
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrophobic mixed solvent induced PLGA-based in situ forming systems for smooth long-lasting delivery of Radix Ophiopogonis polysaccharide in rats
LiNa Wangab,
Xiao Lin*a,
YanLong Hong*b,
Lan Shena and
Yi Fengb
aCollege of Chinese Materia Medica, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China. E-mail: duotang@163.com
bEngineering Research Center of Modern Preparation Technology of TCM of Ministry of Education, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China. E-mail: hfuir@163.com
First published on 17th January 2017
Abstract
To overcome the deficient sustained release of bioactive Radix Ophiopogonis polysaccharide (ROP) from hydrophilic solvent-induced PLGA-based in situ forming systems (ISFSs), hydrophobic mixed solvent-induced ISFSs were investigated, including the factors affecting drug release and the anti-myocardial ischemic activity of a representative formulation. The initial release was reduced by 3.7–8.0 times and the plasma level was significantly prolonged from 4 days to 10–15 days as the hydrophilic N-methyl-2-pyrrolidone (NMP) was replaced by the hydrophobic mixed solvent consisting of 90% benzyl benzoate (BB) and 10% co-solvent (benzyl alcohol, triacetin, or NMP). The effect of 10% co-solvent on the phase inversion rate, depot morphology, and ROP release was unexpectedly high, with NMP surprisingly being optimal although having much higher hydrophilicity. Further studies indicated that the release behaviour could be tailored by changing the ratio of BB to NMP, PLGA molecular weight, and PLGA concentration. Linear reductions in Cmax and AUC0–12 h were observed with the increase of the ratio (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 to 9:1), PLGA MW (20 to 40 kDa), and PLGA level (20% to 40%), respectively. Moreover, it is possible to achieve nearly zero-order release by different formulation combinations. The bioactivity data suggested that thanks to sustained stable release of ROP, the ISFS studied showed an obvious advantage over aqueous solution in the treatment of myocardial ischemia. Therefore, ISFSs with hydrophobic mixed solvents like BB/NMP appear to be promising and suitable for smooth long-lasting release of herb polysaccharides with low oral bioavailability and short plasma half-life, like ROP.
:3 to 9:1), PLGA MW (20 to 40 kDa), and PLGA level (20% to 40%), respectively. Moreover, it is possible to achieve nearly zero-order release by different formulation combinations. The bioactivity data suggested that thanks to sustained stable release of ROP, the ISFS studied showed an obvious advantage over aqueous solution in the treatment of myocardial ischemia. Therefore, ISFSs with hydrophobic mixed solvents like BB/NMP appear to be promising and suitable for smooth long-lasting release of herb polysaccharides with low oral bioavailability and short plasma half-life, like ROP.
1. Introduction
In recent years, a number of macromolecular drugs, such as protein1,2, peptides3,4, and herb polysaccharides5–8 are being used as bioactive agents in the treatment of a variety of diseases. However, because of their short half-life, repeated administration is required. This together with the injection-only nature makes them inconvenient in the application of clinical therapy. In order to prolong drug release and residence time, reduce administration times, minimize adverse effects, and thus, improve patient compliance, various kinds of drug delivery systems have been investigated, such as liposomes,9 microparticles10,11 and in situ forming systems (ISFSs).12,13ISFSs are fluid formulations that can be injected into the body in a minimally invasive manner prior to transforming into a semi-solid/solid depot within the desired tissue, organ, or body cavity.14 ISFSs are commonly comprised of biodegradable polymer, biocompatible solvent, and drug molecules and/or particles. Drugs loaded in ISFSs are generally released via three stages: (i) an initial burst release attributed to non-entrapped drug molecules and phase inversion of the system; (ii) a slow release of drug caused by diffusion from sol-to-gel induced porous structure; (iii) fast drug release at the onset of erosion and the degradation of polymer.15,16 Among the three stages, phase inversion rate can be altered by varying the physicochemical parameters of each part of composition, thus tailored sustained-release profiles can be obtained. As to solvent-induced ISFSs, once injected into an aqueous environment, with the exchange of solvent and nonsolvent which will diminish the solubility of polymer and lead to phase separation and precipitation of the polymers, the liquid solutions of polymers can be transformed into solid/semisolid drug depots in the injection sites, making the formulation obtain an ability of controlled release.16,17 Owing to the advantages of high drug loadings, high patient compliance, less administration frequency, and the characteristics of biodegradability, biocompatibility, and nontoxicity,18 ISFSs present an attractive alternative to conventional preformed implants for parenteral applications19 and have been successfully used to deliver numerous therapeutic agents,20–25 including insulin, risperidone, and antipsychotic.
Presently, a lot of formulations of solvent-induced ISFSs have been studied, such as poly(D,L-lactide-co-glycolide) (PLGA) or poly(lactide) (PLA) in N-methyl-2-pyrrolidone (NMP),26–29 dimethyl sulfoxide (DMSO),25,30 and benzyl benzoate (BB).23,31 Because of the nontoxicity, biodegradability as well as the approval of American Food and Drug Administration (AFDA), PLGA, a copolymer of D,L-lactic (LA) and glycolide (GA),32 has been widely used in the field of pharmacy.15,19 According to the different ratio of the two polymers and their molecular weights, PLGAs having different degradability and molecular weights can be applied in a variety of pharmaceutical technologies, making it possible to control drug release by varying PLGA parameters.31,33
Radix Ophiopogonis polysaccharide (ROP), a natural herb fraction with a weight-average molecular weight of 4.8 kDa,34 was found to have several bioactivities,35–37 such as anti-myocardial ischemia.8 However, as a highly hydrophilic macromolecule, ROP is rarely absorbed after oral administration (absolute bioavailability ∼2%) and rapidly excreted by the kidneys following intravenous administration (plasma half-life ∼30 minutes).38,39 The short in vivo residence time of ROP not only compromises its efficacy but necessitates frequent injection.
In our previous studies,26,40,41 both hydrophilic and hydrophobic ISFSs, using poloxamer and PLGA as carriers, respectively, were investigated for ROP. It was found that the poloxamer-based ISFSs showed a broad plasma peak and half-day high and full ROP exposure, suggesting that they might be more suitable for short-term acute therapies of myocardial ischemia. The PLGA-based formulations, especially of 40% PLGA30k/NMP and 30% PLGA50k/NMP, showed low but lasting ROP plasma concentrations for about 4 days, which indicates that they might be promising for long-term precaution and/or cure of this disease. However, nearly zero-order, week-long or month-long release seems impossible due to high initial burst release caused by fast exchange of hydrophilic NMP with body fluid. To combat this, hydrophobic solvents were tested for PLGA-based ISFSs in this study. Benzyl benzoate (BB), a highly hydrophobic solvent that has been reported to have a strong ability of controlled release, was chosen as the main solvent. To improve PLGA solubility and formulation homogeneity and also to clarify the effect of solvent species and hydrophobicity, benzyl alcohol (BA), triacetin (TA), and NMP were chosen as the co-solvent to form hydrophobic mixed solvents with BB. The goal of the current investigation was to find out some general principles in the long-lasting stable delivery of polysaccharide loaded in ISFSs through studying the effects of mixed solvent, the ratio of BB to NMP, and the level and molecular weight of PLGA on the release of ROP as well as the anti-myocardial ischemic activity of ROP loaded in a representative ISFS.
2. Materials and methods
2.1 Materials and animals
ROP was prepared according to a previously reported method.34 Poly(D,L-lactide-co-glycolide) copolymer (PLGA) with molar lactide to glycolide ratio being 50/50, molecular weight being ∼20, ∼30, or ∼40 kDa, and free carboxylic acids at the ends of the polymeric backbone chain was purchased from Jinan Daigang Biomaterial Co., Ltd. (Shandong, China). Fluorescein isothiocyanate (FITC) was purchased from Sigma (St. Louis, MO, USA). Extra dry dimethyl sulfoxide (DMSO) and N-methyl-2-pyrrolidone (NMP) were purchased from Acros Organics (Geel, Belgium). Isopropanol, petroleum ether (60–90 °C), ethanol, and Span 80 were purchased from Sinopharm Chemical Reagent Ltd. All the other chemicals were of reagent grade and purchased from commercial sources.Male Sprague-Dawley (SD) rats, weighing 240–280 g, were supplied by Lab Animal Center of Shanghai University of Traditional Chinese Medicine. All procedures of the animal experiments were approved by the Animal Ethical Experimentation Committee of Shanghai University of TCM according to the requirements of the National Act on the Use of Experimental Animals (PR China).
2.2 Preparation of FITC-labeled ROP (FROP)
The labeling of ROP with FITC was carried out according to a previous report.39 ROP (1 g) was dissolved in DMSO (10 mL), followed by the addition of FITC (0.1 g), 0.2 mL of a solution of dibutyltin dilaurate in DMSO (1/4, v/v), and 0.5 mL of pyridine. The mixture was well vortexed and heated for 30 min at 95 °C. After complete reaction, the product was precipitated by a mixture of ether and ethanol (3/1, v/v). The resulting precipitates were recovered by centrifugation, washed ten times with the ether/ethanol mixture to fully remove free FITC and other excess reagents, and then dried in vacuum. The yield rate was about 90%.2.3 Preparation and characterization of FROP microparticles
Microparticles were prepared as previously described.41 Briefly, FROP (1 g) was dissolved in water (8 mL), followed by the addition of 40 mL of ethanol (0–5 °C) containing 0.25% Span 80. After stirring the mixture for 2 h, 40 mL of isopropanol (0–5 °C) was added for dehydration, followed by filtration and washing with isopropanol (0–5 °C) and petroleum ether in turn. Microparticles gained were dried in vacuum overnight and stored in desiccator at room temperature before use. The yield rate was about 80%.2.4 Preparation of ISFSs
Injectable ISFSs were prepared by dispersing different amounts of PLGA (20%, 30%, and 40%, w/w) or PLGA with different molecular weights (∼20, ∼30 and ∼40 kDa) in various kinds of solvents at 60 °C for 24 h with intermittent stirring until clear solutions were formed. Ten formulations differing in PLGA grade and/or solvent were prepared (Table 1). The drug-contained formulations were prepared by dispersing a designated portion of FROP microparticles in a certain volume of PLGA solution in a water bath at 40 °C.| Code | PLGA depot formulation (w/w) | Dose (mg kg−1) | Injection volume (mL kg−1) |
|---|---|---|---|
| a PLGA, poly(D,L-lactide-co-glycolide); FROP, fluorescein isothiocyanate-labeled Radix Ophiopogonis polysaccharide; NMP, N-methyl-2-pyrrolidone; BB, benzyl benzoate; BA, benzyl alcohol; TA, triacetin. | |||
| A | Aqueous solution | 15 | 2 |
| B | 30% PLGA40k/NMP | 200 | 2 |
| C | 30% PLGA30k/(BB:BA 9:1) |
200 | 2 |
| D | 30% PLGA30k/(BB:TA 9:1) |
200 | 2 |
| E | 30% PLGA30k/(BB:NMP 9:1) |
200 | 2 |
| F | 30% PLGA30k/(BB:NMP 8:2) |
200 | 2 |
| G | 30% PLGA30k/(BB:NMP 7:3) |
200 | 2 |
| H | 20% PLGA30k/(BB:NMP 8:2) |
200 | 2 |
| I | 40% PLGA30k/(BB:NMP 8:2) |
200 | 2 |
| J | 30% PLGA20k/(BB:NMP 8:2) |
200 | 2 |
| K | 30% PLGA40k/(BB:NMP 8:2) |
200 | 2 |
2.5 In vitro characterization of ISFSs
The dynamic viscosities of formulations were measured at 20 °C and 37 °C, respectively, using a cone/plate viscometer (Physica MCR-101, Anton Paar, Austria). The shear rate sweeps were measured with increasing the shear rate linearly from 10 to 500 s−1 in 2 min. Viscosity values were obtained by averaging recorded values.To investigate the phase inversion of formulations, 1 mL of each formulation was injected into a test tube containing 5 mL of PBS (pH 7.0).42 12-gauge needles were used in this process. After injection, the test tube was incubated in a shaker water bath at 37 °C and shaken at 60 rpm. The phase inversion was observed at various times (0, 10, 60, and 240 min).
2.6 In vivo studies
Rats were dosed according to a predetermined dosage regimen (Table 1). All the formulations were injected subcutaneously to the same position on the back of rats with 12-gauge needles. After injection, the pinpoint caused by the needle in the skin was sealed with medical glue (5 μL) in case the given formulation leaked out. Blood samples (∼400 μL) were collected into heparin-contained tubes by retro-orbital puncture at the specified time points and then immediately centrifuged at 4000 rpm for 10 minutes. The separated plasma was frozen at −20 °C for subsequent analysis. Each plasma sample (100 μL) was mixed with 40 μL of 1 M perchloric acid to precipitate plasma proteins. After centrifugation at 10000 rpm for 2 minutes, 90 μL of the supernatant was transferred to another clean tube and neutralized using 30 μL of 1 M NaOH. After centrifugation again, the FROP level in the second supernatant was determined by high-performance gel permeation chromatography (HPGPC).43 The assay system consisted of an Agilent 1200 series HPLC (Agilent Technologies, Santa Clara, CA, USA) with a fluorescence detector set at λex 495 nm and λem 515 nm. Samples were separated by HPGPC using a Shodex Sugar KS-802 column. The fluent was 0.1 M phosphate buffer (pH 7.0), delivered at a flow rate of 0.5 mL min−1. The chromatographic procedures were performed at 40 °C and the volume of injection was 30 μL. The stability of ROP loaded in formulations was also evaluated using HPGPC.
Morphology of in situ formed implants was studied with the employment of a scanning electron microscope (SEM), Quanta™ 250 FEG. Samples were harvested at Day 3 and Day 7 after subcutaneous injection. Before transferred into the SEM instrument, samples were freeze fractured, lyophilized, and gold coated.
2.7 Anti-myocardial ischemic activity studies
Male SD rats were randomly divided into three groups, i.e., the model group, the aqueous solution group, and the ISFS ‘30% PLGA30k/(BB:NMP 9:1)’ group. All the groups were subjected to acute myocardial ischemia but treated differently afterwards as Table 2 shows. The model group was administrated with normal saline while the other two groups were given the aqueous solution and ISFS formulation of ROP, respectively. Myocardial ischemia was induced as described before.44 In brief, following being anesthetized by intraperitoneal injection of 10% chloral hydrate, rats were intubated and mechanically ventilated. Left intercostal thoracotomy was performed in the intercostal space to expose the heart. Myocardial ischemia was induced by permanent ligation of the left coronary artery. The ischemic condition was verified by evidence of immediate changes including sudden pallor, distinct dilatation and paralysis of the affected portion of the left ventricle. Finally, the thorax was closed. At 1 hour after surgery, the rats were given drugs (Table 2) and kept on their treatment for 5 days and sacrificed at 1 hour after the last treatment.
| Group | Dose of ROP (mg kg−1) | Volume (mL kg−1) | Administration route | Dosing frequency |
|---|---|---|---|---|
| a The model group was administrated with saline. | ||||
| Model | — | 2 | s.c. | Once a day |
| Aqueous solution | 40 | 2 | s.c. | Once a day |
| 30% PLGA30k/(BB:NMP 9:1) |
200 | 2 | s.c. | Once five days |
Infarct sizes of myocardium were measured after triphenyltetrazolium-chloride (TTC) staining. In detail, the heart was harvested and rinsed with normal saline. The excised left ventricle was frozen at −20 °C for 15 minutes, and then sectioned from apex to base into about 2 mm slices. The slices were incubated in a 1% solution of buffered TTC (pH 7.4) at 37 °C for 15 minutes, and then fixed in 10% formaldehyde. The slices were photographed at the next day by digital camera. The infarct region (TTC unstained), was isolated from the rest cardiac tissues, which were stained red by TTC. The infarct and normal tissues were weighed respectively and the infarct was expressed as a percentage of mass of the left ventricle and the whole heart.
2.8 Data analysis
DAS 2.1.1 pharmacokinetic software (Chinese Pharmacology Society) was used to calculate major pharmacokinetic parameters by noncompartmental analysis. Values are reported as the mean ± standard deviation (SD). One-way analysis of variance was used for comparison among groups. A P-value < 0.05 was considered to be statistically significant.3. Results and discussion
3.1 Viscosity studies
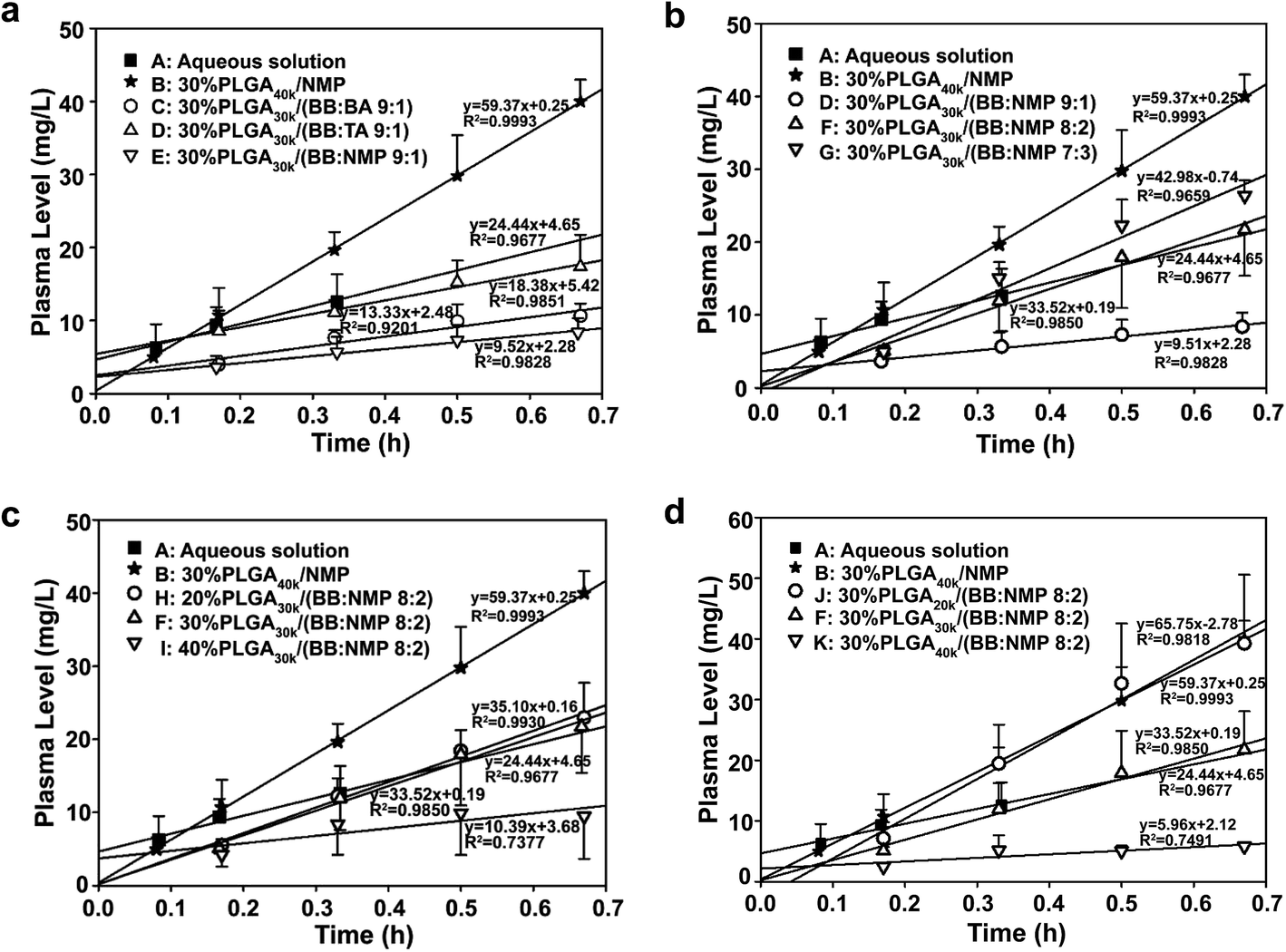
The viscosities of different formulations at 20 °C and 37 °C are shown in Fig. 1, which are related to formulation injectability and early in vivo drug release, respectively. Significant reduction in both viscosity and the effect of all the factors studied on viscosity was observed at 37 °C. That is to say, formulations with high viscosity could be preheated to ∼37 °C before injection to improve their syringeability effectively. Fig. 1a shows the effect of different solvent combination on viscosity, it is notable that the co-solvent system of BB/BA had a relatively low viscosity regardless of its high hydrophobicity, which might be induced by the polymer–solvent interaction. As to the co-solvent systems of BB/NMP and BB/TA, because of predomination of polymer–polymer over polymer–solvent interaction, an increased viscosity could be observed as with the hydrophobicity increasing of the system.19,45 It can be concluded from Fig. 1b–d that with the increasing of the ratio of hydrophobic solvent as well as the molecular weight and the concentration of PLGA used in formulations, the viscosity increased at both 20 °C and 37 °C. Similar to the hydrophilic solvent NMP system,41 abrupt change in viscosity of the hydrophobic co-solvent system (BB:NMP 9:1) also happened at both the PLGA concentration of 30% (w/w) and the PLGA of 30 kDa. Viscosity of ISFSs has been reported to be a major factor that affects the rate of solvent exchange in the first stage of drug release and the rate of diffusion in the second stage. ISFSs with a high viscosity result in a slow rate of diffusion and a high ability of controlled release.
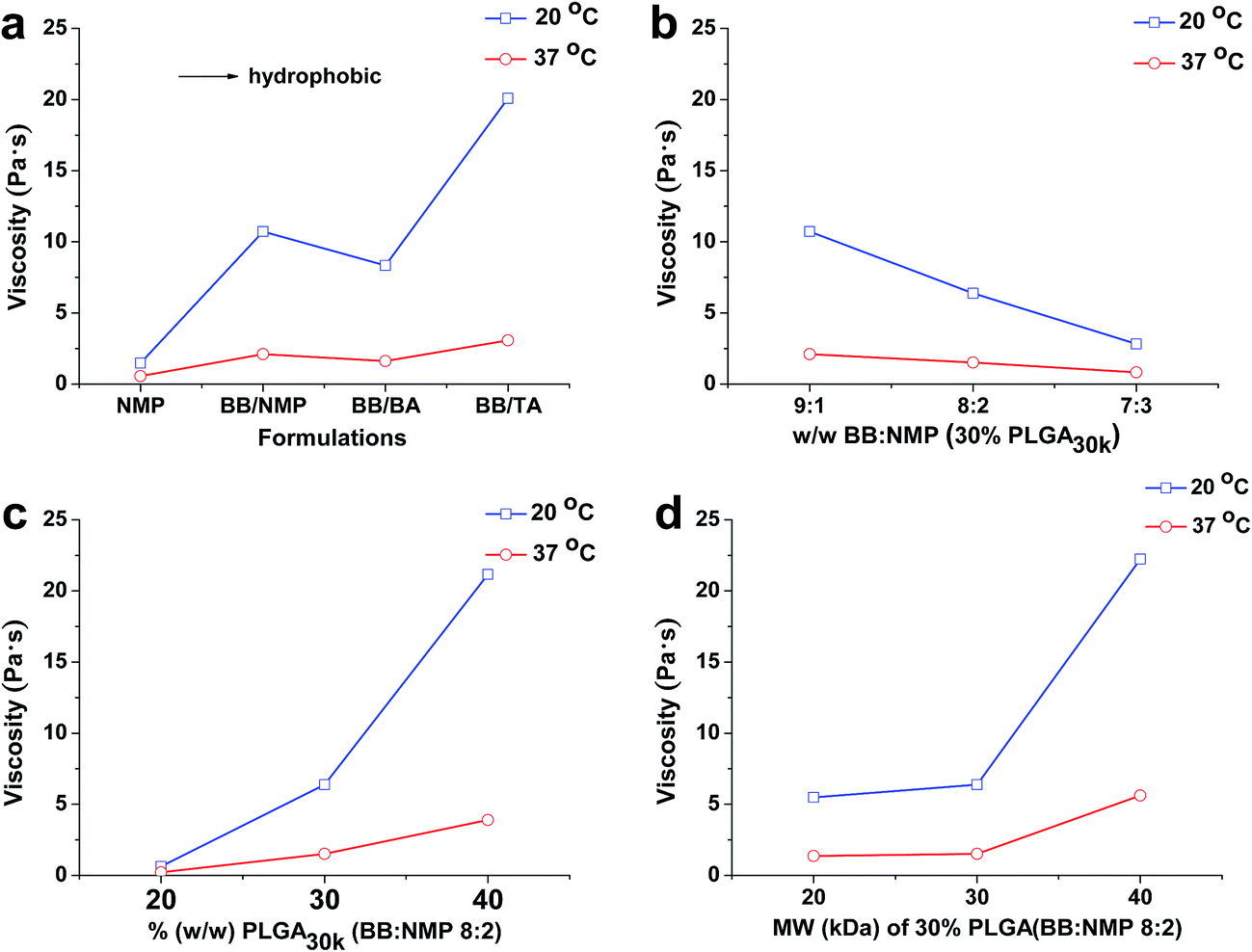 | ||
| Fig. 1 Effect of different mixed solvent (a), ratio of BB to NMP (b), concentration of PLGA (c), and molecular weight of PLGA (d) on the viscosity of PLGA-based in situ forming formulations. Formulations of NMP, BB/NMP, BB/BA, and BB/TA shown in (a) are 30% PLGA40k/NMP, 30% PLGA30k/(BB:BA 9:1), 30% PLGA30k/(BB:TA 9:1), 30% PLGA30k/(BB:NMP 9:1), respectively. PLGA, poly(D,L-lactide-co-glycolide); NMP, N-methyl-2-pyrrolidone; BB, benzyl benzoate; BA, benzyl alcohol; TA, triacetin; MW, molecular weight. | ||
3.2 In vitro implant formation and drug release
As was reported previously,19 the contact of solvent-induced ISFS formulations into the buffer triggered a phase inversion process, which was caused by the solvent and nonsolvent (water) exchange and may cause burst drug release in the initial stage. In terms of the liquid–liquid phase separation process, physical properties of solvent play a critical role. In this study, in vitro implant formation of different solvent formulations loaded with FROP, including 30% PLGA40k/NMP, 30% PLGA30k/(BB:BA 9:1), 30% PLGA30k/(BB:TA 9:1), 30% PLGA30k/(BB:NMP 9:1) was studied and shown in Fig. 2. The formulation of 30% PLGA40k/NMP showed the rapidest phase inversion (which appeared to have been completed within 10 min) and drug release from the depot formed. This is believed to be due to the high water affinity of NMP, which facilitates the phase inversion and the formation of the interconnected polymer-lean phase.46 On the contrary, because of the lower water affinity and slower phase inversion rate, formulations with the hydrophobic solvent mixture of BB/BA, BB/TA, or BB/NMP showed a significantly slow drug release and solid depot formation. The formation rates of solid implants were well consistent with the hydrophobicity of solvent mixtures, with approximately 30%, 70%, and 100% formation at 4 h for the BB/BA, BB/TA, and BB/NMP systems, respectively, indicating that the effect of 10% co-solvent was much more significant than expected from its content. Coincided with its phase inversion rate, the relatively more hydrophilic BB/NMP system showed significantly slower sustained release of FROP than its two counterparts. However, although having significantly different phase inversion rates, the BB/BA and BB/TA systems showed similar drug release behaviours. This indicates that for hydrophobic solvent induced PLGA-based ISFSs, both the phase inversion rate and the microstructure of the solid formed have significant effect on drug release. As a representative formulation, 30% PLGA30k/(BB:NMP 9:1) combined advantages of both hydrophilic and hydrophobic systems. Once being injected into water environment, because of high water miscibility of NMP, a quick exchange between NMP and water was triggered, causing a fast solidification of the water-immiscible polymer. As a result of a more efficient drug entrapment and a lower water penetrate rate into the depots, a lower initial burst release and a more extended release were accomplished.23,47 On the other hand, with the help of BB, the water that penetrated into the depot has a lower rate of exchange with the solvent, thus reducing the release rate of FROP and improving the controlled-release ability of the formulation. So, both the hydrophilicity of NMP and the hydrophobicity of BB are factors contributing to the uniform sustained release of ROP from the depot.
 | ||
| Fig. 2 In vitro phase inversion of formulations with different solvents at various times (0, 10, 60, and 240 min). B, C, D, and E represent the formulations of 30% PLGA40k/NMP, 30% PLGA30k/(BB:BA 9:1), 30% PLGA30k/(BB:TA 9:1), 30% PLGA30k/(BB:NMP 9:1), respectively. PLGA, poly(D,L-lactide-co-glycolide); NMP, N-methyl-2-pyrrolidone; BB, benzyl benzoate; BA, benzyl alcohol; TA, triacetin. | ||
3.3 Morphology of in situ formed PLGA implants
The morphology of ISFSs harvested at Day 3 and Day 7 after injection is shown at two different magnifications in Fig. 3. In general, different microstructures were observed for the four formulations studied. The loosest and most porous structure was formed by the formulation B with pure NMP as the solvent, due to fast and uniform exchange between water-miscible NMP and water. Although also porous, the structure formed by the formulation E with 10% NMP as the polarity-modifying agent of BB was much denser and with pores much smaller in size, due to significantly reduced diffusion rate and flux of NMP. Compared to E, the formulation D formed a looser structure with similar porosity. The least porous and most compact structure was formed by the most hydrophobic formulation C. These remarkable differences in morphology exemplify the unexpected effect of 10% co-solvent again. With the in situ time changing from 3 days to 7 days, significant increase in pore size was observed for all the implants. This is believed to be mainly attributed to drug dissolution rather than PLGA degradation, considering the relatively short time and the relatively high molecular weight of PLGA used. In addition, nearly homogeneous structure of interconnected network throughout the implant was found as the mixed solvent employed in the formulation comes to BB:NMP 9:1, suggesting that the formulation E might be the most optimal with an ability of uniformly controlled release.
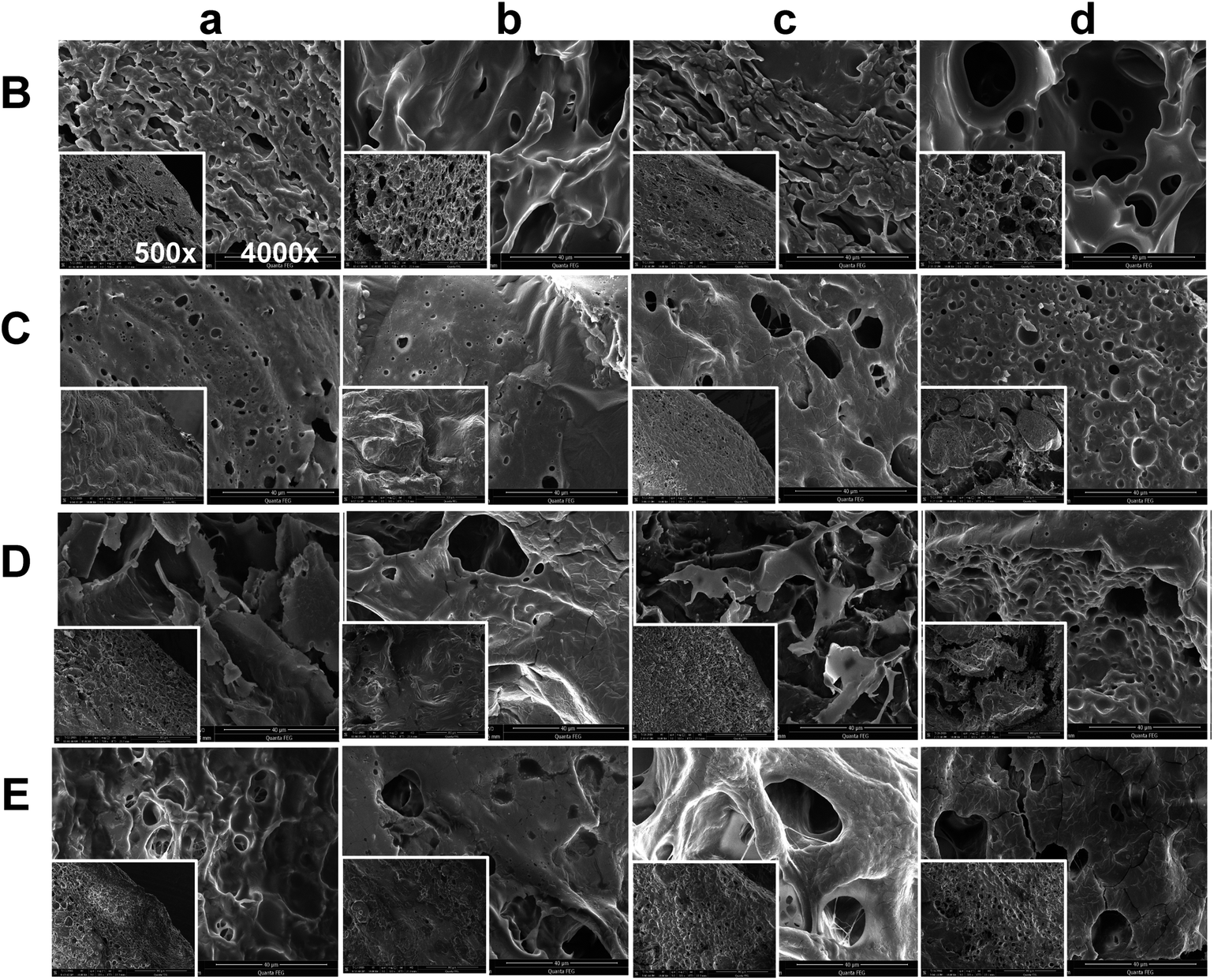 | ||
| Fig. 3 Scanning electron microscopy images of implants harvested at Day 3 (a, b) and Day 7 (c, d) after subcutaneous administration. Column a and c represent images of the shell; column b and d are images of the center of the implants. B, C, D, and E represent the formulations of 30% PLGA40k/NMP, 30% PLGA30k/(BB:BA 9:1), 30% PLGA30k/(BB:TA 9:1), 30% PLGA30k/(BB:NMP 9:1), respectively. PLGA, poly(D,L-lactide-co-glycolide); NMP, N-methyl-2-pyrrolidone; BB, benzyl benzoate; BA, benzyl alcohol; TA, triacetin. | ||
3.4 In vivo studies
For solvent-induced ISFSs, it is vital to choose a “good” solvent with proper solvent strength and aqueous miscibility to achieve expected controlled release of drugs. A variety of solvents, including NMP, DMSO, triacetin (TA), ethyl benzoate, benzyl benzoate (BB), benzyl alcohol (BA), and PEG500DME, can be used. As a representative of hydrophilic solvents, NMP has been widely used in ISFSs and achieved a suitable controlled release for both hydrophilic and hydrophobic drugs, such as ropivacaine,24 protein48 and risperidone.49 BB has also been employed in ISFSs, because of its high hydrophobicity. With limited water miscibility, it can slow the dynamics of phase inversion and solidification process. Furthermore, it can influence the diffusion path that drug molecules take as they leave the implant and alter the resultant morphology of the injected depot system.24,50 To achieve a preferable drug release behaviour, mixed solvent systems, such as BB/BA, DMSO/TA, NMP/BB, NMP/TA, NMP/PEG (one as the main solvent and the other as the property-modifying agent), provide a new choice for ISFSs.23,24,46,51In our previous studies, ISFSs based on PLGA and NMP have been used to achieve a sustain release of ROP for at least several days, while the burst release in the first release stage is still high, due to the highly hydrophilic nature of ROP and its relatively small molecular size as a polysaccharide (weight-average molecular weight: 4.8 kDa).41 Since the polymer PLGA is water-insoluble, upon being exposed to aqueous environment, phase inversion can be triggered, resulting in a two-phase, gelled implant.16 In order to obtain a more stable and longer sustained release of ROP with less initial burst release, hydrophobic BB was investigated as the main solvent for PLGA-based ISFSs in this study. Furthermore, BA, TA, and NMP, as mostly used solvent in PLGA-based ISFSs, were employed as a co-solvent and polarity-modifying agent of BB to improve the miscibility of PLGA with BB and tailor drug release behaviours. The formulation of 30% PLGA40k/NMP, which could be regarded as a representative one for the NMP systems,41 was used as a control. As shown in Fig. 4, there were no observable changes in the chromatographic peak profile and retention time of ROP, indicating the excellent stability of ROP during its in vivo release and absorption from ISFSs. The in vivo release order of the four ISFSs (Fig. 5) coincided well with that observed in vitro (Fig. 2). The maximum concentration (Cmax) of ROP in plasma caused by the hydrophobic mixed solvent (90% BB and 10% co-solvent)-based systems was reduced by 3.7–8.0 times compared to the NMP control (8.71–18.97 mg L−1 vs. 70.12 mg L−1) and the initial release rate of ROP from the systems during 0 to 0.7 hour after subcutaneous injection was significantly reduced as the release slopes decreased from 59.37 to 9.52 (Table 4, Fig. 9). The AUC0–12 h of the formulations with hydrophobic mixed solvent was also significantly (3.77–6.35 times) lower than that of the NMP control and the Cmax/Cs values were remarkably reduced (5.24–11.16 vs. 32.08), too (Table 3). The most profound reduction was achieved by the BB/NMP system, whose Cmax and the initial release rate (slope0–0.7 h = 9.52, r2 = 0.9828) were even 40% and 60% lower than those of aqueous solution of 15 mg kg−1, respectively (Tables 3 and 4), indicating that the release rate of ROP from the BB/NMP system was slower than the others. Besides, much longer and more stable plasma sustained time of ROP was also successfully achieved by the hydrophobic mixed solvent systems (10–15 days vs. 4 days). As a whole, the above-mentioned shortcomings of the NMP systems were effectively overcame by the hydrophobic mixed solvent systems, among which, the BB/NMP system showed a preferable sustained release of ROP due to the fastest phase inversion and the most stable long-lasting drug release. Factors that affect drug release from this system were therefore further investigated, including the ratio of BB to NMP, PLGA concentration, and PLGA molecular weight.
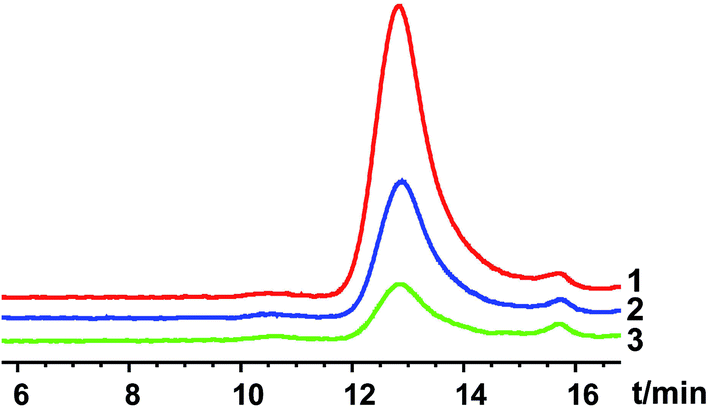 | ||
| Fig. 4 Representative chromatograms for the determination of FITC-labeled ROP released from the in situ forming systems and absorbed into the blood by high-performance gel permeation chromatography (HPGPC) ((1) 10 minutes after injection; (2) 12 hours after injection; (3) 168 hours after injection). | ||
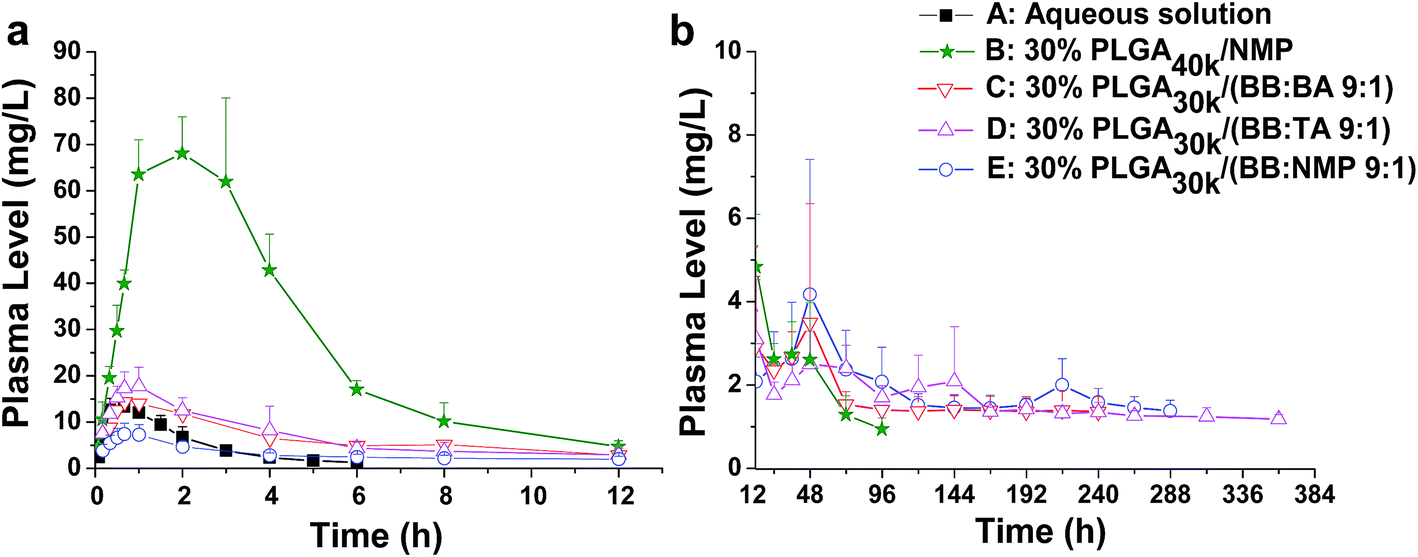 | ||
| Fig. 5 Effect of different solvents on the release of FROP from PLGA-based ISFSs. (a) Plasma FROP level–time profile of 0–12 h. (b) Plasma FROP level–time profile of 12–384 h. All the data not shown in the figures are those that could not be detected by the assay method used. The dose for all the ISFSs was 200 mg kg−1, while that for the control aqueous solution was 15 mg kg−1. PLGA, poly(D,L-lactide-co-glycolide); FROP, fluorescein isothiocyanate labeled Radix Ophiopogonis polysaccharide; NMP, N-methyl-2-pyrrolidone; BB, benzyl benzoate; BA, benzyl alcohol; TA, triacetin. | ||
| Code | Formulation | Parameters | ||||||
|---|---|---|---|---|---|---|---|---|
| AUC(0–12 h) (mg L−1 h−1) | AUC(0–t)a (mg L−1 h−1) | Tmax (h) | Cmax (mg L−1) | Cmax/Csb | MRT (h) | Fc (%) | ||
| a For the aqueous solution, t = ∞; for the other formulations, t is the last time point at which FROP was still detectable.b The steady state concentration (Cs) is the average value of the plasma level from Day 1 to Day 3 for formulation B and from Day 3 to Day 10 for formulations C–K.c F = 200 × AUC0–t(ISFS)/(15 × AUC0–∞(solution)). **P < 0.01 and *P < 0.05, compared with aqueous solution; ##P < 0.01 and #P < 0.05, compared with the formulation of 30% PLGA40k/NMP. Data are presented as mean ± standard deviation. FROP, fluorescein isothiocyanate-labeled Radix Ophiopogonis polysaccharide; NMP, N-methyl-2-pyrrolidone; BB, benzyl benzoate; BA, benzyl alcohol; TA, triacetin; PLGA, poly(D,L-lactide-co-glycolide); AUC, area under the concentration–time curve; MRT, mean residence time; Cmax, maximum concentration in plasma; Tmax, time taken to reach Cmax. | ||||||||
| A | Aqueous solution | — | 37.74 ± 11.05 | 0.33 ± 0.00 | 14.44 ± 2.14 | — | 2.315 ± 0.26 | — |
| B | 30% PLGA40k/NMP | 331.95 ± 4.84 | 514.84 ± 87.52 | 2.00 ± 1.00** | 70.12 ± 11.04** | 32.08 ± 10.19 | 17.48 ± 3.37 | 102.31 |
| C | 30% PLGA30k/(BB:BA 9:1) |
52.06 ± 14.27## | 463.75 ± 81.84 | 1.11 ± 0.77 | 11.74 ± 0.54## | 8.18 ± 1.85## | 97.47 ± 9.91**## | 92.16 |
| D | 30% PLGA30k/(BB:TA 9:1) |
87.73 ± 29.11## | 526.788 ± 108.99 | 1.33 ± 0.58 | 18.97 ± 4.48## | 11.16 ± 4.08## | 140.94 ± 15.67**## | 104.69 |
| E | 30% PLGA30k/(BB:NMP 9:1) |
39.90 ± 8.59## | 558.57 ± 156.17 | 0.89 ± 0.19 | 8.71 ± 1.69## | 5.24 ± 0.99## | 127.55 ± 20.90**## | 111.00 |
| F | 30% PLGA30k/(BB:NMP 8:2) |
91.91 ± 27.09## | 530.99 ± 64.62 | 0.89 ± 0.19 | 22.17 ± 5.80## | 13.11 ± 2.42## | 107.54 ± 10.48**## | 105.52 |
| G | 30% PLGA30k/(BB:NMP 7:3) |
171.28 ± 26.75## | 613.65 ± 82.89 | 1.00 ± 0.00 | 39.04 ± 12.87**## | 21.62 ± 6.57# | 80.81 ± 5.60**## | 121.95 |
| H | 20% PLGA30k/(BB:NMP 8:2) |
196.36 ± 80.01## | 483.79 ± 117.25 | 2.22 ± 1.68** | 32.18 ± 12.25**## | 18.32 ± 5.58## | 41.02 ± 6.13**## | 96.14 |
| I | 40% PLGA30k/(BB:NMP 8:2) |
33.91 ± 20.21## | 408.82 ± 90.45 | 0.50 ± 0.17# | 9.97 ± 5.70## | 6.66 ± 3.80## | 107.59 ± 13.16**## | 81.24 |
| J | 30% PLGA20k/(BB:NMP 8:2) |
156.77 ± 28.46## | 520.07 ± 30.15 | 0.89 ± 0.19 | 42.95 ± 8.95**## | 29.63 ± 1.98 | 65.89 ± 3.29**## | 103.35 |
| K | 30% PLGA40k/(BB:NMP 8:2) |
23.64 ± 3.27## | 449.07 ± 31.40 | 0.56 ± 0.19 | 6.67 ± 1.18## | 6.16 ± 1.37## | 190.58 ± 8.51**## | 89.24 |
| Formulations | A | B | C | D | E | F | G | H | I | J | K |
| Slope | 24.44 | 59.37 | 13.33 | 18.38 | 9.52 | 35.52 | 42.98 | 35.10 | 10.39 | 65.75 | 5.96 |
| r2 | 0.9677 | 0.9993 | 0.9210 | 0.9851 | 0.9828 | 0.9850 | 0.9659 | 0.9930 | 0.7377 | 0.9818 | 0.7491 |
Solvents with different strength and water miscibility used in ISFSs have different contribution to drug release from the depot. Some researchers have reported that by replacing part or total of a hydrophilic solvent with a water immiscible one, phase inversion rate can be decreased and, thus, a profile that is more uniform to zero-order release pattern over an extended release time can be achieved.52,53 This can be attributed to change in the depot morphology from a porous and loose structure to a less porous and more dense structure.54 To know the effect of the co-solvent NMP on drug release from the hydrophobic BB system, three ratios of BB to NMP (9:1, 8:2, and 7:3) were investigated with the same level of PLGA30k (30%, w/w). As is shown in Fig. 6 and 9, initial burst release increased (slope0–0.7 h increased from 9.51 to 42.58) with the decrease of BB to NMP ratio in the formulation as with the decrease in hydrophobicity of the formulation. With comparable Tmax (0.9–1.0 h), linear increases in Cmax (8.71, 22.17, and 39.04 mg L−1, r = 0.9979) and AUC0–12 h (39.90, 91.91, and 171.18 mg L−1 h−1, r = 0.9858) as well as a linear decrease in MRT (127.55, 107.54, and 80.81 h, r = 0.9966) were observed as the ratio changed from 9:1 to 7:3, suggesting a good correlation between hydrophobicity and controlled-release ability of the BB/NMP system. As the hydrophobicity decreased, the formulation possessed a higher water affinity and a faster leaching out of the drug, resulting in a negative contribution to controlling drug release from in situ forming depots.23
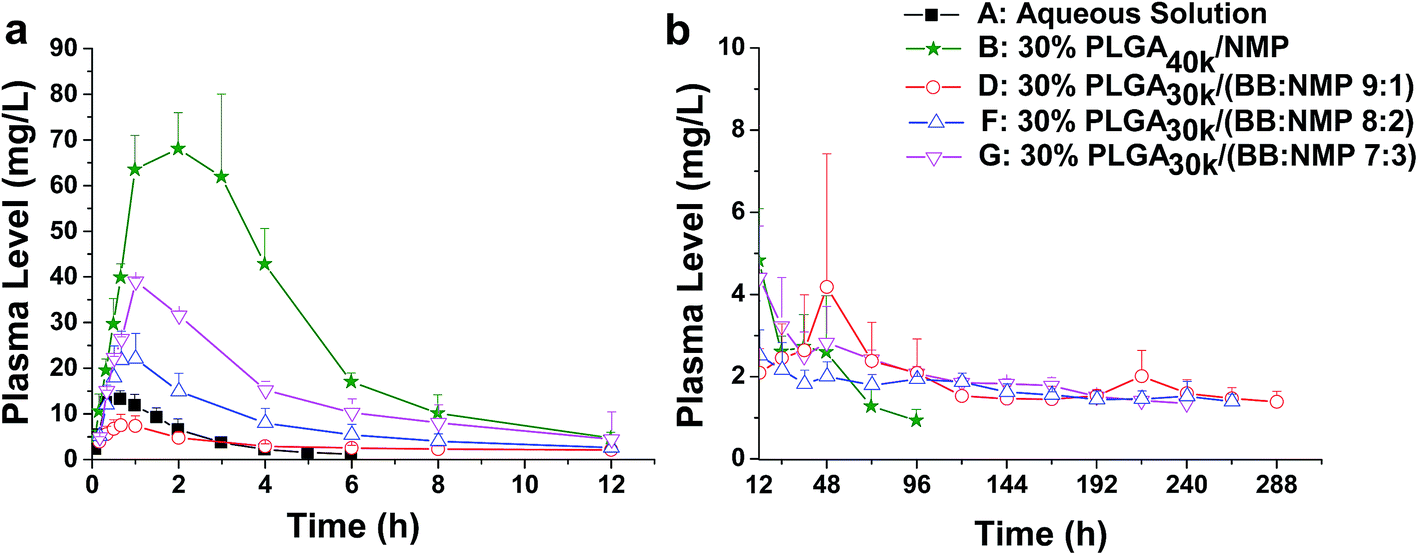 | ||
| Fig. 6 Effect of different ratio of BB to NMP on the release of FROP from PLGA-based ISFSs. (a) Plasma FROP level–time profile of 0–12 h. (b) Plasma FROP level–time profile of 12–288 h. All the data not shown in the figures are those that could not be detected by the assay method used. The dose for all the ISFSs was 200 mg kg−1, while that for the control aqueous solution was 15 mg kg−1. PLGA, poly(D,L-lactide-co-glycolide); FROP, fluorescein isothiocyanate labeled Radix Ophiopogonis polysaccharide; NMP, N-methyl-2-pyrrolidone. | ||
PLGA is a commonly used biodegradable polymer in solvent-induced ISFSs. Its characteristics strongly impact the degradation and hence not only the erosion of the matrix but also the phase inversion dynamics.19 At present, PLGA grades with different lactide/glycolide molar ratios ranging from 100:0 to 0:100 and molecular weights from less than 10 kDa up to 200 kDa are available. PLGAs with different lactide/glycolide molar ratios have different hydrophobicity. Through modulating the polymer hydrophobicity, initial burst release can be modified by influencing the exchange rate of solvent and non-solvent as well as the hydrolysis rate of polymers. For extended drug delivery of more than 2 months, PLGA 50:50 is the mostly used.55 Besides, PLGAs with different end-capping (e.g., carboxyl or hydroxyl) that has different contribution to the polymer degradation can be obtained. In this study, PLGAs with a free carboxyl at the end of the backbone chain and a lactide/glycolide molar ratio of 50:50 were selected as the ISFS matrix, considering both their moderate properties and the planed delivery period of ROP.
The effect of PLGA concentrations (20%, 30%, and 40%) on the release of ROP was investigated with the application of the formulation ‘PLGA30k/(BB:NMP 8:2)’. The results showed that the effect was significant (Fig. 7 and 9 and Table 3). As a whole, initial burst release was decreased with the increase of PLGA concentration as the release slopes of formulations changed from 35.10 to 10.39. As the PLGA level increased from 20% to 40%, linear decreases in Cmax (32.18, 22.17, and 9.97 mg L−1, r = 0.9984) and AUC0–12 h (196.36, 91.91, and 33.91 mg L−1 h−1, r = 0.9735) were observed and the Tmax appeared to be shortened from 2.22 h to 0.50 h. The fast formation of dense solid PLGA shell on the depot surface is believed to contribute to the shortened Tmax at high PLGA levels. Although high plasma levels were observed for the 20% PLGA30k system within the first 12 hours, it still achieved reductions in Cmax (32.18 mg L−1 vs. 70.12 mg L−1) and initial release rate (slope0–0.7 h = 35.10 vs. 59.37), and obtained an increase in MRT by 2.3 times compared to the control that contains 30% PLGA40k and uses NMP alone as the solvent. Nearly zero-order release of ROP with a relatively low ratio value of Cmax/Cs was observed as the concentration of PLGA increased to 40%. This should be attributed to both the high viscosity of the formulation and the fast in situ formation of dense structure, resulting in a cut of diffusion rate of drug to aqueous environment. It is intriguing that with the application of the formulation 40% PLGA30k/(BB:NMP 8:2), Cmax of ROP in plasma was even lower than that achieved by the aqueous solution that contained 13.33-fold lower dose of ROP. Also, the plasma sustained time of ROP was extended by 60 folds, from 0.25 days to 15 days, by the ISFS.
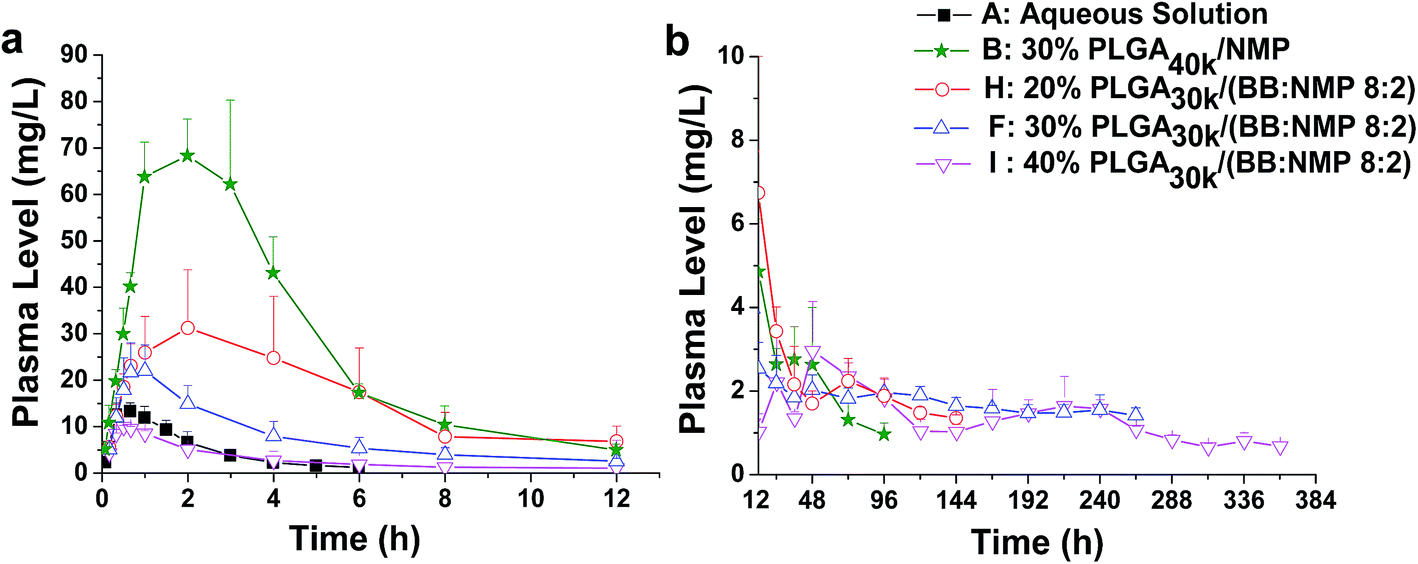 | ||
| Fig. 7 Effect of different PLGA concentrations on the release of FROP from PLGA-based ISFSs. (a) Plasma FROP level–time profile of 0–12 h. (b) Plasma FROP level–time profile of 12–384 h. All the data not shown in the figures are those that could not be detected by the assay method used. The dose for all the ISFSs was 200 mg kg−1, while that for the control aqueous solution was 15 mg kg−1. PLGA, poly(D,L-lactide-co-glycolide); FROP, fluorescein isothiocyanate labeled Radix Ophiopogonis polysaccharide; NMP, N-methyl-2-pyrrolidone. | ||
Polymer molecular weight (MW) is an extremely important property. It can affect many other key polymer properties such as solubility, viscosity, diffusivity, degradability, and glass transition temperature. According to some reports, the MW of PLGA has been pointed out to be one of the key factors that affect matrix erosion and also the initial drug release.19,56 Therefore, the effect of PLGA MW on the release of ROP was investigated by using the formulation 30% PLGA/(BB:NMP 8:2) and three PLGA MWs (20, 30, and 40 kDa). The results showed that drug release rate was significantly reduced by increasing the MW of PLGA as the release slopes during 0 to 0.7 hour after subcutaneous injection decreased sharply from 65.75 to 5.96 (Fig. 8 and 9). Linear decreases in Cmax (42.95, 22.17, and 6.67 mg L−1, r = 0.9965) and AUC0–12 h (156.77, 91.91, 23.64 mg L−1 h−1, r = 0.9998) were also observed with the PLGA MW increasing from 20 to 40 kDa and nearly zero-order release of ROP was achieved by 40 kDa PLGA as with the lowest ratio value of Cmax/Cs. The higher polymer MW means an increase in viscosity (Fig. 1d) and hydrophobicity, causing slower release of organic solvent and, thus, slower phase transition and lower burst release. Similar results were reported in the literatures.57,58 For example, the ISFS prepared with the 48 kDa PLGA was found to have a significantly lower initial release of leuprolide acetate with the first 24 h compared to the lower polymer MW implant (i.e., 12 and/or 34 kDa).57 Among the formulations studied in this work, the lowest initial release (slope0–0.7 h = 5.96), together with the longest (MRT = 190.6 h) and the steadiest sustained release (Cmax/Cs = 6.16), was achieved by the formulation ‘30% PLGA40k/(BB:NMP 8:2)’. It resulted in a Cmax that is only about half of that achieved by the aqueous solution with 13.33-fold lower dose of ROP and is 10.5-fold lower than that caused by the control system containing the same PLGA level and MW but using NMP alone as the solvent. It also caused a MRT that is about 82.3- and 10.9-fold longer than those of the aqueous solution and the control system, respectively. It is also notable that the initial release rate of ROP from this formulation was significantly reduced by 76% (slope0–0.7 h = 5.96 vs. 24.44) and 90% (slope0–0.7 h = 5.96 vs. 59.37) compared to aqueous solution (15 mg kg−1) and 30% PLGA40k/NMP (200 mg kg−1), respectively.
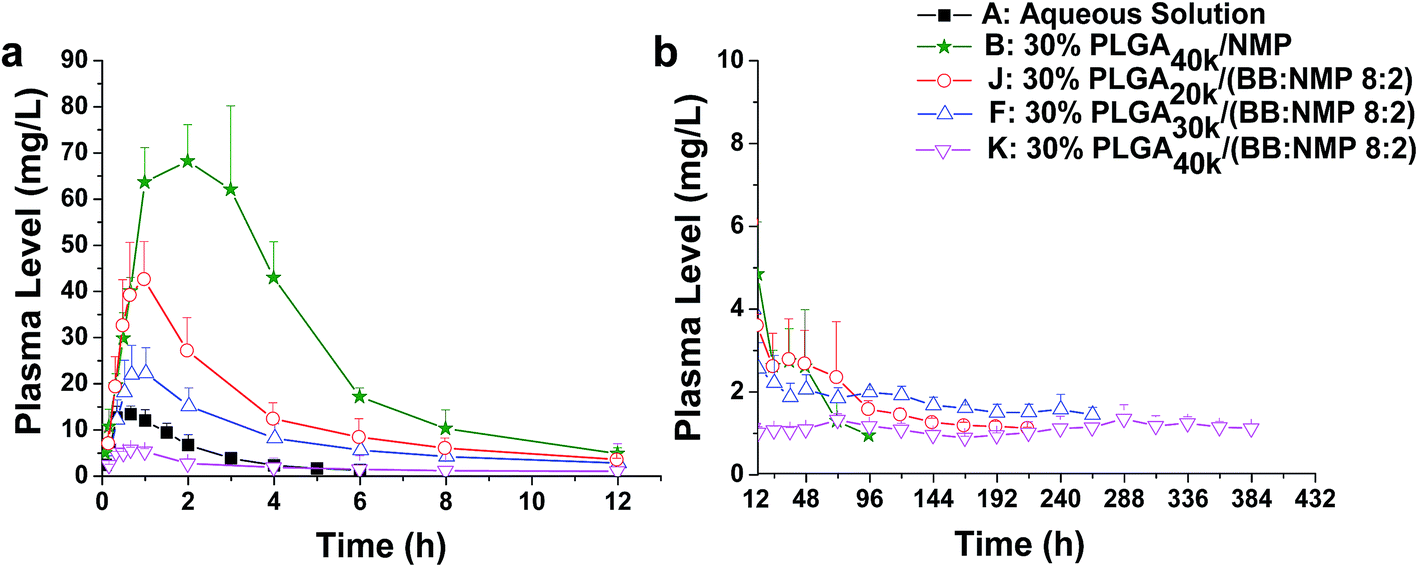 | ||
| Fig. 8 Effect of different molecular weights on the release of FROP from PLGA-based ISFSs. (a) Plasma FROP level–time profile of 0–12 h. (b) Plasma FROP level–time profile of 12–384 h. All the data not shown in the figures are those that could not be detected by the assay method used. The dose for all the ISFSs was 200 mg kg−1, while that for the control aqueous solution was 15 mg kg−1. PLGA, poly(D,L-lactide-co-glycolide); FROP, fluorescein isothiocyanate labeled Radix Ophiopogonis polysaccharide; NMP, N-methyl-2-pyrrolidone. | ||
 | ||
| Fig. 9 Effects of different mixed solvent (a), ratio of BB to NMP (b), PLGA concentration (c), and molecular weight of PLGA (d) on the initial release rate of PLGA-based in situ forming formulations during 0 and 0.7 hour after subcutaneous injection. | ||
As a whole, the release behaviour of ROP from ISFSs can be tailored by varying the solvent composition and PLGA parameters. All the nine hydrophobic mixed solvent-based ISFSs studied showed lower Cmax (1.63 to 10.5 times) and longer MRT (2.35 to 10.9 times) than the 30% PLGA40k/NMP system. Moreover, all of their F values are around 100%, indicating that the release of the incorporated drug from these ISFSs approached 100%.
3.5 Anti-myocardial ischemia activity
To investigate the cardioprotective effect of ROP-loaded ISFSs, the formulation ‘30% PLGA30k/(BB:NMP 9:1)’ was choice as a representative due to its relatively stable release of ROP from the depot (slope0–0.7 h = 9.52, Cmax/Cs = 5.24). Significant reduction of infarct sizes in the groups of aqueous solution and the ISFS was found when compared to the model group (Table 5), suggesting that both the aqueous solution of ROP and the ROP-loaded ISFS played a critical role in the treatment of myocardial ischemia in this study. Also, it is noteworthy that thanks to its long-lasting and stable release of ROP, the ISFS with significantly reduced administration frequency showed a better bioactivity in anti-myocardial ischemia than the aqueous solution. The representative photographs of transverse slices following the TTC staining were shown in Fig. 10.
| Group | Infarct size of the left ventricle (%) | Infarct size of the whole heart (%) |
|---|---|---|
| a * and #P < 0.05 vs. the model group; ** and ##P < 0.01 vs. the model group. | ||
| Model | 23.13 ± 3.58 | 17.58 ± 2.53 |
| Aqueous solution | 17.22 ± 1.89** | 13.37 ± 1.48# |
| 30% PLGA30k/(BB:NMP 9:1) |
15.60 ± 2.72** | 11.96 ± 1.99# |
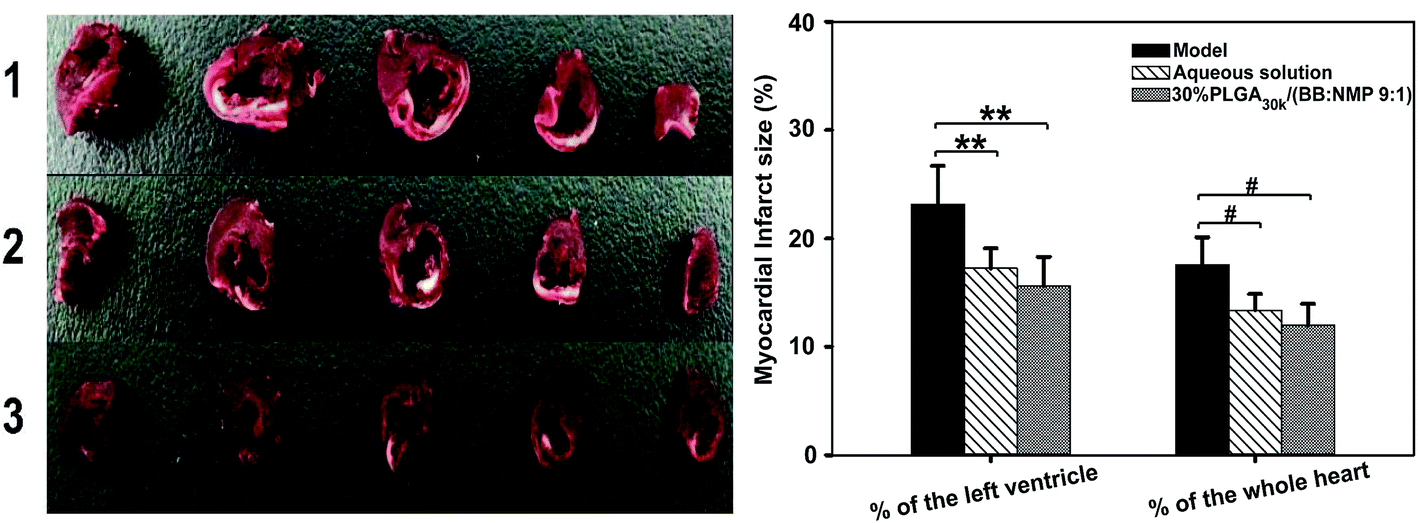 | ||
| Fig. 10 Effects of ROP on myocardial infarct size in rats with myocardial ischemia induced by permanent ligation of the left coronary artery (n = 5; (1) model, (2) aqueous solution of ROP, (3) ROP-loaded ISFS ‘30% PLGA30k/(BB:NMP 9:1)’; * and #P < 0.05 vs. the model group; ** and ##P < 0.01 vs. the model group). PLGA, poly(D,L-lactide-co-glycolide); BB, benzyl benzoate; NMP, N-methyl-2-pyrrolidone. | ||
3.6 Some general formulation and application principles
In this study, it was observed that the ROP release from ISFSs was effectively controlled by varying either solvent polarity or polymer parameters without sacrificing its bioactivity. Moreover, different type-and-level combinations of solvent and polymer could be chosen to achieve comparable release behaviours. This provides flexibility in formulation design to achieve specific aims and/or fit with drugs with specific properties. For example, three different combinations, i.e., 30% PLGA30k/(BB:NMP 9:1), 40% PLGA30k/(BB:NMP 8:2), and 30% PLGA40k/(BB:NMP 8:2) all achieved an at least 12 Day-long plasma sustained time of ROP and a Cmax lower than that caused by the aqueous solution with 13.33-fold lower dose of ROP. Faster release could be achieved by the reduction of PLGA MW, PLGA level, and/or solvent hydrophobicity. For ROP, release behaviours with Cmax/Cs ranging from 5.24 to 32.1 and MRT from 190 h to 17.5 h could be achieved by the reduction of PLGA MW from 40 kDa to 20 kDa, PLGA level from 40% to 20%, and/or solvent hydrophobicity from (BB:NMP 9:1) to pure NMP. If much short-term and high exposure of drugs is expected, hydrogel-based ISFSs become the choice. For example, half-day high and full exposure of ROP was achieved by the application of poloxamer-based ISFSs, such as ‘20% P407’ and ‘24% P407/10% P188’.41 As it is urgent to find a proper form of administration for herb polysaccharides with properties of low oral bioavailability and short plasma half-life, like ROP, lentinan,59 polysaccharide of cistanche deserticola,60 tremella polysacchredes,61 and laminarin,62 ISFSs seem to be a versatile and promising choice to make full use of their bioactivities over a prolonged period of time. In addition, ISFS formulations should be chosen with the consideration of both the disease and the action nature of the polysaccharide used. In general, a loading dose followed by a sustained release at lower concentration may be desirable especially for anti-cancer therapy19 or prevention and/or cure of chronic diseases; namely, the formulation with a relatively lower slope in the initial release and a lower ratio value of Cmax/Cs is preferable. While formulations with higher both initial release rates and peak values of plasma concentration are promising for acute therapies.
4. Conclusion
Initial burst release of ROP, a highly hydrophilic and low MW polysaccharide, was markedly reduced and its sustained-release period was significantly prolonged as the solvent for PLGA-based ISFSs was changed from hydrophilic NMP to hydrophobic mixed solvent consisting of 90% BB and 10% co-solvent (BA, TA, or NMP). The effect of the 10% co-solvent on phase inversion rate, morphology of in situ formed depots, and ROP release was more significant than expected from its content. Among the co-solvents investigated, NMP surprisingly showed to be the most effective in reducing the initial burst release of ROP although having much higher polarity than the others. This should be attributed to a suitable phase inversion behaviour (both rate and morphology) induced by it. Further studies indicated that the release behaviour could be tailored by changing the ratio of BB to NMP, PLGA MW, and PLGA concentration. Linear reductions in Cmax and AUC0–12 h with different slopes was observed with the increase of the ratio of BB to NMP (7:3 to 9:1), PLGA MW (20 kDa to 40 kDa), and PLGA level (20% to 40%), respectively. Moreover, it is possible to achieve nearly zero-order release with at least 12 Day-long plasma sustained time of ROP by different formulation combination without sacrificing its bioactivity of anti-myocardial ischemia. ROP-loaded ISFSs showed an obvious advantage over aqueous solution in terms of the ability of long-lasting and stable delivery and thus significantly reduced frequency of administration. Therefore, ISFSs with hydrophobic mixed solvent like BB/NMP, as a whole, appear to be promising and suitable for long-term and smooth sustained release of herb polysaccharides with properties of low oral bioavailability and short plasma half-life as ROP.
Conflict of interest
The authors report no conflicts of interest in this work.Acknowledgements
This work was supported by Program for New Century Excellent Talents in University (NCET-13-0906), the National Natural Science Foundation of China (81073065), the funds from Shanghai Municipal Commission of Health and Family Planning (ZY3-CCCX-3-5001) and Science and Technology Commission of Shanghai Municipality (15DZ2292000), the Science and Technology Development Fund of Pudong New Area (PKF-2013-003), and the “085” Project (085ZY1219) of Shanghai University of TCM.References
- D. H. Paik and S. W. Choi, Macromol. Rapid Commun., 2014, 35, 1033–1038 CrossRef CAS PubMed.
- N. K. Bejugam, S. G. Gayakwad, A. N. Uddin and M. J. D'Souza, J. Microencapsulation, 2013, 30, 274–282 CrossRef CAS PubMed.
- A. Patel, M. Patel, X. Y. Yang and A. K. Mitra, Protein Pept. Lett., 2014, 21, 1102–1120 CrossRef CAS PubMed.
- A. Gaowa, T. Horibe, M. Kohno, K. Sato, H. Harada, M. Hiraoka, Y. Tabata and K. Kawakami, J. Controlled Release, 2014, 176, 1–7 CrossRef CAS PubMed.
- M. Sun, W. Zhao, Q. Xie, Y. Zhan and B. Wu, J. Surg. Oncol., 2015, 24, 28–34 CrossRef PubMed.
- Z. Liu, J. Xing, S. Zheng, R. Bo, L. Luo, Y. Huang, Y. Niu, Z. Li, D. Wang, Y. Hu, J. Liu and Y. Wu, Carbohydr. Polym., 2016, 142, 141–148 CrossRef CAS PubMed.
- K. K. Auyeung, Q. B. Han and J. K. Ko, Am. J. Chin. Med., 2016, 44, 1–22 CrossRef PubMed.
- L. Wang, C. Yao, F. Wu, X. Lin, L. Shen and Y. Feng, Int. J. Nanomed., 2015, 10, 5729–5737 CAS.
- J. V. Natarajan, A. Darwitan, V. A. Barathi, M. Ang, H. M. Htoon, F. Boey, K. C. Tam, T. T. Wong and S. S. Venkatraman, ACS Nano, 2014, 8, 19–29 CrossRef PubMed.
- K. Park, J. Controlled Release, 2016, 226, 268 CrossRef CAS PubMed.
- A. H. Ghassemi, M. J. van Steenbergen, H. Talsma, C. F. van Nostrum, W. Jiskoot, D. J. Crommelin and W. E. Hennink, J. Controlled Release, 2009, 138, 57–63 CrossRef CAS PubMed.
- R. B. Patel, L. Solorio, H. Wu, T. Krupka and A. A. Exner, J. Controlled Release, 2010, 147, 350–358 CrossRef CAS PubMed.
- A. A. Ghahremankhani, F. Dorkoosh and R. Dinarvand, Pharm. Dev. Technol., 2008, 13, 49–55 CrossRef CAS PubMed.
- J. C. Wright, in Long acting injections and implants: In Situ Forming Systems (Depots). ed. J. C. Wright and D. J. Burgess, Springer, London, 2012, vol. 8, pp. 153–166 Search PubMed.
- P. Agarwal and I. D. Rupenthal, Drug Discovery Today, 2013, 18, 337–349 CrossRef CAS PubMed.
- K. J. Brodbeck, J. R. DesNoyer and A. J. McHugh, J. Controlled Release, 1999, 62, 333–344 CrossRef CAS PubMed.
- L. Solorio, D. Sundarapandiyan, A. Olear and A. A. Exner, J. Pharm. Sci., 2015, 104, 3471–3480 CrossRef CAS PubMed.
- R. E. Eliaz and J. Kost, J. Biomed. Mater. Res., 2000, 50, 388–396 CrossRef CAS PubMed.
- M. Parent, C. Nouvel, M. Koerber, A. Sapin, P. Maincent and A. Boudier, J. Controlled Release, 2013, 172, 292–304 CrossRef CAS PubMed.
- S. Chhabra, V. Sachdeva and S. Singh, Int. J. Pharm., 2007, 342, 72–77 CrossRef CAS PubMed.
- T. Zhang, Q. Peng, F. Y. San, J. W. Luo, M. X. Wang, W. Q. Wu, T. Gong and Z. R. Zhang, Biomaterials, 2015, 45, 1–9 CrossRef CAS PubMed.
- C. Raman and A. J. McHugh, J. Controlled Release, 2005, 102, 145–157 CrossRef CAS PubMed.
- S. Dhawan, R. Kapil and D. N. Kapoor, J. Biomater. Appl., 2011, 25, 699–720 CrossRef CAS PubMed.
- X. Lin, S. Yang, J. Gou, M. Zhao, Y. Zhang, N. Qi, H. He, C. Cai, X. Tang and P. Guo, J. Mater. Sci.: Mater. Med., 2012, 23, 443–455 CrossRef CAS PubMed.
- L. Wang, A. Wang, X. Zhao, X. Liu, D. Wang, F. Sun and Y. Li, Int. J. Pharm., 2012, 427, 284–292 CrossRef CAS PubMed.
- X. Shi, X. Lin, X. Zheng, Y. Feng and L. Shen, Int. J. Nanomed., 2014, 9, 5555–5563 CAS.
- L. Solorio and A. A. Exner, J. Pharm. Sci., 2015, 104, 4322–4328 CrossRef CAS PubMed.
- L. Solorio, B. M. Babin, R. B. Patel, J. Mach, N. Azar and A. A. Exner, J. Controlled Release, 2010, 143, 183–190 CrossRef CAS PubMed.
- S. Chen and J. Singh, Pharm. Dev. Technol., 2005, 10, 319–325 CrossRef CAS PubMed.
- A. M. Avachat and S. S. Kapure, Int. J. Pharm., 2014, 477, 64–72 CrossRef CAS PubMed.
- L. Wang, S. Venkatraman and L. Kleiner, J. Controlled Release, 2004, 99, 207–216 CrossRef CAS PubMed.
- M. Okada, Prog. Polym. Sci., 2002, 27, 87–133 CrossRef CAS.
- R. A. Jain, C. T. Rhodes, A. M. Railkar, A. W. Malick and N. H. Shah, Eur. J. Pharm. Biopharm., 2000, 50, 257–262 CrossRef CAS PubMed.
- D. S. Xu, Y. Feng, X. Lin, H. L. Deng, J. N. Fang and Q. Dong, Yaoxue Xuebao, 2005, 40, 636–639 CAS.
- Y. Wang, Y. Zhu, K. Ruan, H. Wei and Y. Feng, Carbohydr. Polym., 2014, 114, 183–189 CrossRef CAS PubMed.
- W. L. Lin, W. W. Su, X. Y. Cai, L. K. Luo, P. B. Li and Y. G. Wang, Int. J. Biol. Macromol., 2011, 49, 194–200 CrossRef CAS PubMed.
- S. Wang, X. Lin, L. Y. Wang, K. F. Ruan, Y. Feng and X. Y. Li, Int. J. Biol. Macromol., 2012, 50, 734–740 CrossRef CAS PubMed.
- X. Lin, D. S. Xu, Y. Feng and L. Shen, Anal. Biochem., 2005, 342, 179–185 CrossRef CAS PubMed.
- X. Lin, D. S. Xu, Y. Feng, S. M. Li, Z. L. Lu and L. Shen, J. Pharm. Sci., 2006, 95, 2534–2542 CrossRef CAS PubMed.
- K. Zhang, X. Shi, X. Lin, C. Yao, L. Shen and Y. Feng, Drug Delivery, 2015, 22, 375–382 CrossRef CAS PubMed.
- X. Shi, X. Lin, C. Yao, L. Shen and Y. Feng, Int. J. Biol. Macromol., 2015, 72, 553–559 CrossRef CAS PubMed.
- T. Phaechamud and J. Mahadlek, Int. J. Pharm., 2015, 494, 381–392 CrossRef CAS PubMed.
- Z. L. Lu, X. Lin, Y. Feng and D. S. Xu, China Pharm., 2008, 11, 768–770 CAS.
- J. Ye, L. Yang, R. Sethi, J. Copps, B. Ramjiawan, R. Summers and R. Deslauriers, Mol. Cell. Biochem., 1997, 176, 227–233 CrossRef CAS PubMed.
- J. A. Camargo, A. Sapin, C. Nouvel, D. Daloz, M. Leonard, F. Bonneaux, J. L. Six and P. Maincent, Drug Dev. Ind. Pharm., 2013, 39, 146–155 CrossRef CAS PubMed.
- J. R. DesNoyer and A. J. McHugh, J. Controlled Release, 2001, 70, 285–294 CrossRef CAS PubMed.
- M. Kilicarslan, M. Koerber and R. Bodmeier, Drug Dev. Ind. Pharm., 2014, 40, 619–624 CrossRef CAS PubMed.
- R. E. Eliaz and F. C. Szoka Jr, Gene Ther., 2002, 9, 1230–1237 CrossRef CAS PubMed.
- S. Dong, S. Wang, C. Zheng, W. Liang and Y. Huang, Soft Matter, 2011, 7, 5873 RSC.
- S. Chen and J. Singh, Int. J. Pharm., 2005, 295, 183–190 CrossRef CAS PubMed.
- D. N. Kapoor, O. P. Katare and S. Dhawan, Int. J. Pharm., 2012, 426, 132–143 CrossRef CAS PubMed.
- K. Al-Tahami, A. Meyer and J. Singh, Pharm. Dev. Technol., 2006, 11, 79–86 CrossRef CAS PubMed.
- R. Astaneh, M. Erfan, J. Barzin, H. Mobedi and H. R. Moghimi, Adv. Polym. Technol., 2008, 27, 17–26 CrossRef CAS.
- A. J. McHugh, J. Controlled Release, 2005, 109, 211–221 CrossRef CAS PubMed.
- X. S. Wu and N. Wang, J. Biomater. Sci., Polym. Ed., 2001, 12, 21–34 CrossRef CAS PubMed.
- R. B. Patel, A. N. Carlson, L. Solorio and A. A. Exner, J. Biomed. Mater. Res., Part A, 2010, 94, 476–484 Search PubMed.
- R. Astaneh, M. Erfan, H. Moghimi and H. Mobedi, J. Pharm. Sci., 2009, 98, 135–145 CrossRef CAS PubMed.
- Q. Liu, H. Zhang, G. Zhou, S. Xie, H. Zou, Y. Yu, G. Li, D. Sun, G. Zhang, Y. Lu and Y. Zhong, Int. J. Pharmacol., 2010, 397, 122–129 CrossRef CAS PubMed.
- S. S. Zhang, Studies on pharmacokinetics and tissue distribution of Lentinan liposome, Huazhong University of Science and Technology, 2010 Search PubMed.
- X. Y. Wang, Study on the immune pharmacology and absorption character of polysaccharides of Cistanche deserticola, Peking Union Medical College Hospital, 2009 Search PubMed.
- H. L. Xie, Effect of different routes of Tremella polysaccharides on Attenuation and Synergy in antitumor treatment with cyclophosphamide, Jilin University, 2012 Search PubMed.
- Y. H. Xu, Y. L. Yang, H. L. Peng and R. M. Fan, Her. Med., 2007, 26, 60–62 CAS.
| This journal is © The Royal Society of Chemistry 2017 |