
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ring opening of decalin over bifunctional Ni–W carbide/Al2O3–USY catalysts and monofunctional acid Ni–W oxide/Al2O3–USY
Lijing Yuan ab,
Shaoqing Guoc,
Zhenrong Li*a,
Haitao Cuia,
Hongyu Donga,
Liangfu Zhao*a and
Junwei Wanga
ab,
Shaoqing Guoc,
Zhenrong Li*a,
Haitao Cuia,
Hongyu Donga,
Liangfu Zhao*a and
Junwei Wanga
aInstitute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, PR China. E-mail: lfzhao@sxicc.ac.cn
bGraduate University of the Chinese Academy of Sciences, Beijing 100039, PR China
cTaiyuan University of Science and Technology, Taiyuan 030024, PR China
First published on 31st January 2017
Abstract
The ring opening reaction of decalin was studied over two types of catalyst, including bifunctional catalysts (Ni–W carbide/Al2O3–USY) and monofunctional acid catalysts (Ni–W oxide/Al2O3–USY) with different amounts of metal loading. The catalysts were characterized by elemental analysis, X-ray diffraction, BET surface area measurement, element analysis, pulsed CO chemisorption, temperature-programmed oxidation, pyridine IR, temperature-programmed reduction and transmission electron microscopy. The two types of catalyst showed different performance on the ring opening reaction of decalin. The bifunctional catalysts showed high conservation of decalin, high selectivity of ring opening products and low selectivity of cracking products, while the monofunctional acid catalysts showed the opposite. Two different reaction routes during the ring opening reactions of decalin were put forward to further illustrate the bifunctional mechanism and monofunctional acid mechanism.
1. Introduction
Due to the higher requirement for oil and the decrease of petroleum reserves, oil of poor quality should be used more efficiently, such as light cycle oil (LCO) from the fluid catalytic cracking process. LCO is significantly different from diesel oil because of its lower cetane number, higher sulfur content and higher polyaromatic content, which cannot meet the requirement of current international diesel specifications.Hydrocracking and deep hydrotreatment are practical ways to improve fuel quality.1–4 However, hydrocracking can increase the cetane number but it is always accompanied by the carbon number reduction of fuel.5 Meanwhile, oil from the deep hydrotreatment cannot meet the diesel demand because there are still many naphthenes in the diesel fuel.6 Therefore, the ring opening of naphthenes in fuel is a promising method to further increase the cetane number of fuel.3,7
Some studies on ring opening of naphthenes have been published in the past years. The monofunctional acid catalyst, monofunctional metallic catalysts and bifunctional catalysts were used in this reaction. The studies about monofunctional acid catalysts3,6,8 showed that the ring contraction was the first step on ring opening of decalin, and then followed by ring opening and cracking processes. And the monofunctional metallic catalysts6,7,9,10 showed different catalytic mechanism with monofunctional acid catalysts and three different ring opening mechanisms were proposed,11,12 including the multiplet mechanism, the dicarbene mechanism and the metallocyclobutane mechanism.
Recently, the bifunctional catalysts1,7,8,10,12–30 has been used in the ring opening of naphthenes which showed good performance because of its more acid sites and higher hydrogenation on metal sites. The studies about bifunctional catalysts mainly focused on some noble metal catalyst,1,8–10,12,14–16,18–21,24–26,28–31 which are easier sulfur poisoning.7,32,33 It is reported that transition metal carbide catalysts have shown excellent hydrogenation activity for hydrodenitrogenation (HDN) and hydrodesulfurization (HDS) due to its higher hydrogenation on transition metal sites.34–36 Therefore, the higher hydrogenation on transition metal sites possibly facilitate the hydrogenation activity for the ring opening of decalin, but few studies have focused on transition metal carbide catalysts for such a reaction.
In addition to the metal sites, the support is also critical for the catalysts. It is generally accepted that Al2O3 is an attractive support for industrial use because of excellent mechanical and textural property. However it does not have enough strong Brönsted acid sites.3,37 Ultrastable Y (USY) zeolites can provide suitable acidity for catalysts,5 but it is easier to deactivate and lacks the mechanical property. It is speculated that the combination of USY zeolites and Al2O3 probably is an alternative way to provide the suitable acidity and mechanical property for the bifunctional catalysts.5,38
In this paper, USY zeolites were added into Al2O3 to support the Ni–W carbide catalysts (named as Ni–W carbide/Al2O3–USY). As auxiliary, Ni was added to improve the dispersion of W species. Also, the Ni–W oxide catalysts (Ni–W oxide/Al2O3–USY) were also studied for comparison. According to the previous works,6,24 Ni–W carbide/Al2O3–USY was named as bifunctional catalysts and Ni–W oxide/Al2O3–USY was named as monofunctional acid catalysts. Decalin was chosen as the model reactant for the ring opening of naphthenes and hydrogenation.
2. Experimental
2.1 Catalyst preparation
The supports were prepared by mixing USY zeolites (Si/Al = 42) and pseudo-boehmite. It contained 30% USY zeolites and 70% pseudo-boehmite. The physically mixed compounds were extruded to form a cylinder with a diameter of 1.6 mm, which was dried at 100 °C for 12 hours, and then calcined at 550 °C for 2 hours in the muffle. The Ni/W phases (6–32 wt% WO3) were deposited by co-impregnation method with incipient wetness impregnation of nickel nitrate and ammonium metatungstate in the atomic ratio Ni/(W + Ni) of 0.44. The monofunctional acid catalysts were named as CAT-Wx (CAT-W0, CAT-W6, CAT-W12, CAT-W18, CAT-W25 and CAT-W32), while the bifunctional catalysts were named as CAT-WxC (CAT-W6C, CAT-W12C, CAT-W18C, CAT-W25C and CAT-W32C), where x corresponds with the amount of WO3 loading. The catalysts were dried overnight at room temperature, and then dried for 10 hours at 100 °C in the dryer, followed by calcining for 4 hours at 550 °C in the muffle.The bifunctional catalysts were pretreated in the stainless steel tube under a gas mixture (H2/CH4 volumetric ratio of 4) flow of 80 cm3 min−1. Firstly, the catalyst was heated from room temperature to 350 °C at a heating rate of 10 °C min−1. Next, the sample was heated from 350 °C to 500 °C at a heating rate of 1 °C min−1. Then it was heated from 500 °C to 700 °C at a heating rate of 0.5 °C min−1 and holding at 700 °C until the CO and CO2 in outlet gases from the reactor were not detected by using a gas chromatography (GC 9560). The reaction was continued until no carbon oxide formed. At last, the sample cooled down to the reaction temperature. All the carbide samples for characterization were passivated by using 2% O2 in He (v/v) for 5 h to avoid the bulk oxidation.
The monofunctional acid catalysts were pretreated in the stainless steel tube under air flow of 80 cm3 min−1. Same programmed temperature for bifunctional catalysts was used on the monofunctional acid catalysts. The air was flowed to activate the catalysts until the catalysts was cooled to the reaction temperature. All the oxide samples for characterization were activated by the same temperature program.
2.2 Catalyst characterization
The X-ray powder diffraction (XRD) analysis was recorded with a Rigaku D/max-2500 diffractometer, using the Cu Kα radiation at 10 mA and 30 kV. The angle (2θ) was varied from 5° to 80° in 0.02 steps with scanning rate of 8° min−1.The Ni and W contents in the catalysts were determined by an Inductive Couple Plasma atomic emission spectrometer (ICP-AES, Thermo ICAP 6300).
Carbon content of the bifunctional catalysts were analyzed by element analysis (Varion EL).
The specific surface area of all the catalysts was determined by N2 low-temperature physical adsorption method and the BET method.
