
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of soap-free emulsion with high solid content by differential dripping RAFT polymerization-induced self-assembly
Juanjuan Wu,
Chun Tian,
Lifen Zhang*,
Zhenping Cheng * and
Xiulin Zhu*
* and
Xiulin Zhu*
Suzhou Key Laboratory of Macromolecular Design and Precision Synthesis, Jiangsu Key Laboratory of Advanced Functional Polymer Design and Application, State and Local Joint Engineering Laboratory for Novel Functional Polymeric Materials, Department of Polymer Science and Engineering, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China. E-mail: chengzhenping@suda.edu.cn; zhanglifen@suda.edu.cn; xlzhu@suda.edu.cn; Fax: +86-512-65882787
First published on 10th January 2017
Abstract
Polymerization induced self-assembly (PISA) has been a facile and effective approach to prepare highly concentrated block copolymer nano-objects in situ. In this work, a soap-free emulsion with high solid content (60%) was successfully prepared in a semi-batch monomer addition manner. High monomer conversion can also be obtained by adjusting the dripping time. At first, a hydrophilic polymer, poly(poly(ethylene glycol)monomethyl ether methacrylate) (PPEGMA) was synthesized at almost complete monomer conversion in order to be used as a macroRAFT agent without any purification in the following step. Then PPEGMA was chain extended by the second monomer, methyl methacrylate (MMA), added at a very slow rate to form diblock copolymers and further self-assembled into nanoparticles in situ. The resulting latexes were very stable and the particle sizes remained at the nanoscale.
1. Introduction
Since the advent of reversible addition–fragmentation chain transfer (RAFT) polymerization in 1998,1 it has been an effective and robust method to prepare not only well-controlled polymers with pre-designed molecular weight and narrow polydispersity, but also varied topologies, such as block copolymers,2 grafting polymers,3 star polymers,4 etc. RAFT polymerization is a popular method because of its high tolerance to different conditions and convenient operation to carry out heterogeneous reactions, such as suspension, dispersion, emulsion,5 miniemulsion,6 and microemulsion7 polymerization.In the initial work of RAFT mediated emulsion polymerization, only hydrophobic control agents with low chain transfer constants (dithiocarbonate) can lead to successful synthesis (i.e., good control over the polymerization along with formation of stable latexes) while more reactive chain transfer agents proved to be unsuccessful.8 Significant headway hadn't been made until strategies employing macromolecular chain transfer agents were put forward. Charleux et al. first reported an approach to carry out miniemulsion polymerization by using amphiphilic diblock copolymer to emulsify the monomer phase in water, stabilize the particles and polymerize a third monomer for the preparation of triblock copolymers.9
In recent years, polymerization-induced self-assembly (PISA) first proposed by Hawkett et al.10 attracts many scientists' attention since it has excellent advantages, such as high polymer content at a low viscosity, convenient operation and producing diblock copolymer nano-objects with controlled size in the form of either spheres,11 worms12 or vesicles.13 PISA synthesis can be conducted by any type of living radical polymerization, but in fact the majority of literature examples reported are based on reversible addition–fragmentation chain transfer (RAFT) polymerization.14 In general, a RAFT mediated emulsion PISA process includes two steps: (i) the preparation of a soluble polymer which will act as a stabilizer and macromolecular chain transfer agent in second step; (ii) polymerization with second monomer to become block copolymer which will aggregate to form nanoparticles in situ when the chain length exceeds to a certain critical value.15 The macromolecular RAFT agents were originally prepared in organic solvent which need to be removed. Charleux et al.16 simplified it by conducting a two-step, one-pot aqueous procedure. The hydrophilic macroRAFT is prepared in water approximately reaching to full monomer conversion and is directly followed by addition of the hydrophobic monomer in the same reactor. This simple, robust and time-saving strategy has been successfully applied to the polymerization of methacrylic acid (MAA),17 acrylic acid,18 and a mixture of MAA and poly(ethylene oxide)methyl ether methacrylate (PEOMA).19
PISA conducted in aqueous media is normally divided into two types, namely aqueous dispersion polymerization and aqueous emulsion polymerization. The main difference between them is that in aqueous dispersion PISA, a water-miscible monomer that forms a water-insoluble polymer when polymerized or a mutually soluble solvent, such as alcohol is adopted. Some literatures have reported polymerization system at high solids up to 50% obtained by aqueous dispersion PISA while few reports about that by aqueous emulsion PISA. However, only few vinyl monomers are amenable to aqueous dispersion polymerization, including N-isopropylacrylamide (NIPAM),20 N,N-diethylacrylamide (DEAA),21 2-methoxyethyl acrylate (MEA),22 2-hydroxypropyl methacrylate (HPMA)23 and di(ethylene glycol)methyl ether methacrylate (DEGMA).24 Therefore aqueous emulsion PISA is more extensive with respect to monomer species and it is highly appreciated to develop high solids aqueous emulsion PISA. Semi-batch or monomer starved addition manner have been normally used in conventional emulsion to achieve small particle size at a low content of surfactant. The advantage of this addition manner can keep low concentration of monomer at the system during the polymerization.
In this work, we adopted semi-batch addition manner in the PISA process to prepare soap-free emulsion with high solid content and high monomer conversion. Hydrophilic PPEGMA was firstly prepared and then acted as macroRAFT agent to mediate the polymerization of MMA whose addition rate was precisely controlled by a dosing pump. Herein, the conversion of PEGMA in the first step reached nearly 100%, so no purification was needed, which dramatically simplified the synthetic process.
2. Experimental section
2.1. Materials
Poly(ethylene glycol)monomethyl ether methacrylate (PEGMA, average molecular weight is 500 g mol−1, 99%, Sigma-Aldrich) and methyl methacrylate (MMA, 99%, Shanghai Chemical Reagents Co. Ltd (Shanghai, China)) were passed through a neutral alumina column to remove inhibitor. 4-Cyano-4(thiobenzoylthio)pentanoic acid (CTBCOOH) was purchased from Longrepharm Laboratories Ltd (China) and used as received. 2,2′-Azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (AIBI) was purchased from Energy Chemical Co. and used as received. Phosphotungstic acid hydrate was purchased from J&K Chemical Co. Pure water (H2O, Hangzhou Wahaha Group Co. Ltd) was obtained from the supermarket. Tetrahydrofuran (THF, analytical reagent), n-hexane (analytical reagent) and all other chemicals were purchased from Shanghai Chemical Reagents Co. Ltd and used as received.2.2. Synthesis of PPEGMA macroRAFT agents
The typical procedure to prepare PPEGMA with a molar ratio of [PEGMA]0/[CTBCOOH]0/[AIBI]0 = 20/1/0.2 was as follows: CTBCOOH (90.6 mg, 0.325 mmol), AIBI (21.0 mg, 0.065 mmol), PEGMA (3.0 mL, 6.48 mmol) and water (3.0 mL) were added in a flask with a magnetic stir bar. After three freeze–pump–thaw cycles, the flask was put into an oil bath with temperature held at 70 °C. After 1.5 h, the polymerization was quenched by immersing the flask in an iced water bath. The monomer conversion was determined by 1H NMR spectroscopy in D2O. A little amount of sample was freeze-dried and then dissolved in THF, and precipitated out from n-hexane. The molecular weight and dispersity values of polymers were determined by GPC in THF.2.3. Synthesis of PPEGMA-b-PMMA amphiphilic diblock copolymer nanoparticles by RAFT emulsion polymerization
The typical procedure with a molar ratio of [MMA]0/[PPEGMA]0/[AIBI]0 = 100/1/0.2 was as follows: a little amount of MMA (0.14 mL, 1.32 mmol), and AIBI (4.2 mg, 0.013 mmol) were added to a solution of previously synthesized PPEGMA macroRAFT (Mn = 6900 g mol−1, Mw/Mn = 1.29) agent and the amount of water was adjusted according to different solid contents. The reaction mixture was placed in a rubber-sealed flask with a magnetic stir bar. The remainder monomer (0.56 mL, 5.28 mmol) was purged with argon for 15 min to remove the dissolved oxygen prior to addition. After purged with argon for 30 min in an ice bath, the flask was heated at 60 °C in a thermostated oil bath under stirring, and at the same time, the remainder monomer was added into the flask at a given rate monitored by a syringe pump (TJ-3A, Baoding Longerpump Co. Ltd). A balloon filled with argon was linked to the flask to provide only slight overpressure. When monomer was added completely, the polymerization was quenched by immersing the flask in an iced water bath. A small amount of sample was freeze-dried, dissolved in 2 mL of THF, and then precipitated out from n-hexane. The monomer conversion was determined by gravimetric analysis. The molecular weight and molecular weight distribution were determined by GPC with THF as a mobile phase.2.4. Characterization
Number average molecular weight (Mn,GPC) and molecular weight distribution (Mw/Mn) of resulting polymers were determined by TOSOH HLC-8320 gel permeation chromatography (GPC) comprised of a Waters 717plus autosampler, a refractive index detector (TOSOH), TSKgel guard column SuperMP-N (4.6 × 20 mm) and two TSKgel SupermultiporeHZ-N (4.6 × 150 mm). THF was used as the mobile phase, eluting at 0.35 mL min−1 at 40 °C. The narrow PMMA standards with molecular weights ranging from 5 × 102 g mol−1 to 5 × 105 g mol−1 were used as calibration. 1H NMR spectra of the synthesized polymers were recorded on a Bruker 300 MHz nuclear magnetic resonance (NMR) instrument using D2O or DMSO as the solvent. Dynamic light scattering (DLS, Nano ZS from Malvern Instruments) was used to determine the intensity-average hydrodynamic diameter (dh) and the dispersity in size of latex (indicated by the polydispersity index, PDI) at 25 °C. The latex was diluted in deionized water and data were averaged over three measurements. The nanoparticles images were taken by using transmission electron microscopy (TEM, Hitachi H-7000) at an acceleration of 120 kV. Diluted solution was dropped on a copper grid and excess solution was drawn off the edge of the grid with tissue paper. Then the grid was stained with 1% (w/v) phosphotungstic acid and dried under air before observation.3. Results and discussion
3.1. Synthesis of PPEGMA macroRAFT agent
The RAFT polymerizations of PEGMA were conducted in water using in all cases CTB–COOH as the control agent and AIBI as the initiator to synthesize macroRAFT agents (PPEGMA), which were subsequently chain extended using MMA as the monomer under PISA conditions. As shown in Table 1, PPEGMAs with different molecular weights were prepared by changing molar ratios of [PEGMA]0/[CTB–COOH]0. In order to use the polymer solution of PPEGMA directly without further purification in the subsequent PISA process, all the polymerizations were not stopped until the monomer conversion reached close to 100% (at least >96%). It is also found that the values of Mn,GPC by GPC analysis are close to the theoretical Mn,th ones and the values of molecular weight distribution (MWD) are rather low which indicate a well-controlled polymerization process. Furthermore, 1H NMR was also employed to calculate the molar mass of the polymer chains (Mn,NMR) and in all cases, the values of Mn,NMR are very close to the theoretical Mn,th.| Entry | PPEGMA code | Rb | Time (min) | Conv.c (%) | Mn,thd (g mol−1) | Mn,NMR (g mol−1) | Mn,GPC (g mol−1) | Mw/Mn |
