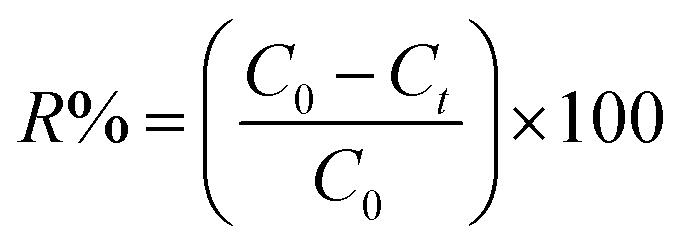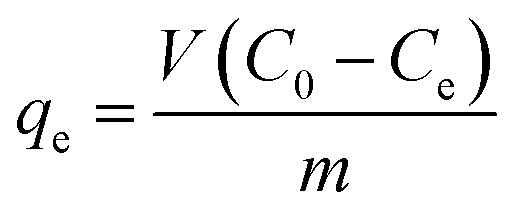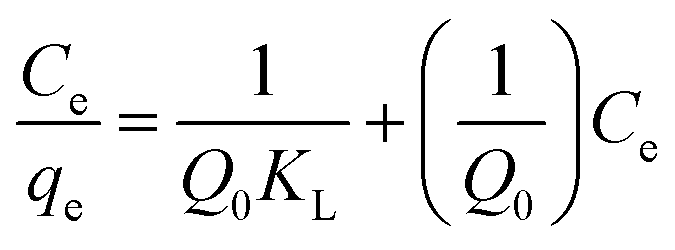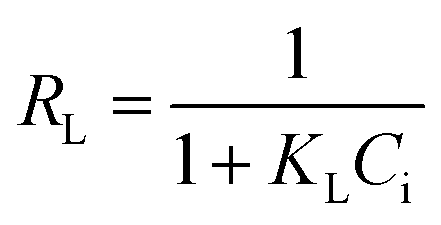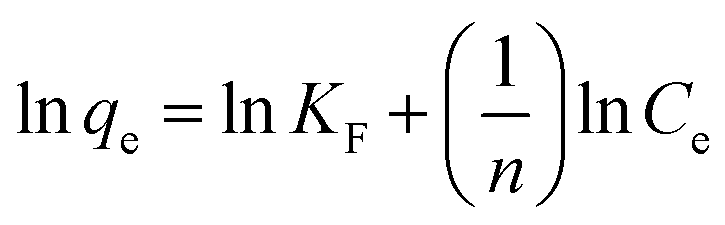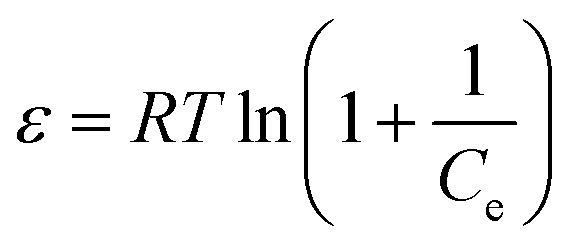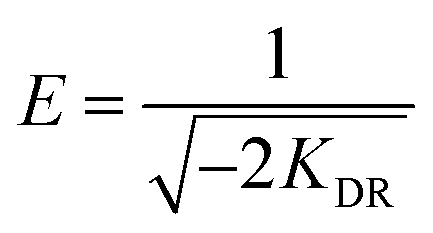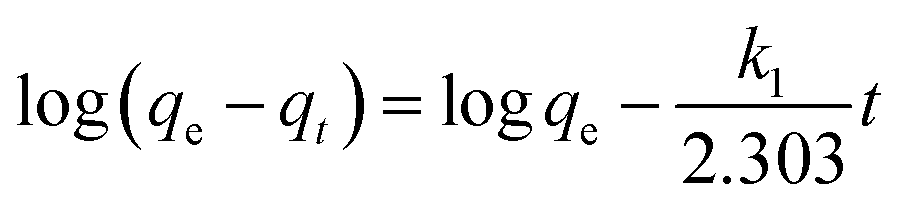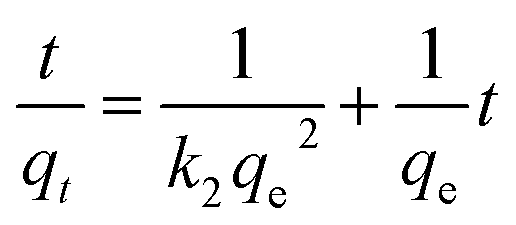DOI:
10.1039/C6RA27782A
(Paper)
RSC Adv., 2017,
7, 5790-5799
Highly effective removal of malachite green from aqueous solution by hydrochar derived from phycocyanin-extracted algal bloom residues through hydrothermal carbonization
Received
5th December 2016
, Accepted 5th January 2017
First published on 17th January 2017
Abstract
Huge volumes of harmful algal bloom residues (ABR) are collected during emergency treatment of cyanobacteria blooms, and phycocyanin-extracted algal bloom residues (PE-ABR) are produced after extraction of phycocyanin from ABR. To utilize them in a more efficient way, we propose a new approach to prepare hydrochars from PE-ABR through hydrothermal carbonization (HTC). The physical characters of as-prepared hydrochars were analyzed by SEM, FTIR and Raman spectroscopy. The PE-ABR hydrochar showed remarkable adsorption capability for removal malachite green (MG), with the maximum adsorption capacity of PE-ABR hydrochar for MG was 89.05 mg g−1. The adsorption equilibrium is consistent with Langmuir's model, and the adsorption kinetics follows the pseudo-second-order equation. Therefore, this work have demonstrated that harmful ABR can be utilized not only as a potential material for production of phycocyanin, but also an excellent source for hydrochar for removal dyes in contaminated wastewater with high adsorption efficiency.
Introduction
Colored effluents are usually discharged from the textile, tannery, food, paper/pulp, printing, cosmetics, plastic and pharmaceutical industries.1 Dyes and pigments in colored wastewaters have posed constant threat against the aquatic environment because they can reduce light penetration and so significantly influence photosynthetic activity of aquatic life.2 Malachite green (MG) as one such dye has also been extensively used as a bactericide, fungicide and parasiticide in aquaculture industries worldwide and may cause toxic effect such as carcinogenesis, mutagenesis and respiratory destruction.3 It has been reported that MG discharged into receiving water bodies even at low concentrations can affect the aquatic life and cause detrimental effects in liver, gill, kidney, intestine and gonads.4 Therefore, nowadays MG is not allowed to be applied in aquaculture in many countries including the United States, China, Canada, and the European Union,5 and the environmental quality standard limit of concentration of MG in water is set in the range of 0.5–100 μg L−1.6 To remove dyes in dye-contaminated wastewater with high concentration, the conventional methods are employed which include aerobic and anaerobic microbial remediation, chemical coagulation, reverse osmosis, oxidation, ion exchange, membrane filtration and electrochemical approaches. But generally, these methods either require high cost of exploitation or are incapable of treating large volumes of effluent.2,6
Alternatively, it is also effective to employ the adsorption method by using solid as adsorbents in treatment of dye-contaminated wastewater. In application of this method, the most widely used adsorbent is activated carbon (AC). However, up to now, production of commercial activated carbon is still an expensive process. Therefore, it is of immense interest to search for a more cost-effective production process of AC materials.7 In this regard, people have tried the methods of pyrolysis of biomasses for production of char material, such as the dry pyrolysis and wet pyrolysis processing approaches. The wet pyrolysis processing is also known as hydrothermal carbonization (HTC), and it has been established as a fast, simple and carbon dioxide negative conversion technique to produce hydrochars from a variety of biomasses. In fact, the technique of HTC has opened up the field of potential feedstocks for char production to a range of nontraditional renewable and plentiful wet agricultural residues and municipal wastes in the recent years.8,9
However, in order to produce hydrochars through HTC in a more cost-effective way, it is still a challenge to acquire the most suitable materials from the nature directly. For this purpose, therefore, in this work we paid special attention to algal bloom residues (ABR), or, cyanobacterial blooms residues. It has been reported that cyanobacterial blooms caused by anthropogenic nutrient inputs to aquatic ecosystems have been expanding worldwide. Over the recent decades, the occurrence of harmful algal blooms has increased globally,7 while refloatation and collection of algae from lakes are considered as the most efficient approaches to reduce the threat of blue algae.10 For example, Chao-Lake in our Anhui province, which is the fifth largest freshwater lake in China, generated a total of 0.29 million tons of algae during bloom season in 2015. The huge volumes of harmful ABR could result in a serious secondary pollution,10 so it has become an urgent task to dispose the toxic algae bloom residues. The methods such as anaerobic digestion to produce biogas,11 anaerobic co-digestion with corn straw,12 pyrolysis of blue-green algae blooms to produce biogas and bio-oil and biochar13,14 have been tried to convert harmful algal blooms into useful materials such as high performance electrodes for sodium ion batteries15 and pure phycobiliproteins.16 And especially, Zhang et al. have succeeded in extraction of phycocyanin from ABR collected from Chao-Lake.17 However, after extraction of phycocyanin, there are still a large amount of ABR, called “phycocyanin extracted algal blooms residues (PE-ABR)”, and these PE-ABR are still rich in carbohydrate, protein or lipid which may be further utilized to produce high values of usage.18 In this work, we intended to employ the technique of HTC to produce hydrochars. Because HTC is an exothermic process used to convert wet feedstocks into carbonaceous solid, it can operate at relatively low temperature compared to other thermal methods.8,19 Actually, the HTC technique has been proved to be effective in many feedstock materials20 including cellulose, lignin, and lignocellulosic materials, as well as a variety of waste-biomasses such as municipal solid waste, distiller's grains, and corncob residues.19,21,22 Therefore, in this work, we initiated the application of HTC in transformation of ABR and especially PE-ABR to hydrochars, and anticipated that they could be used as the good adsorbents for removal of dyes. In the study, we investigated the adsorption properties of the hydrochars as functions of varied operational conditions, and examined the materials using scanning electron microscopy (SEM), Raman spectroscopy and Fourier transform infrared spectroscopy (FTIR). The involved adsorption mechanisms were discussed by evaluating the adsorption kinetics via experimental study and modeling simulation. Our results have showed that the hydrochar derived from PE-ABR has extraordinary high efficiency for removal of MG so that it may become a very promising adsorbent material for treatment of dye-contaminated wastewater.
Materials and methods
Materials
MG hydrochloride (C.I. = 42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, chemical formula = C23H26N2Cl, molecular weight 364.92; absorbance maximum (λmax): 617 nm) was supplied by Sinopharm Chemical Reagent Company, Shanghai, China. The stock solutions (200 mg L−1) of MG were prepared by dissolving accurately weighed amount of the dyes in distilled water. All the chemicals used throughout this study were of analytical-grade. All working solutions of required concentrations were obtained by diluting the stock solution with distilled water.
000, chemical formula = C23H26N2Cl, molecular weight 364.92; absorbance maximum (λmax): 617 nm) was supplied by Sinopharm Chemical Reagent Company, Shanghai, China. The stock solutions (200 mg L−1) of MG were prepared by dissolving accurately weighed amount of the dyes in distilled water. All the chemicals used throughout this study were of analytical-grade. All working solutions of required concentrations were obtained by diluting the stock solution with distilled water.
Adsorbent preparation and characterization
Adsorbent preparation. The harmful algal blooms residue (ABR) was collected from the north shore (31°40′53.61′′N, 117°17′57.06′′E) of the fifth largest freshwater lake in China, namely, Chao-Lake, which is located near Hefei City in Anhui province, during the outbreak of cyanobacteria blooms in summer. The collected ABR were stored at 4 °C before usage. After extraction of phycocyanin by freeze–thaw with salt precipitation method (the extraction and purification of phycocyanin were performed by the combined use of two-step salt precipitation and two-step column chromatography, achieving the final purity of phycocyanin greater than 4 after the four-step purification process),17 the algal residue called phycocyanin-extracted algal bloom residues (PE-ABR) were left and stored at 4 °C before use. To produce hydrochars, both ABR (before phycocyanin extraction) and PE-ABR (after phycocyanin extraction) were centrifuged for 30 minutes with 8000 rpm to collect precipitation. The HTC process was conducted as follows:23 a mixture of ABR or PE-ABR (30 g) and deionized water (15 mL) were placed into an autoclave; next, the reactors was programmed to heat until reaching a predefined peak temperature (200 °C) and then hold for the specified retention time (10 hours); after the reaction, the hydrochar products were washed several times with deionized water; and finally, these samples were separated from water using filtration/centrifugation and dried at 60 °C for further analysis.
Surface characterization. The surface morphology of hydrochar from ABR or PE-ABR was visualized by a scanning electronic microscope (JEOL-2100F, Japan). FTIR spectroscopy was used to identify the chemicals present in the adsorbent. The FTIR spectra of the control samples (hydrochar before adsorption of MG) and the MG-loaded samples (adsorbent 4 g L−1, 100 mg L−1 MG for 24 h) were obtained using a FTIR spectrometer (Alpha-T, Bruker) accumulating 64 scans at a spectral resolution of 1 cm−1. The Raman spectra of MG and hydrochars from ABR and PE-ABR before and after loading MG (adsorbent 4 g L−1, 100 mg L−1 MG for 24 h) were recorded using an Xplora Raman microspectrometer (Horiba Jobin Yvon) with a 532 nm laser and an Olympus 50× long working distance lens. The beam size was about 0.7 μm in diameter and worked with a power of approximately 0.2 mW. The acquisition time of 5 s was used for the Raman measurements at each point.
Batch adsorption experiments. Batch adsorption experiments were conducted by shaking varies masses of hydrochar produced from ABR and hydrochar from PE-ABR with 50 mL aqueous solution of MG of known concentration (50, 75 and 100 mg L−1) in 100 mL flasks placed in a temperature controlled shaker at 25 °C and with adsorbent doses between 1 and 8 g L−1 at a constant shaking rate of 200 rpm. After the desired contact time, the samples were withdrawn from the mixture and centrifuged for 15 min at 8000 rpm. After centrifugation, the concentration of MG in solution was measured at 617 nm using a UV-vis spectrophotometer (Shimadzu, UV-2550, Japan). Controls were measured using adsorbent in distilled water blank and adsorbent-free MG. The percentage removal of MG was calculated by using the following equation:| |
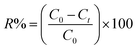 | (1) |
where C0 and Ct are the MG concentrations (mg L−1) initially and at a given time t, respectively. The amount of MG taken by adsorbent (qe) was calculated by:| |
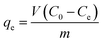 | (2) |
where V is the solution volume (L), m is the amount of adsorbent (g), and C0 and Ce are the initial and equilibrium concentrations of MG (mg L−1), respectively. Three replicates per sample were available for obtaining the average in the evaluation.
Adsorption isotherm. Four isotherm models including Langmuir, Freundlich, Dubinin–Radushkevich and Temkin models were tested for equilibrium description at ambient temperature. Langmuir model can be described by the following equation:| |
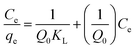 | (3) |
where qe and Ce correspond to the amount adsorbed per gram of adsorbent (mg g−1) and the solute concentration (mg L−1) in the aqueous solution in equilibrium, respectively. Q0 and KL are constants related to the maximum adsorption capacity (mg g−1) and the adsorption energy (L mg−1), respectively.24 The essential characteristics of Langmuir isotherm can be explained in terms of dimensionless constant separation factor (RL) which is expressed as:7,25| |
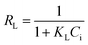 | (4) |
where Ci is initial concentration of MG.Freundlich's model can be described by the following equation:
| |
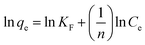 | (5) |
where
KF (mg g
−1) is the adsorption capacity of the adsorbent and
n gives an index for the effectiveness of the adsorption process.
25
Dubinin and Radushkevich (D–R) equation26 is represented by the following equation:
| | |
lnqe = lnQ0 − KDRε2
| (6) |
where
KDR (mol
2 kJ
−2) is a constant related to mean adsorption energy; and
ε is the Polanyi potential, which can be calculated from equation:
| |
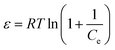 | (7) |
The slope of the plot of lnqe versus ε2 gives KDR (mol2 kJ−2) and the intercept yields the maximum adsorption capacity, Q0 (mg g−1). T is the absolute temperature in K and R is the universal gas constant (8.314 J mol−1 K−1). The mean adsorption energy (E) can also be worked out using the following relationship:
| |
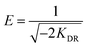 | (8) |
The Temkin equation is given by the following equation:
where
BT =
RT/
bT,
bT is related to the heat of adsorption,
KT is the equilibrium binding constant corresponding to the maximum binding energy.
27
Adsorption kinetics. The controlling mechanism of the adsorption process was investigated by fitting the experimental data with pseudo-first-order, pseudo-second-order kinetic models or the intra-particle diffusion model, respectively. For the pseudo-first-order and pseudo-second-order kinetic models, the kinetics equations are defined as follows:| |
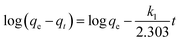 | (10) |
| |
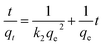 | (11) |
where qt (mg g−1) is the amount of adsorbed MG on hydrochar at time t (min). k1 (1/min) and k2 (g mg−1 min−1) are adsorption rate constants of pseudo-first-order and pseudo-second-order models, respectively. The adsorption rate constant (k1) for MG adsorption was calculated from the slope of the linear plot of ln(qe − qt) versus t. In the latter case, kinetic data were plotted as t/qt against t.25,28According to the intra-particle diffusion model proposed by Weber and Morris,7,29 the initial rate of intra-particle diffusion is calculated by
where
Kw (mg g
−1 min
−1/2) represents intra-particle diffusion rate constant,
C is the intercept and
t denotes contact time (min). The values of
qt versus t1/2 and the rate constants
Kw are directly evaluated from the slope of the regression line.
Results and discussion
Characterization of adsorbent
SEM characterization. The surface characterization was conducted through SEM (Fig. 1). Fig. 1a illustrates the morphological feature of the ABR–hydrochar, showing that part of algal cells could still maintain the morphology after HTC. Fig. 1b displays the hydrochar made from PE-ABR, indicating that the algae cells lost their inherent surface morphology, and most of them were converted into nonuniform micro or nanoscale particles, they contained the higher porosity than ABR–hydrochar. This ultra-structural property may provide favorable for the dye molecules to be trapped and adsorbed by hydrochar derided from PE-ABR compared with that from ABR as we will discuss more in the following sections.
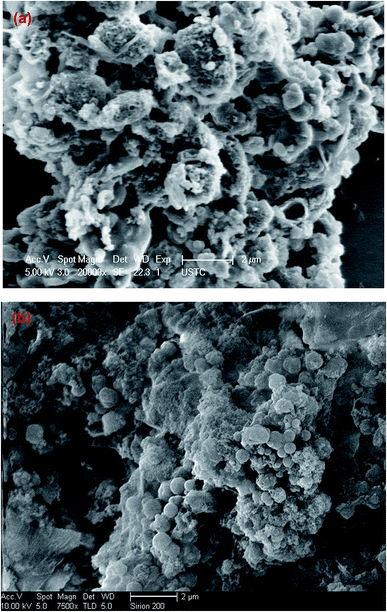 |
| | Fig. 1 Scanning electron micrographs of the surfaces of hydrochars deviated from ABR and PE-ABR by HTC ((a) hydrochar–ABR; (b) hydrochar–PE-ABR). | |
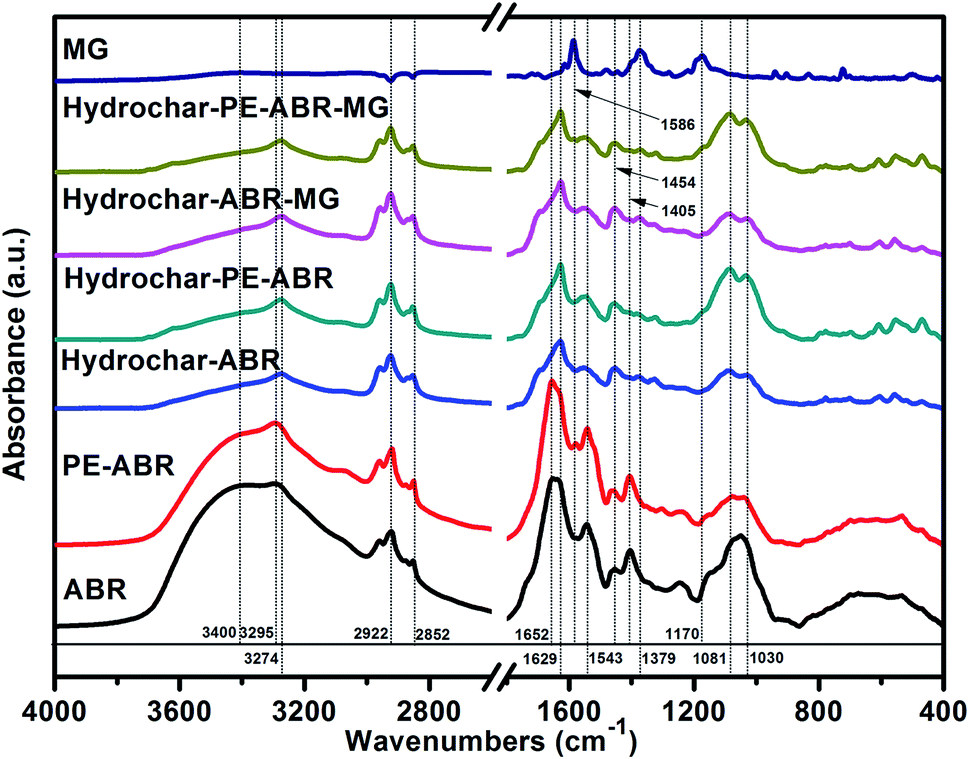
Spectroscopic analysis of ABR and hydrochars by FTIR and Raman spectroscopy. Vibrational spectroscopy such as Raman and FTIR spectroscopy can be very useful in the study of interactions involved in adsorption process. Here, FTIR spectroscopy was employed to analyze the composition of the hydrochars converted from ABR or PE-ABR and the possible interaction between the hydrochars and MG. The FTIR spectra of samples are presented in Fig. 2. It can be seen that the broad region centered at about 3400 cm−1, which can be attributed to stretching vibration of bonded hydroxyl groups and N–H stretching vibration in amino compounds, are highly affected by the HTC processing. The –OH absorption peaks at 3400 and 3393 cm−1 of ABR and PE-ABR are diminished and the sharp peaks are shifted from 3295 and 3294 cm−1 to 3274 and 3276 cm−1 after the HTC treatment (Fig. 2, Table 1). This is most likely due to dehydration of ABR and PE-ABR during the HTC process.30 After MG adsorption, the spectra show the changes in intensity and shift in position. The –OH absorption peaks are shifted from 3274 and 3276 cm−1 to 3273 and 3275 cm−1 for ABR–hydrochar and PE-ABR–hydrochar, respectively. The peaks at 2922 and 2852 cm−1 correspond to asymmetric and symmetric C–H stretching vibrations in aliphatic compounds. The intensities of peaks around 2924 and 2853 cm−1 are increased in the hydrochars. Previous study has shown that these peaks are clearly visible in biomass as well as in the biochars produced at low temperature, but gradually diminished and indiscernible when HTC temperature is above 450 °C.31 In the present study, the temperature of HTC was 200 °C, so the C–H stretching vibrations were still present in the samples before and after HTC treatment. There are no obvious shifts of these two peaks with adsorption of MG onto the both hydrochars. The amide I bands at 1652 and 1656 cm−1 of ABR and PE-ABR are assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations, and they are shifted to 1629 and 1628 cm−1 in the hydrochars. The peak around 1600 cm−1 corresponds to the CC stretching vibration in aromatic compounds.10 The peak shifts indicate that the destruction of the protein in feedstocks leads to more exposure of aromatic compounds in the hydrochar samples. After MG-adsorption onto the hydrochars, these peaks are shifted to 1627 cm−1, indicating the involvement of CC of in hydrochar samples during the adsorption process. The peaks at 1543 and 1542 cm−1 corresponding to C–N stretching (amide II) in ABR and PE-ABR are shifted to 1555 and 1545 cm−1 in hydrochars, and they are shifted to 1553 and 1544 cm−1 with adsorption of MG. The aromatic C–H groups with in-plane bending vibration can be observed at 1454 and 1463 cm−1, and the positions of these peaks are shifted to 1453 and 1457 cm−1 with intensity increase as well in the hydrochars, suggesting that the new functional group are formed by the HTC treatment. These peaks are also visible in the spectra of biochars produced by pyrolysis.31 Obvious shifts to 1454 and 1453 cm−1 are also present in the hydrochars with the adsorption of MG, confirming the interaction between the aromatic C–H groups and MG. A broad peak at about 1080 cm−1 corresponding to C–O stretching are separated into two peaks at about 1081 and 1030 cm−1 after the HTC treatment. The peak at 1030 cm−1 is assigned to Si–O stretching,32 so it indicates that SiO2 is formed in the HTC treatment. The bands shifts of C–O stretching after adsorption of MG by hydrochars also suggests the involvement of C–O group and Si–O stretching in binding MG. The new peaks at 1586 cm−1 indicate CC stretching of the benzene ring of MG; and the peak at 1169 cm−1 indicates C–N stretching vibrations of MG. The intensity of peak at 1586 cm−1 of PE-ABR–hydrochar after adsorption of MG is higher than that of ABR–hydrochar, indicating that higher adsorption capability of MG by PE-ABR–hydrochar. All these observations indicate the interaction between MG and the hydrochars and the results are summarised in Fig. 2 and Table 1.
O stretching vibrations, and they are shifted to 1629 and 1628 cm−1 in the hydrochars. The peak around 1600 cm−1 corresponds to the CC stretching vibration in aromatic compounds.10 The peak shifts indicate that the destruction of the protein in feedstocks leads to more exposure of aromatic compounds in the hydrochar samples. After MG-adsorption onto the hydrochars, these peaks are shifted to 1627 cm−1, indicating the involvement of CC of in hydrochar samples during the adsorption process. The peaks at 1543 and 1542 cm−1 corresponding to C–N stretching (amide II) in ABR and PE-ABR are shifted to 1555 and 1545 cm−1 in hydrochars, and they are shifted to 1553 and 1544 cm−1 with adsorption of MG. The aromatic C–H groups with in-plane bending vibration can be observed at 1454 and 1463 cm−1, and the positions of these peaks are shifted to 1453 and 1457 cm−1 with intensity increase as well in the hydrochars, suggesting that the new functional group are formed by the HTC treatment. These peaks are also visible in the spectra of biochars produced by pyrolysis.31 Obvious shifts to 1454 and 1453 cm−1 are also present in the hydrochars with the adsorption of MG, confirming the interaction between the aromatic C–H groups and MG. A broad peak at about 1080 cm−1 corresponding to C–O stretching are separated into two peaks at about 1081 and 1030 cm−1 after the HTC treatment. The peak at 1030 cm−1 is assigned to Si–O stretching,32 so it indicates that SiO2 is formed in the HTC treatment. The bands shifts of C–O stretching after adsorption of MG by hydrochars also suggests the involvement of C–O group and Si–O stretching in binding MG. The new peaks at 1586 cm−1 indicate CC stretching of the benzene ring of MG; and the peak at 1169 cm−1 indicates C–N stretching vibrations of MG. The intensity of peak at 1586 cm−1 of PE-ABR–hydrochar after adsorption of MG is higher than that of ABR–hydrochar, indicating that higher adsorption capability of MG by PE-ABR–hydrochar. All these observations indicate the interaction between MG and the hydrochars and the results are summarised in Fig. 2 and Table 1.
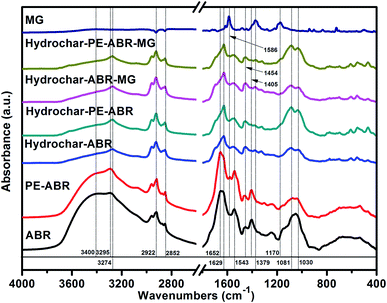 |
| | Fig. 2 FTIR spectra of MG, ABR, PE-ABR, hydrochar–ABR, hydrochar–PE-ABR and MG loaded hydrochar (MG concentration = 100 mg L−1; adsorbent dosage = 4 g L−1; temperature = 25 °C; contact time = 24 h; stirring speed = 200 rpm). | |
Table 1 The FTIR spectral characteristics of hydrochars and their feedstocks, and hydrochars before and after adsorption of MG
| Wavelength assignment |
Feedstocks |
Hydrochar |
Hydrochar after MG-loaded |
| ABR (cm−1) |
PE-ABR (cm−1) |
Hydrochar–ABR (cm−1) |
Hydrochar–PE-ABR (cm−1) |
Hydrochar–ABR–MG (cm−1) |
Hydrochar–PE-ABR–MG (cm−1) |
| –OH or N–H stretching |
3400 |
3393 |
3274 |
3276 |
3273 |
3275 |
| 3295 |
3294 |
|
|
|
|
| C–H and –CH2 asymmetric stretching |
2922 |
2919 |
2924 |
2923 |
2924 |
2923 |
| 2852 |
2850 |
2853 |
2852 |
2853 |
2852 |
| CC or CO stretching (amide I) |
1652 |
1656 |
1629 |
1628 |
1627 |
1627 |
| CC stretching of the benzene ring |
— |
— |
— |
— |
— |
1586 |
| C–N stretching (amide II) |
1543 |
1542 |
1555 |
1545 |
1553 |
1544 |
| –CH2 scissoring or in-plane bend of carbonyl |
1454 |
1463 |
1453 |
1457 |
1454 |
1453 |
| COO– stretching |
1405 |
1406 |
— |
— |
— |
— |
| Symmetric C–H bending |
— |
— |
1379 |
1384 |
1377 |
1371 |
| Aromatic C–N stretching vibrations |
— |
— |
— |
— |
1170 |
1169 |
| C–O stretching |
1081 |
1078 |
1086 |
1086 |
1087 |
1085 |
| Si–O group or C–O bending |
1051 |
1045 |
1030 |
1033 |
1031 |
1033 |
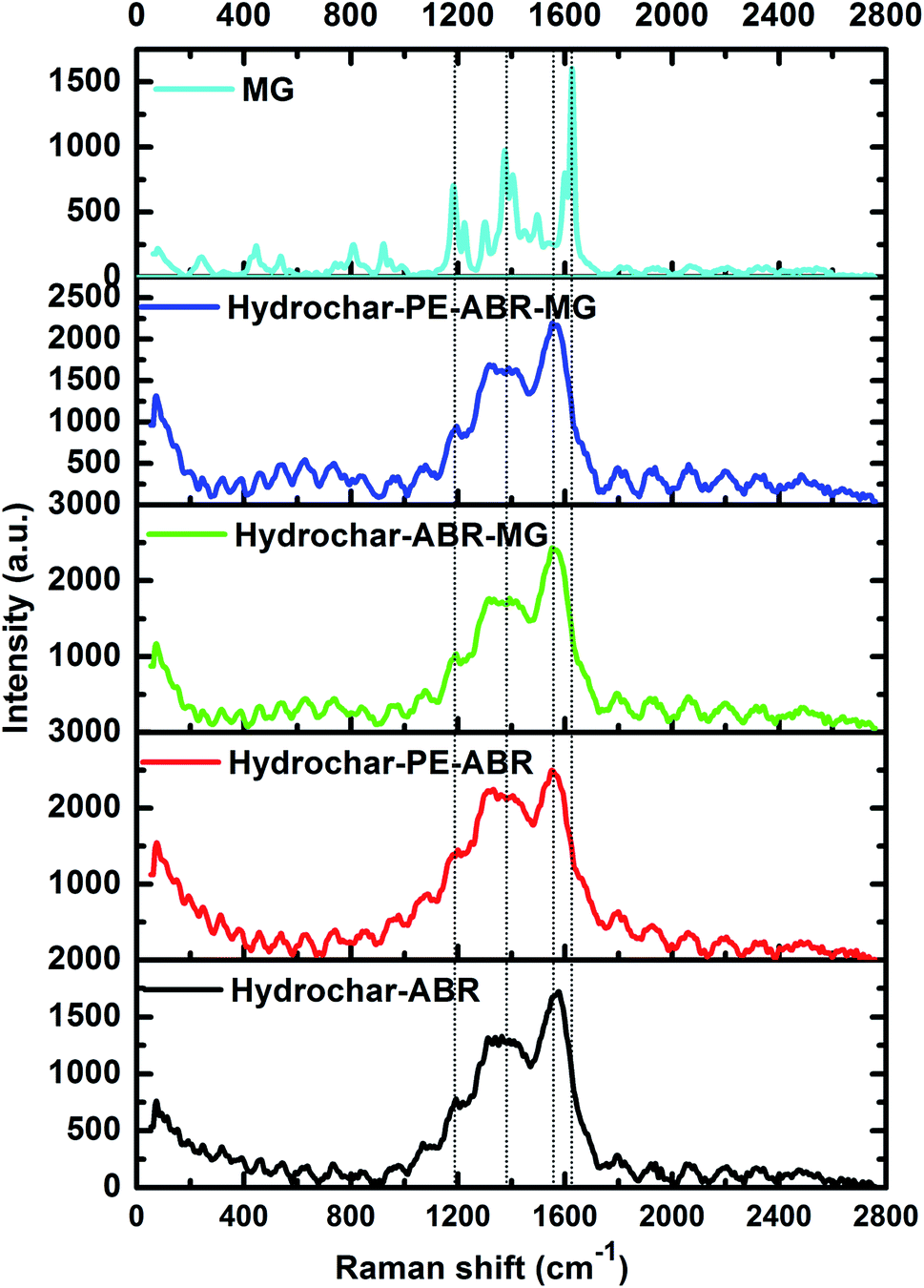
The Raman spectrum of carbonaceous material is typically characterized by two bands: the G band (about 1580 cm−1) is associated with sp2 C atoms and the D band (about 1350 cm−1) corresponds to sp3 C atoms, deriving from the defects and/or disorders of graphite crystal.33 Generally, the relative intensity ratio of D to G bands (ID/IG) is a measure of disorder degree and average size of the sp2 domains in graphitized structure.34 The Raman spectra of two types of hydrochars exhibit two characteristic peaks related to the D band and G band, respectively, implying the formation of carbonaceous materials after ABR and PE-ABR treated by HTC (Fig. 3). The ID/IG intensity ratio of PE-ABR hydrochar is 0.87, which is higher than that of ABR–hydrochar, suggesting that the higher disorder degree of the sp3 domains of PE-ABR hydrochar. After the adsorption of MG, the ID/IG intensity ratios decrease respectively from 0.78 and 0.87 to 0.71 and 0.74 for ABR–hydrochar and PE-ABR–hydrochar, the decrease of the ID/IG intensity ratios means interactions between amorphous structures in the carbon structures and MG. The new peak at 1200 cm−1 appears in the MG adsorbed hydrochar samples, and the intensity of this peak in PE-ABR–hydrochar–MG samples is significantly higher than that of the ABR–hydrochar–MG samples, implying more favorable adsorption of MG on PE-ABR–hydrochar than ABR–hydrochar (Fig. 3). These results also imply the interaction between MG and the hydrochars, which are consistent with the above FTIR analysis and the foregoing morphology analysis results given by the SEM observation.
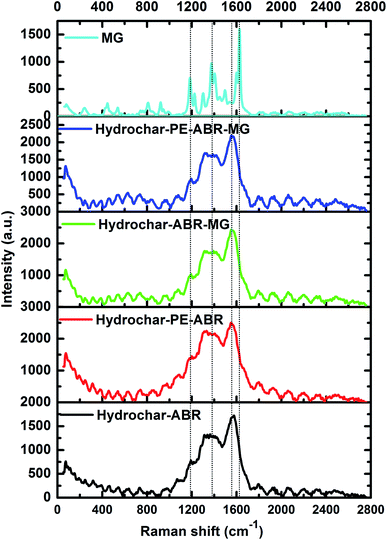 |
| | Fig. 3 Raman spectra of hydrochar–ABR and hydrochar–PE-ABR before and after MG adsorption (MG concentration = 100 mg L−1; adsorbent dosage = 4 g L−1; temperature = 25 °C; contact time = 24 h; stirring speed = 200 rpm). | |
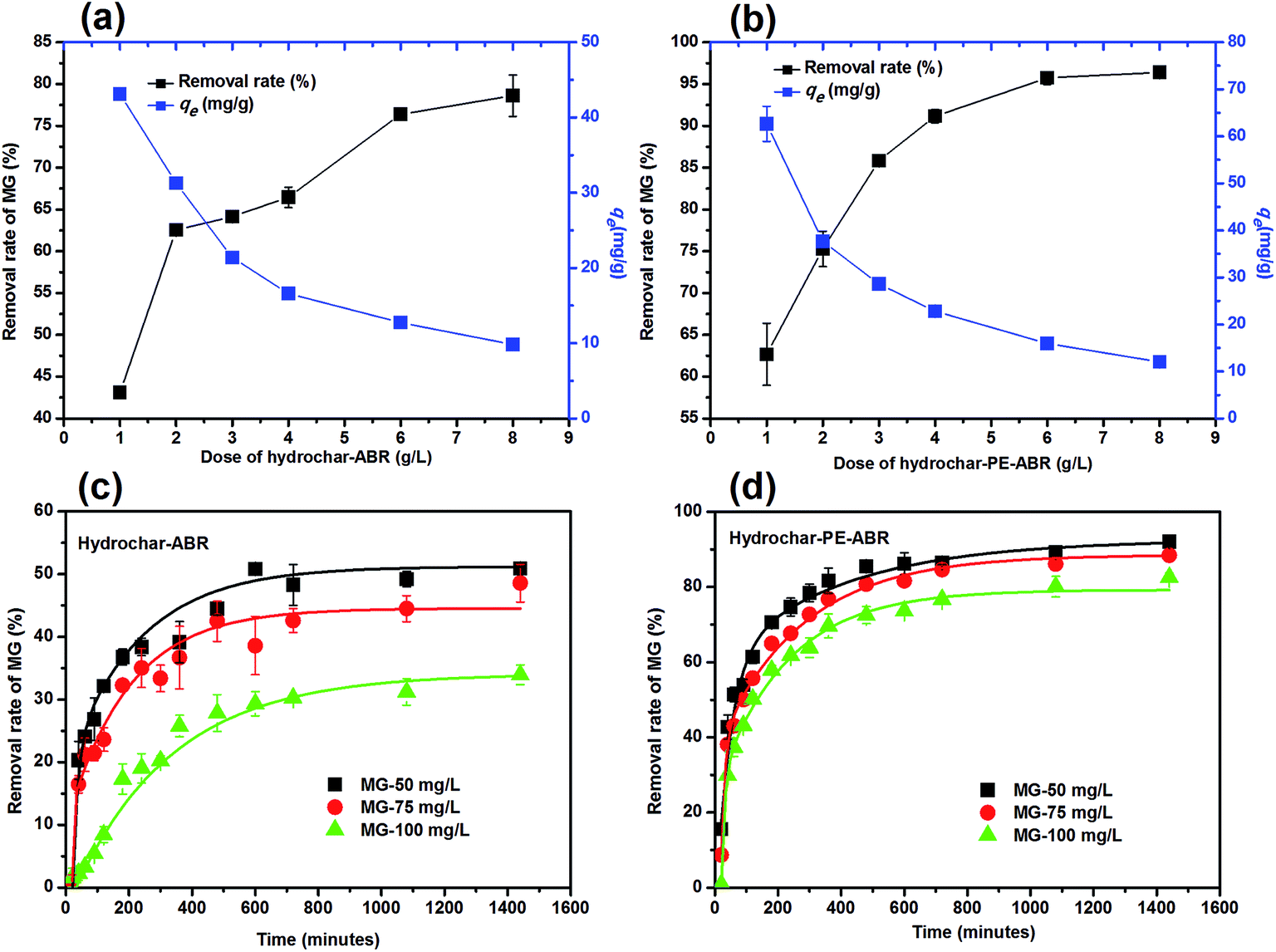
Influence of adsorbent dosage and initial MG concentration on adsorption efficiency. The influence of adsorbent dosage and initial concentration of MG on the efficiency of MG removal was also investigated. As shown in Fig. 4a, the percent removal of MG increased from 43.1% to 78.6% in ABR–hydrochar concentration from 1 to 8 g L−1 after 48 h contact. The removal rates of MG by PE-ABR–hydrochar were increased from 62.7% to 92.4% with increase dosage from 1 to 8 g L−1 (Fig. 4b). The results suggest that the PE-ABR–hydrochar has higher adsorption capability of MG than ABR–hydrochar, and this can be explained by that the PE-ABR hydrochar has more rough and irregular surface morphology than the ABR hydrochar as mentioned in the former section. Effects of initial concentration on MG removal by the ABR–hydrochar and the PE-ABR–hydrochar were studied by carrying out the experiments at different initial concentrations (50, 75 and 100 mg L−1). The percent MG removal decreased from 50.9% to 33.9% after the treatment by the ABR–hydrochar and from 92.1% to 82.6% after the treatment by the PE-ABR–hydrochar as the initial concentration of MG increases from 50 to 100 mg L−1 for 4 g L−1 of adsorbent at equilibrium contact time of 24 h (Fig. 4c and d). At low concentration, the ratio of available surface to the initial MG concentration is larger, so the removal may become higher. As the limited active sites on hydrochars become saturated at a relatively higher concentration, the increment in adsorption capacity become restricted, so the percentage removal become lesser.35
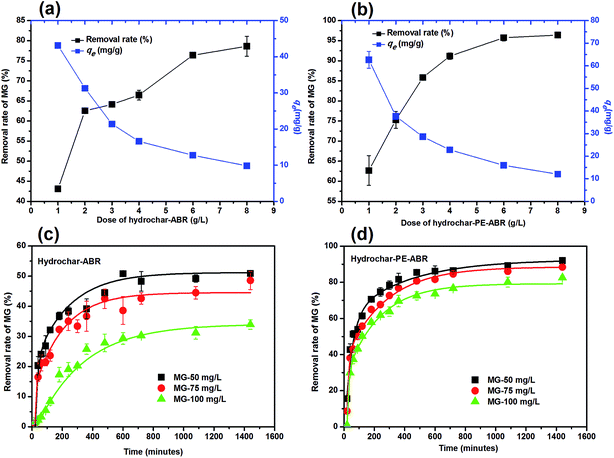 |
| | Fig. 4 Effects of hydrochar dosages and the initiation concentrations of MG on the adsorption of MG. (a) Dose of hydrochar–ABR; (b) dose of hydrochar–PE-ABR; (c) effects of initiation concentrations of MG on adsorption by hydrochar–ABR; and (d) adsorption by hydrochar–PE-ABR. | |
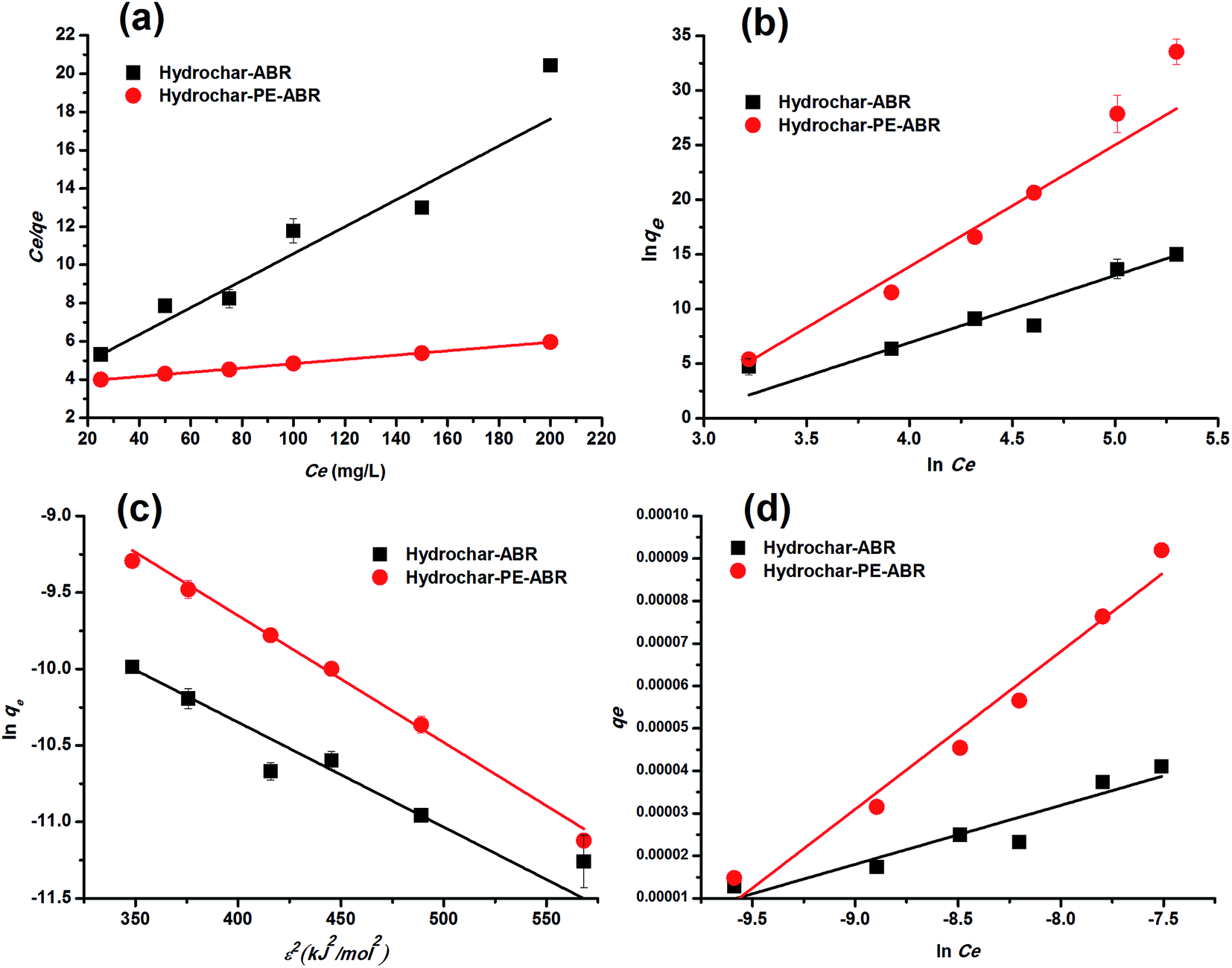
Adsorption isotherm. To examine the relationship between adsorbent and adsorbate at equilibrium, and to search for the maximum adsorption capacity, four adsorption isotherm models including Langmuir, Freundlich, D–R and Temkin isotherms were applied and evaluated. The plots of Ce/qe versus Ce, lnqe versus lnCe, lnqe versus ε2 and qe versus lnCe turn out to be linear (Fig. 5). The values of Q0, KL, KF, KDR, KT, n and the regression correlation coefficients were calculated from the slope and intercept of these plots and reported in Table 2. These values of regression coefficients were used as the fitting criteria to find out the applicability of the foregoing four isotherms. It is found that the plot depicts the linear form of the Langmuir isotherm with extremely high correlation coefficients for MG adsorption by the hydrochar–ABR and the hydrochar–PE-ABR (R2 = 0.979 and 0.999), indicating that the adsorption of MG on hydrochars was likely to be monolayer and the distribution of active sites on the adsorbent was homogeneous.27 RL is a positive number whose magnitude determines the feasibility of the adsorption process.7,26 The RL values for each of the different initial concentrations are between 0 and 1, indicating favorable adsorption of MG onto the hydrochars. Freundlich model can also fit well with the experimental data (Table 2). The values of heterogeneity factor n of Freundlich model were more than 1 for the two hydrochars at different temperatures, indicating that the adsorption was favorable.27 Both Langmuir and Freundlich isotherms fit well with the experimental data indicating both monolayer and heterogeneous surface conditions may exist.
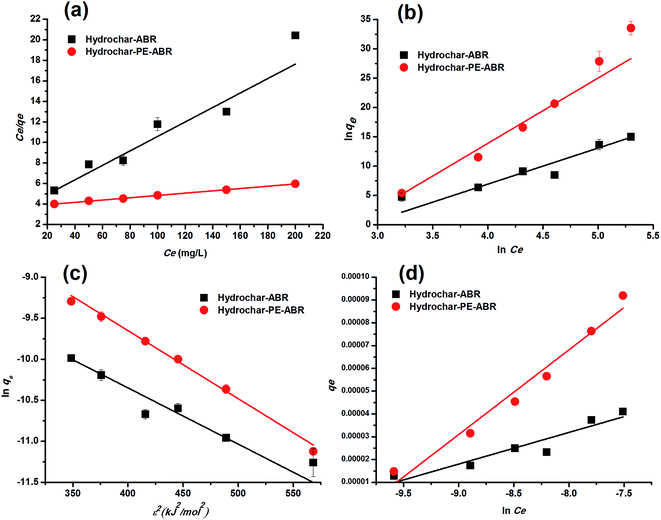 |
| | Fig. 5 Langmuir (a), Freundlich (b), D–R (c) and Temkin (d) fitting isotherms of MG on hydrochar–ABR and hydrochar–PE-ABR (adsorbent dosage = 4 g L−1; contact time = 24 h; stirring speed = 200 rpm). | |
Table 2 Isotherm parameters for removal of MG by hydrochar–ABR and hydrochar–PE-ABR (time = 24 h; adsorbent dosage = 4 g L−1; stirring speed = 200 rpm)
| Models |
Parameters |
Hyrochar–ABR |
Hydrochar–PE-ABR |
| Langmuir model |
Q0 (mg g−1) |
43.113 |
89.047 |
| KL (L mg−1) |
0.015 |
0.003 |
| R2 |
0.979 |
0.999 |
| RL |
0.247–0.725 |
0.623–0.930 |
| Freundlich model |
KF (mg g−1) |
11.925 |
17.796 |
| n |
1.796 |
1.159 |
| R2 |
0.983 |
0.984 |
| D–R model |
Q0 (mg g−1) |
181.045 |
646.093 |
| KDR (mol2 kJ2 × 10−3) |
6.85 |
8.29 |
| R2 |
0.976 |
0.991 |
| E (kJ mol−1) |
8.544 |
7.766 |
| Temkin model |
bT (105 kJ mol−1) |
1.792 |
6.698 |
| KT (L mg−1) |
81.514 |
51.206 |
| R2 |
0.973 |
0.992 |
Dubinin and Radushkevich have proposed another isotherm which is applied to estimate the mean free energy of adsorption. D–R isotherm constant (KDR) can be used to determine the mean free energy of adsorption per mole of the adsorbate (E, kJ mol−1), which in turn gives an idea about the type of adsorption i.e. physisorption or chemisorptions.26 If the value of E is between 8 and 16 kJ mol−1 then the adsorption process follows by chemical ion-exchange, and if E < 8 kJ mol−1 the adsorption process is of physical nature, whereas if the value is more than 16 kJ mol−1, then chemisorption prevails.7,36 Our results showed that E value was 5.84 and 7.766 kJ mol−1 below 8 kJ mol−1 (Table 2), indicating that the physical adsorption was the main process for the adsorption of MG by hydrochars in this work. The positive values of E indicate that the sorption process was endothermic and that higher solution temperature would favor the sorption process.7
Temkin isotherm contains a factor that explicitly takes into account adsorbing species–adsorbate interactions. This model assumes that the energy of the molecule adsorption will decrease linearly with coverage because of the adsorbate and adsorbent interactions.37 So, we applied Temkin isotherm as well to evaluate the adsorption potentials of the adsorbents for adsorbate ions. The value of Temkin adsorption potential constant KT is 81.51, and 51.21 L mg−1 for adsorption MG by hydrochar–ABR and hydrochar PE-ABR hydrochar. The values of Temkin constant bT that is related to the heat of MG adsorption onto the two hydrochars were shown in Table 2, confirming a strong interaction between MG ions and the adsorbents surface.
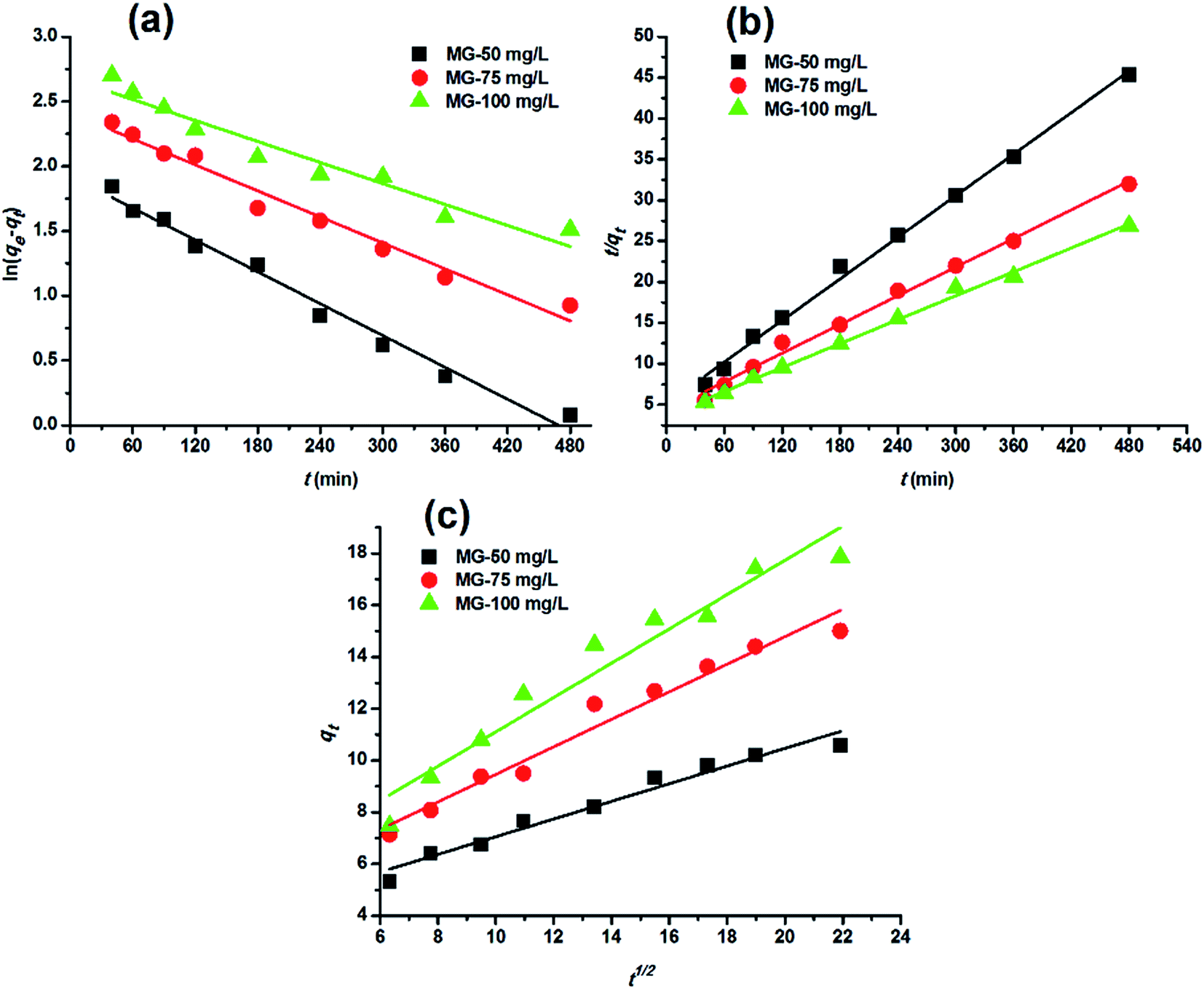
Adsorption kinetics. In order to analyze the adsorption kinetics of MG onto the hydrochars, the pseudo-first-order, pseudo-second-order and intra-particle diffusion models were applied to the experimental data. The adsorption kinetics described by the relationship between contact time and MG uptake by 4 g L−1 the two hydrochars for three initial MG concentrations (50, 75 and 100 mg L−1) were discussed later. The linear fitting results of PE-ABR hydrochar (log(qe − qt) versus t, (t/qt) versus t and qt versus t1/2) are presented in Fig. 6. To quantify the applicability of each model, the correlation coefficient R2 was calculated from these plots. Comparison of the correlation coefficients R2 shows that the pseudo-second-order model fits better the experimental data (R2 > 0.943 for hydrochar–ABR and R2 > 0.993 for hydrochar–PE-ABR) than the pseudo-first-order model (R2 is in the range of 0.735–0.982) (Table 3). The better fit with pseudo-second-order model implies the adsorption process is interaction controlled with chemisorptions involved. But at the same time, the initial rapid phase within the first 480 minutes may involve physical adsorption or ion exchange at hydrochar surface. Therefore, there are at least two mechanisms involved in the actual process. Similar results have been observed in the adsorption of malachite green onto palm flower based activated carbon.29 Contribution of intra-particle diffusion mechanism can be tested by applying the Weber and Morris equation as described earlier. It is clear that the relationships between hydrochars and MG for different concentrations at a particular adsorbent do not fit well and do not pass through the origin (Fig. 6). The correlation coefficients R2 of the hydrochar derived from PE-ABR is higher than that of the hydrochar–ABR, which indicates that intra-particle diffusion is favorable for PE-ABR–hydrochar and more than one process affects the adsorption of MG by the hydrochars. The slope of the initial linear portion has been used to derive the intra-particle rate constant Kw.36 The various values of Kw along with the values of C are shown in Table 3. The rate constant for intra-particle diffusion of PE-ABR–hydrochar increases with increase of MG concentration which indicates an increasing boundary layer effect.
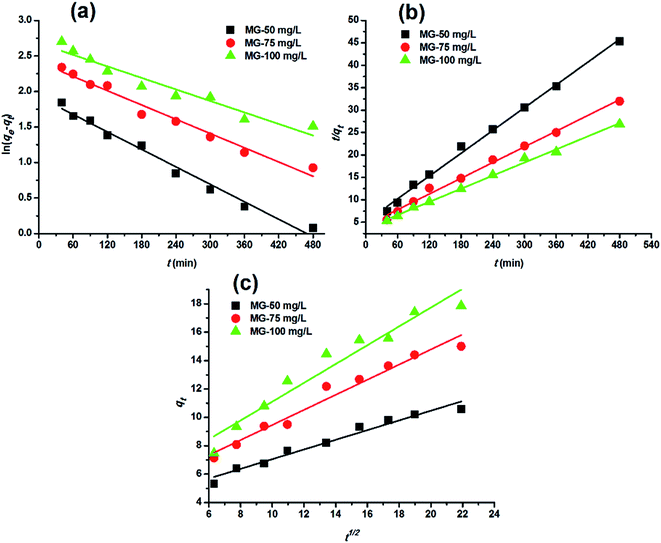 |
| | Fig. 6 Adsorption kinetic plots for adsorption of MG on hydrochar–PE-ABR. (a) Pseudo-first-order kinetics; (b) pseudo-second-order kinetics; (c) intra-particle diffusion kinetics (adsorbent dosage = 4 g L−1; stirring speed = 200 rpm). | |
Table 3 Comparison of rate constants calculated based on respective pseudo-first order, pseudo-second order and intra-particle diffusion kinetic models (hydrochar–ABR and hydrochar–PE-ABR dosage = 4 g L−1; stirring speed = 200 rpm)
| Samples |
Parameters |
| First-order kinetics |
Second-order kinetics |
Intra-particle diffusion kinetics |
| Initial Conc. (mg L−1) |
k1 (1/min) |
qe (mg g−1) |
R2 |
k2 (g mg−1 min−1) |
qe (mg g−1) |
R2 |
Kw (mg g−1 min−1/2) |
C (mg g−1) |
R2 |
| Hydrochar–ABR |
50 |
0.0047 |
5.73 |
0.735 |
0.0029 |
5.938 |
0.993 |
0.181 |
1.744 |
0.884 |
| 75 |
0.0050 |
8.86 |
0.858 |
0.0012 |
8.861 |
0.978 |
0.292 |
1.504 |
0.939 |
| 100 |
0.0045 |
11.26 |
0.910 |
0.0006 |
9.127 |
0.943 |
0.380 |
1.381 |
0.841 |
| Hydrochar–PE-ABR |
50 |
0.0094 |
6.84 |
0.982 |
0.0014 |
11.799 |
0.996 |
0.341 |
3.648 |
0.964 |
| 75 |
0.0077 |
11.15 |
0.972 |
0.0008 |
17.117 |
0.993 |
0.532 |
4.150 |
0.965 |
| 100 |
0.0062 |
14.55 |
0.935 |
0.0006 |
20.517 |
0.996 |
0.663 |
4.479 |
0.941 |
Comparison with other adsorbents. The maximum uptake capacities of the hydrochars derived from ABR and PE-ABR towards MG together with those of other hydrochars, biochars or other carbonaceous materials reported in the literature are listed in Table 4. The data suggest that the maximum adsorption capacity obtained in the present study is higher than most of other carbonaceous materials. Our previous work using pine wood after decayed by brown-rot fungi for removal rate of MG showed the maximum adsorption capacity reach to 42.63 mg g−1,36 was also less than that of the present work result. To increase oxygen-containing surface functional groups of hydrochar, particularly carboxylic group, hydrogen peroxide could be used to oxidize carbonized surfaces of hydrochar from pine needles to increase the adsorption capacity of MG (increased from 52.91 mg g−1 to 97.08 mg g−1).6 In present study, owing to the algal cell rupture or deformation during the extraction process of phycocyanin, the hydrochar–PE-ABR has richer structure and surface functional groups compared to the hydrochar–ABR, we achieved therefore increased the maximum adsorption capacity.
Table 4 Comparison of adsorption capacities of various adsorbents used for MG removal
| Adsorbent |
Temperature (°C) |
Adsorption capacity (mg g−1) |
References |
| Rice husk biochar |
35 |
67.6 |
27 |
| Hydrothermal carbonization of pine needles (HTC-PN) |
25 |
52.91 |
6 |
| Rambutan peel based activated carbon (RPAC) |
30 |
329.49 |
25 |
| Activated carbon laboratory grade |
30 |
42.18 |
38 |
| Activated carbon derived from Borassus aethiopum flower |
30 |
48.48 |
29 |
| Activated carbon prepared from the epicarp of Ricinus communis |
27 |
27.78 |
39 |
| Biochars produced by liquefaction of sewage sludge |
30 |
49.3 |
35 |
| Pine wood decayed by brown-rot fungi (BRW) |
30 |
42.63 |
36 |
| Hydrochar–ABR |
25 |
43.11 |
This study |
| Hydrochar–PE-ABR |
25 |
89.05 |
This study |
Conclusions
This work presents a novel approach that makes better use of phycocyanin-extracted algal bloom residue by converting it to hydrochars through hydrothermal carbonization. The PE-ABR–hydrochar shows the maximum adsorption capacity of 89.05 mg g−1 for initial MG concentration of 100 mg L−1 and hydrochars dosages of 4 g L−1, much higher than that of ABR–hydrochar (43.11 mg g−1). The mechanism of the MG adsorption is ascribing to the interactions between the various functional groups of the hydrochar interacting with MG. Therefore, we have claimed a more effective and economic way to deal with the waste microalgae biomass from blooms.
Acknowledgements
This work was supported partly by the Natural Science Foundation of China (No. 11635013, No. 21207137 and No. 11475217), and the National Basic Research Program of China (Grant No. 2014CB932002, No. 2013CB934304).
References
- D. Das and A. Pal, Chem. Eng. J., 2016, 290, 371–380 CrossRef CAS
 .
. - A. Salima, B. Benaouda, B. Noureddine and L. Duclaux, Water Res., 2013, 47, 3375–3388 CrossRef CAS PubMed .
- L. Ai, H. Huang, Z. Chen, X. Wei and J. Jiang, Chem. Eng. J., 2010, 156, 243–249 CrossRef CAS .
- T. Bhagavathi Pushpa, J. Vijayaraghavan, S. J. Sardhar Basha, V. Sekaran, K. Vijayaraghavan and J. Jegan, Ecotoxicol. Environ. Saf., 2015, 118, 177–182 CrossRef CAS PubMed .
- J.-X. Dong, C. Xu, H. Wang, Z.-L. Xiao, S. J. Gee, Z.-F. Li, F. Wang, W.-J. Wu, Y.-D. Shen, J.-Y. Yang, Y.-M. Sun and B. D. Hammock, J. Agric. Food Chem., 2014, 62, 8752–8758 CrossRef CAS PubMed .
- H. H. Hammud, A. Shmait and N. Hourani, RSC Adv., 2015, 5, 7909–7920 RSC .
- H. Zhang, Y. Tang, D. Cai, X. Liu, X. Wang, Q. Huang and Z. Yu, J. Hazard. Mater., 2010, 181, 801–808 CrossRef CAS PubMed .
- J. A. Libra, K. S. Ro, C. Kammann, A. Funke, N. D. Berge, Y. Neubauer, M.-M. Titirici, C. Fühner, O. Bens, J. Kern and K.-H. Emmerich, Biofuels, 2011, 2, 71–106 CrossRef CAS .
- S. M. Heilmann, H. T. Davis, L. R. Jader, P. A. Lefebvre, M. J. Sadowsky, F. J. Schendel, M. G. von Keitz and K. J. Valentas, Biomass Bioenergy, 2010, 34, 875–882 CrossRef CAS .
- X.-Z. Yuan, X.-S. Shi, C.-X. Yuan, Y.-P. Wang, Y.-L. Qiu, R.-B. Guo and L.-S. Wang, Water Res., 2014, 49, 113–123 CrossRef CAS PubMed .
- M. D. Marsolek, E. Kendall, P. L. Thompson and T. R. Shuman, Bioresour. Technol., 2014, 151, 373–377 CrossRef CAS PubMed .
- W. Zhong, L. Chi, Y. Luo, Z. Zhang, Z. Zhang and W.-M. Wu, Bioresour. Technol., 2013, 134, 264–270 CrossRef CAS PubMed .
- B. Maddi, S. Viamajala and S. Varanasi, Bioresour. Technol., 2011, 102, 11018–11026 CrossRef CAS PubMed .
- Z. Hu, Y. Zheng, F. Yan, B. Xiao and S. Liu, Energy, 2013, 52, 119–125 CrossRef CAS .
- X. Meng, P. E. Savage and D. Deng, Environ. Sci. Technol., 2015, 49, 12543–12550 CrossRef CAS PubMed .
- M. P. Padgett and D. W. Krogmann, Photosynth. Res., 1987, 11, 225–235 CrossRef CAS PubMed .
- F. Zhang, B. Zhao, J. Cai, M. Yuan, J. Sheng and J. Wang, J. Environ. Eng. Technol, 2015, 5, 499–503 CAS .
- F. Deniz and R. A. Kepekci, Desalin. Water Treat., 2015, 57, 12257–12263 CrossRef .
- L. Zhang, Q. Wang, B. Wang, G. Yang, L. A. Lucia and J. Chen, Energy Fuels, 2015, 29, 872–876 CrossRef CAS .
- B. Weiner, J. Poerschmann, H. Wedwitschka, R. Koehler and F.-D. Kopinke, ACS Sustainable Chem. Eng., 2014, 2, 2165–2171 CrossRef CAS .
- A. Broch, U. Jena, S. K. Hoekman and J. Langford, Energies, 2013, 7, 62–79 CrossRef .
- N. D. Berge, K. S. Ro, J. Mao, J. R. V. Flora, M. A. Chappell and S. Bae, Environ. Sci. Technol., 2011, 45, 5696–5703 CrossRef CAS PubMed .
- X. Zhu, Y. Liu, F. Qian, S. Zhang and J. Chen, Energy Fuels, 2015, 29, 5222–5230 CrossRef CAS .
- T. L. Silva, A. Ronix, O. Pezoti, L. S. Souza, P. K. T. Leandro, K. C. Bedin, K. K. Beltrame, A. L. Cazetta and V. C. Almeida, Chem. Eng. J., 2016, 303, 467–476 CrossRef CAS .
- M. A. Ahmad and R. Alrozi, Chem. Eng. J., 2011, 171, 510–516 CrossRef CAS .
- G. K. Parshetti, S. Chowdhury and R. Balasubramanian, Bioresour. Technol., 2014, 161, 310–319 CrossRef CAS PubMed .
- L. Leng, X. Yuan, G. Zeng, J. Shao, X. Chen, Z. Wu, H. Wang and X. Peng, Fuel, 2015, 155, 77–85 CrossRef CAS .
- B. H. Hameed and M. I. El-Khaiary, J. Hazard. Mater., 2008, 159, 574–579 CrossRef CAS PubMed .
- S. Nethaji, A. Sivasamy, G. Thennarasu and S. Saravanan, J. Hazard. Mater., 2010, 181, 271–280 CrossRef CAS PubMed .
- J. Petrović, N. Perišić, J. D. Maksimović, V. Maksimović, M. Kragović, M. Stojanović, M. Laušević and M. Mihajlović, J. Anal. Appl. Pyrolysis, 2016, 118, 267–277 CrossRef .
- G. Bekiaris, C. Peltre, L. S. Jensen and S. Bruun, Spectrochim. Acta, Part A, 2016, 168, 29–36 CrossRef CAS PubMed .
- J. H. Yuan, R. K. Xu and H. Zhang, Bioresour. Technol., 2011, 102, 3488–3497 CrossRef CAS PubMed .
- C.-W. Huang, S.-C. Chiu, W.-H. Lin and Y.-Y. Li, J. Phys. Chem. C, 2008, 112, 926–931 CAS .
- F. Lian, G. Cui, Z. Liu, L. Duo, G. Zhang and B. Xing, J. Environ. Manage., 2016, 176, 61–68 CrossRef CAS PubMed .
- L. Leng, X. Yuan, H. Huang, J. Shao, H. Wang, X. Chen and G. Zeng, Appl. Surf. Sci., 2015, 346, 223–231 CrossRef CAS .
- H. Zhang, Y. Tang, X. Liu, Z. Ke, X. Su, D. Cai, X. Wang, Y. Liu, Q. Huang and Z. Yu, Desalination, 2011, 274, 97–104 CrossRef CAS .
- D. K. Mahmoud, M. A. M. Salleh, W. A. W. A. Karim, A. Idris and Z. Z. Abidin, Chem. Eng. J., 2012, 181–182, 449–457 CrossRef CAS .
- I. D. Mall, V. C. Srivastava, N. K. Agarwal and I. M. Mishra, Colloids Surf., A, 2005, 264, 17–28 CrossRef CAS .
- T. Santhi, S. Manonmani and T. Smitha, J. Hazard. Mater., 2010, 179, 178–186 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *abc
*abc