Temperature programmed oxidation (TPO) was carried out on a MS instrument (OMNISTAR). 100 mg of sample was pretreated in Ar at 100 °C for 2 h. Then the sample was heated from 100 °C to 900 °C at a rate of 5 °C min−1. The 10% O2/Ar (v/v) was used as oxidation gas with a flow rate of 30 ml min−1.
The Brönsted and Lewis acidities of the catalysts, as determined by pyridine adsorption infrared spectroscopy (pyridine IR), were analyzed by Thermo scientific (Nicolet 380). All samples were pressed into self-supporting wafers with a diameter of 13 mm. The samples were purified in the sample cell for 1 h at 450 °C prior to pyridine adsorption experiment. Next, a pyridine vapor was introduced into the sample cell for 30 min and then the weakly bonded pyridine was removed under vacuum. Different desorbed temperature (250 °C, 350 °C and 450 °C) was set to identify the Brönsted acid sites and Lewis acid sites.
Hydrogen temperature programmed reduction (H2-TPR) was analyzed by Quantachrome Instruments (Chem BET Pulsar). The U-shaped vessel contained 0.1 g sample (20–40 mush) and heated from 100 °C to 900 °C at a 10 °C min−1 heating rate. The 10% H2/He (v/v) was used as reducing gas with a flow rate of 30 ml min−1.
Carbon monoxide chemisorption (CO uptakes) was used to titrate the number of accessible surface metal atoms on alumina-supported tungsten carbides by Quantachrome Instruments (Chem BET Pulsar).39 Titration was carried out by U-shaped vessel contained 0.2 g sample (20–40 mush). The carbide catalyst was reduced in flowing gas mixture (H2/He volumetric ratio of 4, 30 cm3 min−1) at 500 °C for 2 h, purged in flowing He for 1 h, and then quenched at 30 °C.
Transmission electron microscopy (TEM) was performed with a JEOL (JEM 2100) high-resolution transmission electron microscopy. The samples were dispersed in ethanol and suspend on a 3.5 mm diameter Cu grid.
2.3 Catalytic evaluation
The experiments were performed in a continuous flow fixed-bed microreactor. The reaction conditions were carried out at hydrogen pressure of 5 MPa, temperature of 295–345 °C and catalyst loading of 3 g with catalyst size of 10–20 mesh. The feed flow is 4 ml h−1 and the H2/oil = 1000 (v/v). The decalin (trans/cis ratio of 1.5) was pumped into the reactor by a constant flow pump (CM 6000).The liquid reaction products were collected four times in eight hours. The samples were analyzed with a gas chromatograph (Agilent 7890 A) equipped with a capillary column (HP-5MS) and a flame ionization detector (FID). Helium was used as a carrier gas. The following temperature program was applied: initial temperature 50 °C holding for 5 min, heating rate of 3.3 °C min−1 to 130 °C, then heating rate of 5 °C min−1 to 200 °C, and dwelling at 200 °C for 5 min. The same condition for GC was used to quantify the products. Due to the complicated products were obtain for such a reaction, the isomerization products and ring opening products were identified by GC-MS technique (Agilent 7890-5975) and quantified by GC (Agilent 7890 A).
The products from decalin ring opening reaction were classified in three groups as follows: molecular weight lower than decalin were named as the cracking products (CP), all the alkyl-substituted monocyclic C10, linear and branched C10 were referred to the ring opening products (ROP); and any other bicyclical C10 products (except for decalin) were denoted as isomer (ISO).
The decalin conversion (XD), the yield of ROP (YROP) and the selectivity of ROP (SROP) on the catalysts were calculated using the following equations: 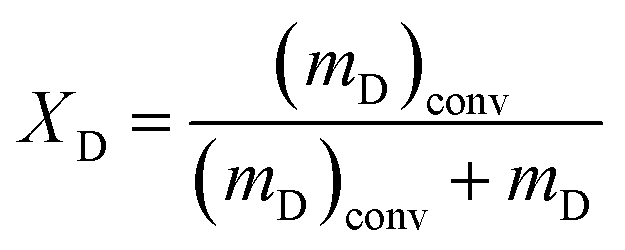 ,
, 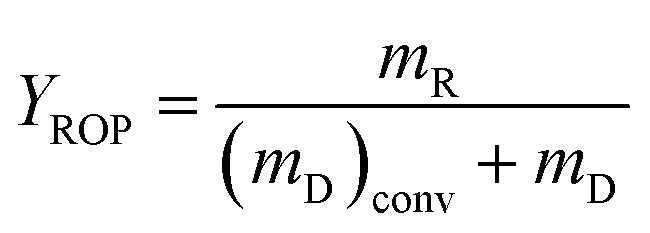 ,
, 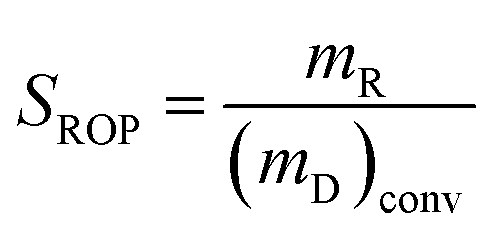 , where the mD is the mass of the decalin, (mD)conv is equal to the mass of decalin which has been converted into the products. mR is the mass of the ROP. Same method was used to calculate the yield and selectivity of ISO and CP.
, where the mD is the mass of the decalin, (mD)conv is equal to the mass of decalin which has been converted into the products. mR is the mass of the ROP. Same method was used to calculate the yield and selectivity of ISO and CP.