|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions: VPEGMA = 3.0 mL, VPEGMA/Vwater = 1/1 (v/v), T = 70 °C.b R = [PEGMA]0/[CTB–COOH]0/[AIBI]0.c Calculated from the data of 1H NMR spectra.d Mn,th = ([PEGMA]0/[CTB–COOH]0) × Mw,PPEGMA × conversion% + Mw,CTB–COOH. | ||||||||
| 1 | PPEGMA-1 | 15/1/0.2 | 140 | 98.0 | 7600 | 8500 | 7800 | 1.11 |
| 2 | PPEGMA-2 | 20/1/0.2 | 90 | 99.0 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 100 |
10200 |
9200 | 1.10 |
| 3 | PPEGMA-3 | 25/1/0.2 | 140 | 99.3 | 12680 |
13700 |
11000 |
1.09 |
| 4 | PPEGMA-4 | 35/1/0.2 | 150 | 99.6 | 17700 |
17500 |
14000 |
1.07 |
3.2. Soap-free emulsion RAFT polymerization in semi-batch way
Emulsion polymerizations were carried out with MMA as a hydrophobic monomer, AIBI as a water-soluble initiator and PPEGMA as macroRAFT agent. The polymer solution of PPEGMA was used directly followed by the addition of calculated amount of water according to the desired solid content of the system. At first, one fifth of total monomer was added to the system, so that hydrophilic chain of PPEGMA could react with MMA to form block copolymer, PPEGMA-b-PMMA, which may facilitate the formation of nanoparticles in the subsequent PISA process. The other part of monomer was injected slowly to the system in a semi-batch way with dripping rate precisely monitored, which can prevent high viscosity of emulsion and ensure the polymerization goes smoothly. As a consequence, the monomer was gradually consumed to form longer block copolymer chains upon its addition and when the chain reached its critical length, it would become insoluble in the water and gradually self-assemble to amphiphilic nanoparticles just abiding the rules of polymerization induced self-assembly. Additionally, it's expected that once finishing the monomer addition, the monomer conversion can approximately get to 100%.As shown in Table 2, the effect of addition time to the monomer conversion was investigated when the solid content was 10 per cent at 60 °C and the monomer conversion appeared to increase first and then decrease along with the extension of dripping time. Furthermore, it can be found that 110 min is the most appropriate addition time at which monomer conversion can reach over 95%. This may be because of the longer dripping time making more MMA have time to polymerize while it had adverse effect on conversion when the dripping time was over 110 min, which may be caused by less amount of initiator was decomposable, so that the number of active radicals decreased in the system. The molecular weights of the copolymers determined by GPC are in agreement with their corresponding theoretical ones (Mn,th) and the ones calculated by 1H NMR (Mn,NMR). Besides, the molecular weight distributions of copolymers in all cases are relatively narrow, indicating well control of this polymerization system. GPC traces of PPEGMA-b-PMMA (Fig. 1) also showed an obvious shift compared with that of original macroRAFT agent, PPEGMA. The structure of resultant copolymer was analyzed by 1H NMR spectroscopy. From Fig. 2, it can be observed the peaks of methyl ester groups of PMMA segment (b in Fig. 2) were overlapped with methylene protons of PPEGMA block (b in Fig. 2) with chemical shift ranging from 3.37 to 4.25 ppm. The chemical shifts at 3.20–3.28 ppm (c in Fig. 2) corresponded to the methyl ester groups in PPEGMA. The chemical shifts at 0.40–2.11 ppm (a in Fig. 2) were attributed to methylene protons and methyl protons of PPEGMA-b-PMMA. The chemical shifts at 7.35 to 8.05 ppm (d in Fig. 2) belonged to the phenyl protons of the end group (–SC(S)Ph).
| Entry | Dripping time (min) | Conv. (%) | Mn,thb (g mol−1) | Mn,NMR (g mol−1) | Mn,GPC (g mol−1) | Mw/Mn | dhc (nm) | PDIc |
|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions: [MMA]0/[PPEGMA-2]0/[AIBI]0 = 100/1/0.2, VMMA = 0.7 mL, solid content = 10%, T = 60 °C.b Mn,th = ([MMA]0/[PPEGMA-2]0) × Mw,MMA × conversion% + Mn,NMR(PPEGMA-2).c dh and PDI were obtained by dynamic light scattering (DLS). | ||||||||
| 1 | 30 | 19.0 | 12100 |
11200 |
10000 |
1.16 | 173 | 0.10 |
| 2 | 90 | 82.2 | 18400 |
16400 |
16300 |
1.26 | 23 | 0.19 |
| 3 | 100 | 92.9 | 19400 |
18700 |
15600 |
1.33 | 34 | 0.13 |
| 4 | 110 | 95.3 | 19700 |
16500 |
16200 |
1.30 | 36 | 0.17 |
| 5 | 120 | 93.0 | 19500 |
16300 |
17400 |
1.29 | 31 | 0.15 |
| 6 | 140 | 79.4 | 18100 |
18200 |
12800 |
1.25 | 29 | 0.20 |
| 7 | 150 | 73.4 | 17500 |
15600 |
16000 |
1.16 | 19 | 0.09 |
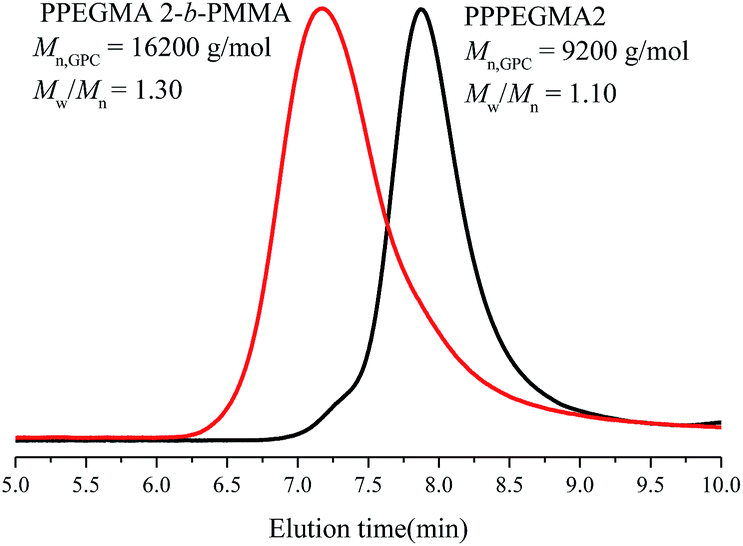 | ||
| Fig. 1 GPC curves of macroRAFT agent, PPEGMA-2 and block copolymer, PPEGMA-2-b-PMMA. | ||
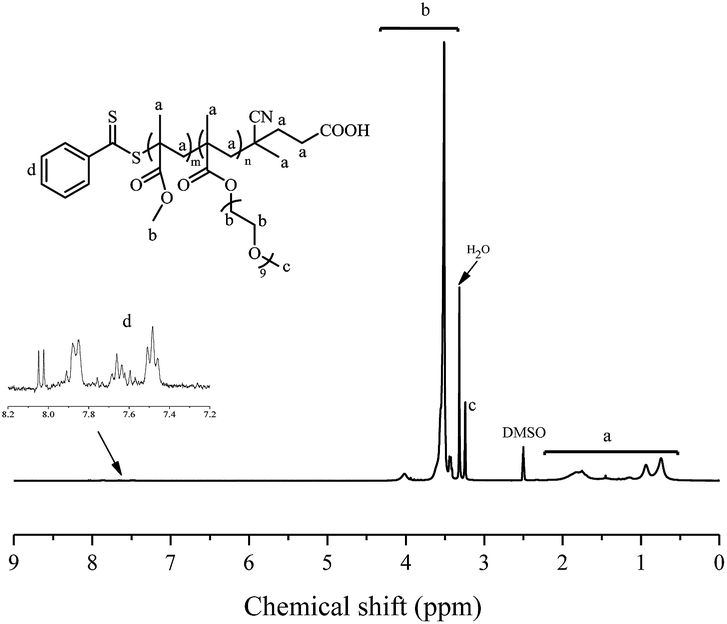 | ||
| Fig. 2 1H NMR spectrum (300 MHz, DMSO-d6) of PPEGMA-b-PMMA prepared by emulsion RAFT polymerization in semi-batch way. | ||
In all cases, stable latexes were formed. The nanoparticles sizes determined by DLS in Table 2 are all around 30 nm except latex labelled in entry 1. Only latex at dripping time of 30 min was turbid while the others were transparent. Since the monomer conversion is rather low, it can be inferred that big droplets derived from many unreacted monomers may attribute for the problem. Moreover, the particle size distribution is relatively narrow which indicate uniformity of obtained latex. Micrograph of self-assembled block copolymers at the conversion of 95.3% is shown in Fig. 3(A) and shows the nanoparticles size is around 20 nm, which is slightly broad than that obtained by DLS because of the swelling of the hydrophilic PPEGMA segment in water.
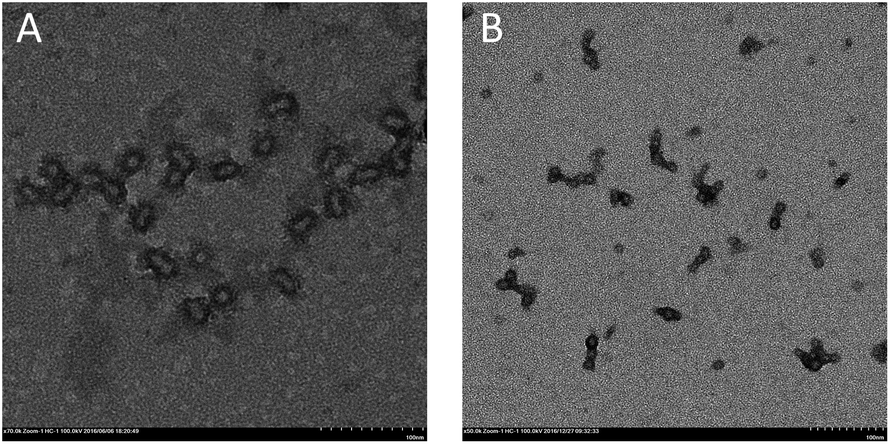 | ||
| Fig. 3 TEM figure image of latex corresponding to entry 4 in Table 2 (A) and entry 6 in Table 3 (B). | ||
In order to exert the advantage of semi-batch addition manner, emulsions with high solid content from 20 to 60 per cent were also prepared. As can be seen from Table 3, high monomer conversion can still be obtained at dripping time of 110 min when solid content varied from 10% to 30%. However, monomer conversions were far from satisfaction at the solid content of 40% and 60%. This is because the higher the solid content, the higher the viscosity of the polymerization system, so shorter reaction time is needed to obtain high monomer conversion. When reducing the addition time to 60 min at 50% solid content, monomer conversion (entry 5 in Table 3) can be reached to 93%, which confirmed the above speculation. Besides, the values of Mn,GPC corresponded to theoretical ones (Mn,th) and the ones calculated by 1H NMR (Mn,NMR). The values of Mw/Mn were also very narrow in each case. The nanoparticles sizes were all around 30 nm except that at 60% solid content. Additionally, stable latexes with transparent appearance were obtained except that macroscopic turbidity was observed for entry 6 in Table 3. The TEM image of latex at 60% solid content is shown in Fig. 3(B) and aggregation between nanoparticles is obvious. In fact, the size of every nanoparticle no matter in free or aggregation situation is about 20 nm, which indicates that there is no big nanoparticle formation, so nanoparticles can't be distributed uniformly at high solid content can explain this phenomenon.
| Entry | Solid content (%) | Conv. (%) | Mn,thb (g mol−1) | Mn,NMR (g mol−1) | Mn,GPC (g mol−1) | Mw/Mn | dhc (nm) | PDIc |
|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions: [MMA]0/[PPEGMA-2]0/[AIBI]0 = 100/1/0.2, dripping time = 110 min, VMMA = 0.7 mL, T = 60 °C.b Mn,th = ([MMA]0/[PPEGMA-2]0) × Mw,MMA × conversion% + Mn,NMR(PPEGMA-2).c dh and PDI are were obtained by dynamic light scattering (DLS).d Dripping time = 60 min. | ||||||||
| 1 | 10 | 95.3 | 20900 |
16500 |
16200 |
1.30 | 36 | 0.17 |
| 2 | 20 | 93.4 | 20700 |
18100 |
15700 |
1.17 | 29 | 0.17 |
| 3 | 30 | 90.2 | 20400 |
17800 |
15700 |
1.14 | 27 | 0.19 |
| 4 | 40 | 65.6 | 17900 |
16000 |
14000 |
1.16 | 30 | 0.23 |
| 5d | 50 | 93.0 | 19500 |
20700 |
16200 |
1.22 | 36 | 0.24 |
| 6 | 60 | 74.3 | 18800 |
13300 |
13100 |
1.14 | 79 | 0.34 |
In order to investigate the extensity of semi-batch addition manner, varied molecular weights of PPEGMAs were used as macroRAFT agents. From Table 4, it can be observed that the molar mass of the copolymers analyzed by GPC are close to the theoretical ones the ones calculated by 1H NMR (Mn,NMR). Besides, the dispersity values are low, which indicating well controlled effect of the macroRAFT agents. Moreover, stable latexes with transparent appearance were obtained in all cases and the average diameter of nanoparticles analyzed by DLS are all around 30 nm and the particles dispersities keep at a low level.
| Entry | PPEGMA code | Conv. (%) | Mn,thb (g mol−1) | Mn,NMR (g mol−1) | Mn,GPC (g mol−1) | Mw/Mn | dhc (nm) | PDIc |
|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions: [MMA]0/[PPEGMA-2]0/[AIBI]0 = 100/1/0.2, dripping time = 110 min, VMMA = 0.7 mL, solid content = 10%, T = 60 °C.b Mn,th = ([MMA]0/[PPEGMA]0) × Mw,MMA × conversion% + Mn,NMR(PPEGMA).c dh and PDI were obtained by dynamic light scattering (DLS). | ||||||||
| 1 | PPEGMA-1 | 58.8 | 14300 |
11900 |
12500 |
1.12 | 19 | 0.12 |
| 2 | PPEGMA-2 | 95.3 | 19700 |
16500 |
16200 |
1.30 | 36 | 0.17 |
| 3 | PPEGMA-3 | 74.8 | 21100 |
18600 |
15300 |
1.14 | 20 | 0.09 |
| 4 | PPEGMA-4 | 85.5 | 26000 |
28600 |
18600 |
1.16 | 27 | 0.16 |
4. Conclusions
Soap-free emulsion with solid content up to 60% was prepared in differential semi-batch monomer addition way by polymerization induced self-assembly (PISA) process from PPEGMA-b-PMMA. The obtained latex was very stable with particle size almost invariably at 20 nm at 10–50% solid content and narrow particle size distribution. This approach may provide an alternative way for preparing high solid content soap-free emulsion by a novel strategy of differential semi-batch monomer addition manner. At the same time, high monomer conversion (>95%) can be achieved by just adjusting the addition time, which facilitates to decrease the residue of monomer after a PISA process, and therefore facilitates its practical application.Acknowledgements
The financial support from the Project of Science and Technology Development Planning of Jiangsu Province (No. BK20141192), the National Natural Science Foundation of China (No. 21674071) and the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) is gratefully acknowledged.Notes and references
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- (a) Z. Li, W. Chen, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2015, 6, 5030–5035 RSC; (b) Y. Ma and T. P. Lodge, Macromolecules, 2016, 49, 3639–3646 CrossRef CAS; (c) C.-Y. Kuo, T.-M. Don, S.-C. Hsu, C.-F. Lee, W.-Y. Chiu and C.-Y. Huang, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1109–1118 CrossRef CAS; (d) Z. Cheng, X. Zhu, E. T. Kang and K. G. Neoh, Langmuir, 2005, 21, 7180–7185 CrossRef CAS PubMed; (e) Z. Cheng, X. Zhu, G. D. Fu, E. T. Kang and K. G. Neoh, Macromolecules, 2005, 38, 7187–7192 CrossRef CAS; (f) X. Jiang, X. Du, L. Zhang and Z. Cheng, Macromol. Chem. Phys., 2013, 214, 654–663 CrossRef CAS; (g) S. L. Chen, P. F. Shi and W. Q. Zhang, Chinese J. Polym. Sci., 2017, 35, 455–479 CrossRef CAS.
- (a) M. Barsbay, O. Güven, M. H. Stenzel, T. P. Davis, C. Barner-Kowollik and L. Barner, Macromolecules, 2007, 40, 7140–7147 CrossRef CAS; (b) Y. Hou, J. Jiang, K. Li, Y. Zhang and J. Liu, J. Phys. Chem. B, 2014, 118, 1962–1967 CrossRef CAS PubMed; (c) N. Zammarelli, M. Luksin, H. Raschke, R. Hergenröder and R. Weberskirch, Langmuir, 2013, 29, 12834–12843 CrossRef CAS PubMed.
- (a) X. T. Cao, C. L. Zhang, S. B. Wu and Z. S. An, Polym. Chem., 2014, 5, 4277–4284 RSC; (b) X. F. Shi, W. Zhou, Q. Qiu and Z. S. An, Chem. Commun., 2012, 48, 7389–7391 RSC.
- (a) D. Tumnantong, G. L. Rempel and P. Prasassarakich, Eur. Polym. J., 2016, 80, 145–157 CrossRef CAS; (b) S. Xu, J. Huang, S. Xu and Y. Luo, Polymer, 2013, 54, 1779–1785 CrossRef CAS; (c) W. Zhang, F. D'agosto, P.-Y. Dugas, J. Rieger and B. Charleux, Polymer, 2013, 54, 2011–2019 CrossRef CAS; (d) Z. Li, W. Chen, Z. Zhang, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2015, 6, 1937–1943 RSC.
- (a) S. Dong, S. Cheng and P. B. Zetterlund, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2104–2109 CrossRef CAS; (b) A. J. P. Van Zyl, R. F. P. Bosch, J. B. Mcleary, R. D. Sanderson and B. Klumperman, Polymer, 2005, 46, 3607–3615 CrossRef CAS; (c) L. Yang, Y. Luo and B. Li, Polymer, 2006, 47, 751–762 CrossRef CAS.
- (a) K. D. Hermanson, S. Liu and E. W. Kaler, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6055–6070 CrossRef CAS; (b) A. Sogabe and C. L. Mccormick, Macromolecules, 2009, 42, 5043–5052 CrossRef CAS; (c) B. Kumru and N. Bicak, RSC Adv., 2015, 5, 30936–30942 RSC.
- M. J. Monteiro and J. De Barbeyrac, Macromolecules, 2001, 34, 4416–4423 CrossRef CAS.
- (a) L. Houillot, C. Bui, M. Save, B. Charleux, C. Farcet, C. Moire, J.-A. Raust and I. Rodriguez, Macromolecules, 2007, 40, 6500–6509 CrossRef CAS; (b) F. Stoffelbach, B. Belardi, J. M. R. C. A. Santos, L. Tessier, K. Matyjaszewski and B. Charleux, Macromolecules, 2007, 40, 8813–8816 CrossRef CAS.
- C. J. Ferguson, R. J. Hughes, D. Nguyen, B. T. T. Pham, R. G. Gilbert, A. K. Serelis, C. H. Such and B. S. Hawkett, Macromolecules, 2005, 38, 2191–2204 CrossRef CAS.
- (a) J. Wu, H. Jiang, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2016, 7, 2486–2491 RSC; (b) M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2013, 29, 7416–7424 CrossRef CAS PubMed.
- (a) A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS; (b) L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- (a) Q. Zhang and S. Zhu, ACS Macro Lett., 2015, 4, 755–758 CrossRef CAS; (b) Y. T. Li and S. P. Armes, Angew. Chem., Int. Ed., 2010, 49, 4042–4046 CrossRef CAS PubMed.
- (a) B. Charleux, G. Delaittre, J. Rieger and F. D'agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS; (b) M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- (a) J. T. Sun, C. Y. Hong and C. Y. Pan, Soft Matter, 2012, 8, 7753–7767 RSC; (b) J. T. Sun, C. Y. Hong and C. Y. Pan, Polym. Chem., 2013, 4, 873–881 RSC.
- I. Chaduc, W. Zhang, J. Rieger, M. Lansalot, F. D'agosto and B. Charleux, Macromol. Rapid Commun., 2011, 32, 1270–1276 CrossRef CAS PubMed.
- I. Chaduc, M. Girod, R. Antoine, B. Charleux, F. D'agosto and M. Lansalot, Macromolecules, 2012, 45, 5881–5893 CrossRef CAS.
- I. Chaduc, A. Crepet, O. Boyron, B. Charleux, F. D'agosto and M. Lansalot, Macromolecules, 2013, 46, 6013–6023 CrossRef CAS.
- (a) W. Zhang, F. D'agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2012, 45, 4075–4084 CrossRef CAS; (b) W. Zhang, F. D'agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 7584–7593 CrossRef CAS.
- Z. An, Q. Shi, W. Tang, C.-K. Tsung, C. J. Hawker and G. D. Stucky, J. Am. Chem. Soc., 2007, 129, 14493–14499 CrossRef CAS PubMed.
- (a) C. Grazon, J. Rieger, N. Sanson and B. Charleux, Soft Matter, 2011, 7, 3482–3490 RSC; (b) G. Delaittre, M. Save and B. Charleux, Macromol. Rapid Commun., 2007, 28, 1528–1533 CrossRef CAS.
- (a) G. Liu, Q. Qiu and Z. An, Polym. Chem., 2012, 3, 504–513 RSC; (b) G. Liu, Q. Qiu, W. Shen and Z. An, Macromolecules, 2011, 44, 5237–5245 CrossRef CAS.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- W. Shen, Y. Chang, G. Liu, H. Wang, A. Cao and Z. An, Macromolecules, 2011, 44, 2524–2530 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |