
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermoresponsive polyamic acid-conjugated gold nanocarrier for enhanced light-triggered 5-fluorouracil release†
Pa Fan Hsiaoabc,
Rajeshkumar Anbazhagand,
Chou Hsiao-Yingd,
Adhishankar Vadivelmurugand and
Hsieh-Chih Tsai *d
*d
aDepartment of Dermatology, Mackay Memorial Hospital, Taipei, Taiwan
bMackay Medicine, Nursing and Management College, Taipei, Taiwan
cMackay Medical College, New Taipei City, Taiwan
dGraduate Institute of Applied Science and Technology, National Taiwan University of Science and Technology, Taipei 106, Taiwan. E-mail: h.c.tsai@mail.ntust.edu.tw
First published on 25th January 2017
Abstract
In this study, a light-responsive 5-fluorouracil (5-FU) drug carrier system was developed by combining gold nanoparticles with thermosensitive hydrophilic polyamic acid. The hydrophilic polyamic acid was cyclized when the synthesized gold polymer (GP) nanocluster was exposed to heat, and gold acted as a photothermal transfer agent, converting light energy into heat energy. After polyamic acid modification, the size of the GP nanocluster increased to 100 nm. The increase in the size of the GP nanocluster might be due to multiple chemical bonds between gold and the thiol group present in polyamic acid. The size of the GP nanocluster decreased after exposure to a temperature of approximately 30 °C, which changed polyamic acid into cyclized polyimide and cleaved the gold–thiol bonds. The light-induced successful release of the anticancer drug 5-FU from the GP nanocluster was confirmed through UV-vis spectroscopy. In addition, no toxicity was observed in cells treated with the GP loaded with 5-FU (GP–FU) under dark conditions. This result indicates the biocompatibility of the GP cluster and thus demonstrates the safety of the carrier when the anticancer drug was loaded inside the cluster. HeLa cells irradiated with light apparently died, indicating that light successfully induced drug release. These findings suggest that the synthesized thermosensitive nanocluster can be used as a nanocarrier.
1. Introduction
The development and tailoring of multifunctional nanoparticles with combined properties, such as stability in biological media, absorption, emission, and sensitivity to external stimuli (pH, light, or temperature), are of major interest in biomedical and bioengineering applications.1 Because of the remarkable physicochemical properties of nanomaterials, their application to medicine can provide possible solutions for various challenges currently encountered in cancer treatments.2,3 The present study aims to investigate a multifunctional nanoparticle containing an external stimuli (such as light or heat)-responsive polymer.4,5A gold nanoparticle (GNP) is a unique and safe material for biomedical application. Because of their tunable surface plasmon resonance and photothermal effects, GNPs are a promising candidate for various theranostic and thermo chemotherapeutic applications.6,7 Among other optical probes, GNPs have excellent advantages such as noncytotoxicity, excellent biocompatibility, easy synthesis and surface functionalization, strong light absorption and scattering effect, and high photothermal conversion rate and photostability.8,9 To enhance drug loading and delivery, the surface of GNPs is often tailored with polymers and some small organic molecules.10,11 Furthermore, the nanoparticle accumulation in the tumor region can be controlled by external stimuli such as the magnetic field, electrical field, light, and ultrasound.12–14 The local application of external heat can also trigger the site-specific accumulation of intravenously injected thermal-responsive polymers and their cargo.15 In addition to available applications, using light as an external stimulus and enabling spatiotemporal control of cargo delivery have attracted considerable interest. In recent applications, drug-encapsulated carriers measuring 100–500 nm in size and containing a photoswitch for payload delivery have been developed.16 These new strategies can effectively amplify the selectivity of particular effects, because the timing and intensity of stimuli can be precisely controlled.
When anticancer drugs are encapsulated within a polymeric matrix, the drug carrier must have low toxicity. Therefore, the design, synthesis, and tailoring of polymer vesicles for a broad range of applications, ranging from material science to biomedical science, have attracted considerable interest. Xia et al. successfully demonstrated that gold nanocages covered with a thermally responsive polymer, poly (N-isopropylacrylamide), could release anticancer drugs upon laser irradiation.17 Therefore, GNPs could act as a highly light-responsive drug carrier through the translocation of light energy into heat. The thermally sensitive P(NIPAM) polymer exhibits a hydrophobic and hydrophilic transition at approximately 32 °C.18 This type of phase transition behavior would cause difficulty in loading and releasing the anticancer drug. When the drug loaded in P(NIPAM) is exposed to heat exceeds 32 °C, the original hydrophilic polymer chain starts to collapse and becomes hydrophobic. Consequently, stacking the loaded drug inside the collapsed polymer becomes easier than releasing it. Therefore, to design a new thermal responsive polymer system to provide the highest therapeutic effect is desirable.
Poly(amic acid) (PAA) is a class of negatively charged copolymers synthesized through a polycondensation reaction between diamines and dianhydrides.19 PAA contains amide and carboxylic groups in its backbone. However, they are commonly recognized as intermediate polymers in the formation of polyimides. Unlike aromatic PAA and aromatic polyimides, aliphatic PAA is soluble in various dipolar aprotic solvents. Furthermore, the presence of carbonyl groups allows PAA to interact with nearby –NH2 groups, commonly observed in biomaterials, through hydrogen bonding. Nevertheless, aromatic PAA has less biomedical applications because of its poor solubility in water.20,21 We particularly focused on PAA as a potential candidate for drug and gene delivery applications. In our previous study, we synthesized a PAA for ion exchange-induced (calcium ion) DNA delivery.22 We synthesized the PAA through a polycondensation reaction between aliphatic diamine and aliphatic dianhydride. Moreover, we observed that the synthesized PAA cyclized to form polyimides upon exposure to heat or light, and the resultant polyimide product exhibited excellent biocompatibility.23 Therefore, the synthesized PAA can be utilized as a thermoresponsive nanocarrier in biomedical applications.
In this study, GNPs tailored with light and thermosensitive polyamic acid chemically conjugated to a gold nanomaterial were prepared and then loaded with 5-fluorouracil (5-FU), an anticancer drug. The prepared GP nanocluster was identified through Fourier transform infrared spectroscopy (FT-IR), proton nuclear magnetic resonance (1H NMR), and ultraviolet-visible spectroscopy (UV-visible) to confirm its photophysical properties. In addition, a dynamic light scattering (DLS) instrument was used to determine the size of the synthesized nanocluster. The thermosensitive property of the synthesized GNPs was confirmed using temperature-dependent UV and DLS measurements. The morphology of the synthesized nanocluster was evaluated through HRTEM microscopy. Furthermore, the MTT assay was performed to demonstrate the biocompatibility of the nanocluster. Fluorescence microscopy images were taken to identify the light-triggered drug delivery of the GP–FU nanocluster.
2. Experimental section
2.1. Materials
Hexamethylenediamine, diethylenetriaminepentaacetic dianhydride, and biological water were acquired from Sigma-Aldrich. Dimethyl sulfoxide was obtained from Macron Fine Chemicals. Regenerated cellulose tubular membrane with 1000 nominal value was purchased from Orange Scientific. All solutions and reagents were used without further purification.2.2. Synthesis of GNPs
The synthesis scheme depended on the reduction of Au(III) by using sodium borohydride in the presence of pure reduced glutathione. In a typical reaction, 30 μmol of a HAuCl4 was dissolved in 20 mL of deionized water containing 50 μL of 2 M NaOH, followed by the addition of 200 μL of 50 mM HAuCl4 aqueous solution (ratio of Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :glutathione = 1:3). The mixture was stirred for 5 min. Subsequently, 400 μL of 50 mM NaBH4 was added dropwise. The reaction mixture was stirred for 15 h at room temperature and then purified from free ligands by applying three cycles of centrifugation; the resulting dispersions were stored at 4 °C for further use. Size analysis confirmed the synthesized GNPs has particle size about 15 nm.
:glutathione = 1:3). The mixture was stirred for 5 min. Subsequently, 400 μL of 50 mM NaBH4 was added dropwise. The reaction mixture was stirred for 15 h at room temperature and then purified from free ligands by applying three cycles of centrifugation; the resulting dispersions were stored at 4 °C for further use. Size analysis confirmed the synthesized GNPs has particle size about 15 nm.
2.3. Synthesis of methylenediamine–diethylenetriaminepentaacetic dianhydride polymer
The polymer was synthesized by conjugating the amine group of hexamethylenediamine (HMDA) to the carboxylic group of diethylenetriaminepentaacetic dianhydride (DTPA), as described in our previous study.22 Briefly, DTPA in anhydrous DMSO was slowly added to a 10 mL solution of HMDA in anhydrous DMSO, and the reaction was stirred at room temperature for 12 h under nitrogen atmosphere to produce the HMDA–DTPA polymer. The product was collected after dialysis against deionized water and freeze drying, and the resultant sponge-like polyamic acid polymer was stored at 0 °C for further use.2.4. Synthesis of the thiol group-terminated HMDA–DTPA–cysteamine polymer
The HMDA–DTPA polymer was dispersed in 10 mL of deionized water, and 100 and 250 μL of 100 mM EDC and NHS solutions were then added to the polymer solution, respectively. Carboxylic groups on the polymer surface were activated to form reactive NHS ester intermediates. After 30 min, cysteamine (Cys) (molar ratio of Cys to HMDA–DTPA is 1:1) was added to the carboxyl group-activated polymer solution, and the resultant solution was stirred for 4 h to complete amine coupling with the activated carboxylic group. The thiol group-terminated polymer product was collected after dialysis against deionized water and freeze drying, and the resultant product was stored at 4 °C for further use.
2.5. Synthesis of a polymer-functionalized GNP
5 mg HMDA–DTPA–Cys–SH was added to 5 mL of GNPs in water. The mixtures were maintained under mild stirring overnight at room temperature. The resulting suspensions were centrifuged at 10000 rpm for 30 min to achieve GP sedimentation, and the supernatant was discarded. The resulting residue was redissolved in 1 mL of double distilled water, repeatedly washed for at least three times to remove the excess polyamic acid polymer, and stored at 4 °C for further use. The successful functionalization was confirmed through DLS, UV, FT-IR, H1 NMR, and TEM instruments.
2.6. FU loading in GP
5-FU was loaded in GPs by stirring 2 mL of a 5-FU (1 mg mL−1) solution in DMSO solution with 2 mL of GPs (2 mg mL−1) for 12 h at room temperature. The dispersion was then centrifuged (10000 rpm for 30 min) to collect 5-FU-loaded GPs (GP–FU). The concentration of 5-FU in the supernatant was determined using a UV-vis spectrophotometer at 265 nm and the weight of the drug entrapped in the nanoparticles was calculated with a calibration curve.
2.7. Drug release experiment
We added 1.0 mL of a GPF solution in a dialysis membrane (1000 Da), and the resulting membrane was covered with 15 mL of PBS or water in a 20 mL vial. The resulting setup was irradiated with UV light at different time durations (0–20 min). After irradiation, the desired amount of PBS or water was taken out and replaced with the same amount of PBS or water in the experiment to maintain the same amount of liquid. The released drug in the filtrate was determined by measuring the absorption at 270 nm by using a UV-vis spectrometer.2.8. Cell viability assay
A HeLa cell line was cultured and maintained in DMEM (Wisent Inc., USA) supplemented with 10% fetal bovine serum (Wisent Inc.) and 1% sodium pyruvate (Wisent Inc.) at 37 °C in a humidified atmosphere of 5% CO2. To determine the cytotoxicity of the synthesized nanomaterials, the cells were plated in triplicate at a density of 2 × 104 cells per well in a 96-well plate. After incubation for 24 h, the cells were exposed to 1, 2, 3, 4, and 5 μg of 5-FU or an equivalent dose of GP–FU or the nanocluster alone. Cytotoxicity was assessed 24 h after treatment by using the standard MTT (5 mg mL−1) assay.2.9. In vitro light-triggered drug release cytotoxicity
The HeLa cells in each well were cultured on a 96-well plate at a density of 2 × 104 cells for 24 h, followed by treatment with 5-FU, GP, or GP–FU (at a desired concentration of 5-FU) with a new medium. For laser treatment, the cells were irradiated for 5 min (365 nm) followed by incubation for 24. To investigate the light-triggered cytotoxicity, the old medium was replaced with a fresh medium containing the MTT reagent (5 mg mL−1), and the cells were allowed to incubate for 2–4 h. After formazan dye formation, the MTT solution was removed followed by the addition of DMSO. A microplate reader was used to determine cell viability by evaluating the absorbance measurement of purple formazan formation.2.10. Cell apoptosis imaging
HeLa cells were cultured in a confocal dish at a density of 2 × 105 and grown for 24 h in an incubator at 37 °C in a humidified atmosphere of 5% CO2. Subsequently, the cells were split into two groups: one group for staining with Alexa Fluor 488 annexin V to identify the early stage of apoptosis and staining with propidium iodide (PI) to identify dead cells, and the other group for dark toxicity examination. The staining with Alexa Fluor 488 annexin V was performed according to the manufacturer's protocol. Briefly, the old medium in the confocal dish was removed, cells were washed three times with PBS, and 100 μL of 10 mM annexin-binding buffer (50 mM Alexa Fluor binding buffer diluted with five times of double distilled water) containing 5 μL of Alexa Fluor 488 annexin V was added to the cells. The cells were incubated for 15 min at room temperature and then washed three times with PBS. For PI staining, 100 μL of HEPES buffer containing 0.1 μg mL−1 of PI was subsequently used to stain the cells for 5–10 min at room temperature.3. Results and discussion
Thermoresponsive thiol group-terminated polyamic acid was synthesized through the anhydride ring opening followed by condensation with diamine, according to our previously reported method with little modifications, as shown in Scheme 1. The HMDA–DTPA thermoresponsive polymer was characterized using various techniques such as FTIR and 1H NMR to corroborate the polymer formation. The successful conjugation of HMDA to DTPA was confirmed through FT-IR spectroscopy; as illustrated in Fig. S1,† the peaks appearing at 1684 and 1650 cm−1 correspond to the carbonyl stretching vibration and the bending vibration of amide, respectively. These typical peaks confirm the successful conjugation of DTPA to HMDA through amide bond formation (HMDA–DTPA). Finally, Cys was conjugated to the HMDA–DTPA polymer through EDC/NHS chemistry, and the corresponding primary amine of Cys appeared at 3215 cm−1. The product was collected through dialysis against double distilled water followed by freeze drying.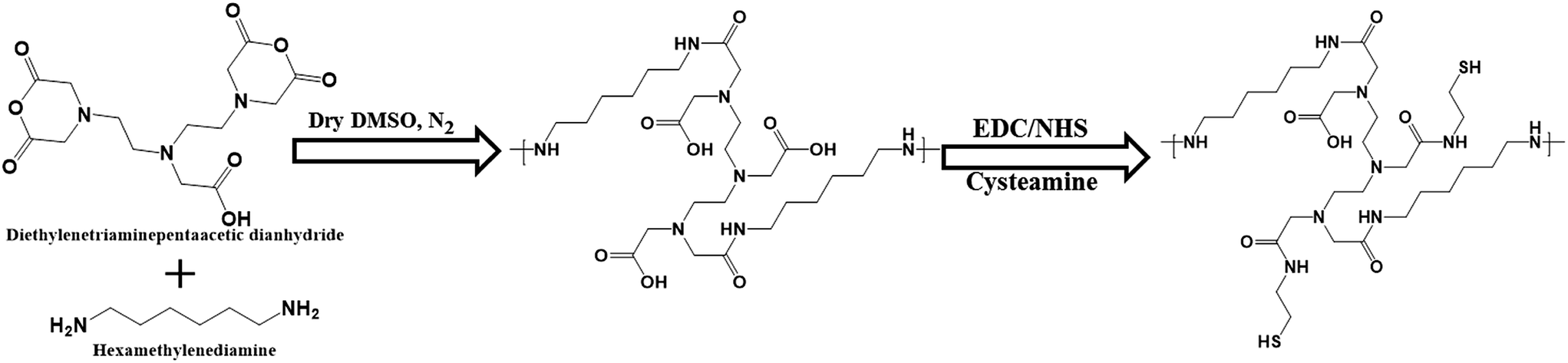 | ||
| Scheme 1 Synthetic approach used to synthesize the thermoresponsive polyamic acid polymer tailored with thiol group. | ||
The 1H NMR spectrum presented in Fig. 1a reveals the absence of a primary amine peak, thus confirming the formation of HMDA–DTPA polymer. Moreover, the carboxylic hydroxyl proton disappeared, and this was due to the proton exchange between D2O and the carboxyl group. Except for this, all DTPA and HMDA protons were resolved correctly. After the functionalization of Cys with the synthesized HMDA–DTPA polymer, the CH2 proton from Cys near the amide bond appeared at 3.6 ppm, clearly confirming the successful conjugation of Cys to the HMDA–DTPA polymer (Fig. 1b). The grafting ratio of Cys in HMDA–DTPA polymer was calculated around 58% in the polymer chain. GNPs were synthesized following a previously reported protocol.24 UV-vis spectroscopy was used to identify the optical properties of the synthesized GNPs. The dominant surface plasmon resonance showed a peak at 516 nm, which specified the formation of GNPs with a particle size of approximately 15 nm (Fig. 2a and c) and the corresponding zeta potential of approximately −25 mV (Fig. S2†).25 Transmission electron microscopy (TEM) revealed the uniform distribution of synthesized nanomaterials (Fig. 2d). In addition, thermoresponsive PAA was chemically modified on the GNP surface through the thiol group; the thiol group forms a covalent-like bond with GNPs. PAA conjugation significantly changed the optical properties of the GNPs in the near-infrared region (Fig. 2a), which is very useful for deep skin penetration. Furthermore, TEM revealed cluster formation after polymer conjugation (Fig. 2e and f) and a cluster-like structure, thus increasing the size of the GNPs to 100 nm (Fig. 2b) and the corresponding zeta potentials to −33 mV (Fig. S3†). The increased size of the GP nanocluster caused by the multiple chemical bonding between gold and thiol group present in polyamic acid resulted in the formation of a cluster with an increased particle size. These size distribution results are consistent with DLS results.
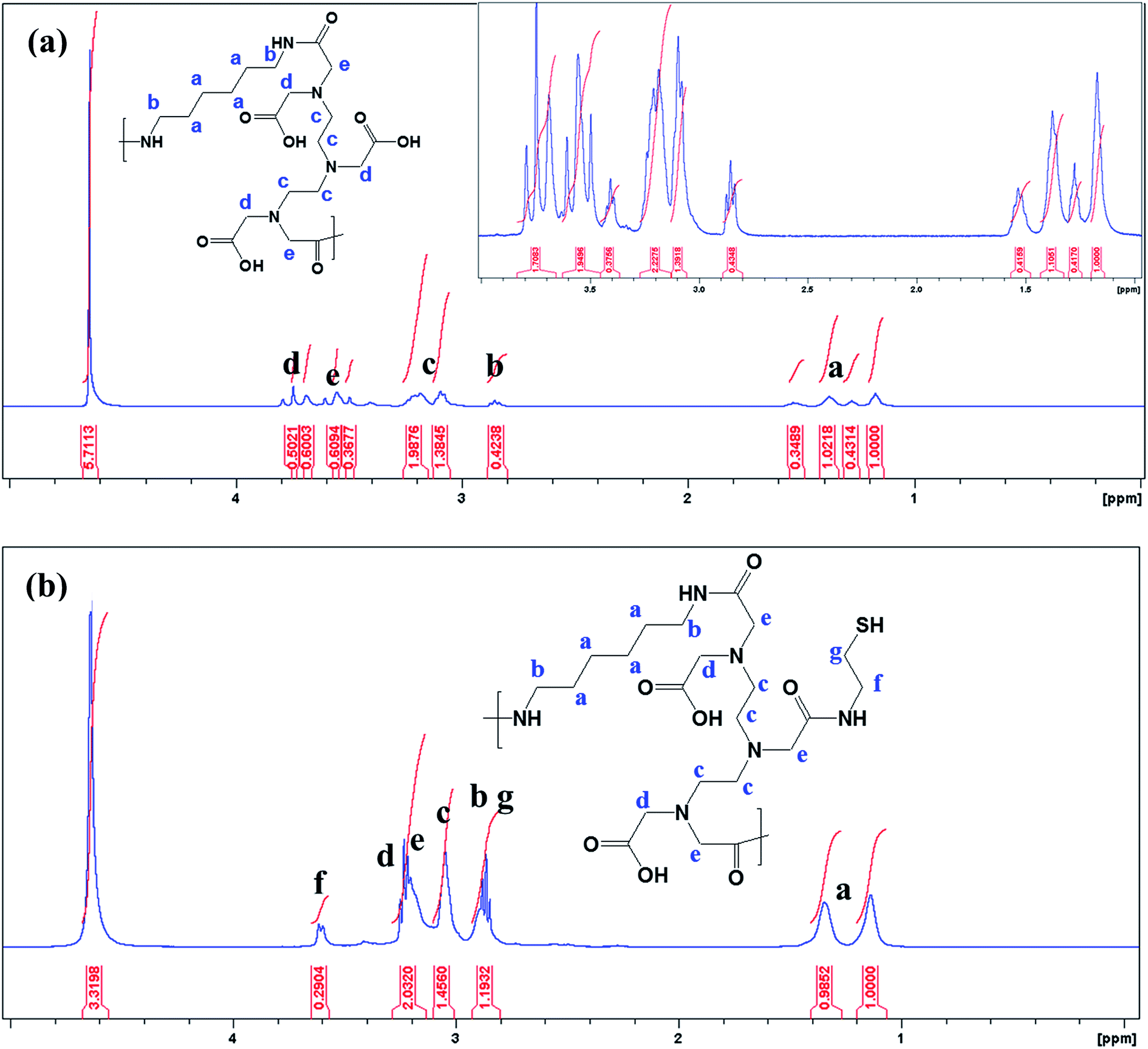 | ||
| Fig. 1 1H NMR spectrum of synthesized HMDA–DTPA (a) and thiol group terminated HMDA–DTPA polymer (b). | ||
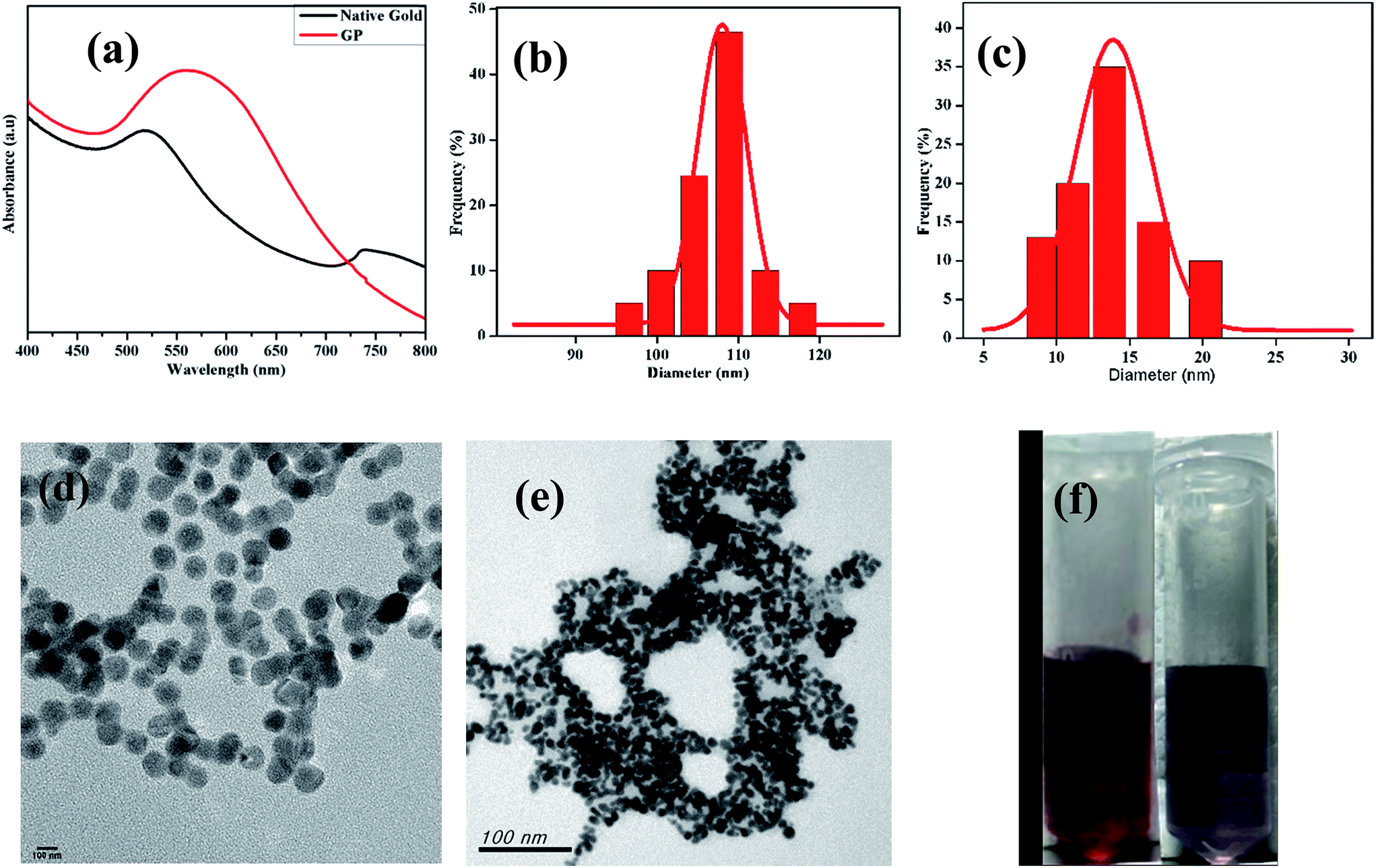 | ||
| Fig. 2 UV-visible spectrum of synthesized gold nanoparticle, GP (a) and corresponding particle size (b) and (c). TEM images of synthesized gold (d), and GP nanoparticle (e). Optical images of synthesized gold (left) and GP nanoparticle (right) (f). | ||
The energy-dispersive X-ray measurements indicated that the GP nanocluster was composed of gold, carbon, nitrogen, and oxygen (Fig. S4†). Furthermore, to investigate the thermoresponsive property of the GP nanocluster, the GP nanocluster was exposed to heat or light. The temperature-dependent absorption property of the synthesized nanocluster was evaluated, and the absorption properties of the GP nanocluster were inversely proportional to the applied temperature (Fig. 3a and b). The optical properties of the GP nanocluster significantly changed upon exposure to temperature. In particular, increasing the temperature to above 25 °C caused a major change in the UV absorption, indicating that there might be some changes in the morphology of GP nanocluster. The stability of the GP nanocluster was measured using UV-vis spectra for 2 weeks in water at 37 °C. No significant change in the absorption was observed (data not shown).
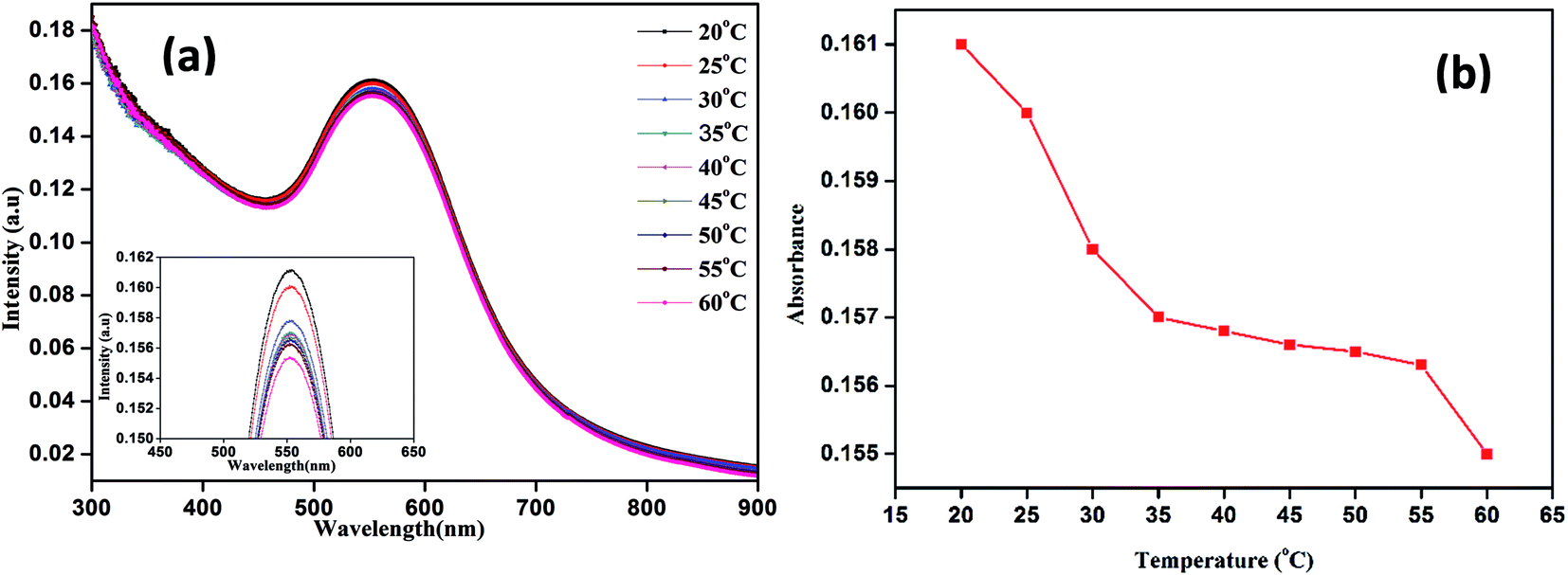 | ||
| Fig. 3 Temperature dependent UV-vis absorption properties of nanocluster (GP) (a) UV-vis absorption intensity versus wavelength, (b) UV-vis absorption intensity versus temperature. | ||
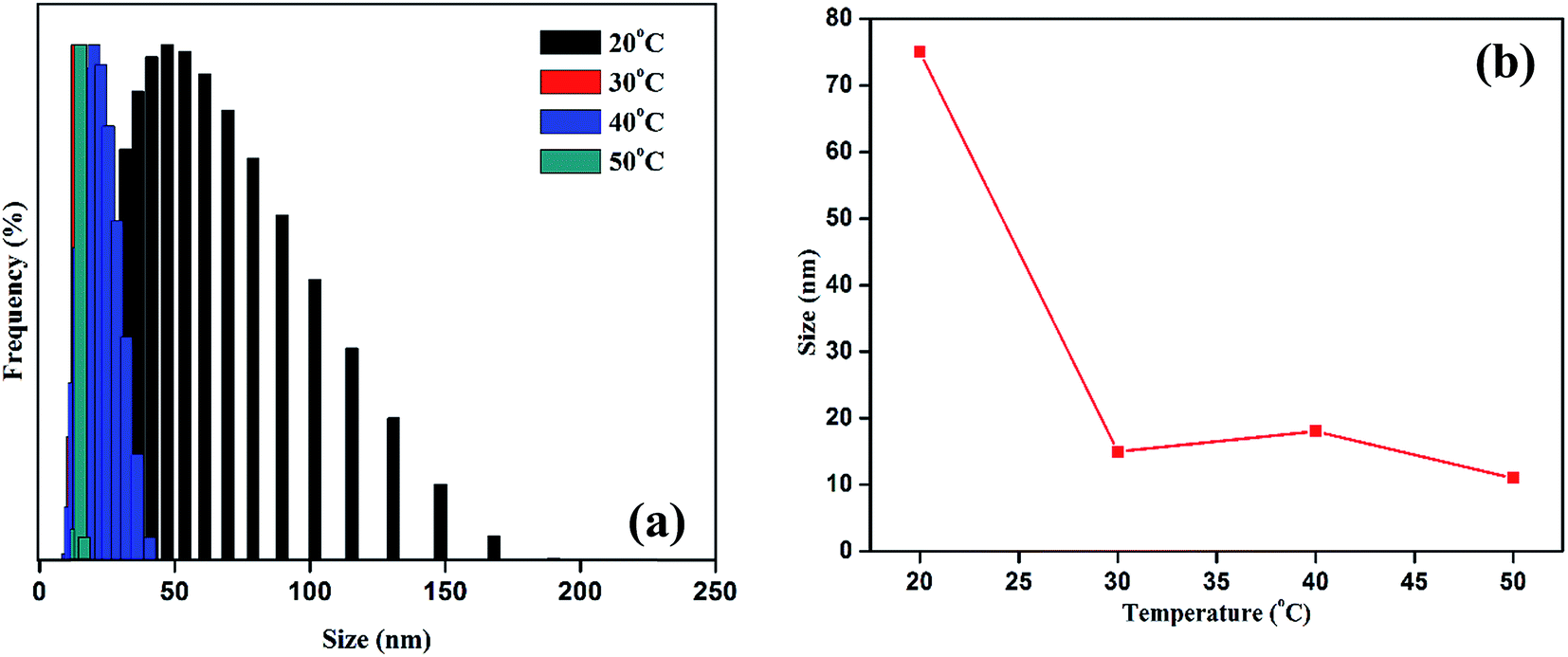 | ||
| Fig. 4 The temperature dependent hydrodynamic size of synthesized GP nanocluster (a) the size distribution of nanocluster at various temperature (b) the Z-average diameter of nanocluster at various temperature. | ||
DLS was used to investigate the thermoresponsive behavior of the polymer shell. The DLS results were similar to those of the UV measurement. As presented in Fig. 4a and b, the average hydrodynamic diameter of the GP nanocluster decreased with increasing temperature. The hydrodynamic diameter of the GP nanocluster at 20 °C was approximately 100 nm, suggesting that the GP nanocluster was stable at 20–25 °C. However, the hydrodynamic diameter changed considerably when the temperature increased from 25 °C to 50 °C at a pH of 7. The decrease in the size was due to the ring closure mechanism of polyamic acid22 and also cleavage of gold thiol bond at the high temperature,26,27 which could be identified by a change in the hydrodynamic diameter. However, the cyclization process is irreversible due to the formation of more stable polyimides, and formed polyimides also has less toxicity. Moreover, there was no significant size change was observed in GP nanocluster at pH 2 to pH 12 confirming that the stability of synthesized nanocluster. By contrast, in unconjugated GNPs, no significant changes were observed in both size and optical properties upon exposure to temperature or light. These results confirm the thermoresponsive property of the GP nanocluster (Scheme 2).
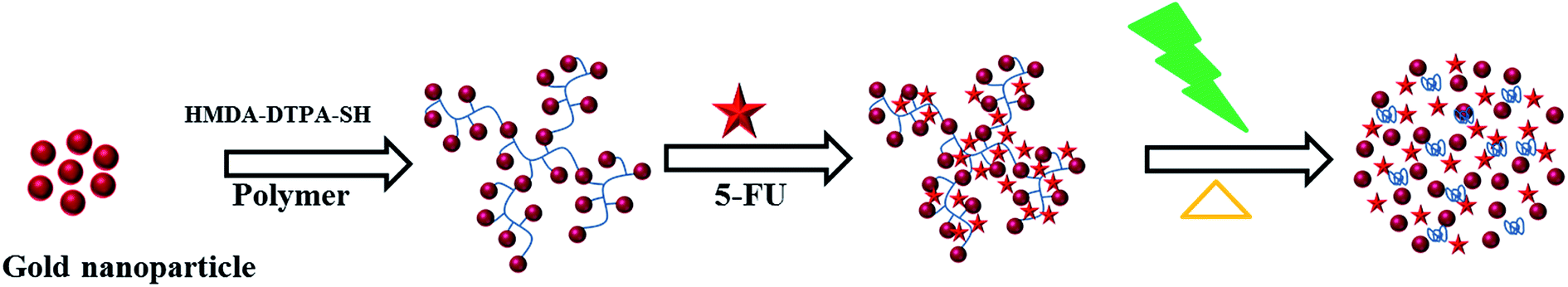 | ||
| Scheme 2 Polymer conjugation and light triggered FU anticancer drug release. | ||
We investigated the drug loading and releasing abilities of the GP nanocluster. 5-FU, a commonly used anticancer drug, was selected as the model drug to evaluate the drug loading property (Fig. 5a and b). The loading level of 5-FU was estimated to be approximately 3.8% in weight, which was calculated from calibration curve. To evaluate the light-controlled drug release, the efficacy of chemotherapy with the help of the GP nanocluster was investigated by determining the absorption in the UV range. Five sets of the GP nanocluster solution were irradiated with only UV light for different time durations (0–20 min) (Fig. 6a and b). A determined amount of released drug solution was removed from the irradiated sample, and UV absorption measurement was then performed at 265 nm. 5-FU release was proportional to the laser irradiation time, which could be confirmed by its UV absorption peak at 265 nm. After 5 min of irradiation, 5-FU started to drain from the GP nanocluster, and its maximum release was observed after 20 min of irradiation.
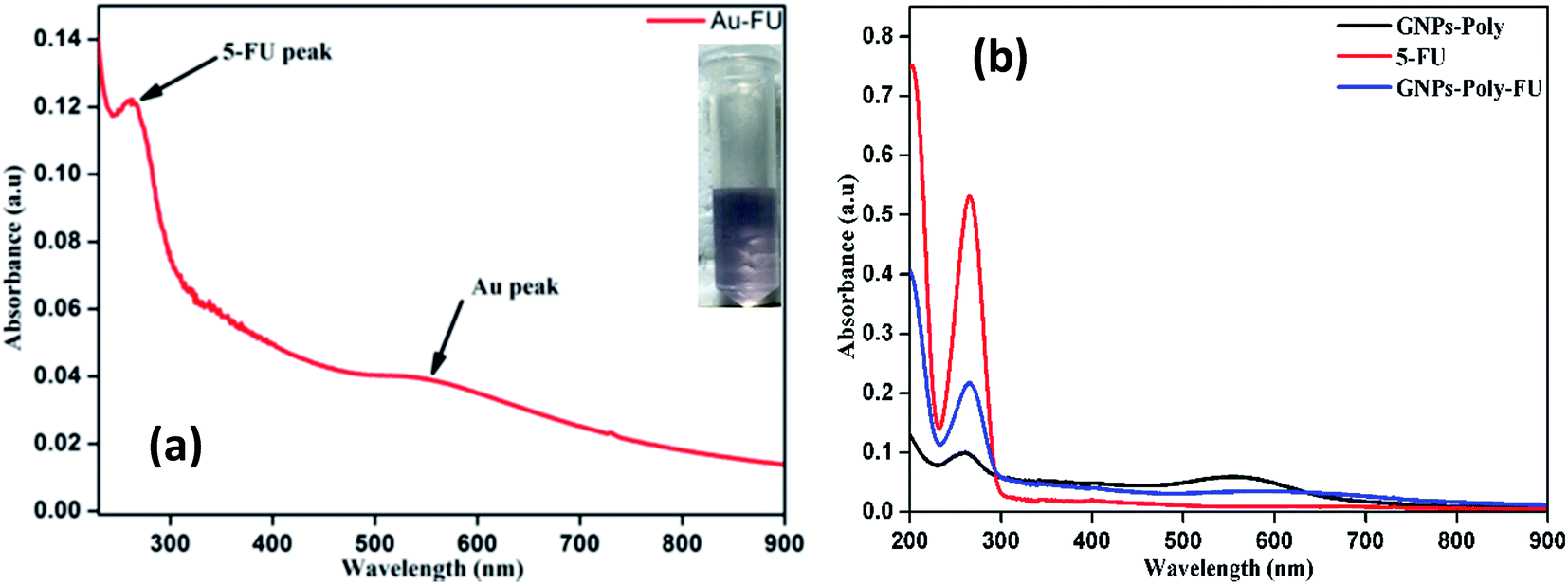 | ||
| Fig. 5 UV absorption of GP–FU (a) and corresponding comparative drug loading profile (b). | ||
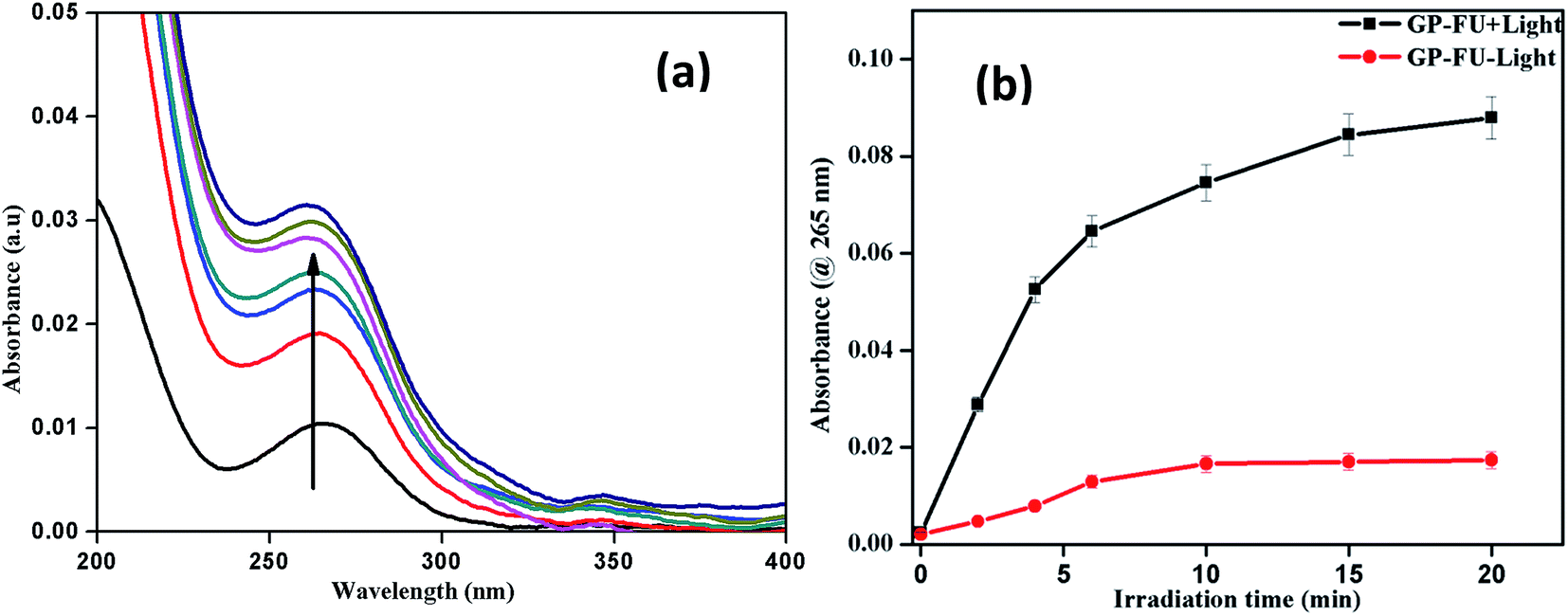 | ||
| Fig. 6 UV-visible spectrum of 5-FU release upon irradiation of 365 nm laser (a) and corresponding cumulative 5-FU drug release profile of GP–FU absorbance @ 265 nm vs. irradiation time (b). | ||
The cytotoxicity levels of GP–FU and GP were determined using the standard MTT assay protocol. Free 5-FU and GP–FU with an equivalent dose of 5-FU exhibited different cytotoxic effects upon exposure to HeLa cancer cells in a dose-dependent manner, whereas GP and GP–FU alone showed negligible cytotoxicity levels at various concentrations (Fig. 7a). These results are quite reasonable because all synthesized GNPs functionalized with the thermoresponsive polymer were previously reported to exhibit low cytotoxicity and high biocompatibility.28 Furthermore, we evaluated the use of the GP–FU nanocluster as a photo-controlled drug delivery system by performing HeLa cell culture experiments. Free 5-FU, the GP nanocluster, and the GP–FU nanocluster dispersed in cell culture media were added to the cells, which were then exposed to 365 nm of light for 5 min. After 24 h of incubation, no significant toxicity was observed in cells irradiated with only light. Similarly, no significant cell death was observed in cells treated with only the GP or GP–FU nanocluster. These results indicate the lack of toxicity from the light or the carrier. Taken together, these findings indicate that the GP–FU nanocluster can be used as a nanocarrier as well as a caging group for the anticancer drug 5-FU. A vital advantage of external control is the possibility of externally monitoring drug dosing. To validate this drug release property of the GP–FU nanocluster, HeLa cells were first treated with 1, 2, 3, 4, and 5 μg solutions of the GP–FU nanocluster and then exposed to 365 nm of UV light for 5 min (Fig. 7b). The cytotoxicity analysis after 24 h of incubation demonstrated that the toxicity increased with the concentrations of the GP–FU nanocluster. The dramatic change in cell viability effectively correlated with the dose of the released drug upon exposure to light. However, at the same dose, 5-FU killed a lower number of cells than did the GP–FU nanocluster. All these results support that GP–FU nanoclusters effectively release 5-FU from the nanocluster. To further investigate the intracellular delivery of 5-FU by the carrier, the GP–FU nanocluster was treated with live HeLa cells and observed through fluorescence microscopy (Fig. 8). The HeLa cells were incubated with an appropriate carrier for 12 h after light irradiation, followed by staining with annexin V and PI. If cells are in the early stage of apoptosis, annexin V binds to the membrane of the cells and emits green fluorescence, which could be identified through fluorescence microscopy. Similarly, if cells are in the later stage of apoptosis, PI binds to the nucleus of the cells and emits red fluorescence, which can also be identified through fluorescence microscopy. The cells incubated with free 5-FU exhibited a green color in the membrane and a red color in the nucleus, suggesting the rapid diffusion and early internalization of 5-FU, leading to cell death. However, we observed a hyperintense green color in the membrane and a red color in the nucleus of the cells treated with GP–FU, indicating successful internalization of GP–FU following the release of the anticancer drug 5-FU upon light irradiation. In addition, the therapeutic efficiency may be due to the synergistic effect of the anticancer drug and the photothermal effect of the GNPs, which was three times higher than that of 5-FU alone.29 However, cells treated with the GP nanocluster did not exhibit a strong green color, which may be attributed to the photothermal effect of the GNPs. In the control group, no significant color was observed. Similar to the results of light-induced cell viability, these results confirm the vital role of the nanocluster in 5-FU drug release in live HeLa cells.
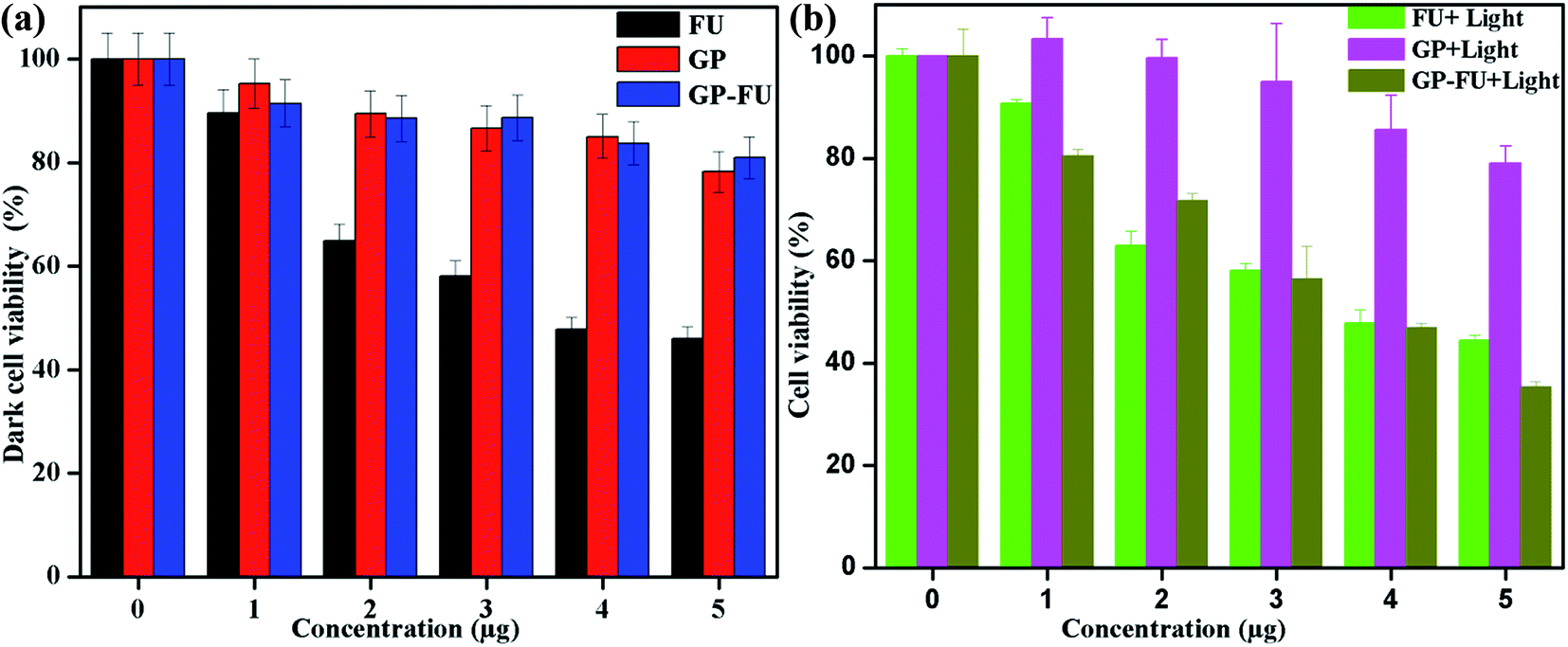 | ||
| Fig. 7 In vitro concentration-dependent cell viability of HeLa (a) and light triggered drug released cell viability of HeLa (b) incubated for 24 h. | ||
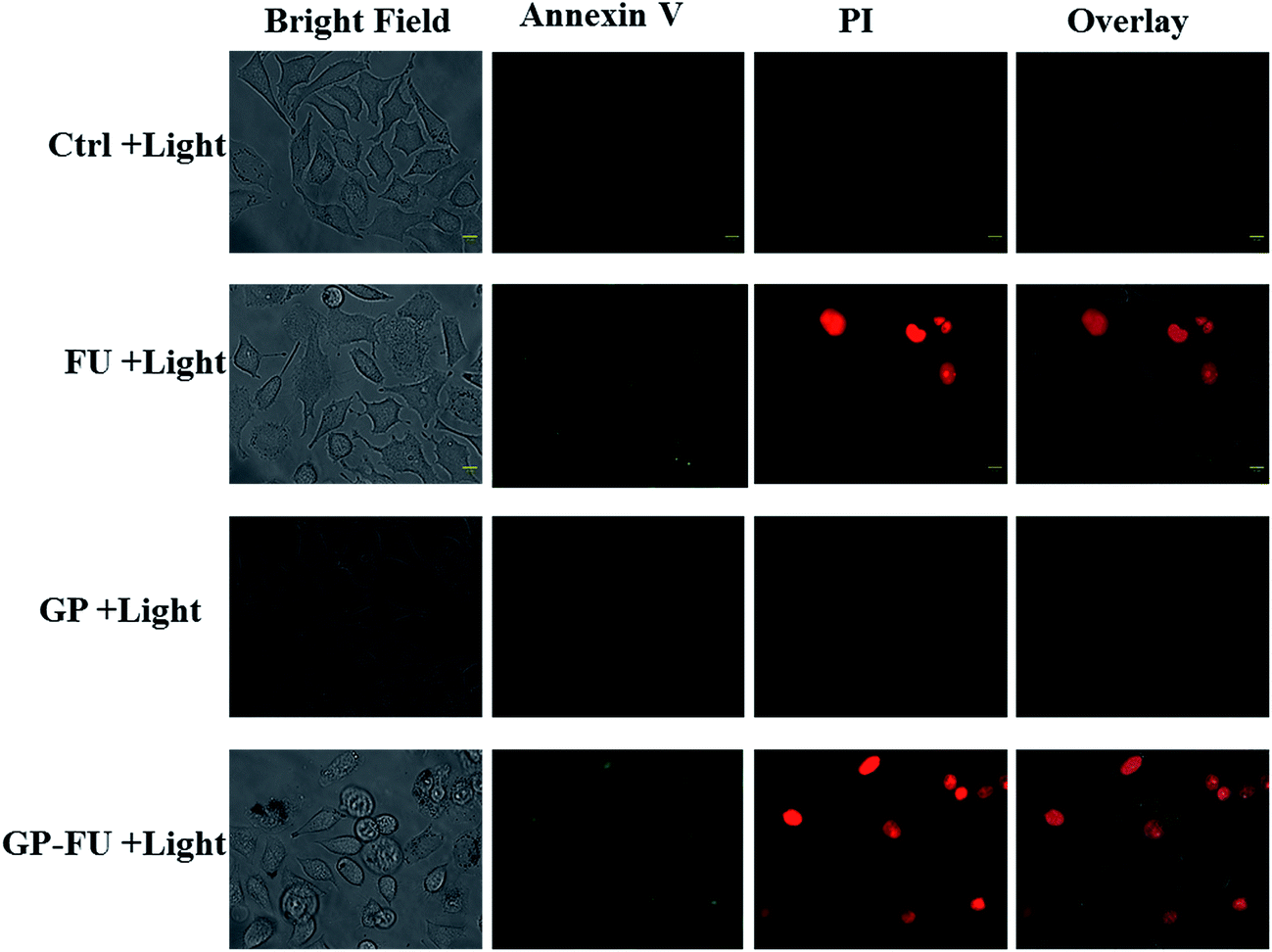 | ||
| Fig. 8 Intracellular release of FU anticancer drug in HeLa cell from GP–FU stained with annexin followed by propidium iodide (scale bar 10 μm). | ||
4. Conclusion
Thiol-terminated thermoresponsive PAA was synthesized and conjugated to GNPs. The chemical conjugation on the GNP surface was confirmed using various techniques including NMR and IR. The light-responsive property of the GP nanocluster was successfully demonstrated using DLS and UV. Furthermore, the results of light-irradiated drug release confirmed the light-responsive property of the GP–FU nanocluster. The water-soluble polymer enhanced the stability of the GP nanocluster in water for over 1 month. Thus, the drug-loaded GP nanocluster exhibited high water solubility, t cell viability in dark conditions, light-triggered drug release, and effective toxicity to HeLa cells upon light irradiation. All these results suggest that the nanocluster can be used as an effective stimulus nanocarrier in the future.References
- X. Xue, F. Wang and X. Liu, J. Mater. Chem., 2011, 21, 13107 RSC.
- J. H. Park, G. V. Maltzahn, L. Ong, A. Centrone, T. A. Hatton, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Adv. Mater., 2010, 22, 880–885 CrossRef CAS PubMed.
- J. H. Park, G. V. Maltzahn, M. J. Xu, V. Fogal, V. R. Kotamraju, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 981–986 CrossRef CAS PubMed.
- M. Liras, O. García, N. Guarrotxena, M. Palacios-Cuesta and I. Quijada-Garrido, Polym. Chem., 2013, 4, 5751 RSC.
- M. Liras, I. Quijada-Garrido, M. Palacios-Cuesta, S. Muñoz-Durieux and O. García, Polym. Chem., 2014, 5, 433 RSC.
- L. Xu, Y. Liu, Z. Chen, W. Li, L. Wang, X. Wu, Y. Ji, Y. Zhao, L. Ma, Y. Shao and C. Chen, Nano Lett., 2012, 12, 2003–2012 CrossRef CAS PubMed.
- H. Chen, L. Shao, Q. Li and J. Wang, Chem. Soc. Rev., 2013, 42, 2679–2724 RSC.
- X. Y. Shi, S. H. Wang, S. Meshinchi, M. E. V. Antwerp, X. D. Bi, I. Lee and J. R. Baker, Small, 2007, 3, 1245–1252 CrossRef CAS PubMed.
- A. C. Curry, M. Crow and A. Wax, J. Biomed. Opt., 2008, 13, 1–7 CrossRef PubMed.
- Y. Cheng, J. D. Meyers, A. M. Broome, M. E. Kenney, J. P. Basilion and C. Burda, J. Am. Chem. Soc., 2011, 133, 2583–2591 CrossRef CAS PubMed.
- K. Huang, H. Ma, J. Liu, S. Huo, A. Kumar, T. Wei, X. Zhang, S. Jin, Y. Gan, P. C. Wang, S. He, X. Zhang and X. J. Liang, ACS Nano, 2012, 6, 4483–4493 CrossRef CAS PubMed.
- O. Veiseh, J. W. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62, 284–304 CrossRef CAS PubMed.
- M. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 993–1003 CrossRef PubMed.
- S. F. Medeiros, A. M. Santos, H. Fessi and A. Elaissari, Int. J. Pharmacol., 2011, 403, 139–161 CrossRef CAS PubMed.
- C. Chen Weihsu, X. Zhang Andrew and S.-D. Li, Eur. J. Nanomed., 2012, 4, 89–93 CrossRef PubMed.
- Z. Zhang, J. Wang, X. Nie, T. Wen, Y. Ji, X. Wu, Y. Zhao and C. Chen, J. Am. Chem. Soc., 2014, 136, 7317–7326 CrossRef CAS PubMed.
- M. S. Yavuz, Y. Cheng, J. Chen, C. M. Cobley, Q. Zhang, M. Rycenga, J. Xie, C. Kim, K. H. Song, A. G. Schwartz, L. V. Wang and Y. Xia, Nat. Mater., 2009, 8, 935–939 CrossRef CAS PubMed.
- A. Prasannan, H. C. Tsai, Y. S. Chen and G. H. Hsiue, J. Mater. Chem. B, 2014, 2, 1988–1997 RSC.
- J. A. Kreuz and J. R. Edman, Adv. Mater., 1998, 10, 1229–1232 CrossRef CAS.
- A. S. Mathews, I. Kim and C. S. Ha, J. Appl. Polym. Sci., 2006, 102, 3316–3326 CrossRef CAS.
- A. Srividhya and B. S. R. Reddy, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1707–1726 CrossRef.
- S. Y. Wu, L. T. Chang, S. Peng and H. C. Tsai, Int. J. Nanomed., 2015, 10, 1637–1647 CAS.
- P. Starr, C. M. Agrawal and S. Bailey, J. Biomed. Mater. Res., Part A, 2016, 104, 406–412 CrossRef CAS PubMed.
- C. Zhang, Z. Zhou, Q. Qian, G. Gao, C. Li, L. Feng, Q. Wang and D. Cui, J. Mater. Chem. B, 2013, 1, 5045–5053 RSC.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 4212–4217 CrossRef CAS.
- A. R. Herdt, S. M. Drawz, Y. Kang and A. T. Taton, Colloids Surf., B, 2006, 51, 130–139 CrossRef CAS PubMed.
- M. Borzenkov, G. Chirico, L. D'Alfonso, L. Sironi, M. Collini, E. Cabrini, G. Dacarro, C. Milanese, P. Pallavicini, A. Taglietti, C. Bernhard and F. Denat, Langmuir, 2015, 31, 8081–8091 CrossRef CAS PubMed.
- O. S. Muddineti, B. Ghosh and S. Biswas, Int. J. Pharm., 2015, 484, 252–267 CrossRef CAS PubMed.
- X. Huang and M. A. El-Sayed, J. Adv. Res., 2010, 1, 13–28 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27563j |
| This journal is © The Royal Society of Chemistry 2017 |