
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Continuous flow synthesis of citrate capped gold nanoparticles using UV induced nucleation†
H. du Toita,
T. J. Macdonaldb,
H. Huanga,
I. P. Parkinb and
A. Gavriilidis *a
*a
aDepartment of Chemical Engineering, University College London, Torrington Place, London, WC1E 7JE, UK. E-mail: a.gavriilidis@ucl.ac.uk
bDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK
First published on 1st February 2017
Abstract
A new approach for synthesising gold nanoparticles of controlled size in the presence of trisodium citrate is presented. UV light is employed as a photoinitiator for the reduction of Au(III) by citrate. The UV induced nucleation takes place in a glass capillary tube (0.8 mm internal diameter) illuminated by a series of germicidal UVC lamps. This has been coupled sequentially with a heated coil to accelerate growth. In this way the processes of nucleation and growth are effectively separated. Slug flow is utilised in order to avoid tube fouling using heptane as segmenting fluid. Increasing UV intensity and temperature of the growth section lead to decrease of nanoparticle size, whilst varying UV exposure time results in a nonmonotonic effect on particle size. By varying UV intensity from 0 to 1461 mW cm−2 at a contant exposure time of 10 s, and a constant growth time of 20 min, the nanoparticles obtained range in size from 9.5 ± 1.3 nm to 36.1 ± 6.9 nm at a low growth temperature of 60 °C, and 6.6 ± 0.8 nm to 14.2 ± 6.4 nm at a high growth temperature of 100 °C.
Introduction
Applications of gold nanoparticles (AuNPs) are greatly dependant on their size, as well as the capping agent and adorned functional groups.1,2 Though much success has been achieved producing small AuNPs with tightly controlled size using thiol capping (such as in the Brust–Schiffrin method)3 the attachment of thiols is often irreversible and thus potential applications are limited.4Significant focus has been given to methods which do not employ such strongly bound capping agents and instead rely on weaker capping agents or surfactants which can easily be removed or exchanged such as in the classical Turkevich method,5 or techniques utilising oleylamine.6 In these cases particle size is instead determined by parameters such as reaction temperature or the concentration of the reducing agent.5–9 Since both temperature and concentration are inherently variable in a dynamic reactor system, largely due to imperfect mixing,10 batch processes often result in variability in the particle sizes produced, as well as high polydispersity. In order to improve on these techniques many researchers have opted for the use of microfluidic continuous flow systems.11–13 Such continuous flow systems provide great benefits such as controlled mixing and narrow residence time distribution (particularly in segmented flow systems), which can reduce the variation in particle size observed between batches.13
Using light to induce the reduction of gold with instantaneous and uniform exposure to light, may offer faster initiation of nucleation as compared to exposure to heat or a chemical reagent. Light can also be switched on or off at will to precisely control when reduction is initiated and when it stops.
Researchers have employed high energy UV light to promote the ‘clean’ reduction (reduction without the use of a chemical reducing agent) of Au(III) to form AuNPs14,15 which has been speculated to occur in stages as follows:14
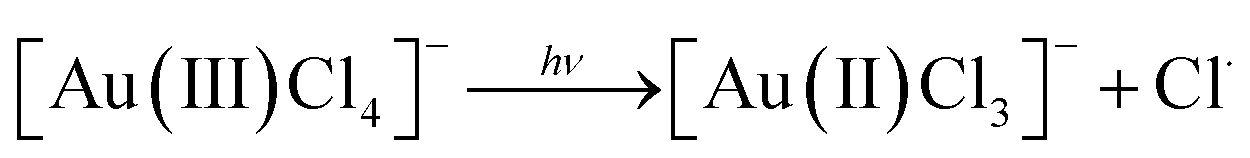
| 2[Au(II)Cl3]− → [Au(III)Cl4]− + [Au(I)Cl2]− |
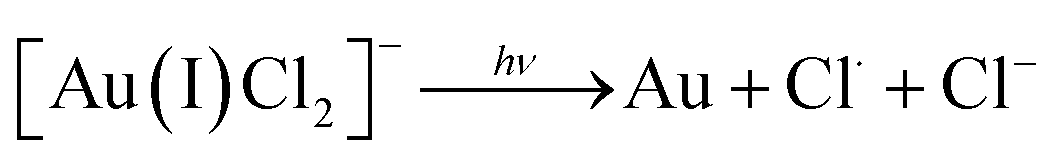
Studies which utilise UV reduction of tetrachloroauric acid have predominantly focused on batch systems, largely due to the fact that UV light is relatively weak at promoting reduction, and thus long exposure times (several hours or even days) are required.16–18
The use of photo activated chemical reducing agents coupled with UV light eliminates the variability originating from improper mixing. In this case all reactants can be mixed perfectly before the reducing agent is ‘switched on’ using UV light. Since the process of activating the reductant with light is relatively fast, systems of this type have often been employed in batch and continuous flow systems.19,20 This approach has been used to create AuNPs,21 gold-silver core–shell nanoparticles20 as well as a host of other platinum group metallic nanoparticles.22 In all cases a photosensitive reagent was employed which rapidly decays to form ketyl radicals in the presence of UV light. These radicals then go on to reduce Au(III) to its metallic state. Though such systems benefit from controlled activation of the reaction, they do not have the ability to instantaneously ‘switch-off’ the reduction process since, once activated, the reducing agents cannot be deactivated with similar ease. They also require the addition of extra stabilising components, such as polyvinylpyrrolidone,21 and thus the resulting AuNP solutions can be complex. This may limit the potential applications of particles produced in such a way, since the presence of photosensitive reducing agents, their resulting ketyl radicals, and added stabilisers may require additional downstream processing in order to remove them from solution prior to use.