3. Results and discussion
3.1 Catalyst characterization
The XRD pattern of two types of catalysts shown in Fig. 1 presented the main reflections lines of the USY zeolite (2θ = 6.31°, 10.34°, 12.08°, 15.95°, 19.01°, 20.71°, 24.05°, 27.55° and 31.99°) and γ-alumina (2θ = 45.78°, 54.87° and 66.67°). With the increase of metal loading amount, the diffraction peaks of the zeolites and γ-alumina decreased obviously. Generally, the increasing metal loaded will cover part of the support surface and lead to the decreasing of the diffraction peaks.40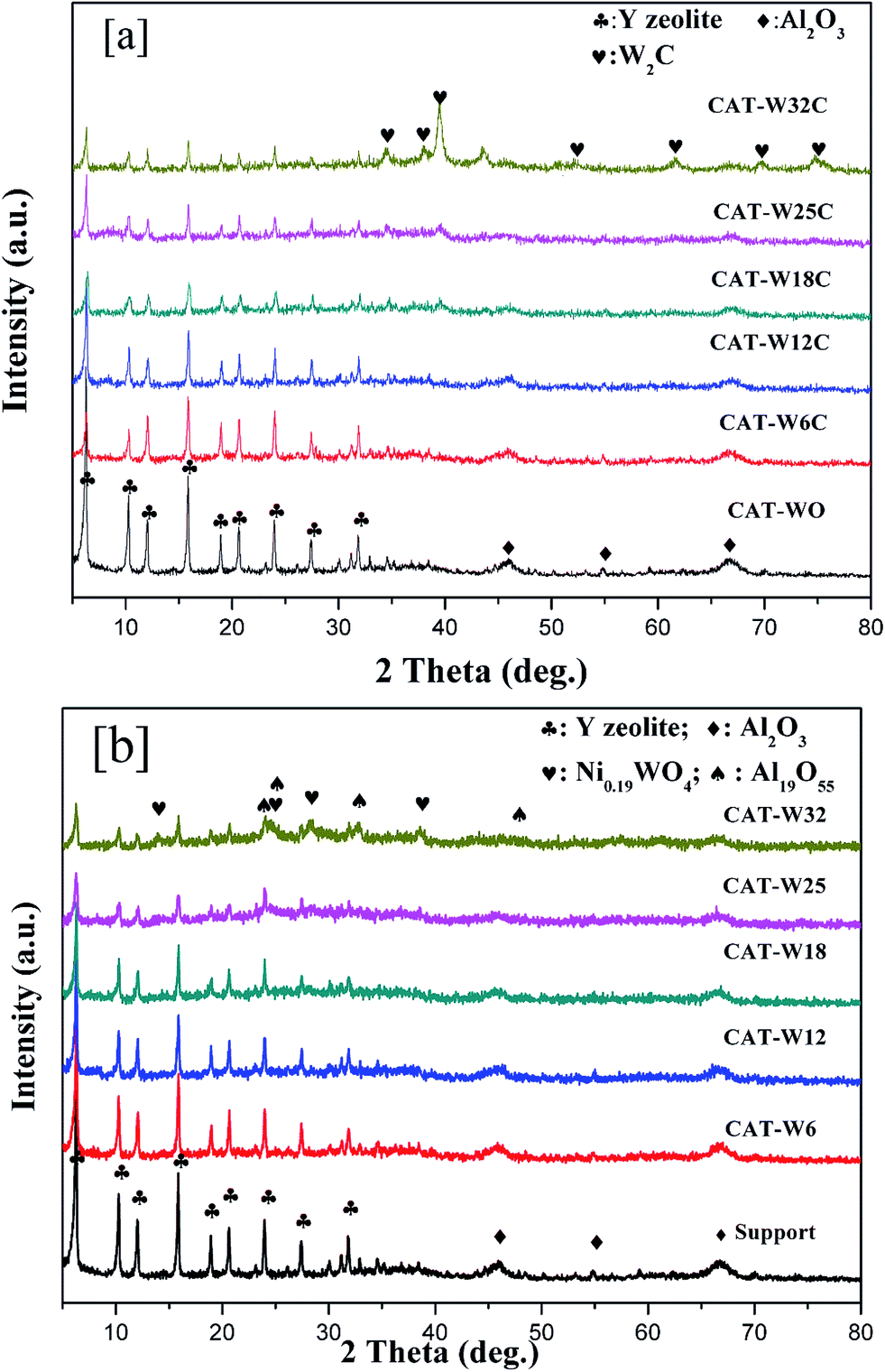 | ||
| Fig. 1 XRD patterns of the Ni–W catalysts with different content of metal loading. (a) Bifunctional catalysts and (b) monofunctional acid catalysts. | ||
In Fig. 1a, it can be seen that the bifunctional catalysts of CAT-W6C, CAT-W12C and CAT-W18C showed no tungsten carbide phases, which implied that W and Ni phases were well dispersed on the surface of the support. For CAT-W25C and CAT-W32C, some new diffraction peaks (at 34.52°, 38.03°, 39.57°, 52.3°, 61.86°, 69.77°, 74.98° and 75.98°) can be found which were possibly attributed to W2C (Pdf no. 35-776).
Fig. 1b shows that the W and Ni phases on the monofunctional acid catalysts of CAT-W6, CAT-W12 and CAT-W18 were also well dispersed on the surface of the support. The new diffraction peaks can be found for CAT-W25 and CAT-W32. The peaks at 13.98°, 28.3° and 38.5° were attributed to Ni0.19WO4 (Pdf no. 40-311) and the peaks at 24.02°, 24.3°, 32.66° and 47.64° were attributed to W19O55 (Pdf no. 45-167).
BET surface area (SBET, m2 g−1), Ni/W loading (examined by ICP), W/C ratio (W/C molar ratio) and CO uptakes (μmol g−1) of the bifunctional and the monofunctional acid catalysts were listed in Tables 1 and 2. The SBET of the two types of catalysts both decreased with increasing metal loading amount, which probably ascribe to the coverage of metal phases. The SBET has no significant difference between the two types catalysts with the same metal loading amount. This indicates that carbide pretreatment did not affect the surface area at the experimental condition used in this study.
| Sample | CAT-W6C | CAT-W12C | CAT-W18C | CAT-W25C | CAT-W32C |
| Ni (wt%) | 1.2 | 2.4 | 3.6 | 4.8 | 6.0 |
| W (wt%) | 5.2 | 10.1 | 14.7 | 19.7 | 25.2 |
| W/C ratio | 2.1 | 1.8 | 2.3 | 1.9 | 2.4 |
| SBET (m2 g−1) | 359 | 325 | 292 | 258 | 224 |
| CO uptakes (μmol g−1) | 15.1 | 35.2 | 44.0 | 43.5 | 40.5 |
| Sample | CAT-W0 | CAT-W6 | CAT-W12 | CAT-W18 | CAT-W25 | CAT-W32 |
| Ni (wt%) | 0 | 1.2 | 2.4 | 3.6 | 4.8 | 6.0 |
| W (wt%) | 0 | 5.2 | 10.2 | 14.8 | 19.9 | 25.4 |
| WO3 (wt%) | 0 | 6.5 | 12.8 | 18.6 | 25 | 32 |
| SBET (m2 g−1) | 393 | 364 | 329 | 295 | 263 | 227 |
As shown in Table 1, the W/C ratio of the bifunctional catalysts was approximately equal to 2 and close to the stoichiometric value of W2C, which is in accordance with the XRD results on the CAT-W25C and CAT-W32C. Similar result was observed on the previous studies on the Mo2C catalysts.7,41 According to the literatures, the W2C might be the main active phase in the bifunctional catalysts.42–44
The CO uptakes was used to characterize the surface metal atoms and to provide an estimation of the surface metal atoms on the carbide catalysts which is also shown in Table 1. The CO uptakes sharply increased to the maximum of 44 μmol g−1 and decreased gradually with increase of the metal loading amount. It probably indicated a well dispersion of metal on the catalyst of CAT-W6C, CAT-W12C and CAT-W18C. Also, it implied crystallite agglomeration on the catalysts of CAT-W25C and CAT-W32C according to the lower CO uptakes,45 which was consistent with the XRD results.
There are two forms of carbon in the bifunctional catalysts. One is lattice carbon, which exists in the form of W2C. The other one is carbonaceous residue, which deposits on the surface of the bifunctional catalysts. TPO was carried out to distinguish the difference of the two carbon forms. As shown in Fig. 2, the bifunctional catalysts showed two broad peaks below 650 °C, which were attributed to the oxidation of lattice carbon in the Ni carbide and W carbide.41,46 All the bifunctional catalysts showed a broad stage of oxidation which might be attributed to the limitation of the pore structure. A slow peak was observed above 700 °C on the CAT-W25C and CAT-W32C, which should be ascribed to the oxidation of carbon residues.41,47 This indicates that two forms of carbon exist in the bifunctional catalysts. One was incorporated into the lattice of bifunctional catalysts as the main active phase, while the other one was deposited as carbon residues on the surface of the bifunctional catalysts.
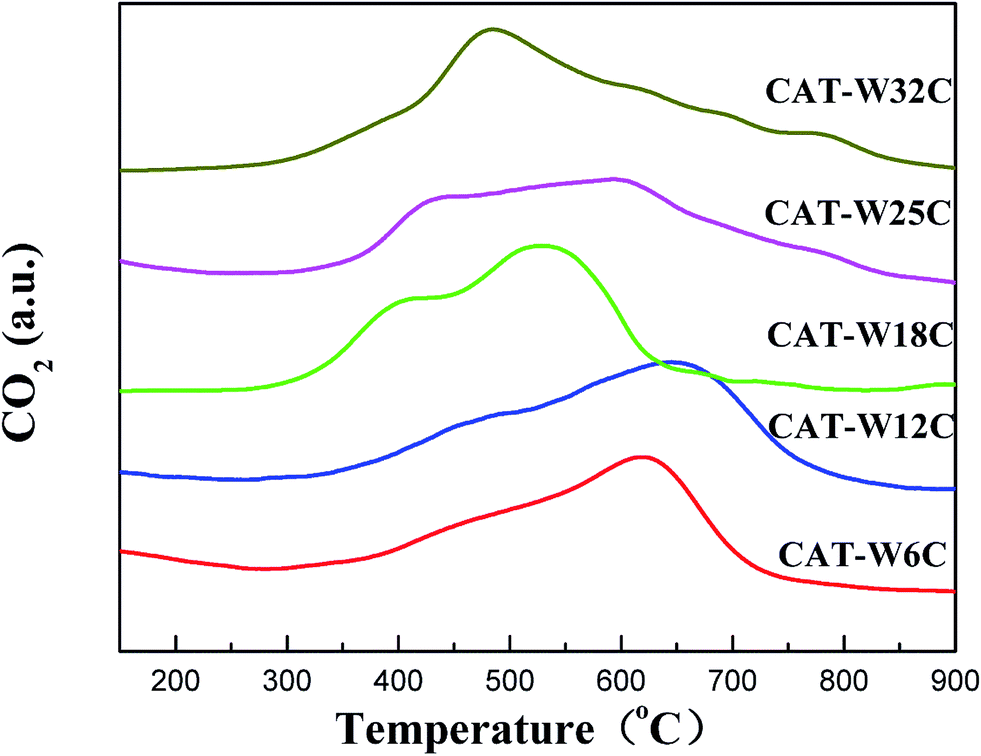 | ||
| Fig. 2 TPO profiles of the bifunctional catalysts. | ||
The Brönsted and Lewis acidities of the catalysts were measured by Py-IR and the results are listed in Tables 3 and 4. For the bifunctional catalysts, with the increasing of the metal loading, the amount of Lewis acid sites and weak Brönsted acid sites were decreased. It may be caused by the fact that the surface of the supports was covered by the W deposition.6,48,49 The strong Brönsted acid sites did not change obviously with the change of the metal loading amount. But it was lower than that for the monofunctional acid catalysts with the same metal loading amount, which was possibly due to that the W oxide phase was reduced by the carbide pretreatment, resulting in the decrease of the W–O–Al phases and the formation of W2C. Similar results were observed by J.-M. Manoli et al. on Mo2C/Al2O3 catalysts and Mingqiang Shao on NiWS/MCM48 catalyst.34,50
| Catalyst | Brönsted acid sites, μmol mg−1 | Lewis acid sites, μmol mg−1 | ||||
|---|---|---|---|---|---|---|
| 250 °C (weak) | 350 °C (moderate) | 450 °C (strong) | 250 °C (weak) | 350 °C (moderate) | 450 °C (strong) | |
| CAT-W6C | 33.7 | 1.7 | 1.3 | 145.4 | 65.3 | 25.3 |
| CAT-W12C | 26.5 | 4.5 | 1.3 | 167.9 | 79.9 | 30.2 |
| CAT-W18C | 10.7 | 2.0 | 1.6 | 137.0 | 70.5 | 30.5 |
| CAT-W25C | 14.9 | 1.4 | 1.3 | 35.9 | 12.6 | 1.0 |
| CAT-W32C | 10.6 | 1.4 | 1.0 | 31.5 | 13.8 | 0.9 |
| Catalyst | Brönsted acid sites, μmol mg−1 | Lewis acid sites, μmol mg−1 | ||||
|---|---|---|---|---|---|---|
| 250 °C (weak) | 350 °C (moderate) | 450 °C (strong) | 250 °C (weak) | 350 °C (moderate) | 450 °C (strong) | |
| CAT-WO | 54.1 | 23.8 | 1.2 | 194.4 | 121.0 | 88.4 |
| CAT-W6 | 35.9 | 18.2 | 1.3 | 179.9 | 117.1 | 40.4 |
| CAT-W12 | 43.9 | 19.63 | 2.5 | 173.9 | 97.0 | 54.5 |
| CAT-W18 | 39.6 | 12.2 | 4.2 | 122.5 | 63.0 | 29.4 |
| CAT-W25 | 27.4 | 15.1 | 4.1 | 98.2 | 44.9 | 13.4 |
| CAT-W32 | 24.8 | 10.2 | 3.8 | 82.1 | 36.9 | 12.1 |
For the monofunctional acid catalysts, with the increasing of the metal loading, the amount of Lewis acid sites and weak Brönsted acid sites were decreased. At the same time, the strong Brönsted acid sites increased obviously when the WO3 loading is less than 18%. It should be attributed to the W–O–Al phases formed by the metal oxide and proton units privileged configuration.49,51 However, small part of the strong Brönsted acid sites was possibly covered by bulk metal phases when WO3 loading is higher than 18%, leading to a slightly decrease of the strong Brönsted acid sites for CAT-W25 and CAT-W32.
Fig. 3 shows the result of H2-TPR of the catalysts ((a): bifunctional catalysts, (b): monofunctional acid catalyst). It can be seen that all the bifunctional catalysts showed a broad peak below 600 °C, which should be attributed to the reduction of surface oxygen during the passivation process.7,41 The peaks area at 600–900 °C on the bifunctional catalysts was lower obviously than that on the monofunctional acid catalysts. It might be attributed to the reduction of the NiO–WO3 phase on the bifunctional catalysts during the carbide pretreatment, similar results has been observed on the previous studies when compared the Ni–Mo oxide and Ni–Mo carbide catalysts.41
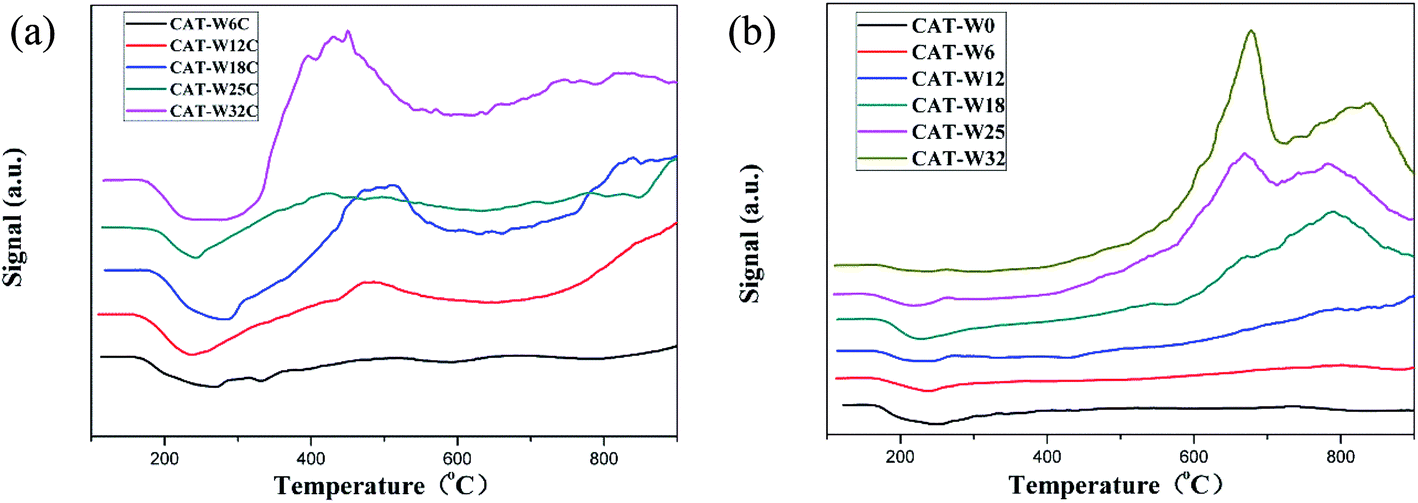 | ||
| Fig. 3 H2-TPR of the catalysts with different metal loading amount: (a) bifunctional catalysts and (b) monofunctional acid catalysts. | ||
For the monofunctional acid catalysts, the peak of CAT-W12, CAT-W18, CAT-W25 and CAT-W32 at 540 °C should be attributed to the NiO phase.40,52,53 The peak at 780 °C for all the monofunctional acid catalysts (except for the CAT-W0) should be attributed to the NiO–WO3 phase.38,53 Meanwhile, no peak was observed below 400 °C, which implied that NiO cannot be reduced at the reaction temperature (295 °C, 320 °C and 345 °C) and there exists a strong metal–support interaction. Similar results were observed by previous studies.38 At the same time, a new peak was observed at 670 °C for CAT-W25 and CAT-W32, which might be attributed to the reduction of NiWO4 or nickel polytungstate like phase.38,53
The transmission electron micrographs of the samples (CAT-W6C, CAT-W18C, CAT-W32C, CAT-W6, CAT-W18 and CAT-W32) are shown in Fig. 4a–f. With the metal loading increasing, the coverage of the metal increased obviously. Meanwhile, the average particle size increased from 3.9 nm (CAT-W6C) to 7.5 nm (CAT-W32C) for the bifunctional catalysts and from 3.8 nm (CAT-W6) to 7.9 nm (CAT-W32) for the monofunctional acid catalysts. At the same time, similar average particle size can be found for the two types of catalysts with the same metal loading amount.
 | ||
| Fig. 4 TEM micrographs of the samples: (a) CAT-W6C, (b) CAT-W18C, (c) CAT-W32C, (d) CAT-W6, (e) CAT-W18 and (f) CAT-W32. Arrows indicate the metal particles while ovals indicate the supports. | ||
3.2 Conversion of decalin
The conversion of decalin with the catalysts at different temperature (295 °C, 320 °C, 345 °C) is presented in Fig. 5. For two types of catalysts, the conversion of decalin increased with the increasing of the reaction temperature and was almost 100% at 345 °C for some catalysts. It is interesting to find that the decalin conversion increased obviously for low amount of metal loading (WO3 loading < 18%) while a slow decrease was observed for high amount of metal loading (WO3 loading > 18%) at the same temperature.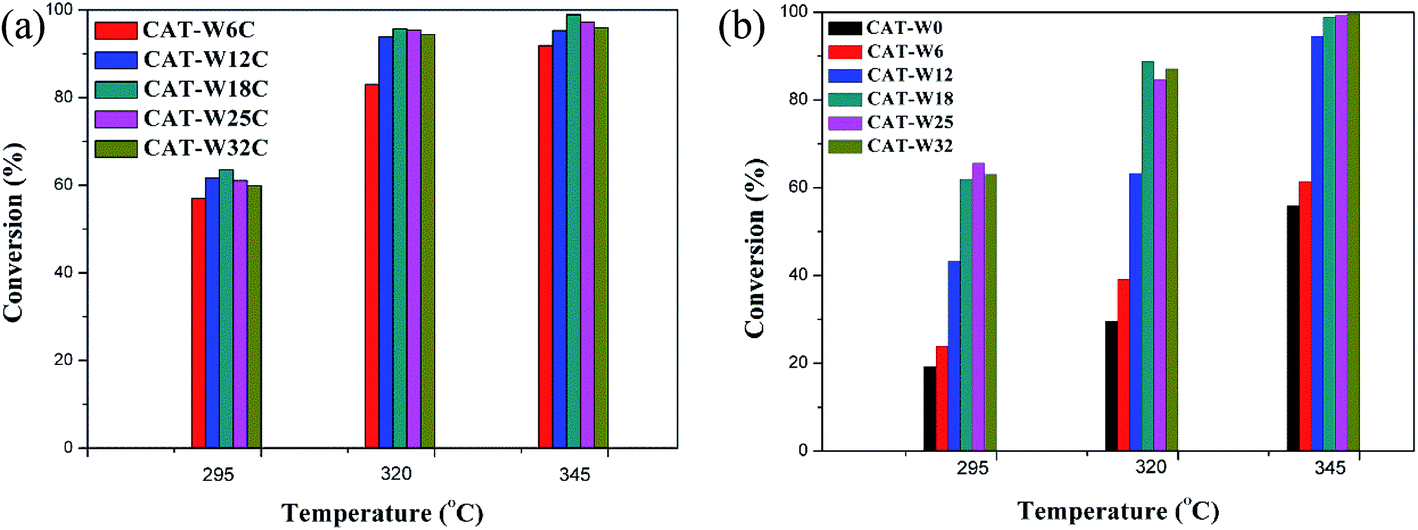 | ||
| Fig. 5 The conversion of two types of catalysts at different temperature: (a) bifunctional catalysts, (b) monofunctional acid catalysts. Treaction = 295, 320 and 345 °C. | ||
Although the conversion trend was similar for the two types catalysts, the reaction paths should be different. For the bifunctional catalysts, the conversion trend was consistent with the result of CO uptakes of the catalysts. Since the amount of the strong Brönsted acid sites for the bifunctional catalysts was almost constant at different amount of metal loading (see Table 3), the increased conversion should be attributed to the metal sites. For the monofunctional acid catalyst, it should possibly be attributed to the abundant strong Brönsted acid sites since the amount of the strong Brönsted acid sites increased with the amount of metal loading (see Table 4).
It also can be seen that the conversion of bifunctional catalysts is higher obviously than that for the monofunctional acid catalysts for the same amount of metal loading (WO3 loading < 18%) at the same temperature, although the amount of the strong Brönsted acid sites and the average particle size was change slowly. It indicated that the bifunctional catalysts were better than the monofunctional catalysts on the conversion of decalin, which implied the importance of the metal sites over the bifunctional catalysts.
3.3 Reaction products distribution
To illustrate the reaction products distribution, the ring opening of decalin was carried out at 320 °C because the yield of ROP was highest at 320 °C for most of the catalysts. And the corresponding products yield as well as the conversion of decalin was shown in Fig. 6. For two types of catalysts, the conversion of decalin and yield of ROP increased obviously for low amount of metal loading (WO3 loading < 18%) while a slow decrease was observed for high amount of metal loading (WO3 loading > 18%). Meanwhile, it could be clearly seen that both the conversion of decalin and the yield of ROP under the monofunctional acid catalysts were lower than that under of the bifunctional catalysts at the same metal loading amount.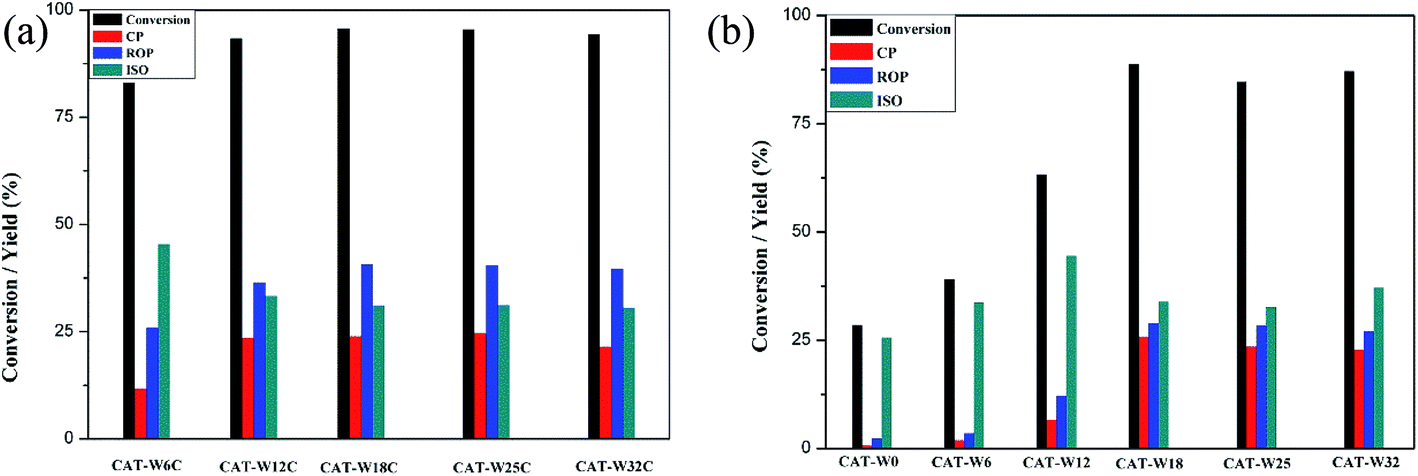 | ||
| Fig. 6 The conversion and products yield of two types of catalysts at different temperature: (a) bifunctional catalysts, (b) monofunctional acid catalysts. Treaction = 320 °C. | ||
Considering the conversion for CAT-W18C and CAT-W18 catalysts was highest on the two types of catalysts at 320 °C (see Fig. 6), the CAT-W18C and CAT-W18 catalyst were chose to illustrate the products distribution. High yield of ROP (YROP = 40.7), low yield of CP (YCP = 23.9) and ISO (YISO = 31.1) was obtained on the CAT-W18C, opposite trends were observed on the CAT-W18 (YROP = 28.9, YCP = 25.8 and YISO = 33.9). The higher YROP for the CAT-W18C should be caused by the fact that high hydrogenation activity on metal sites of bifunctional catalysts makes olefinic intermediates formed after β-scission easier hydrogenating to form ROP rather than CP.30 In addition, the lower strong Brönsted acid sites on bifunctional catalysts can avoid the subsequent cracking reaction of ROP.8
At the same time, the yield of CP for the monofunctional acid catalysts shown in Fig. 6b with the amount of metal loading at the same conditions, which is consistent with the amount of strong Brönsted acid sites (see Table 4). This implied that the catalysts with strong Brönsted acid sites is easier to form CP, which is consistent with D. Kubička et al. report.3 As mentioned above, the conversion of decalin and the yield of ROP over the bifunctional catalyst were higher than that of under the monofunctional catalysts. It is easier to generate the ROP over the bifunctional catalysts while the CP over the monofunctional catalysts.
The products selectivity for ring opening of decalin on two types of catalysts at different reaction temperature is shown in Fig. 7 ((a): bifunctional catalysts, (b): monofunctional acid catalysts). Fig. 7 showed that the selectivity of ISO was highest at low conversion whereas a low selectivity of ROP and CP was observed which indicated that ROP and CP were secondary reaction compounds while the ISO was the first reaction products.3,6
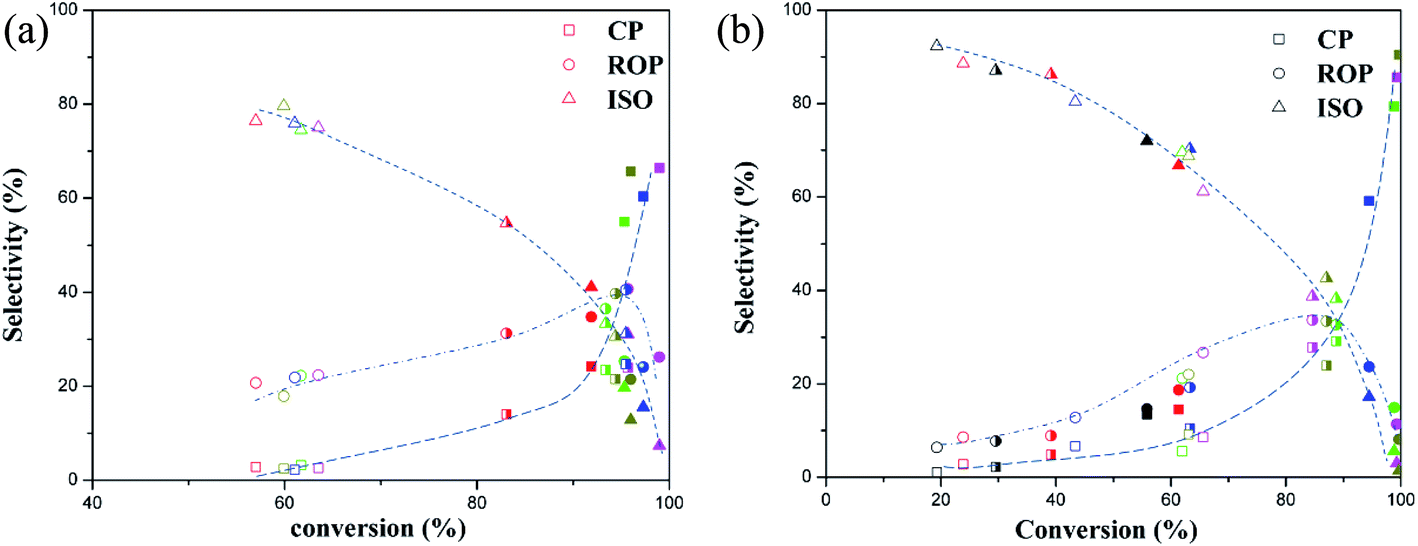 | ||
| Fig. 7 Products selectivity obtained with the two types of catalysts: (a) bifunctional catalysts, (b) monofunctional acid catalysts Treaction = 295 (open symbol), 320 (half-filled symbol) and 345 °C (filled symbol) (black symbol: CAT-W0; red symbol: CAT-W6C/CAT-W6; blue symbol: CAT-W12C/CAT-W12; green symbol: CAT-W18C/CAT-W18; magenta symbol: CAT-W25C/CAT-W25; yellow symbol: CAT-W32C/CAT-W32). | ||
Fig. 7a also showed that the selectivity of ROP and CP increased with increasing conversion, which should be attributed to the further reaction of ISO. Meanwhile, the high selectivity of CP was observed and the selectivity of ROP decreased at high conversion, which should be caused by the cracking of ISO and ROP.6 The decalin conversion and ROP yield was accordance with the CO uptakes, which implied that the metal sites on the bifunctional catalysts played an important role in the production of ROP.
It can be proposed that the ring opening of decalin over bifunctional catalysts obeys bifunctional mechanism.12,30 Firstly, the dehydrogenation of decalin on the metal sites formed a carbenium ion, and then the produced carbenium ion underwent the skeletal isomerization or the ring opening on acid sites by β-scission to form an olefin. Lastly, the corresponding olefin was hydrogenated on the metal sites. At the same time, β-scission was occurred on the isomer and ring opening products resulting in the production of CP.
As shown in Fig. 7b, all the monofunctional catalysts showed similar trends with bifunctional catalysts on products distribution for this reaction. At the same time, the decalin conversion and ROP yield was in keeping with the change of the strong Brönsted acid sites at the same temperature. This agrees well with Kubicka,3 who observed similar phenomena on ring opening of decalin over proton-form zeolites. Based on this, the ring opening of decalin mechanism can be proposed as follows according to acid mechanism.3,6 The initial step is protolytic dehydrogenation and forming a carbenium ion on acid sites. Next the carbenium ion formed on acid sites is skeletal isomerization or its ring is opened by β-scission, producing the ISO. And then, the corresponding ISO on acid sites was hydrogenated, producing the ROP or CP.3,6 At the same time, the ISO and ROP after protonation can be involved in another β-scission which leads to the formation of CP.3
As mentioned above, the dehydrogenation/hydrogenation occurred over the metal sites and β-scission occurred over the acid sites for the bifunctional catalysts, while the dehydrogenation/hydrogenation and β-scission occurred over the acid sites for the monofunctional acid catalysts. The proposed mechanisms for ring opening of decalin on two types of catalysts were presented in Fig. 8 ((a): bifunctional catalysts, (b): monofunctional acid catalysts).
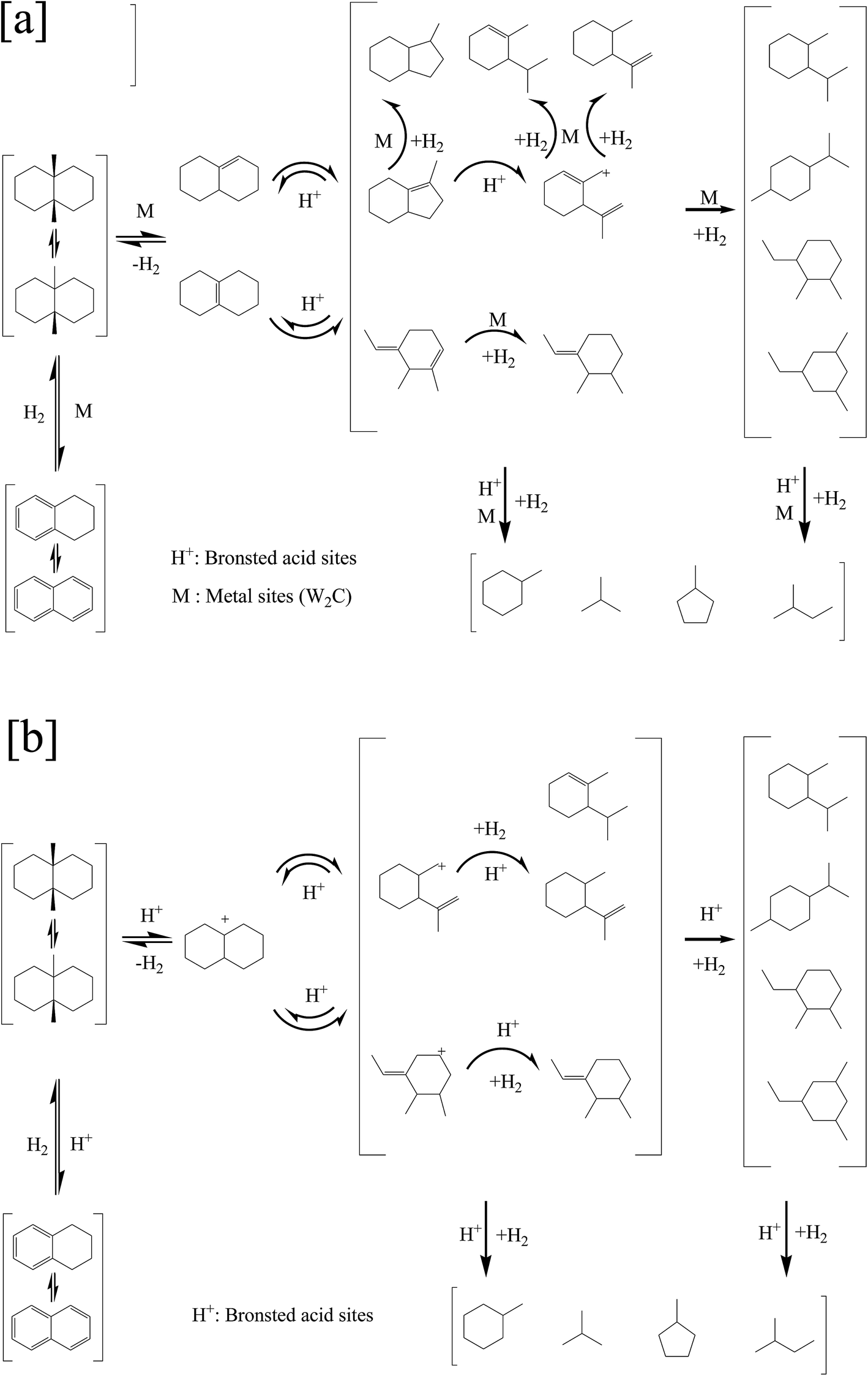 | ||
| Fig. 8 Proposed mechanism for ring opening of decalin: (a) bifunctional mechanism, (b) monofunctional acid mechanism. | ||
4. Conclusion
The ring opening reaction of decalin over the Ni–W bifunctional catalysts and Ni–W monofunctional acid catalysts has been studied.Compared with the monofunctional acid catalysts, the bifunctional catalysts had lower amount of strong Brönsted acid sites, but they showed better performance with higher conversion, higher selectivity of ROP and lower selectivity of CP in the ring opening reaction of decalin, which should be attributed to the metal sites on the bifunctional catalysts. For the monofunctional acid catalysts, the strong Brönsted acid sites on the catalysts played an important role on the catalytic performance. The ring opening of decalin on the Ni–W bifunctional catalysts possibly obey the bifunctional mechanism, while Ni–W monofunctional acid catalysts possibly obey the monofunctional acid mechanism. Generally, both the bifunctional catalysts and the monofunctional acid catalysts worked well in the ring opening reaction of decalin. However, the bifunctional catalysts were more suitable for this reaction and the CAT-W18C showed the best performance at 320 °C.
Acknowledgements
Supported by the “Strategic Priority Research Program” Demonstration of Key Technologies for Clean and Efficient Utilization of Low-rank Coal (GraXDA07020200) as well as the National Natural Science Foundation of China (Grant No. 41372350).References
- G. McVicker, J. Catal., 2002, 210, 137–148 CrossRef CAS.
- C. Song and X. Ma, Appl. Catal., B, 2003, 41, 207–238 CrossRef CAS.
- D. Kubička, N. Kumar, P. Mäki-Arvela, M. Tiitta, V. Niemi, T. Salmi and D. Y. Murzin, J. Catal., 2004, 222, 65–79 CrossRef.
- S.-C. Qi, L. Zhang, X.-Y. Wei, J.-i. Hayashi, Z.-M. Zong and L.-L. Guo, RSC Adv., 2014, 4, 17105 RSC.
- L. Wang, B. Shen, F. Fang, F. Wang, R. Tian, Z. Zhang and L. Cui, Catal. Today, 2010, 158, 343–347 CrossRef CAS.
- R. Moraes, K. Thomas, S. Thomas, S. Van Donk, G. Grasso, J.-P. Gilson and M. Houalla, J. Catal., 2012, 286, 62–77 CrossRef CAS.
- K. C. Mouli, V. Sundaramurthy and A. K. Dalai, J. Mol. Catal. A: Chem., 2009, 304, 77–84 CrossRef CAS.
- M. Santikunaporn, J. Herrera, S. Jongpatiwut, D. Resasco, W. Alvarez and E. Sughrue, J. Catal., 2004, 228, 100–113 CrossRef CAS.
- A. Haas, S. Rabl, M. Ferrari, V. Calemma and J. Weitkamp, Appl. Catal., A, 2012, 425–426, 97–109 CrossRef CAS.
- T. He, Y. Wang, P. Miao, J. Li, J. Wu and Y. Fang, Fuel, 2013, 106, 365–371 CrossRef CAS.
- F. Gault, Adv. Catal., 1981, 30, 1 CAS.
- D. Kubička, N. Kumar, P. Mäki-Arvela, M. Tiitta, V. Niemi, H. Karhu, T. Salmi and D. Y. Murzin, J. Catal., 2004, 227, 313–327 CrossRef.
- S. Lecarpentier, J. Vangestel, K. Thomas, J. Gilson and M. Houalla, J. Catal., 2008, 254, 49–63 CrossRef CAS.
- D. Kubička, T. Salmi, M. Tiitta and D. Y. Murzin, Fuel, 2009, 88, 366–373 CrossRef.
- N. Kumar, D. Kubicka, A. L. Garay, P. Mäki-Arvela, T. Heikkilä, T. Salmi and D. Y. Murzin, Top. Catal., 2009, 52, 380–386 CrossRef CAS.
- M. Du, Z. Qin, H. Ge, X. Li, Z. Lü and J. Wang, Fuel Process. Technol., 2010, 91, 1655–1661 CrossRef CAS.
- M. Kangas, D. Kubička, T. Salmi and D. Y. Murzin, Top. Catal., 2010, 53, 1172–1175 CrossRef CAS.
- D. Kubička, M. Kangas, N. Kumar, M. Tiitta, M. Lindblad and D. Y. Murzin, Top. Catal., 2010, 53, 1438–1445 CrossRef.
- J.-W. Park, K. Thomas, J. van Gestel, J.-P. Gilson, C. Collet, J.-P. Dath and M. Houalla, Appl. Catal., A, 2010, 388, 37–44 CrossRef CAS.
- S. Rabl, D. Santi, A. Haas, M. Ferrari, V. Calemma, G. Bellussi and J. Weitkamp, Microporous Mesoporous Mater., 2011, 146, 190–200 CrossRef CAS.
- S. A. D'Ippolito, L. B. Gutierrez and C. L. Pieck, Appl. Catal., A, 2012, 445–446, 195–203 CrossRef.
- K. Jaroszewska, A. Masalska, K. Bączkowska and J. R. Grzechowiak, Catal. Today, 2012, 196, 110–118 CrossRef CAS.
- N. Kumar, P. Mäki-Arvela, N. Musakka, D. Kubicka, M. Kangas, M. Tiitta, H. Österholm, A. R. Leino, K. Kordás, T. Heikkilä, T. Salmi and D. Y. Murzin, Catal. Ind., 2013, 5, 105–122 CrossRef.
- R. Moraes, K. Thomas, S. Thomas, S. Van Donk, G. Grasso, J.-P. Gilson and M. Houalla, J. Catal., 2013, 299, 30–43 CrossRef CAS.
- D. Santi, T. Holl, V. Calemma and J. Weitkamp, Appl. Catal., A, 2013, 455, 46–57 CrossRef CAS.
- D. P. Upare, B. J. Song and C. W. Lee, J. Nanomater., 2013, 2013, 1–6 CrossRef.
- K. Arve, P. Mäki-Arvela, K. Eränen, M. Tiitta, T. Salmi and D. Y. Murzin, Chem. Eng. J., 2014, 238, 3–8 CrossRef CAS.
- S. A. D'Ippolito, C. Especel, L. Vivier, F. Epron and C. L. Pieck, Appl. Catal., A, 2014, 469, 532–540 CrossRef.
- S. A. D'Ippolito, C. Especel, L. Vivier, F. Epron and C. L. Pieck, Appl. Catal., A, 2014, 469, 541–549 CrossRef.
- C. A. A. Monteiro, D. Costa, J. L. Zotin and D. Cardoso, Fuel, 2015, 160, 71–79 CrossRef CAS.
- M. A. Vicerich, M. Oportus, V. M. Benitez, P. Reyes and C. L. Pieck, Appl. Catal., A, 2014, 480, 42–49 CrossRef CAS.
- S. Nassreddine, L. Massin, M. Aouine, C. Geantet and L. Piccolo, J. Catal., 2011, 278, 253–265 CrossRef CAS.
- M. A. A. Arribas, P. Concepción and A. N. Martínez, Appl. Catal., A, 2004, 267, 111–119 CrossRef CAS.
- J. M. Manoli, P. Da Costa, M. Brun, M. Vrinat, F. Maugé and C. Potvin, J. Catal., 2004, 221, 365–377 CrossRef CAS.
- H. A. Al-Megren, S. L. González-Cortés, T. Xiao and M. L. H. Green, Appl. Catal., A, 2007, 329, 36–45 CrossRef CAS.
- V. Sundaramurthy, A. K. Dalai and J. Adjaye, Catal. Today, 2007, 125, 239–247 CrossRef CAS.
- S.-C. Qi, X.-Y. Wei, Z.-M. Zong and Y.-K. Wang, RSC Adv., 2013, 3, 14219 RSC.
- G. Cui, J. Wang, H. Fan, X. Sun, Y. Jiang, S. Wang, D. Liu and J. Gui, Fuel Process. Technol., 2011, 92, 2320–2327 CrossRef CAS.
- P. Da Costa, J. L. Lemberton, C. Potvin, J. M. Manoli, G. Perot, M. Breysse and G. Djega-Mariadassou, Catal. Today, 2001, 65, 195–200 CrossRef CAS.
- S. Bendezú, R. Cid, J. Fierro and A. L. Agudo, Appl. Catal., A, 2000, 197, 47–60 CrossRef.
- W. Zhang, Y. Zhang, L. Zhao and W. Wei, Energy Fuels, 2010, 24, 2052–2059 CrossRef CAS.
- H. H. Hwu and J. G. G. Chen, Chem. Rev., 2005, 105, 185–212 CrossRef CAS PubMed.
- J. Sun, M. Zheng, X. Wang, A. Wang, R. Cheng, T. Li and T. Zhang, Catal. Lett., 2008, 123, 150–155 CrossRef CAS.
- R. B. Levy and M. Boudart, Science, 1973, 181, 547–549 CAS.
- S. T. Oyama, X. Wang, Y. K. Lee and W. J. Chun, J. Catal., 2004, 221, 263–273 CrossRef CAS.
- C. A. Ribeiro, W. R. de Souza, M. S. Crespi, J. A. G. Neto and F. L. Fertonani, J. Therm. Anal. Calorim., 2007, 90, 801–805 CrossRef CAS.
- C. Bouchy, C. Pham-Huu, B. Heinrich, E. G. Derouane, S. B. Derouane-Abd Hamid and M. J. Ledoux, Appl. Catal., A, 2001, 215, 175–184 CrossRef CAS.
- X. Chen, G. Clet, K. Thomas and M. Houalla, J. Catal., 2010, 273, 236–244 CrossRef CAS.
- A. M. Turek, I. E. Wachs and E. DeCanio, J. Phys. Chem., 1992, 96, 5000–5007 CrossRef CAS.
- M. Shao, H. Cui, S. Guo, L. Zhao and Y. Tan, RSC Adv., 2016, 6, 61747–61757 RSC.
- S. Lecarpentier, J. Vangestel, K. Thomas and M. Houalla, J. Catal., 2007, 245, 45–54 CrossRef CAS.
- E. Rodríguez-Castellón, A. Jiménez-López and D. Eliche-Quesada, Fuel, 2008, 87, 1195–1206 CrossRef.
- W. L. Y. Choong-Hyon Kim, I. C. Lee and S. I. Woo, Appl. Catal., A, 1996, 144, 159–175 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |