
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Detection of Janus-activated kinase-1 and its interacting proteins by the method of luminescent oxygen channeling†
Xin-Xin Guoa,
Han-Tao Wua,
Si-Hui Zhuangac,
Zhen-Hua Chena,
Rong-Liang Lianga,
Yao Chena,
Ying-Song Wu*a and
Tian-Cai Liu *ab
*ab
aState Key Laboratory of Organ Failure, Institute of Antibody Engineering, School of Laboratory Medicine and Biotechnology, Southern Medical University, Guangzhou 510515, Guangdong, P. R. China. E-mail: wg@smu.edu.cn; Fax: +86-20-37247604; Tel: +86-20-62789355
bGuangdong Provincial Key Laboratory of Tropical Disease Research, School of Public Health, Southern Medical University, Guangzhou 510515, Guangdong, P. R. China. E-mail: liutc@smu.edu.cn; Fax: +86-20-37247604; Tel: +86-20-62789355
cCentral People's Hospital of Zhanjiang, Zhanjiang, P. R. China
First published on 1st February 2017
Abstract
Janus-activated kinase-1 (JAK1) plays an important role in many signaling pathways, including the JAK–STAT and SOCS pathways. The activation of the JAK1–STAT3 pathway is a key role in tumor inflammation and immunity by promoting inflammation. The detection of its interacting proteins is of great significance in many diseases. Although luminescent oxygen channeling is commonly used in immunoassay technologies, its use in the detection of JAK1 and interacting proteins has not been reported to date. We constructed eukaryotic overexpression plasmids pENTER–JAK1-His and pENTER–STAT3-His and, following single transfection and co-transfection, measured their expression by western blotting. Co-immunoprecipitation and luminescent oxygen channeling were then used to identify the protein interactions. JAK1 and STAT3 proteins were shown to be successfully overexpressed, and co-immunoprecipitation identified their interaction in vitro. We also detected protein interactions between JAK1 and interleukin 4 receptor (IL4R), JAK1 and growth hormone receptor (GHR). In conclusion, we verified that JAK1 and STAT3 interact in vitro using co-immunoprecipitation, and demonstrated for the first time the potential application of luminescent oxygen channeling for detecting JAK1 and its interacting proteins. Also we think this assay could help other researchers find unreported proteins that also have interactions in vitro.
Introduction
Janus-activated kinase-1 (JAK1), a member of the Janus family of kinases, is expressed in various tissues and cells. Current studies suggest that many signal transduction events in the body require the participation of JAK1 protein, and that mutation of JAK1 and its excessive expression in cells is associated with the development of various diseases, such as endometrial cancers, gastric cancer.1,2 STAT3 is a member of the STAT family of transcription factors and mediates cellular responses to cytokines and growth factors. STAT3 has an important role in tumor inflammation and immunity by promoting pro-oncongenic inflammation pathways, indicating that STAT3 is a crucial target for preventing cancer.3–5 JAK1–STAT3 is an important signaling pathway, involved in cell proliferation in vivo, cell differentiation, apoptosis, cell survival, and the immunomodulatory process. It is induced by different receptors of the innate and adaptive arms of the immune system.6,7 It is reported that the JAK1–STAT3 pathway could be activated by interleukins (IL), such as IL6, 1L10. The activated JAK1 (phosphorylated JAK1) resulted in the activation of STAT3 (phosphorylated STAT3), and the occurrence of tumor inflammation, and the detection of overexpressed JAK1 and STAT3 is of importance.8–11Protein interactions play significant roles in specific biological processes, participating in cellular functions and activities. Different proteins interact to form a protein–protein network, involving biological processes such as the maintenance of cellular integrity, gene expression and regulation, and immune response.12–14 At present, the conventional assays used to identify protein interactions are the yeast two-hybrid assay, tandem affinity purification, co-immunoprecipitation (Co-IP), GST pull-down assay.15–21 These assays have their limitations, however: Co-IP cannot be used quantitatively, GST pull-down assays produce a high rate of false positive results, oligo precipitation is susceptible to non-specific protein interactions, and yeast two-hybrid assay is limited by nucleoprotein.
Immunoassay technologies are of clinical importance in the early detection of disease biomarkers. Such technologies, including enzyme-linked immunosorbent assay and time resolved fluoroimmunoassay, can be used for rapid and precise quantification.22–24 The luminescent oxygen channeling assay (LOCA) is based on an excited and reactive form of singlet oxygen produced from ambient O2 in solution. LOCA is a homogeneous immunoassay and, compared with non-homogeneous immunoassays, does not require the separation of bound and free antigens/antibodies, meaning that incubation time for the assay is greatly reduced to 60 min. LOCA also has unique advantages over other immunoassays, such as a lower quantification limit and easy automation.
Here, we describe a new LOCA assay to detect JAK1 and its interacting proteins, and verify the feasibility of this method by comparison with Co-IP (STAT3 for example). We successfully prepared functional microspheres as acceptor beads that conjugated with anti-STAT3 antibody (anti-STAT3 MPs), and that biotinylated anti-JAK1 antibody could specifically combine with streptavidin coated on the surface of donor beads. This assay could also be applied to the detection of other signal pathway interaction proteins in the future, also this assay could help other researchers finding not reported proteins that also have interactions in vitro.
Materials and methods
Materials
Lipofectamine 2000 (lipo 2000) was purchased from Invitrogen (Carlsbad, CA, USA), aldehyde-modified polystyrene microspheres, Pierce™ BCA Protein Assay Kit were purchased from Thermo Fisher Scientific (Waltham, MA, USA), restriction enzymes AsiSI and MIuI were purchased from BioLabs (New England), T4 DNA ligase were purchase from Takara (Tokyo, Japan). Biotin N-hydroxysuccinimide ester, 2-(N-morpholino) ethanesulfonic acid (MES) and dimethyl sulfoxide was purchased from Sigma-Aldrich Fine Chemicals (St. Louis, MO, USA). Anti-STAT3 antibody (rabbit source, detects endogenous levels of total STAT3 protein) and anti-JAK1 antibody (rabbit source, detects endogenous levels of total JAK1 protein) were purchased from Cell Signaling Technology (Danvers, MA, USA) and MAPP protein was purchased from RayBiotech (Norcross, GA, USA). Anti-His-tag antibody (mouse source), goat anti-rabbit IgG–HRP antibody and goat anti-mouse IgG–HRP antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). GAPDH anti-mouse monoclonal antibody as an internal protein standard was purchased from ProteinTech (Chicago, IL, USA). Streptavidin-coated donor beads were purchased from Beyond (Shanghai, China).Construction of eukaryotic expression plasmids pENTER–JAK1 and pENTER–STAT3
The eukaryotic expression plasmid pENTER has cleavage sites for AsiSI and MIuI restriction enzymes. We obtained human JAK1 and STAT3 cDNAs from Han Lab (Xiamen University, China). PCR primers were designed with restriction sites for AsiSI and MIuI (Table S1†). PCR was performed using the primer pair. Following series of double digestion, T4 DNA ligase enzyme, randomly selected the positive clones, the pENTER–JAK1 and pENTER–STAT3 plasmids were constructed. The plasmids were verified by PCR and double enzyme digestion using AsiSI and MIuI. Sequencing of pENTER–JAK1 and pENTER–STAT3 also showed that both the sequences were corrected and had a His-tag at the carbon terminal as desired.Plasmids single transfection and co-transfection in HEK293 cells
HEK293 human embryonic kidney cells (purchased from the Type Culture Collection of the Chinese Academy of Sciences, Shanghai, China) were grown in DMEM supplemented with 10% fetal bovine serum. HEK293 cells were seeded in 6-well plates 1 day before transient transfection. Transfection was carried out according to the manufacturer's recommendations. 48 h after transfection, the cells were lysate by 200 μL RIPA lysis buffer (Beyotime BioTechnology, Shanghai, China). Plasmid pENTER–STAT3 was transfected as the same way.Single transfection preliminary experiment results told us that the overexpression amount of protein STAT3 was larger than protein JAK1. So we use 2.5 μg pENTER–JAK1 and 0.5 μg pENTER–STAT3 as the amount of co-transfection per well. Next procedures were as the same way above.
Western blotting was used to identify the expression of JAK1 and STAT3 in the samples, with free pENTER plasmids used as negative controls. Here anti-JAK1 antibody, anti-STAT3 antibody, and GAPDH monoclonal antibody were used as single transfection samples primary antibodies, goat anti-rabbit IgG–HRP antibody and goat anti-mouse IgG–HRP antibody were used as secondary antibody. Anti-His tag antibody and GAPDH monoclonal antibody were used as co-transfection primary antibodies, and goat anti-mouse IgG–HRP was used as secondary antibody.
Co-immunoprecipitation (Co-IP) to detect JAK1–STAT3 interacting proteins
We used 2 μg of anti-STAT3 antibody as a combination antibody to detect interaction between STAT3 and JAK1 proteins in 1 mL of lysate prepared as described previously. Empty plasmid lysate was used as a blank. Western blotting was used to identify JAK1 interacting protein, with anti-JAK1 antibody used as primary antibody and goat anti-rabbit IgG–HRP used as secondary antibody.LOCA detection system
LOCA was performed using two kinds of microbeads, donor beads and acceptor beads. The donor beads, containing the photosensitizer phthalocyanine that can be excited at 680 nm, convert ambient triplet oxygen to singlet oxygen, and when two bead types are within 200 nm of each other, acceptor beads can generate chemiluminescence.25,26 Here, LOCA was used to detect JAK1–STAT3 protein interaction in co-transfection cell lysate (Fig. 1). The spectra of the acceptor beads were presented in Fig. 2. The fluorescence and absorption spectra of the acceptor beads were collected on the fluorescence spectrometer (LS-55) and NANODROP 2000c, respectively. The emission spectra of the acceptor beads were 520–620 nm. The fluorescence (λem = 520–650 nm) of the acceptor beads was detected for LOCA.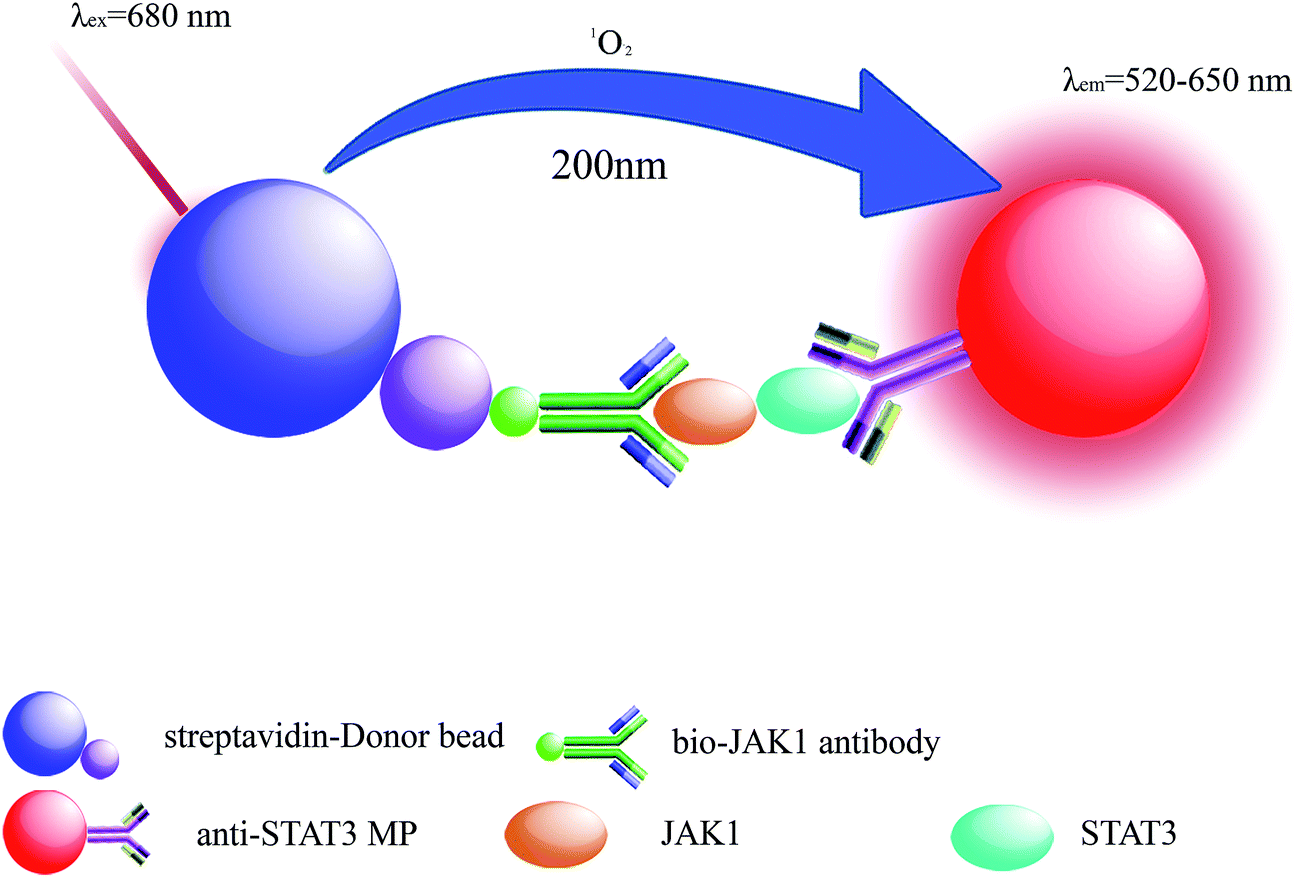 | ||
| Fig. 1 Schematic of the detection of JAK1 and STAT3. The streptavidin-donor beads, containing the photosensitizer phthalocyanine that can be excited at 680 nm, convert ambient triplet oxygen to singlet oxygen, and when the JAK1–STAT3 lysate was added, the corresponding antibodies could bring the two bead types together within 200 nm of each other, acceptor beads can generate chemiluminescence. The fluorescence intensity was measured on the EnSpire® multimode plate reader. | ||
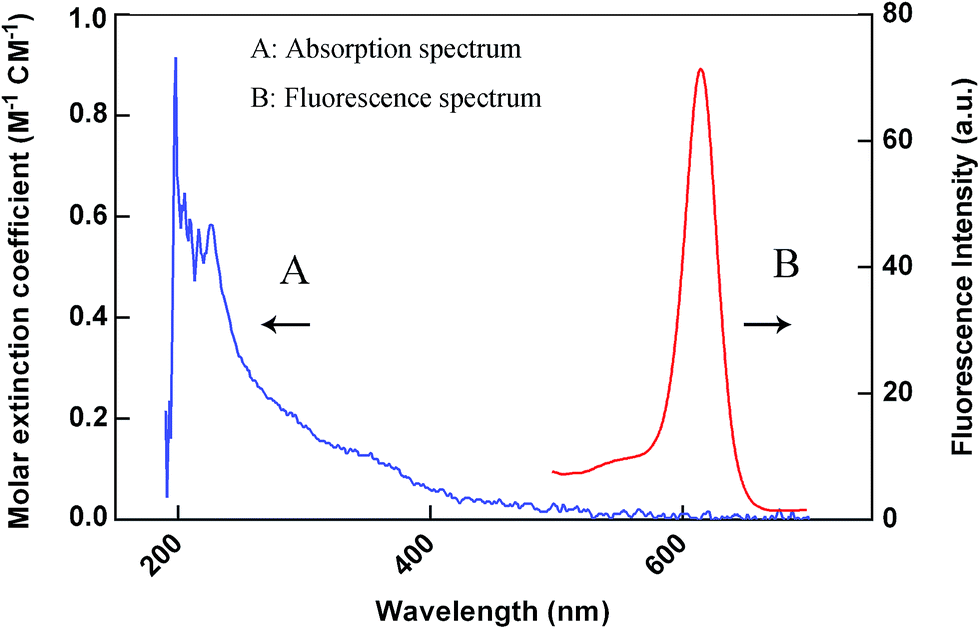 | ||
| Fig. 2 Spectra of the acceptor beads. The curve A and curve B are the absorption and fluorescence spectrum of the acceptor beads, respectively. | ||
Bio-JAK1 antibody and anti-STAT3 microspheres captured JAK1–STAT3 interacting proteins once the streptavidin-coated donor beads were added to the wells and the distance between the two types of beads was within 200 nm. Energy was transferred from donor beads to acceptor beads relying on singlet oxygen around. The fluorescence intensity was measured on the EnSpire® multimode plate reader.
First, for biotin conjugation, anti-JAK1 antibody and biotin N-hydroxysuccinimide ester were combined at a mass ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Lysate containing free biotin was removed by centrifugation and biotinylated (bio)-JAK1 antibody was collected at a concentration of 5 mg mL−1 in 0.13 M PBS, pH 7.4.
:1. Lysate containing free biotin was removed by centrifugation and biotinylated (bio)-JAK1 antibody was collected at a concentration of 5 mg mL−1 in 0.13 M PBS, pH 7.4.
Then, anti-STAT3 antibody was conjugated with aldehyde-modified microspheres. 1 mg quantity of microspheres, 30 μg anti-STAT3 antibody, 10 μL sodium borohydride were mixed and incubated at 37 °C for 24 h. Unbound protein was removed by centrifugation (15000g, 15 min, 4 °C) and microspheres successfully conjugated to anti-STAT3 antibody were stored in 0.13 M PBS (pH 7.4) at a concentration of 10 mg mL−1.
Next, 1 μL bio-JAK1 antibody and 1 μL anti-STAT3 microspheres were added per well in 96 well OptiPlate (Perkin-Elmer, Waltham, MA, USA), and co-transfection cell lysate was added in volumes of 0 μL, 10 μL, 25 μL, 50 μL, and 75 μL, with lysis buffer used to make all samples up to 75 μL as required (basing on the volume limitation, 75 μL lysate was the maximum sample amount). Co-transfected JAK1 and human tyrosine protein kinase (LYN, a protein that has no interaction with JAK1 in vitro) plasmids lysate was used as the negative control, and untreated HEK293 cells were used as the blank control. OptiPlate was incubated with shaking at 37 °C in the dark. Next, 175 μL of streptavidin-coated donor beads was added into the wells and cells were incubated in the dark with shaking at 37 °C. The fluorescence intensity was measured on the EnSpire® multimode plate reader (Perkin-Elmer).
Results and discussion
pENTER–JAK1 and pENTER–STAT3 construction and co-transfection
The constructed plasmids pENTER–JAK1 and pENTER–STAT3 were verified using PCR and double enzyme digestion using restriction enzymes AsiSI and MIuI (Fig. S1†), JAK1 is about 3500 base pairs (bp) in size, and between 3000 bp and 5000 bp, while STAT3 is about 2210 bp, between 2000 bp and 3000 bp. Free pENTER plasmid is about 7500 bp, and above 5000 bp. We obtained a single band by agarose gel electrophoresis from PCR products that contained JAK1 (Fig. S1A†) and STAT3 (Fig. S1C†). Two bands containing pENTER and JAK1 (Fig. S1B†) and pENTER and STAT3 (Fig. S1D†) were observed. The DNA sequencing results therefore verified the expected sequence.Western blotting analyses
The pENTER–JAK1 and pENTER–STAT3 plasmids were next transfected into 293T cells and western blotting was performed to identify the expression of JAK1 and STAT3 proteins. Protein GAPDH, which is a housekeeping gene encoded protein that expressed high levels in organisms, was used as the internal control (36 kD). Significant upregulation of JAK1 and STAT3 was observed, compared with the blank group (Fig. S2†), and the expression of STAT3 (Fig. S2B†) was greater than that of JAK1 (Fig. S2A†). Next, pENTER–JAK1 and pENTER–STAT3 were transfected simultaneously, with each transfected separately as controls. Western blotting showed that JAK1 and STAT3 proteins were successfully co-transfected (Fig. S3,† lane 3) and that the expression level was consistent with those of the plasmids transfected singly (Fig. S3,† lanes 1 and 2).Co-immunoprecipitation (Co-IP) analyses
Here, we used anti-STAT3 antibody and co-transfection cell lysate as the experimental group (lane 1), MAPP protein which does not interact with JAK1 and co-transfection cell lysate as the negative control group (lane 2), and anti-STAT3 antibody and empty plasmid transfection lysate as a blank group (lane 3). Anti-STAT3 antibody was shown to bind STAT3 protein in the cell lysate. JAK1 interacting protein could also be detected by protein G. Western blotting was used to identify protein interaction with JAK1 (Fig. S4†). A purpose band for JAK1 was observed in lane 1, while lanes 2 and 3 had no bands. There were no nonspecific bands observed. This indicated that there was interaction between JAK1 and STAT3.Optimization of the concentration of microspheres and reaction time
In this assay, the concentration of microspheres could affect the reaction. The donor and acceptor beads (MPs) were simply mixed in the 96-well plates with the MPs at concentration of 0.001, 0.01, 0.1, 1 and 5 mg mL−1 (Fig. 3). Calibration was carried out using log–log regression and the best-fit calibration was determined to be described by the following equation, log(Y) = 0.68log(X) + 4.22, with a correlation coefficient of 0.9998. When the concentration of MPs was greater than 0.01 mg mL−1, background noise could not be ignored with the detection limit reached. Hence, to avoid false positives, 0.01 mg mL−1 was selected as the optimum MPs concentration. With the concentrations lower than 0.01 mg mL−1, the results indicated that the fluorescence intensity increased with incubation time showed in Fig. 4. After 40 min incubation the reaction reached a dynamic balance. Then, 40 min was selected as the optimum reaction time.
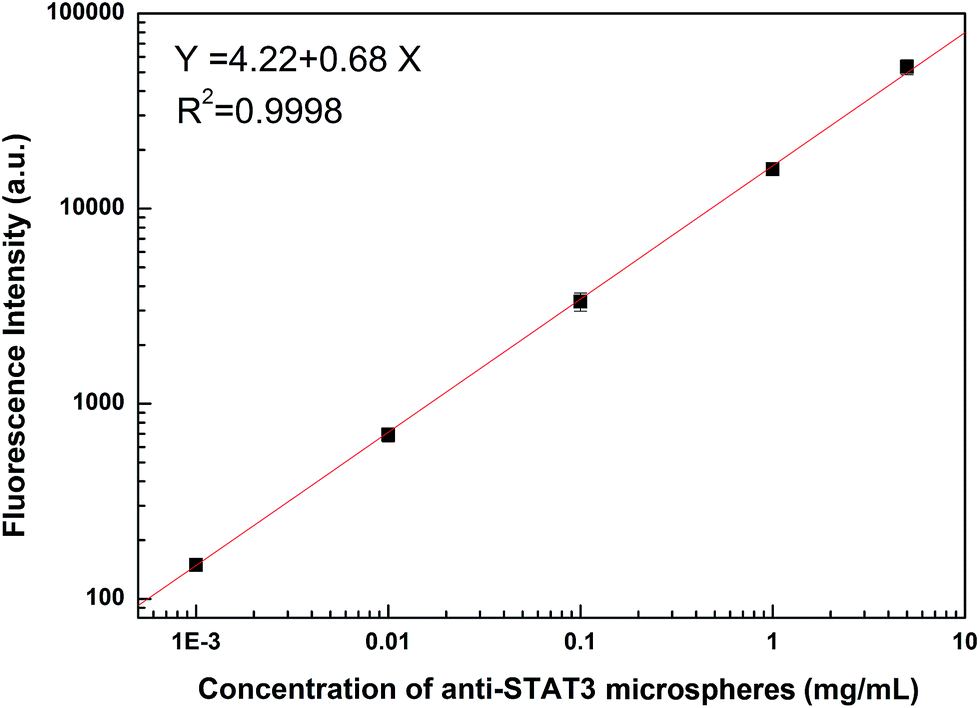 | ||
| Fig. 3 Influence of the concentration of anti-STAT3 microspheres on fluorescence intensity (0.001, 0.01, 0.1, 1, and 5 mg mL−1). | ||
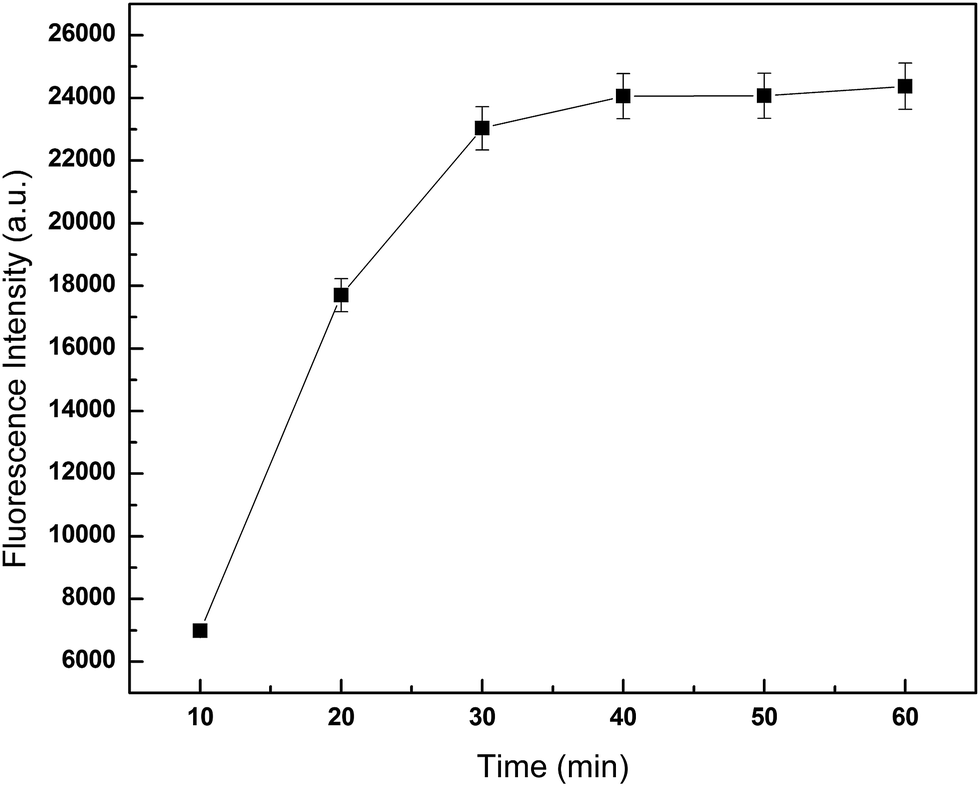 | ||
| Fig. 4 Influence of incubation time on fluorescence intensity. 75 μL JAK1–STAT3 lysate was added per well, and detected fluorescence intensity at different incubation time. | ||
LOCA detection system analyses
LOCA was used to detect JAK1–STAT3 protein interaction in co-transfection cell lysate (Fig. 5). Only the streptavidin–biotin binding could bring the donor beads and acceptor beads together and shortens their distance so much that singlet oxygen could diffuse into the acceptor beads. The fluorescence intensity was measured and increased according to the volume of cell lysate, indicating increasing amounts of interacting proteins. The fluorescence intensity of overexpressed JAK1–STAT3 was noticeably higher than that of the negative and blank controls. This indicated that LOCA could be applied in the detection of JAK1–STAT3.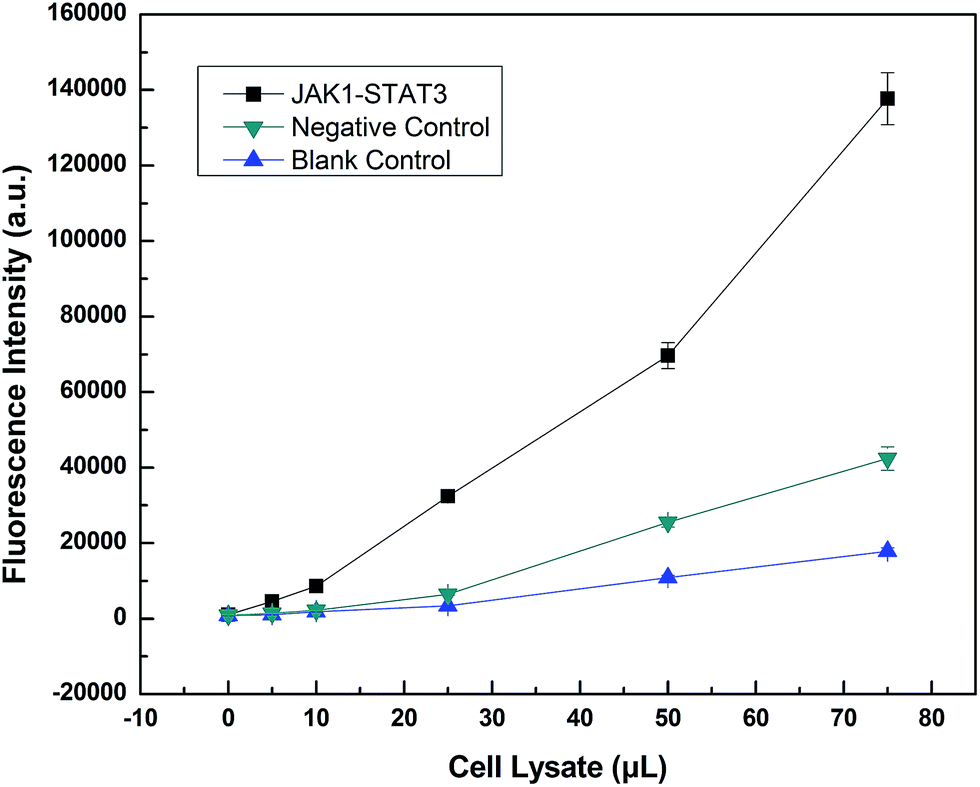 | ||
| Fig. 5 Detection of JAK1–STAT3 by LOCA. | ||
Furthermore, we used LOCA to detect GHR and IL4R, other proteins known to interact with JAK1. The same conjugation method was used as described above, and the results were obtained for JAK1–GHR (Fig. 6A) and JAK1–IL4R (Fig. 6B). Compared with negative and blank controls, JAK1–GHR and JAK1–IL4R groups both showed positive results, and increasing volumes of cell lysate caused higher fluorescence intensity as before. The maximum volume of cell lysate used was 75 μL, significantly less than the 1 mL volume needed for Co-IP. The LOCA assay showed good sensitivity and a rapid detection time, and therefore has potential for application in the detection of JAK1 and other interacting proteins, as well as interacting proteins from other signaling pathways, also this could provide convenience for other researchers finding unknown and that not reported proteins that also have interactions in vitro.
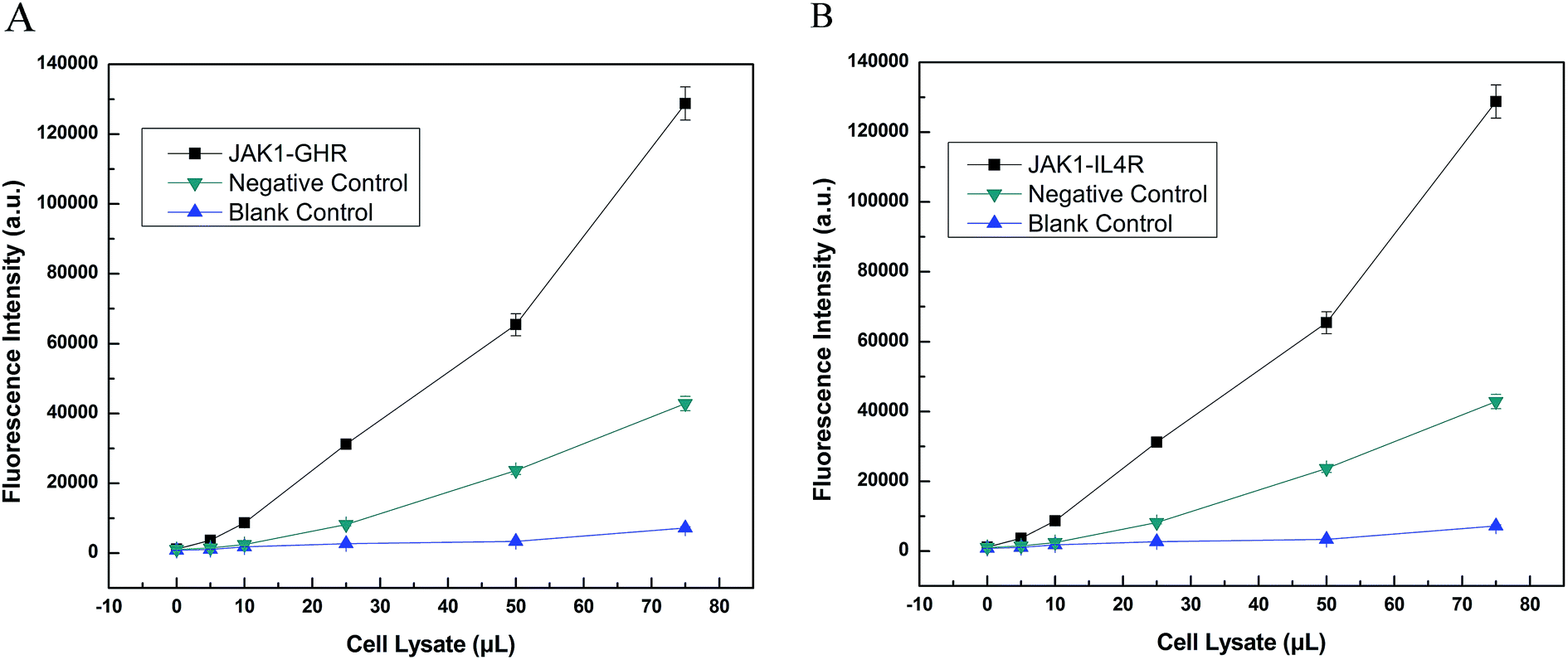 | ||
| Fig. 6 (A). Detection of JAK1–GHR by LOCA. (B) Detection of JAK1–IL4R by LOCA. | ||
Conclusions
JAK1 takes a key role in many signaling events, for example, JAK–STAT pathway, IL-7 signaling pathways, IL-2 pathway, IL-4 pathway, and IL-27-mediated signaling events.27–30 At present, as reported about forty proteins interact with JAK1, which involved most of the signaling pathways. Therefore, the detection of known interacting proteins and the selection of potential interacting proteins are of great significance. But most researchers focus on the action mode and mechanism, we combined interacting protein pairs with immunoassays expecting fewer operational steps and less incubation time.LOCA is easy for automation and high-throughput in routine testing and has been widely applied in molecular diagnosis. LOCA is a homogeneous method without and the required operational steps and the incubation time are reduced. Another, LOCA can be operated in 96/384/1536 well plates where the sample amount could be as low as 5 μL. LOCA has been widely applied in the detection of proteins, polypeptides and small molecules. By LOCA, Zhang and et al. detected small molecules combining affinity recognition of aptamers and steric hindrance effect of thrombin.31,32 Here two DNAs respectively conjugated with Dig and biotin were accessible with thrombin. The streptavidin-donor beads and anti-Dig antibody conjugated-acceptors beads closed to each other forming a complex and resulted in detection signal. Inspired by their work, in this report we combined the complex signaling pathways with LOCA and expected an effective and fast assay in the detection of interacting proteins.
We successfully constructed recombinant plasmids expressing JAK1 and STAT3 proteins and, using Co-IP, identified that JAK1 and STAT3 interact in vitro. This pair of interacting proteins was also detected using a LOCA system, with high sensitivity. In comparison with Co-IP, which typically takes an additional day to perform, the detection time using LOCA can be reduced to within 1 h. Here, we established a new assay to detect the interacting protein pairs JAK1–STAT3, JAK1–GHR, and JAK1–IL4R and performed preliminary quantification of expression. This assay has potential for future detection of other interacting proteins and in other signing pathways, hoping that universal and high-throughput kits could be developed.
Conflict of interest
The authors declare no conflict of interest.Ethical approval
The Ethical Committee of Science and Technology Department of Southern Medical University approved this study (REC number: 20151058B).Guarantor
Tian-Cai Liu.Contributorship
Xin-Xin Guo, Han-Tao Wu, Si-Hui Zhuang and Zhen-Hua Chen contributed to experimental work. Tian-Cai Liu and Ying-Song Wu researched literature and conceived the study. Xin-Xin Guo, Rong-Liang Liang and Yao Chen were involved in protocol development, gaining ethical approval, and data analysis. Xin-Xin Guo wrote the first draft of the manuscript. All authors reviewed and edited the manuscript and approved the final version of the manuscript.Acknowledgements
This work was funded by the National Natural Science Foundation of China (Grant no. 21575058) and the National High Technology Research and Development Program (“863” program) of China (Grant no. 2014AA020904).References
- E. Stelloo, M. A. Versluis, H. W. Nijman, M. D. Bruyn, A. Plat, E. M. Osse, R. H. V. Dijk, R. A. Nout, C. L. Creutzberg and G. H. D. Bock, Oncotarget, 2016, 7, 39885–39893 Search PubMed.
- B. Song, H. Zhan, Q. Bian and J. Gu, Mol. Med. Rep., 2016, 13, 4475–4480 CAS.
- S. Grivennikov, E. Karin, J. Terzic, D. Mucida, G. Y. Yu, S. Vallabhapurapu, J. Scheller, S. Rose-John, H. Cheroutre and L. Eckmann, Cancer Cell, 2009, 15, 103–113 CrossRef CAS PubMed.
- M. Kujawski, M. Kortylewski, H. Lee, A. Herrmann, H. Kay and H. Yu, J. Clin. Invest., 2008, 118, 3367–3377 CrossRef CAS PubMed.
- H. Yu, D. Pardoll and R. Jove, Nat. Rev. Cancer, 2009, 9, 798–809 CrossRef CAS PubMed.
- A. H. Wagner, M. Conzelmann, F. Fitzer, T. Giese, K. Gulow, C. S. Falk, O. H. Kramer, S. Dietrich, M. Hecker and T. Luft, Biochem. Pharmacol., 2015, 96, 52–64 CrossRef CAS PubMed.
- M. van der Zee, A. Sacchetti, M. Cansoy, R. Joosten, M. Teeuwssen, C. Heijmans-Antonissen, P. C. Ewing-Graham, C. W. Burger, L. J. Blok and R. Fodde, Cancer Res., 2015, 75, 3608–3622 CrossRef CAS PubMed.
- L. Tam, L. M. Mcglynn, P. Traynor, R. Mukherjee, J. M. S. Bartlett and J. Edwards, Br. J. Cancer, 2007, 97, 378–383 CrossRef CAS PubMed.
- P. J. Murray, J. Immunol., 2007, 178, 2623–2629 CrossRef CAS.
- P. J. Murray, Curr. Opin. Pharmacol., 2006, 6, 379–386 CrossRef CAS PubMed.
- W. Wen, W. Liang, J. Wu, C. M. Kowolik, R. Buettner, A. Scuto, M. Y. Hsieh, H. Hong, C. E. Brown, S. J. Forman, D. Horne, R. Morgan, M. Wakabayashi, T. H. Dellinger, E. S. Han, J. H. Yim and R. Jove, Mol. Cancer Ther., 2014, 13, 3037–3048 CrossRef CAS PubMed.
- Q. Li, S. Gu, N. Li, Z. Li, W. Lai and Y. Zeng, J. Biomed. Eng., 2015, 32, 1255–1260 CAS.
- E. Schwarz, R. Izmailov, P. Lio and A. Meyer-Lindenberg, Schizophr. Bull., 2016, 42, 1334–1342 CrossRef PubMed.
- H. B. Engin, J. F. Kreisberg and H. Carter, PLoS One, 2016, 11, e152929 Search PubMed.
- Y. Saito, T. Nakagawa, A. Kakihana, Y. Nakamura, T. Nabika, M. Kasai, M. Takamori, N. Yamagishi, T. Kuga, T. Hatayama and Y. Nakayama, J. Cell. Biochem., 2016, 117, 2109–2117 CrossRef CAS PubMed.
- Y. Yu, Y. Li and Y. Zhang, Neurosci. Bull., 2016, 32, 171–176 CrossRef CAS PubMed.
- M. L. Valero, R. Sendra and M. Pamblanco, J. Proteomics, 2016, 136, 183–192 CrossRef CAS PubMed.
- J. Verhelst, D. De Vlieger and X. Saelens, J. Visualized Exp., 2015, 98, e52871 Search PubMed.
- Y. Hu, K. L. Yin, X. Ma and H. F. Xia, PLoS One, 2014, 9, e103695 Search PubMed.
- M. B. Einarson, E. N. Pugacheva and J. R. Orlinick, Cold Spring Harbor Protocols, 2007, 2007, t4757 Search PubMed.
- B. C. Chu and L. E. Orgel, Biochem. Biophys. Res. Commun., 2001, 283, 351–355 CrossRef CAS PubMed.
- Z. H. Chen, Y. S. Wu, M. J. Chen, J. Y. Hou, Z. Q. Ren, D. Sun and T. C. Liu, J. Fluoresc., 2013, 23, 649–657 CrossRef CAS PubMed.
- G. Lin, H. Huang, T. Liu, C. He, J. Liu, S. Chen, J. Hou, Z. Ren, W. Dong and Y. Wu, J. Virol. Methods, 2014, 206, 89–94 CrossRef CAS PubMed.
- Z. Q. Ren, T. C. Liu, J. Y. Hou, M. J. Chen, Z. H. Chen, G. F. Lin and Y. S. Wu, Luminescence, 2014, 29, 591–597 CrossRef CAS PubMed.
- E. F. Ullman, H. Kirakossian, S. Singh, Z. P. Wu, B. R. Irvin, J. S. Pease, A. C. Switchenko, J. D. Irvine, A. Dafforn, C. N. Skold and A. Et, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 5426–5430 CrossRef CAS.
- S. H. Zhuang, X. X. Guo, Y. S. Wu, Z. H. Chen, Y. Chen, Z. Q. Ren and T. C. Liu, J. Fluoresc., 2016, 26, 317–322 CrossRef CAS PubMed.
- J. W. Duan, Z. Lu, B. Jiang, B. V. Yang, L. M. Doweyko, D. S. Nirschl, L. E. Haque, S. Lin, G. Brown and J. Hynes, Bioorg. Med. Chem. Lett., 2014, 24, 5721–5726 CrossRef CAS PubMed.
- R. A. Kirken, H. Rui, M. G. Malabarba, M. Kawamura, J. J. Oshea and W. L. Farrar, J. Cell. Biochem., 1995, 38, 38 Search PubMed.
- K. Cerosaletti and J. H. Buckner, The Review of Diabetic Studies, 2012, 9, 188–200 CrossRef PubMed.
- Y. Koshelnick, M. Ehart, P. Hufnagl, P. C. Heinrich and B. R. Binder, J. Biol. Chem., 1997, 272, 28563–28567 CrossRef CAS PubMed.
- K. Zhang, Q. Yang, X. Zhu and K. Wang, Sens. Actuators, B, 2014, 200, 19–24 CrossRef CAS.
- K. Zhang, K. Wang, X. Zhu and M. Xie, Biosens. Bioelectron., 2015, 77, 264–269 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27424b |
| This journal is © The Royal Society of Chemistry 2017 |