Synthesising AuNPs through the use of UV light for the reduction of tetrachloroauric acid by sodium citrate has also been investigated.23–25 In this case there are two additional photochemical processes that occur which help to accelerate the formation of AuNPs. The first is a well-established process for the photoreduction of metal ions on the surface of colloidal metals.26,27 This occurs when citrate ions are decomposed by high energy UV light to form acetone-1,3-dicarboxylates as follows:
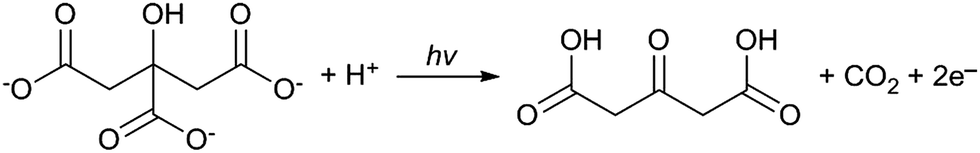
| Au3+ + 3e− → Au0 |
Since this process reportedly occurs on the surface of colloidal metals it can be credited for enhancing the growth of AuNPs after nucleation has occurred. The second process, as proposed by Yang et al., is a complexing and excitation process akin to the classical Turkevich method, except that the reduction of Au(III) by citrate is activated by high intensity UV light instead of heat.23 This is coupled with the formation of H˙ and OH˙ radicals by the irradiation of water with UV light and progresses as follows (* denotes an excited state):
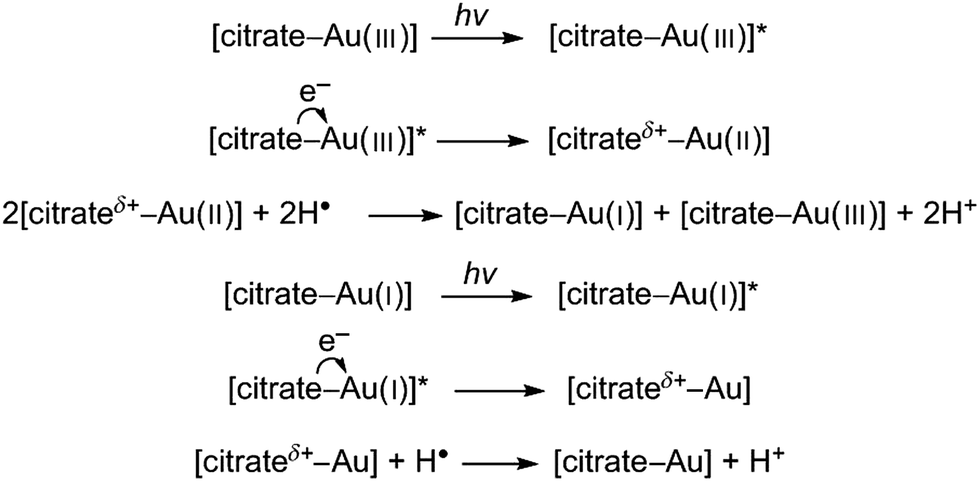
Thus, since UV light not only activates the reducing agent, but also drives the reaction forward, the photoreduction of Au(III) in the presence of citrate has the combined benefits of both UV light strategies discussed previously. Specifically the reducing agent can be activated after being fully mixed, and the rate of reaction can then be controlled by controlling the intensity of the UV light. However, unlike direct UV reduction, this method does not allow for instantaneous arresting of the reduction process since citrate will continue to reduce the gold precursor.28
To the best of our knowledge the reduction of tetrachloroauric acid to form AuNPs using a UV-citrate driven system has never been attempted in a continuous flow system. This would allow us to control particle size by controlling reduction rates with varied intensities of, or exposure times to, UV light. Since the applications of citrate capped particles are already well documented and, crucially, since they are already used in many different healthcare applications, the continuous size-controlled manufacture of such particles would be of potential interest to industry.
Experimental
Materials and nanoparticle characterisation
Heptane, tri-sodium citrate (Na3C6H5O7) and tetrachloroauric acid (HAuCl4) were of analytical grade and purchased from Sigma-Aldrich. All aqueous solutions used were prepared with RO purified water (15 MΩ). Fluorinatedethylenepropylene (FEP) tubing (1 mm ID, VICI Jour) was used for all fluidic pathways and connections. All flows were established using syringe pumps (Legato 270P, KD Scientific).Particle size measurements were performed by TEM imaging (Jeol 2010–200 kV) and differential centrifugal sedimentation (DCS) (CPS 24 000 Disc Centrifuge, CPS Instruments). Particle counts and sizes from TEM micrographs were obtained using the Pebbles software for unbiased particle sizing of TEM micrographs.29 Approximately 300 particles were used for each count. The DCS was calibrated by adjusting the input particle density to 12.3 g cm−3, such that the average particle size obtained by TEM and DCS agreed for a range of test citrate-capped AuNPs ranging between 8 nm and 30 nm.
It is worth noting that both techniques have shortcomings when it comes to analysing metallic nanoparticles. The energy from the electron beam used to capture TEM micrographs has the ability to produce AuNPs from unreacted HAuCl4 without the addition of any reducing agents.7 Some AuNPs observed could thus be artefacts created during analysis. DCS on the other hand relies on the forced sedimentation of particles and is thus limited by the diffusivity of the particles analysed. If the particles are too small they diffuse faster than they sediment and will therefore not be detected. AuNPs less than 3 nm cannot be detected by DCS. Dynamic light scattering (DLS) was also considered, but was discounted as it requires very high AuNP concentrations for small particles, and it is thus also unable to detect AuNPs less than 3 nm at the concentrations employed. Ultimately DCS was considered the best tool since it does not affect AuNP morphology and, unlike DLS, can easily analyse samples with multi-modal distributions.
Experimental setup
The experimental setup is illustrated in Fig. 1. Firstly an aqueous 0.6 mM HAuCl4 solution was mixed with an aqueous 3 mM Na3C6H5O7 solution using a simple Tee assembly (P-632, IDEX Health & Science LLC). Both solutions were delivered by the syringe pumps at an equal flow rate of 0.054 ml min−1. Following a coiled tubing loop which allowed for sufficient mixing, this aqueous stream was subsequently combined with a heptane stream flowing at 0.107 ml min−1 using a second Tee assembly in order to create segmented flow which served to prevent fouling on the tubing walls.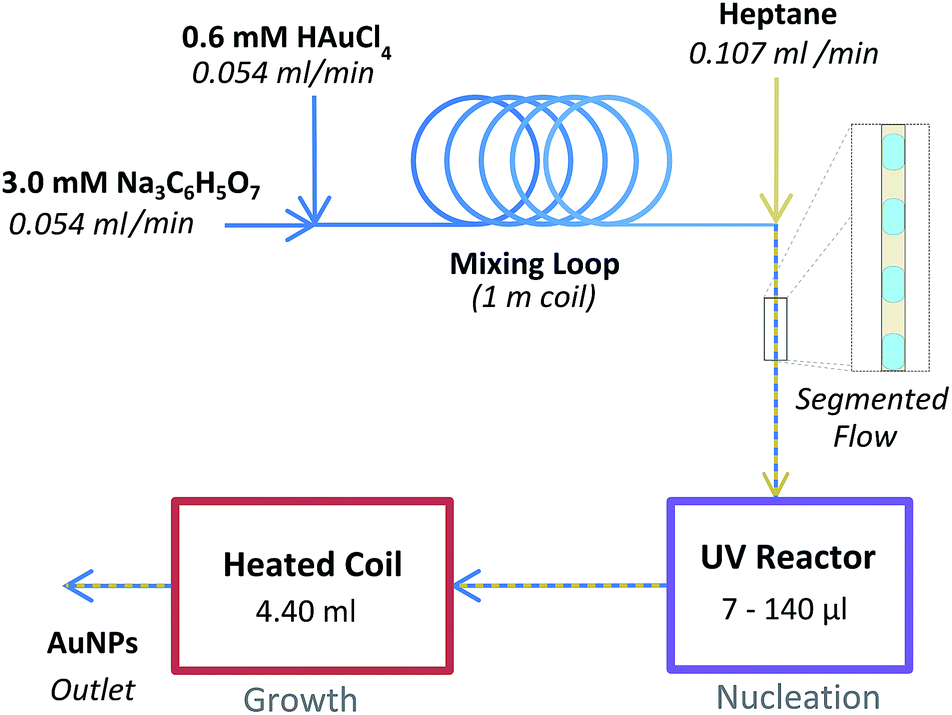 | ||
| Fig. 1 Schematic of reactor setup used. | ||
This stream subsequently flowed into the variable intensity UV reactor which contained a glass capillary (1.0 mm OD, 0.8 mm ID, Hirchmann). By using different capillaries covered with variable lengths of aluminium foil, the reactor volume exposed to UV light was altered (7, 17.5, 35, 70, and 140 μl). The reactor was placed in a 20 cm long hexagonal aluminium chamber polished with phosphoric acid. A 9 W germicidal UVC lamp (Arcadia UK) was mounted on to each of the 6 sides of the chamber. The lamps were connected to a control box of our own construction which allowed for individual control of each of the 6 lamps. The incident UVC power was measured at the centre of the reactor using a UVX Radiometer and UVX-25 sensor (Ultra-Violet Products Ltd). The incident UVC power was measured to be 456, 717, 971, 1122, 1310 and 1461 mW cm−2 for 1, 2, 3, 4, 5 and 6 UVC lamps respectively. The entire chamber was cooled externally by using an incubator (LR69802, Swell UK) such that the temperature of the reactant stream within the UV reactor never exceeded 40 °C during operation (when 3 or fewer lamps were used the temperature in the UV reactor did not exceed 30 °C).
Upon exiting the UV reactor the flow immediately entered a heated tubing coil, which acted as a AuNP growth section. This consisted of a long length of tubing coiled up and submerged in a glycerol bath. The volume of this growth reactor was 4.40 ml which, at the flow rate used, resulted in a heated residence time of 20 min. Finally the flow passed through a 20 psi back pressure regulator (P-791, IDEX Health & Science LLC), which served to prevent evaporation in the growth reactor, and into a sample collection vial. No samples were collected until a stable segmented flow profile was established (with uniform slug sizes of approximately 2 mm in length) and at least one reactor residence time had passed (at least 30 min).
Results and discussion
Preliminary UV study
Prior to the development of the continuous flow system (with sequential UV and thermal reactors), a simple feasibility study was conducted using only UV light as a photoinitiator for the reduction of HAuCl4 by Na3C6H5O7 to synthesise AuNPs. A range of mixtures of HAuCl4 and Na3C6H5O7 were passed through the UV reactor with only one 9 W lamp. The volume of the capillary exposed to UV was fixed at 70 μl and the corresponding residence time was varied using different flow rates. Fig. 2 shows the UV-Vis spectra results of one such experiment, where the concentration of citrate was in large excess (a 0.54 mM HAuCl4 solution was premixed with a 16.2 mM Na3C6H5O7 solution before being loaded into a syringe and pumped through the UV reactor).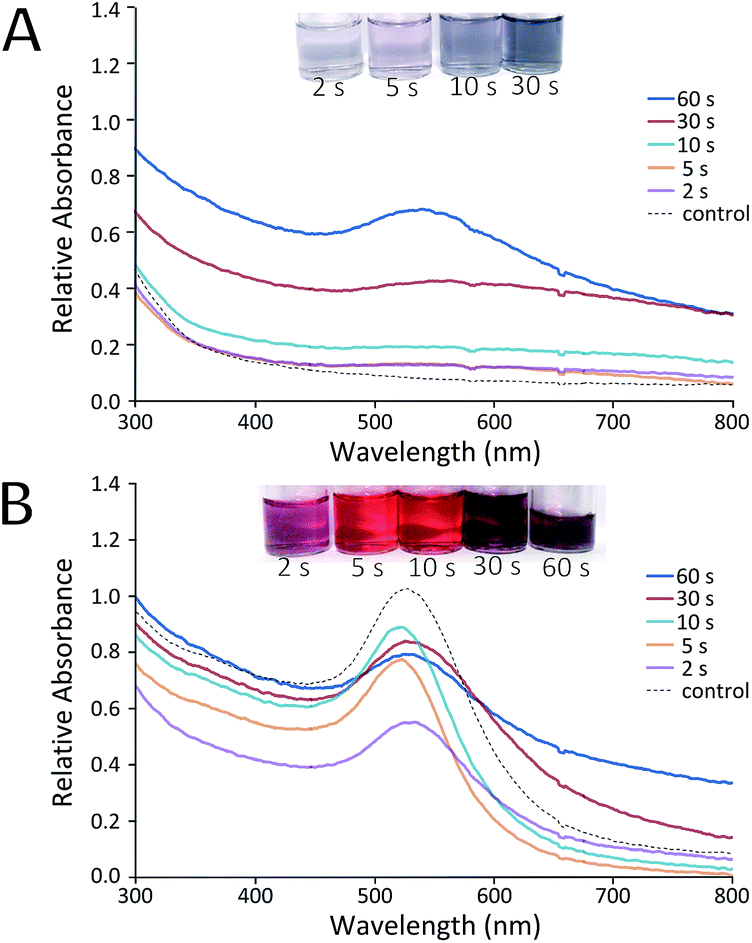 | ||
| Fig. 2 UV-Vis spectra of samples produced with exposure to UV light for indicated times at room temperature. (A) Samples immediately after experiment, (B) samples analysed after 5 days. Control experiments were conducted in the absence of exposure to UV light. | ||
There is a stark difference in the AuNP solutions produced immediately after exiting the UV reactor, and those samples analysed 5 days later. Immediately after reaction (Fig. 2A) most samples showed little evidence of particle formation when compared to a batch control experiment (where the reactants were mixed but not passed through the UV reactor). Only samples with UV exposure residence times of 30 s or more showed notable changes in absorption spectra. This suggests that the UV reactor was unable to complete the reduction of Au(III) within the allotted residence time. Instead, reduction (and thus growth of the AuNPs) continued uncontrolled following the reaction. Even though growth does not seem to have finished in the reactor, it appears that there has been an excessive level of nucleation in some cases. This is evidenced by the fact that samples exposed to 30 s or more of UV light showed significant signs of aggregation (Fig. 2B), producing particles of lower quality than the control. It was postulated that this aggregation was due to a high number of nuclei which were unstable and thus aggregated as they continued to grow.
Taking into account the fact that UV light is seemingly adept at producing nuclei, and the established fact that the thermally driven Turkevich process is capable of producing fully grown and stable AuNPs, it was consequently decided to develop a process which incorporated the best features from both processes. Varied intensity and exposure to UV light was used to control nucleation of AuNPs which were immediately grown to a final stable particle population using a heated growth system. This led to the development of the reactor system described in the Experimental section, which was utilised for all subsequent experiments.
Effect of UV intensity on particle size
The effect of the intensity of UV light used during exposure was investigated using constant UV reactor volume of 35 μl, and a constant growth temperature of 80 °C. Standard reactant compositions and flow conditions were used as described in the experimental setup section. Thus, the residence time under UV and under heat was kept constant at 10 s and 20 min respectively in each case, while UV intensity was varied by selectively switching a different number of UV lamps on. TEM micrographs of the particles, as well as the particle size distributions obtained by DCS analysis in each case, are shown in Fig. 3 (a table summarising the different average particle sizes from all experiments is shown in ESI Table S1†).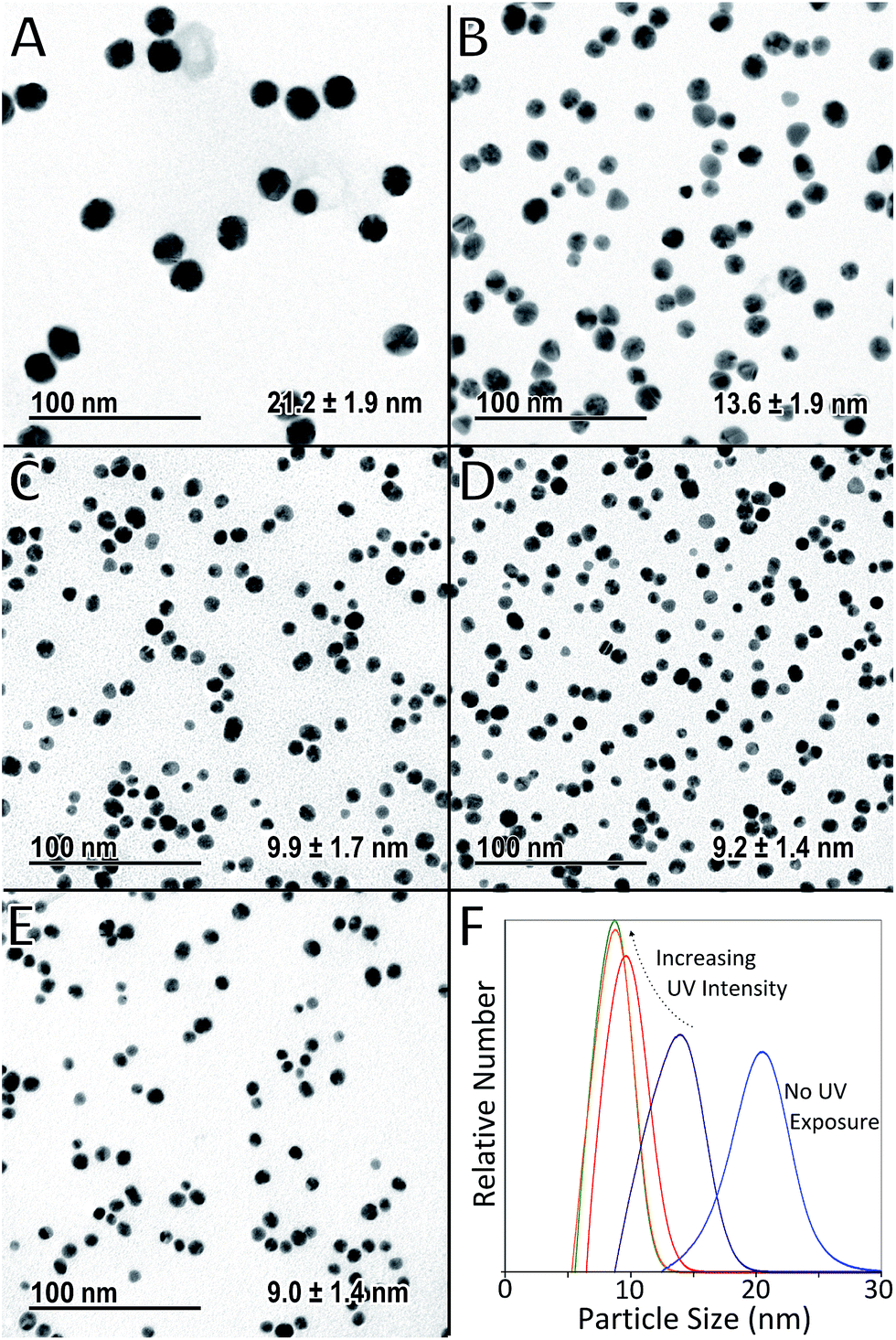 | ||
| Fig. 3 TEM micrographs of AuNPs produced with different intensities of UV light at constant UV residence time (10 s), growth residence time (20 min) and growth temperature (80 °C). (A) No UV lamps used, (B) one lamp, (C) two lamps, (D) three lamps, (E) six lamps and (F) particle size distributions obtained from DCS (normalised by area under each curve). | ||
Fig. 3A shows the particles obtained when the UV reactor remains switched off (i.e. only thermal initiation for the reduction of HAuCl4 by Na3C6H5O7 is used to synthesise particles in the heated coil). In this case the average particle size from TEM was found to be 21.2 nm with a standard deviation of 1.9 nm. This was concurrent with DCS analysis (Fig. 3F) which showed an average size of 20.3 nm with a standard deviation of 2.6 nm. Fig. 3B–E show the particles obtained when the UV reactor was switched on at different intensities (i.e. when UV light and heat were used sequentially as photo and thermal initiators for the reduction of HAuCl4 by Na3C6H5O7 to form AuNPs). The particle sizes determined by TEM were 13.6 ± 1.9 nm, 9.9 ± 1.7 nm, 9.2 ± 1.4 nm and 9.0 ± 1.4 nm for particles synthesised using one, two, three and six UV lamps respectively. These results showed exceptional concurrence with the corresponding particle sizes obtained by DCS which were 13.6 ± 2.1 nm, 9.8 ± 1.5 nm, 8.7 ± 1.4 nm and 8.7 ± 1.4 nm. The strong agreement in particle size obtained using different techniques validated the calibration of the DCS and thus this technique was used preferentially for subsequent particle size measurements.
These results demonstrate that a degree of control of particle size can be achieved using varied UV intensities for a relatively short UV exposure time of just 10 s. With increasing UV intensity both the average particle size and polydispersity are reduced. This suggests that a higher intensity of UV light leads to more nucleation events. Assuming the nuclei are stable and do not aggregate, this increase in the number of nuclei results in an increase of smaller particles, since the concentration of HAuCl4 available for growth becomes limiting. This effect appears to have an upper limit, since there is very little difference in the particles produced when using 3 or 6 UV lamps. Thus, there may be a limit to the amount of nuclei produced possibly dependant on the concentrations of Na3C6H5O7 and HAuCl4 employed.
Effect of growth temperature on particle size
The effect of the growth temperature on the final particle size was investigated using a constant UV reactor volume of 35 μl ensuring the UV exposure time and growth time were constant at 10 s and 20 min respectively. The temperature of the growth reactor was varied between 60 and 100 °C. For comparison, the synthesis was also conducted without any UV exposure. The average particle size and its standard deviation obtained are shown in Fig. 4.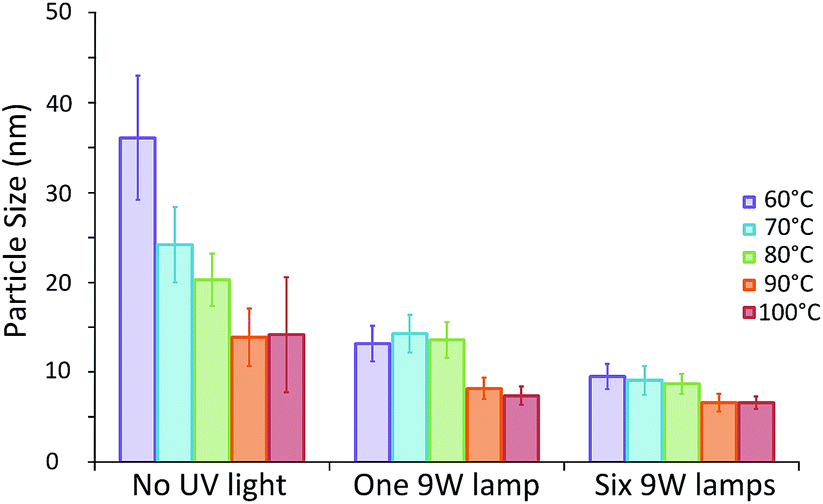 | ||
| Fig. 4 Average particle size of AuNPs produced with 20 min residence time in the thermal reactor at different temperatures following 10 s of low or high intensity UV exposure (one and six UV lamps respectively), as well as particles produced without UV induced nucleation. Data obtained by DCS analysis. Error bars refer to particle size standard deviation. | ||
There is a drastic change in particle sizes synthesised in the absence of UV light with temperatures of up to 90 °C. The average particle size is shown to decrease significantly with increasing temperature down to a minimum of approximately 14 nm for particles synthesised at 90 °C or above (13.9 ± 3.2 and 14.2 ± 6.4 nm for 90 and 100 °C respectively). This indicates that increasing the synthesis temperature leads to more nucleation events, resulting in a smaller average particle size up to a point where no further enhancement is possible. This differs to what was observed in batch by Takiyama30 and Wuithschick et al.7 who found that a minimum particle size was obtained at a synthesis temperature of 60 °C, after which particle size increased with temperature. The difference in trends observed in both of these previous studies and the current study can likely be attributed to different mixing techniques employed. In both studies the Na3C6H5O7 was added to a hot HAuCl4 solution under constant mixing. Thus, the reaction was instantly initiated before the solutions could be adequately mixed. In this case a lower temperature would result in a slower reaction rate. Even though the mixing time would be the same, the nucleation period would be longer and thus possibly less affected by mixing. In our study the citrate and HAuCl4 are already premixed before being rapidly heated. A higher temperature, and therefore a higher reaction rate, allows for more nucleation at an early stage of the process consequently resulting in a smaller average particle size.
In contrast, when the UV reactor is employed prior to the heated growth reactor, the same temperature dependence of particle size is not observed. In fact the average particle size seems to be unaffected by growth reactor temperature, when this is below 80 °C, both when 1 and 6 UV lamps are employed (approximately 13.7 and 9.1 nm respectively). This suggests that in these cases all nucleation occurs in the UV reactor and thus the temperature of the heated section has no effect on the average particle size obtained. This is a promising result, since it implies that the process is robust under these conditions, since it is unaffected by fluctuations in the temperature of the growth reactor.
It is only when temperatures of 90 °C or above are used that the average particle size is affected. When either 90 or 100 °C is used the average particle size is further reduced to approximately 7.8 or 6.6 nm for 1 or 6 UV lamps respectively. This may be due to additional nucleation in the growth reactor resulting in even smaller final particle sizes. The particle sizes observed for both 90 and 100 °C are again similar, suggesting that this enhancement is limited up to a point, as was observed when using the systems in the absence of UV light.
The AuNPs obtained were found to be very stable. Fig. 5 shows a TEM image of particles synthesised with 10 s of high intensity UV exposure (6 lamps) followed by 20 min of growth at 100 °C taken after 28 days of storage at room temperature. The average particle size (obtained by TEM analysis) was 6.8 ± 0.9 nm. This is almost identical to that obtained by DCS immediately after the experiment was completed, which was 6.6 ± 0.8 nm.
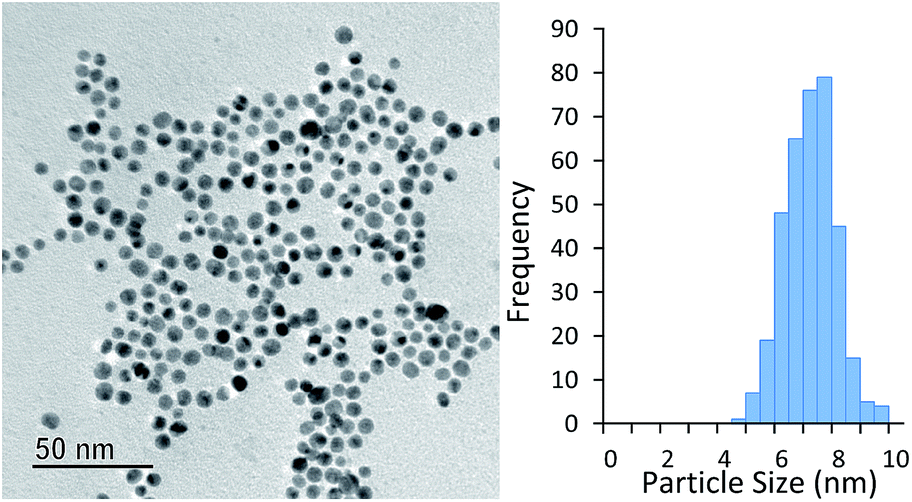 | ||
| Fig. 5 Particles synthesised with 10 s of UV exposure (six UV lamps) and subsequently grown at 100 °C for 20 min. TEM taken 28 days after experiment was conducted. | ||
Effect of UV exposure time on particle size
The final variable investigated was the effect of residence time under UV irradiation. For this study, a series of identical glass capillaries were modified by covering them with different lengths of adhesive aluminium foil tape. This was done such that varied lengths (and volumes) of the capillaries were exposed to UV light, and thus the UV exposure time could be altered, whilst keeping temperature and residence time in the thermal reactor constant at 80 °C and 20 min respectively. Two cases were investigated, specifically under the highest UV intensity (6 UV lamps), and the lowest UV intensity (1 UV lamp). Fig. 6 shows the average particle size and the standard deviation of the particles produced.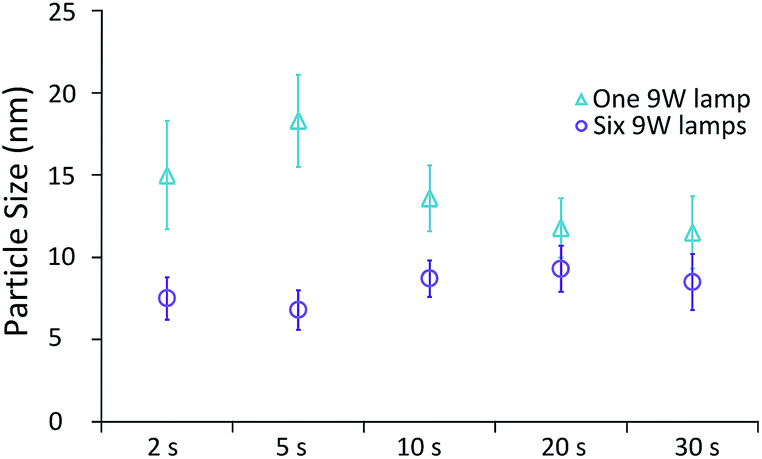 | ||
| Fig. 6 Average particle size of AuNPs produced with different exposure times to low and high intensity UV light (one and six UV lamps respectively). The temperature and residence time in the thermal reactor was 80 °C and 20 min respectively. Data obtained by DCS analysis. Error bars refer to particle size standard deviation. | ||
Under low UV intensity (1 lamp), there appears to be a local maximum of particle size with 5 s of UV irradiation. The existence of this peak was verified by repeat experiments between 2 s and 10 s of UV exposure (see ESI Fig. S1†). At higher irradiation times particle size appears to decrease with increasing irradiation time. Overall the standard deviation (i.e. polydispersity) decreases with exposure time. Conversely, when the highest intensity of UV light is used, particle size appears to be minimised at 5 s of UV irradiation with a slight increase of particle size at longer exposure times. The standard deviation however appears almost constant regardless of exposure time used.
The occurrence of local maximum and minimum in particle size indicates that there may be several processes that affect particle size which are competitive. For example, the different processes for enhancing AuNP formation discussed in the introduction could be opposing each other. Since one process (the degradation of citrate to form acetone-1,3-dicarboxylate) reportedly promotes growth, and the other (excitation and subsequent reduction of citrate–Au complex) can enhance both nucleation and growth there may be competition due to different reaction rates.
Alternatively, the local maximum could be caused by different growth mechanisms as illustrated by Fig. 7. After 5 s of UV exposure, there could be an excess of unstable nuclei formed. These nuclei could then be growing by aggregation during the transit between the UV reactor and the thermal reactor following the mechanism proposed by Polte et al.31 Since less HAuCl4 would be consumed at shorter exposure times further nucleation could occur in the heated coil. When higher intensity UV light or longer exposure times are used the nuclei grow to a more stable, or quasi-stable, size within the UV reactor and thus do not grow by aggregation during transit. This sequence of events would also explain the trend in polydispersity observed with the greatest polydispersity also occurring at short UV exposure times.
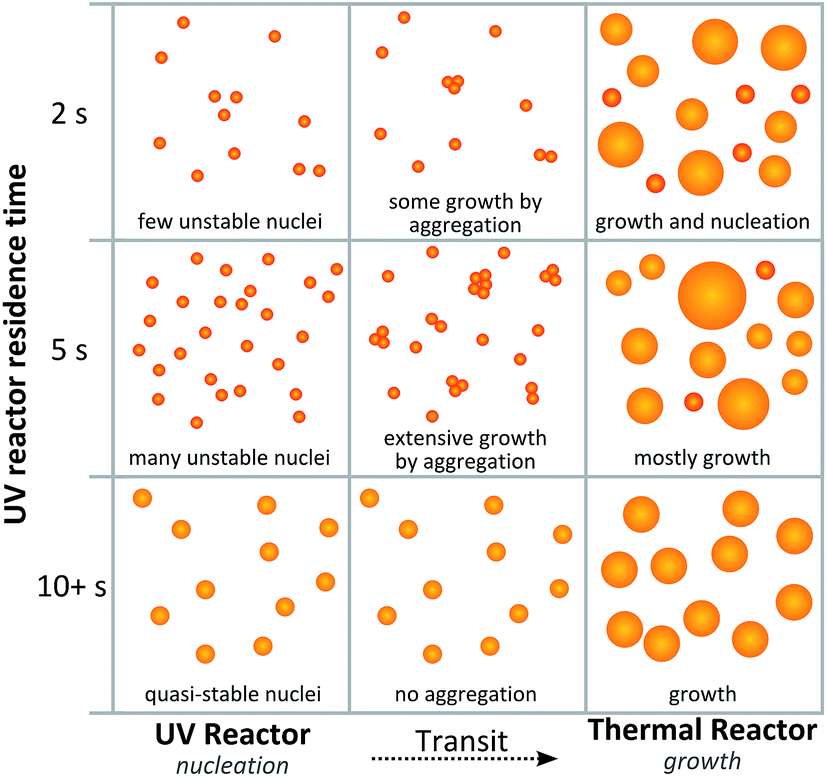 | ||
| Fig. 7 Proposed scheme for particle formation which would result in local maximum particle size at low UV intensities. | ||
Conversely a slight increase in particle size is observed with exposure time when high intensity UV light is used. This effect may be due to complementary effects in the UV reactor and the thermal ‘growth’ reactor. As already determined by the growth temperature dependence of particle size, a degree of nucleation can also occur in the heated coil. When short UV residence times are used the high intensity light allows for the synthesis of quasi-stable nuclei, and at the same time there may still be sufficient unreacted HAuCl4 for further nucleation in the heated coil. Since nucleation could then occur in both reactors, the total number of nuclei would be maximised, resulting in smaller average particle sizes. At longer high intensity UV exposure times, the quasi-stable nuclei may grow to a larger size in the UV reactor, leaving an insufficient amount of HAuCl4 for further nucleation in the heated coil.
Conclusions
We have demonstrated a new approach to the citrate based synthesis of AuNPs. By developing a system which uses both UV and thermal initiation of the reduction of Au(III) by citrate in complementary fashion, we have been able to effectively separate nucleation and growth. We have also demonstrated that particle size can be controlled by manipulating the nucleation stage of AuNP synthesis using UV light and producing reliable results using exposure times as short as a few seconds. This is a significant advancement from typical UV processes which often require hours of exposure for particle synthesis and have thus been unsuitable for continuous flow production.Acknowledgements
The authors thank the EPSRC for financial support (EP/M015157/1) through the Manufacturing Advanced Functional Materials (MAFuMa) scheme, and the MAFuMa team with a special thanks to Prof. Peter Dobson.References
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS PubMed.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802, 10.1039/C39940000801.
- Y. Xue, X. Li, H. Li and W. Zhang, Nat. Commun., 2014, 5, 4348 CAS.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- H. Hiramatsu and F. E. Osterloh, Chem. Mater., 2004, 16, 2509–2511 CrossRef CAS.
- M. Wuithschick, A. Birnbaum, S. Witte, M. Sztucki, U. Vainio, N. Pinna, K. Rademann, F. Emmerling, R. Kraehnert and J. Polte, ACS Nano, 2015, 9, 7052–7071 CrossRef CAS PubMed.
- N. G. Bastús, J. Comenge and V. Puntes, Langmuir, 2011, 27, 11098–11105 CrossRef PubMed.
- C. Ziegler and A. Eychmüller, J. Phys. Chem. C, 2011, 115, 4502–4506 CAS.
- K. S. Elvira, X. C. I. Solvas, R. C. R. Wootton and A. J. deMello, Nat. Chem., 2013, 5, 905–915 CrossRef CAS PubMed.
- J. Ftouni, M. Penhoat, A. Addad, E. Payen, C. Rolando and J.-S. Girardon, Nanoscale, 2012, 4, 4450–4454 RSC.
- S. Duraiswamy and S. A. Khan, Small, 2009, 5, 2828–2834 CrossRef CAS PubMed.
- H. Tsunoyama, N. Ichikuni and T. Tsukuda, Langmuir, 2008, 24, 11327–11330 CrossRef CAS PubMed.
- Y. Shang, C. Min, J. Hu, T. Wang, H. Liu and Y. Hu, Solid State Sci., 2013, 15, 17–23 CrossRef CAS.
- K. Kurihara, J. Kizling, P. Stenius and J. H. Fendler, J. Am. Chem. Soc., 1983, 105, 2574–2579 CrossRef CAS.
- F. Kim, J. H. Song and P. Yang, J. Am. Chem. Soc., 2002, 124, 14316–14317 CrossRef CAS PubMed.
- K. Esumi, J. Hara, N. Aihara, K. Usui and K. Torigoe, J. Colloid Interface Sci., 1998, 208, 578–581 CrossRef CAS PubMed.
- A. Kameo, A. Suzuki, K. Torigoe and K. Esumi, J. Colloid Interface Sci., 2001, 241, 289–292 CrossRef CAS PubMed.
- K. L. McGilvray, M. R. Decan, D. Wang and J. C. Scaiano, J. Am. Chem. Soc., 2006, 128, 15980–15981 CrossRef CAS PubMed.
- C. M. Gonzalez, Y. Liu and J. C. Scaiano, J. Phys. Chem. C, 2009, 113, 11861–11867 CAS.
- L. Hafermann and J. M. Köhler, J. Nanopart. Res., 2015, 17, 1–8 CrossRef CAS.
- L. Hafermann and J. M. Köhler, Chem. Eng. Technol., 2015, 38, 1138–1143 CrossRef CAS.
- S. Yang, Y. Wang, Q. Wang, R. Zhang and B. Ding, Colloids Surf., A, 2007, 301, 174–183 CrossRef CAS.
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plech, J. Phys. Chem. B, 2006, 110, 15700–15707 CrossRef CAS PubMed.
- M. A. Uppal, A. Kafizas, M. B. Ewing and I. P. Parkin, New J. Chem., 2010, 34, 2906–2914 RSC.
- M. Maillard, P. Huang and L. Brus, Nano Lett., 2003, 3, 1611–1615 CrossRef CAS.
- A. M. Ahern and R. L. Garrell, Anal. Chem., 1987, 59, 2813–2816 CrossRef CAS.
- C. Li, D. Li, G. Wan, J. Xu and W. Hou, Nanoscale Res. Lett., 2011, 6, 1–10 Search PubMed.
- S. Mondini, A. M. Ferretti, A. Puglisi and A. Ponti, Nanoscale, 2012, 4, 5356–5372 RSC.
- K. Takiyama, Bull. Chem. Soc. Jpn., 1958, 31, 944–950 CrossRef CAS.
- J. Polte, T. T. Ahner, F. Delissen, S. Sokolov, F. Emmerling, A. F. Thünemann and R. Kraehnert, J. Am. Chem. Soc., 2010, 132, 1296–1301 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27173a |
| This journal is © The Royal Society of Chemistry 2017 |