Data are mean ± SD (n = 3) (**P < 0.01). | ||
Discussion and conclusion
Aptamers are a class of therapeutic oligonucleotides that form specific three dimensional structures dictated by their sequences.21 Besides its high sensitivity and specificity, it has other merits, such as simple and quick preparation procedure, chemical stability, no immunogenicity or toxicity, a wide range of targets and feasibility to be functionally modified.43 Thus, it is an ideal candidate for disease diagnosis and therapy.In this study, a CD20 targeting aptamer, ACDA, has been screening our from “Harved DNA library” containing 1015 random sequences. It is well acknowledged that the selection parameters of SELEX process exert a direct influence on the quantity and quality of the evolved aptamers.44,45 The more stringent the conditions the greater the binding affinity and discriminative capacity of the selected aptamers.44,45 Therefore, for purpose of isolating aptamers with high affinity, Rituximab (a competitive mAb) binding CHO/CD20P cells were employed for positive selection in last 5 rounds (round 10–15) of cell-SELEX. Experimental results revealed that the resultant ACDA exhibits higher binding avidity to surface CD20 than that of Rituximab Fab indicated by smaller dissociation constant (Kd).
Previous publications have demonstrated that Rituximab induction of apoptosis is enhanced when cross-linked via anti-Fc secondary antibodies.46,47 Crosslink of CD20 binding mAbs promotes translocation of the cellular CD20 into lipid rafts, causing inhibition of P38 MAPK & ERK1/2 survival pathways and activation of Caspase cascades.48 This apoptotic mechanism has been utilized in our previous studies to design therapeutic systems utilizing antibody–polymer conjugates (a novel anti-CD20 mAb's nanoarray).8,34,35 For evaluating whether the crosslink of aptamers can have similar effects, an anti-CD20 aptamer nanoarray (P-ACDA) was generated in the present work. Excitingly, P-ACDA can activate intracellular caspase cascades and evoke potent apoptosis in targeting NHL cells as expected. Currently, aptamers have been developed as targeting ligands for specifically delivering diagnostic or therapeutic agents into malignant tissues and cells. However, our present study revealed that the anti-CD20 aptamers can themselves be an effective therapeutic candidate in treating NHL. Comparing with the anti-CD20 mAb nanoarray constructed in our previous studies, the P-ACDA owns the following advantages. Firstly, the anti-drug antibody (ADA) was considered to be one of the important reasons for the drug resistance of mAbs including Rituximab, which might be one of the biggest obstacles for antibody based targeted therapy.49 FDA recommended that the assessment of immunogenicity of biological is a key element of pre- and clinical studies.50 Whereas the non-immunogenicity of ACDA provides a potential tool to solve this problem. Secondly, the penetration of antibody nanoarray might be limited due to it's relatively high molecular weight (MW) to some extent, which can be resolved by using aptamers with much smaller MWs. Thirdly, due to the complexity and diversity, a variety of molecular probes is required to discriminate distinct features of malignant cells. However, it is infeasible to systematically produce a group of mAbs for origin identification in a short time. As an alternative strategy, aptamer libraries usually have larger volume (about 1016), which makes it possible to screening out molecular probes with higher affinity and specificity for specific tumor cells. Thus, aptamer technology is a better tool to accurately and sensitively distinguish malignant cells for the diagnosis and treatment of various cancers.51 However, DNA and RNA are naturally occurring biomolecules, a variety of nuclease enzymes have co-evolved to catalyze their degradation, which pose limitations for the use of aptamers in vivo.52 Post-selection modification (such as 2′-O-Me, 2′-NH2, and 2′-F modifications etc.) of the nucleic acid backbone can greatly reduce nuclease susceptibility, and this strategy has been successfully implemented to enable the use of Macugen (pegaptanib), which has been approved by FDA for treating age-related macular degeneration in 2004, and represents the first aptamer-based commercial therapeutics.53 Besides, an even more powerful approach is directly selecting aptamers from libraries bearing modified backbones or nucleobases.54 In order for further investigation of P-ACDA, we will introduce different modifications during the fabrication of P-ACDA, and their pharmacokinetic parameters would be determined and compared in our further investigation.
Conflicts of interest
No potential conflicts of interest were disclosed.Acknowledgements
This work was supported by General Financial Grant from the China Postdoctoral Science Foundation (2016M592934), National Natural Science Foundation of China (31400778, 8160102545, 81672890) and Shanghai Municipal Commission of Health and Family Planning Fund (20114253).References
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144(5), 646–674 CrossRef CAS PubMed.
- T. M. Allen, Nat. Rev. Cancer, 2002, 2(10), 750–763 CrossRef CAS PubMed.
- A. M. Scott, J. D. Wolchok and L. J. Old, Nat. Rev. Cancer, 2012, 12(4), 278–287 CrossRef CAS PubMed.
- C. T. Ellebrecht, V. G. Bhoj, A. Nace, E. J. Choi, X. Mao, M. J. Cho, G. Di Zenzo, A. Lanzavecchia, J. T. Seykora, G. Cotsarelis, M. C. Milone and A. S. Payne, Science, 2016, 353(6295), 179–184 CrossRef CAS PubMed.
- M. Y. El, Br. J. Nurs., 2015, 24(16), S4–S13 CrossRef PubMed.
- C. Sellmann, A. Doerner, C. Knuehl, N. Rasche, V. Sood, S. Krah, L. Rhiel, A. Messemer, J. Wesolowski, M. Schuette, S. Becker, L. Toleikis, H. Kolmar and B. Hock, J. Biol. Chem., 2016, 291(48), 25106–25119 CrossRef CAS PubMed.
- D. Jagadeesh and M. R. Smith, Current Treatment Options in Oncology, 2016, 17(10), 55 CrossRef PubMed.
- H. Li, G. Zhang, C. Jiang, F. Zhang, C. Ke, H. Zhao, Y. Sun, M. Zhao, D. Chen, X. Zhu, L. Zhang, B. Li, J. Dai and W. Li, Oncotarget, 2015, 6(27), 24192–24204 CrossRef PubMed.
- C. L. Grandjean, F. Montalvao, S. Celli, D. Michonneau, B. Breart, Z. Garcia, M. Perro, O. Freytag, C. A. Gerdes and P. Bousso, Sci. Rep., 2016, 6, 34382 CrossRef CAS PubMed.
- C. Gasperini, S. Haggiag and S. Ruggieri, Expert Opin. Invest. Drugs, 2013, 22(10), 1243–1253 CrossRef CAS PubMed.
- G. Cartron, H. Watier, J. Golay and P. Solal-Celigny, Blood, 2004, 104(9), 2635–2642 CrossRef CAS PubMed.
- F. Zhang, J. Yang, H. Li, M. Liu, J. Zhang, L. Zhao, L. Wang, R. LingHu, F. Feng, X. Gao, B. Dong, X. Liu, J. Zi, W. Zhang, Y. Hu, J. Pan, L. Tian, Y. Hu, Z. Han, H. Zhang, X. Wang and L. Zhao, OncoImmunology, 2016, 5(5), e1143995 CrossRef PubMed.
- S. J. Horning, A. Younes, V. Jain, S. Kroll, J. Lucas, D. Podoloff and M. Goris, J. Clin. Oncol., 2005, 23(4), 712–719 CrossRef CAS PubMed.
- M. Hoffmann, M. Troch, H. Eidherr, T. Traub-Weidinger, C. Jonak, L. Muellauer and M. Raderer, Leuk. Lymphoma, 2011, 52(1), 42–45 CrossRef CAS PubMed.
- J. L. Teeling, W. J. Mackus, L. J. Wiegman, J. H. van den Brakel, S. A. Beers, R. R. French, T. van Meerten, S. Ebeling, T. Vink, J. W. Slootstra, P. W. Parren, M. J. Glennie and J. G. van de Winkel, J. Immunol., 2006, 177(1), 362–371 CrossRef CAS.
- P. Sidaway, Nat. Rev. Clin. Oncol., 2016, 13(8), 466 CrossRef PubMed.
- J. L. Teeling, R. R. French, M. S. Cragg, J. van den Brakel, M. Pluyter, H. Huang, C. Chan, P. W. Parren, C. E. Hack, M. Dechant, T. Valerius, J. G. van de Winkel and M. J. Glennie, Blood, 2004, 104(6), 1793–1800 CrossRef CAS PubMed.
- A. Ivanov, S. A. Beers, C. A. Walshe, J. Honeychurch, W. Alduaij, K. L. Cox, K. N. Potter, S. Murray, C. H. Chan, T. Klymenko, J. Erenpreisa, M. J. Glennie, T. M. Illidge and M. S. Cragg, J. Clin. Invest., 2009, 119(8), 2143–2159 CAS.
- H. Ren, C. Zhang, L. Su, X. Bi, C. Wang, L. Wang and B. Wu, Biochem. Biophys. Res. Commun., 2015, 457(4), 572–577 CrossRef CAS PubMed.
- M. S. Sabatine, JAMA, J. Am. Med. Assoc., 2016, 315(23), 2525–2526 CrossRef CAS PubMed.
- Y. A. Shieh, S. J. Yang, M. F. Wei and M. J. Shieh, ACS Nano, 2010, 4(3), 1433–1442 CrossRef CAS PubMed.
- P. Ray and R. R. White, Pharmaceuticals, 2010, 3(6), 1761–1778 CrossRef CAS PubMed.
- J. S. Vorhies and J. J. Nemunaitis, Biol.: Targets Ther., 2007, 1(4), 367–376 CAS.
- S. D. Jayasena, Clin. Chem., 1999, 45(9), 1628–1650 CAS.
- K. M. Song, S. Lee and C. Ban, Sensors, 2012, 12(1), 612–631 CrossRef PubMed.
- S. C. Gopinath, T. S. Misono, K. Kawasaki, T. Mizuno, M. Imai, T. Odagiri and P. K. Kumar, J. Gen. Virol., 2006, 87(3), 479–487 CrossRef CAS PubMed.
- K. Sefah, Z. W. Tang, D. H. Shangguan, H. Chen, D. Lopez-Colon, Y. Li, P. Parekh, J. Martin, L. Meng, J. A. Phillips, Y. M. Kim and W. H. Tan, Leukemia, 2009, 23(2), 235–244 CrossRef CAS PubMed.
- P. Sundaram, H. Kurniawan, M. E. Byrne and J. Wower, Eur. J. Pharm. Sci., 2013, 48(1–2), 259–271 CrossRef CAS PubMed.
- T. A. Seldon, K. E. Hughes, D. J. Munster, D. Y. Chin and M. L. Jones, J. Biomol. Tech., 2011, 22(2), 50–52 Search PubMed.
- L. Coleman and S. M. Mahler, Protein Expression Purif., 2003, 32(2), 246–251 CrossRef CAS PubMed.
- K. M. Ruff, T. M. Snyder and D. R. Liu, J. Am. Chem. Soc., 2010, 132(27), 9453–9464 CrossRef CAS PubMed.
- N. L. Al-Youssef, S. M. Ghobadloo and M. V. Berezovski, RSC Adv., 2016, 6, 12435–12438 RSC.
- Y. Zhang, L. Wang, X. Chong, X. Yu, Y. Meng, J. Dong, C. Wang, H. Wang, Y. Yang, T. Xia, J. Zhao and B. Li, Biochem. Biophys. Res. Commun., 2016, 477(4), 755–760 CrossRef CAS PubMed.
- H. Li, Y. Sun, D. Chen, H. Zhao, M. Zhao, X. Zhu, C. Ke, G. Zhang, C. Jiang, L. Zhang, F. Zhang, H. Wei and W. Li, Sci. Rep., 2015, 5, 15712 CrossRef CAS PubMed.
- H. F. Li, C. Wu, T. Chen, G. Zhang, H. Zhao, C. H. Ke and Z. Xu, Int. J. Nanomed., 2015, 10, 4783–4796 CrossRef CAS PubMed.
- T. Yoshimura and K. Esumi, J. Colloid Interface Sci., 2004, 276(2), 450–455 CrossRef CAS PubMed.
- M. J. Glennie, R. R. French, M. S. Cragg and R. P. Taylor, Mol. Immunol., 2007, 44(16), 3823–3837 CrossRef CAS PubMed.
- M. Okroj, A. Osterborg and A. M. Blom, Cancer Treat. Rev., 2013, 39(6), 632–639 CrossRef CAS PubMed.
- W. Li, H. Li, J. Li, H. Wang, H. Zhao, L. Zhang, Y. Xia, Z. Ye, J. Gao, J. Dai, H. Wang and Y. Guo, Int. J. Nanomed., 2012, 7, 4661–4677 CrossRef CAS PubMed.
- E. Kuranaga, Genes Cells, 2012, 17(2), 83–97 CrossRef CAS PubMed.
- Z. T. Schafer and S. Kornbluth, Dev. Cell, 2006, 10(5), 549–561 CrossRef CAS PubMed.
- P. Li, D. Nijhawan, I. Budihardjo, S. M. Srinivasula, M. Ahmad, E. S. Alnemri and X. Wang, Cell, 1997, 91(4), 479–489 CrossRef CAS PubMed.
- X. Zhang, J. Zhang, Y. Ma, X. Pei, Q. Liu, B. Lu, L. Jin, J. Wang and J. Liu, Int. J. Biochem. Cell Biol., 2014, 46, 1–8 CrossRef CAS PubMed.
- D. Chen, Y. Orenstein, R. Golodnitsky, M. Pellach, D. Avrahami, C. Wachtel, A. Ovadia-Shochat, H. Shir-Shapira, A. Kedmi, T. Juven-Gershon, R. Shamir and D. Gerber, Sci. Rep., 2016, 6, 33351 CrossRef CAS PubMed.
- S. C. Gopinath, Anal. Bioanal. Chem., 2007, 387(1), 171–182 CrossRef CAS PubMed.
- N. Zhang, L. A. Khawli, P. Hu and A. L. Epstein, Clin. Cancer Res., 2005, 11(16), 5971–5980 CrossRef CAS PubMed.
- S. R. Aluri, P. Shi, J. A. Gustafson, W. Wang, Y. A. Lin, H. Cui, S. Liu, P. S. Conti, Z. Li, P. Hu, A. L. Epstein and J. A. MacKay, ACS Nano, 2014, 8(3), 2064–2076 CrossRef CAS PubMed.
- A. R. Jazirehi and B. Bonavida, Oncogene, 2005, 24(13), 2121–2143 CrossRef CAS PubMed.
- G. Shankar, C. Pendley and K. E. Stein, Nat. Biotechnol., 2007, 25(5), 555–561 CrossRef CAS PubMed.
- I. C. Buttel, P. Chamberlain, Y. Chowers, F. Ehmann, A. Greinacher, R. Jefferis, D. Kramer, H. Kropshofer, P. Lloyd, A. Lubiniecki, R. Krause, A. Mire-Sluis, T. Platts-Mills, J. A. Ragheb, B. M. Reipert, H. Schellekens, R. Seitz, P. Stas, M. Subramanyam, R. Thorpe, J. H. Trouvin, M. Weise, J. Windisch and C. K. Schneider, Biologicals, 2011, 39(2), 100–109 CrossRef CAS PubMed.
- Y. Pu, Z. Zhu, H. Liu, J. Zhang, J. Liu and W. Tan, Anal. Bioanal. Chem., 2010, 397(8), 3225–3233 CrossRef CAS PubMed.
- L. C. Griffin, G. F. Tidmarsh, L. C. Bock, J. J. Toole and L. L. Leung, Blood, 1993, 81(12), 3271–3276 CAS.
- E. W. Ng, D. T. Shima, P. Calias, E. J. Cunningham, D. R. Guyer and A. P. Adamis, Nat. Rev. Drug Discovery, 2006, 5(2), 123–132 CrossRef CAS PubMed.
- K. N. Meek, A. E. Rangel and J. M. Heemstra, Methods, 2016, 106, 29–36 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27154e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |