
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solid-state structure and electronic states of hydrogen-bonded dimer of pyridyl-substituted tetrathiafulvalene salted with PF6−†
Tomoaki Kanetoua,
Ryo Tsunashima *a,
Norihisa Hoshinob and
Tomoyuki Akutagawab
*a,
Norihisa Hoshinob and
Tomoyuki Akutagawab
aGraduate School of Science and Engineering, Yamaguchi University, Yoshida 1677-1, Yamaguchi, 753 8512, Japan. E-mail: ryotsuna@yamaguchi-u.ac.jp
bInstitute of Multidisciplinary Research for Advanced Materials (IMRAM), Tohoku University, Sendai 980-8577, Japan
First published on 18th January 2017
Abstract
Hydrogen bonding was investigated in a newly prepared salt of pyridyl-substituted tetrathiafulvalene (TTFPy) that formed a dimer structure by hydrogen bonding and was salted with PF6− anions, namely, (TTFPy2H)PF6 (1). Structural analysis of the salt by single-crystal X-ray diffraction and temperature-dependent FTIR spectra and measurements of its electrical conductivity and dielectric properties were performed. Thermal fluctuations were investigated for protons between two TTFPy molecules. An anisotropic dielectric anomaly was observed owing to local proton transfer between two pyridyl moieties. This behavior was well explained by a model of Debye-type dipole relaxation. In addition, DFT calculations showed that the dipole originated from local proton transfer, but the dipole moment in a network of TTFPy⋯H+⋯TTFPy dimers was amplified by a factor of 2 owing to coupling with the electronic polarization of TTFPy moieties.
Introduction
Hydrogen bonds are one of the most important forms of intermolecular interaction. These attractive forces occur between proton donors and acceptors and appear not only in chemical systems, but also in biological systems.1,2 A proton can bridge molecules with a weak interactive force in comparison to a covalent bond, so that it enables the assembly of supramolecular systems, where the assembly and disassembly of molecular components are triggered and controlled via external stimuli.Proton dynamics also play an important role in physical properties, for example, structural phase transitions, dielectric properties, proton transfer, and proton conduction.3–8 An electric field induces the displacement of protons by a distance that depends on the degree of structural freedom. The results range from solid-state proton conduction by long-range transport to dielectric fluctuations by the local transfer of protons. Similar behavior also occurs with electrons, which usually have a higher degree of mobility than that of protons owing to their smaller size and lower mass.
Proton-coupled electron transfer systems have received considerable attention. These perform a key process for energy conversion in biological systems and its development for future electronics has been expected. From these points of view, tetrathiafulvalene (TTF) derivatives are promising agents in which the electronic properties and ion dynamics display a strong correlation.9–17 Mori et al. have demonstrated switching of electrical conductivity on the basis of hydrogen bonding dynamics using TTF substituted by the hydrogen bonding site of catechol. Oxidized (open-shell) TTF formed two-dimensional π-electron-conducting layers, which were connected by hydrogen bonds between catechol moieties.13 Deuterated species exhibited a structural phase transition with a charge-ordered electronic structure, which originated from the transfer or displacement of deuterium within the hydrogen bond network. This system is expected in switchable or stimuli-responsive molecular materials.
By analogy with TTF, its pyridyl-substituted analog (TTFPy)12,14–17 is also interesting. In particular, in its hydrogen-bonded dimer TTF–Py⋯H+⋯Py–TTF the electronic and protic states were strongly coupled to each other owing to the conjugated system.12 Protonation of the pyridyl moiety strongly affected its molecular orbitals, because it occupied mainly the lowest unoccupied molecular orbital (LUMO). These insights into TTFPy motivated us to develop it not only for supramolecular systems in which the ion dynamics are strongly correlated with the electronic system, but also for multifunctional systems with dielectric, conducting, magnetic, and/or optical properties. In this paper, we investigate the structural details and electronic system of the dimer in a newly prepared single crystal composed of the TTFPy-based hydrogen-bonded dimer salted with the PF6− anion ((TTFPy2H)PF6 (1)).
Temperature-dependent structural analysis by single-crystal X-ray diffraction (SC-XRD) and Fourier transform infrared spectroscopy (FTIR) revealed that the hydrogen bonds were symmetrical around the midpoint of the two pyridyl moieties. The TTF moiety is not oxidized, so that it exhibits a closed-shell electronic structure. Both DC and AC electrical measurements showed anisotropic characteristics where a Debye-type relaxation of the dipole moment was observed in a direction parallel to the hydrogen bond, which suggested fluctuations of the hydrogen bonding proton. By density functional theory (DFT) calculations, only a slight displacement due to thermal vibrations was suggested; however, a large dipole fluctuation resulted owing to coupling with the electronic system on the TTF moiety. Our results shown here will be interesting for developing supramolecular electronic systems for amplifying dipole moments.
Results and discussion
Crystal structure
A single crystal of 1 was prepared by an evaporation method. The composition of the crystal was determined to be (TTFPy2H)PF6 by SC-XRD analyses, as described below. Because structural SC-XRD analyses were performed at 93 K and 323 K, the structural features were almost independent of temperature except for thermal vibrations.In the crystal, TTFPy molecules formed a dimer structure through intermolecular N⋯H⋯N hydrogen bonds on the pyridyl moieties (Fig. 1). A similar dimer structure has been reported for F4–TCNQ salt by Mori et al.12 In the salt, one side of a TTFPy molecule was protonated and hydrogen-bonded to another molecule, where the intermolecular N⋯N distance (dNN) was reported to be 2.781(3) Å. On the other hand, the values of dNN for 1 were less, namely, 2.645 Å and 2.676 Å at 93 K and 323 K, respectively. In contrast to the asymmetrical dimer structure of F4–TCNQ salt, the crystal of 1 possessed an inversion center at the midpoint of the two N atoms, giving a symmetrical hydrogen-bonded dimer along the crystallographic a + c direction.
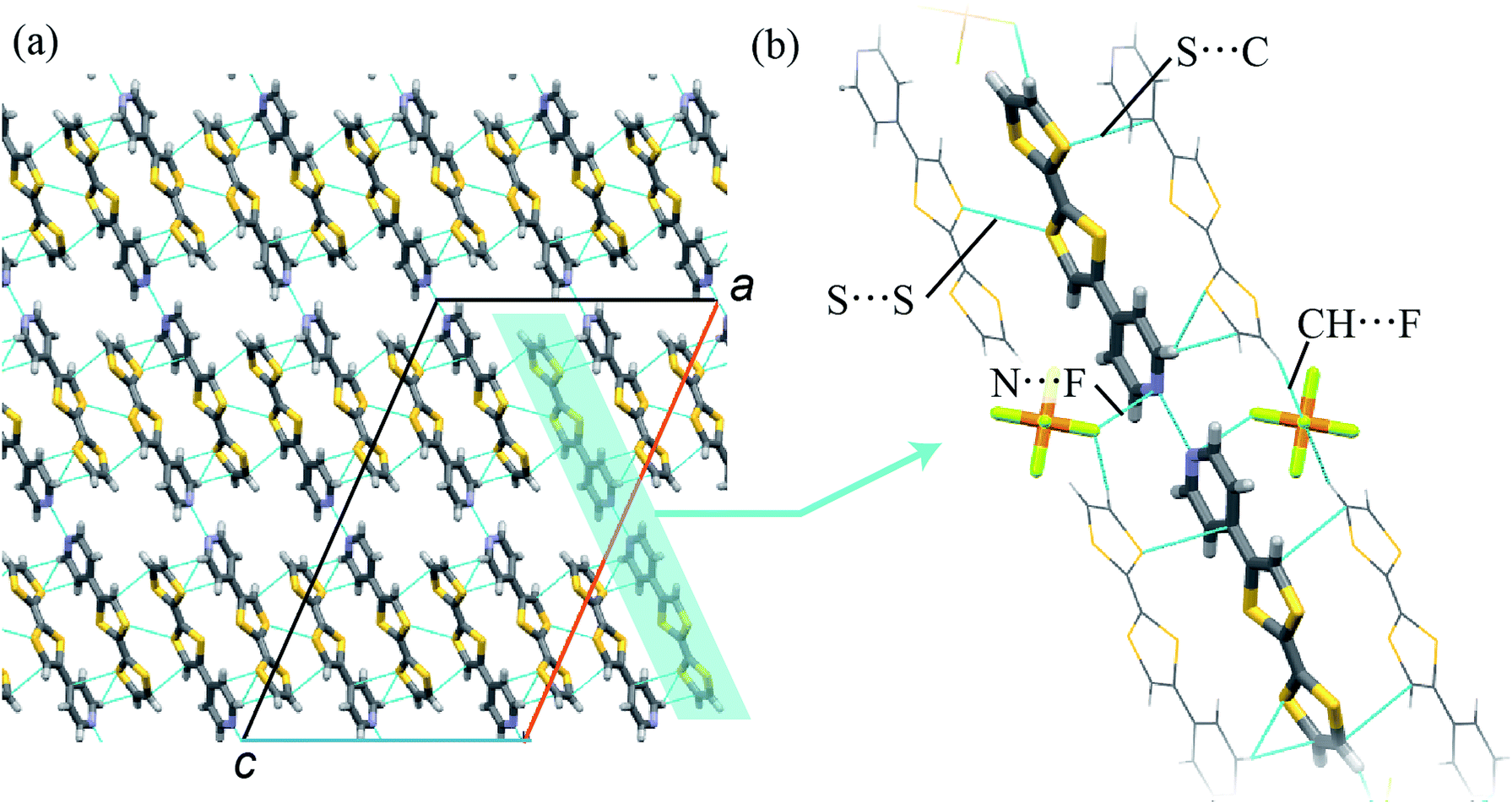 | ||
| Fig. 1 (a) Packing structure and (b) its extended view of 1 (C, H, N, S, P, and F atoms are represented in gray, white, blue, yellow, orange, and light green, respectively, in the stick model), in which anions are not shown. Intermolecular interactions are highlighted by dashed lines, where the interatomic distances are those at 93 K. | ||
To obtain a detailed structural view of the pyridyl moieties, the CNC angle was measured, because this angle differed between the protonated form N–H+ and the hydrogen-bonded form N⋯H+.18 Regarding the angles in the TTFPy molecule, the bond angles were reported to be 115.4° for TTFPy and 121.9° for TTFPyH+.12 The CNC angle in 1 was estimated to be 118.4° (93 K) and 117.9° (323 K). These values corresponded to the average value for the protonated and hydrogen-bonded forms, suggesting that possible structures included: (i) N⋯H+⋯N hydrogen bonding; (ii) glass-like static disorder with N–H+⋯N and N⋯H+–N; and (iii) dynamic disorder (thermal fluctuations). These three situations cannot be distinguished by crystallography alone. We will discuss them later with results obtained from temperature-dependent FTIR measurements.
In addition to hydrogen bonding, weak intermolecular interactions were observed in TTFPy molecules. The shortest S⋯S distance between TTF moieties (dSS) was 3.585 Å (93 K) and the S⋯C distance from one TTF moiety to another pyridyl moiety (dSC) was 3.431 Å (93 K). In addition, TTFPy molecules were connected to PF6− anions via CH⋯F interactions (the C–F distance was 3.275 Å at 93 K) and N⋯F interactions (the N–F distance was 2.966 Å at 93 K). We believe that the electrostatic interaction is also important to stabilize the dimmer structure. The observed intermolecular atomic distances were almost equal to or shorter than the sum of the van der Waals radii of the atoms, namely, F (1.47 Å), S (1.80 Å), N (1.55 Å), and C (1.70 Å).18
It has been reported that the oxidation state of the TTF moiety is correlated with the length of the central C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. Usually, oxidation elongates this bond. Neutral TTFPy had CC bond distances of 1.344(9) Å.12 The length of the central CC bond in 1 (dCC) was observed to be 1.340(3) Å at 93 K, which indicated that the TTFPy molecule in 1 was not oxidized.
C bond. Usually, oxidation elongates this bond. Neutral TTFPy had CC bond distances of 1.344(9) Å.12 The length of the central CC bond in 1 (dCC) was observed to be 1.340(3) Å at 93 K, which indicated that the TTFPy molecule in 1 was not oxidized.
Temperature-dependent FTIR measurements
Next, we investigated temperature-dependent FTIR spectra recorded at 110 K, 300 K, and 393 K (Fig. 2a). The timescale of proton dynamics is slower than that of FTIR measurements. Thus, the structure for both type (ii) and type (iii) disorders will display two types of peak that originated from pyridine and pyridinium moieties. Here TTFPy and TTFPyH+ exhibited different peaks at 1593 cm−1 and 1628 cm−1. These peaks were assigned as originating from pyridyl moieties, where protonation caused a blue shift.19 Compound 1 displayed a single peak at 1614 cm−1, of which the position was less temperature-dependent. Because this peak was observed almost midway between those of TTFPy and TTFPyH+, the hydrogen bonding structure in the crystal was neither type (ii) nor type (iii) but N⋯H+⋯N hydrogen bonding of type (i). The peak width became narrower at low temperatures, which indicated that the thermal displacement (fluctuation) of protons between two N atoms was decreased.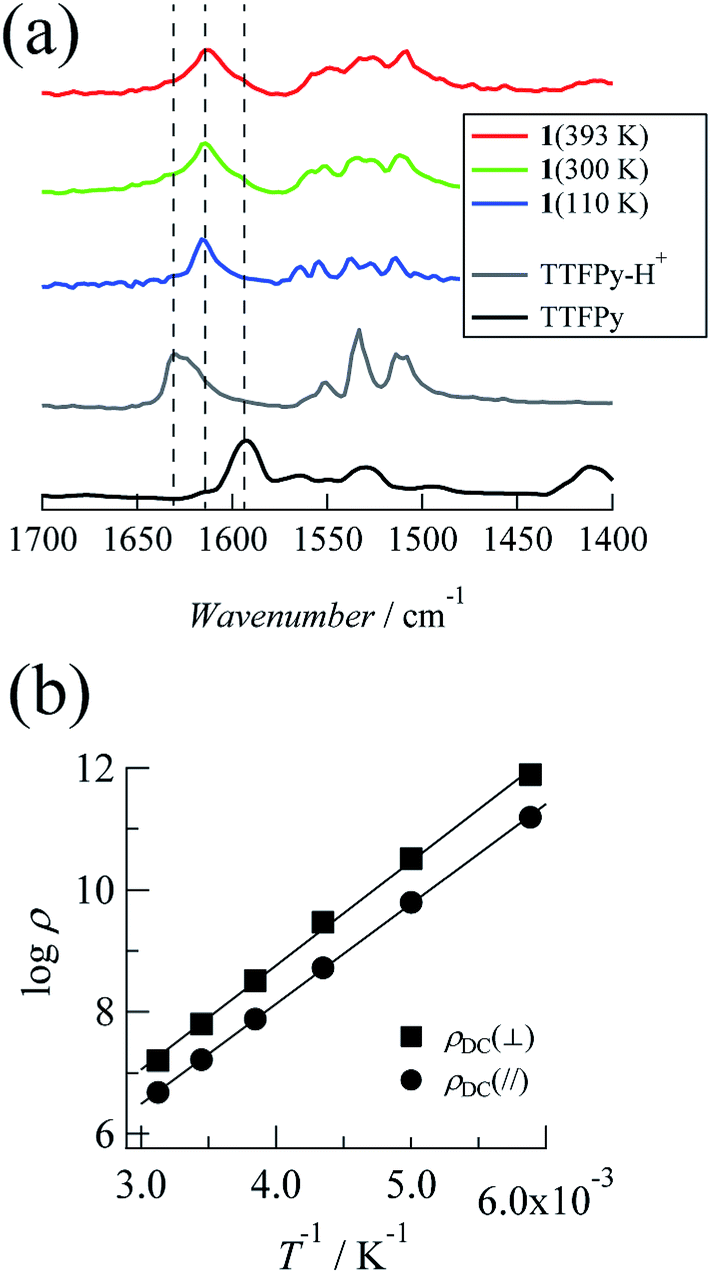 | ||
| Fig. 2 (a) FTIR spectra of 1 (393, 300, and 110 K), TTFPy–H+, and TTFPy. (b) Arrhenius plots of DC resistivity measured along the ac plane (parallel to the N⋯H⋯N hydrogen bonds, plotted with closed circles) and the b axis (perpendicular to the N⋯H⋯N hydrogen bonds, plotted with closed squares) using single crystals of 1. | ||
Electrical properties
The TTFPy molecule in 1 has a closed-shell electronic structure. Only narrow-band conduction or thermally activated hopping conduction is expected. The temperature-dependent DC electrical resistivity and AC impedance (170–320 K) were determined using the same single crystal along the crystallographic a + c direction (parallel to the hydrogen bonds) and the b axis (perpendicular to the hydrogen bonds).DC electrical resistivity
The current–voltage (I–V) characteristics between −5 and +5 V were characterized at the measurement temperatures and almost linear relationships were observed, which indicated ohmic contact between the crystal and the electrode (the plots are shown in the ESI†). Fig. 2b shows Arrhenius plots of DC resistivity (ρDC in Ω cm) estimated from the I–V characteristics for each direction. The resistivity at 320 K was 4.76 × 106 Ω cm in the a + c direction and decreased with temperature. The activation energies (Ea,DC) were determined to be 0.33 eV from the slope of the Arrhenius plots. Similar behavior was observed along the b direction, with a resistivity of 1.60 × 107 Ω cm at 320 K and an activation energy of 0.34 eV. These values are within the range reported for organic conductors with a closed-shell electronic structure.20 In addition, the less anisotropic characteristics corresponded to a structural situation in which strong intermolecular π-interactions that enable conduction were not observed.AC impedance measurements
Complex impedances were also measured using the same crystal after the DC measurements. Fig. 3 shows R–X plots, where R represents the resistance (Ω) and X represents the reactance (Ω), respectively, for the complex impedance Z* = R − jX. Single semicircular arcs were observed for both directions and were expressed as an equivalent circuit consisting of a parallel RC circuit with a resistance Rp (Ω) and a capacitance Cp (F) (where X = 1/ωC).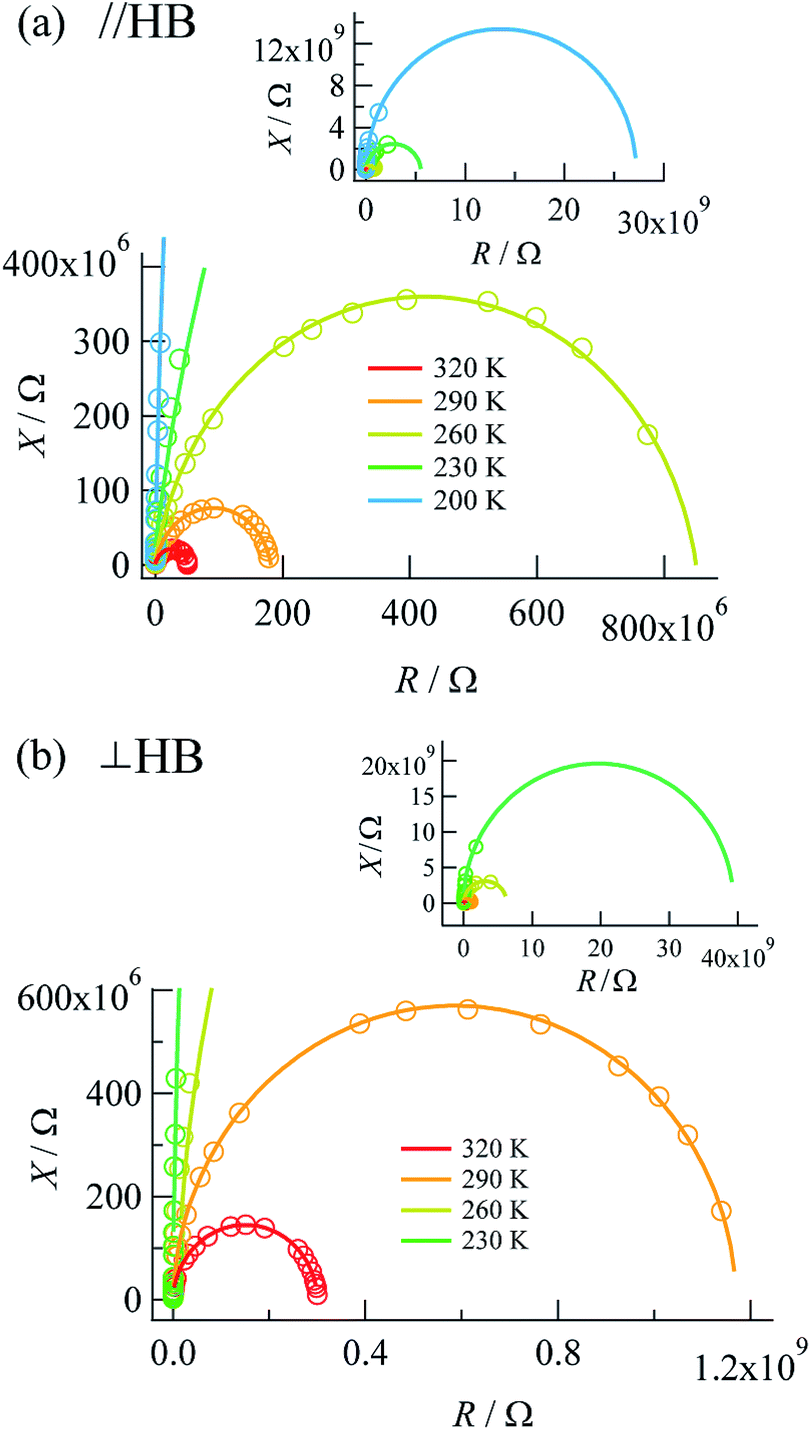 | ||
| Fig. 3 R–X plots measured along (a) the a + c direction (parallel to the N⋯H⋯N hydrogen bonds) and (b) the b axis (perpendicular to the N⋯H⋯N hydrogen bonds). | ||
The value of the resistance Rp was determined from the intercept of the semicircle on the real R axis. The Rp values at the measurement temperatures agreed well with those obtained by DC measurements (Table 1), as defined by the resistivity ρac = Rp × S/d, where S and d are the electrode area and electrode distance, respectively. The activation energies determined by DC and AC measurements were also comparable to each other, which showed that the complex impedance on the lower-frequency side was governed by DC conduction.
| Temperature/K | Direction | |||
|---|---|---|---|---|
| a + c | b | |||
| ρDC/Ω cm | ρAC/Ω cm | ρDC/Ω cm | ρAC/Ω cm | |
| 320 | 4.76 × 106 | 5.11 × 106 | 1.60 × 107 | 1.62 × 107 |
| 290 | 1.64 × 107 | 1.80 × 107 | 6.25 × 107 | 6.46 × 107 |
| 260 | 7.61 × 107 | 8.48 × 107 | 3.23 × 108 | 3.34 × 108 |
| 230 | 5.25 × 108 | 5.22 × 108 | 2.96 × 109 | 2.13 × 109 |
| 200 | 6.19 × 109 | 2.70 × 109 | 3.27 × 1010 | 3.10 × 1010 |
| 170 | 1.56 × 1011 | — | 7.78 × 1011 | — |
| Ea,DC/eV | 0.33 | 0.34 |
| Ea,AC/eV | 0.29 | 0.34 |
Complex impedance can be converted into complex permittivity (ε* = ε1 − jε2) for analysis of the dipole relaxation process. Plots of frequency spectra of the real and imaginary parts of the complex permittivity are shown in Fig. 4. The ε1 value measured along the hydrogen bonding axis at 320 K (Fig. 4a) exhibited sudden decreases at a frequency that decreased with a decrease in temperature. However, the values measured in directions perpendicular to the hydrogen bonds (Fig. 4b) were almost frequency-independent. This anisotropy strongly indicates that the observed dielectric dispersion was accounted for by hydrogen bonds, namely the thermal fluctuation of protons between N atoms that caused dipole relaxation. The frequency dispersion of ε1 in Debye-type dipole relaxation is represented as follows:
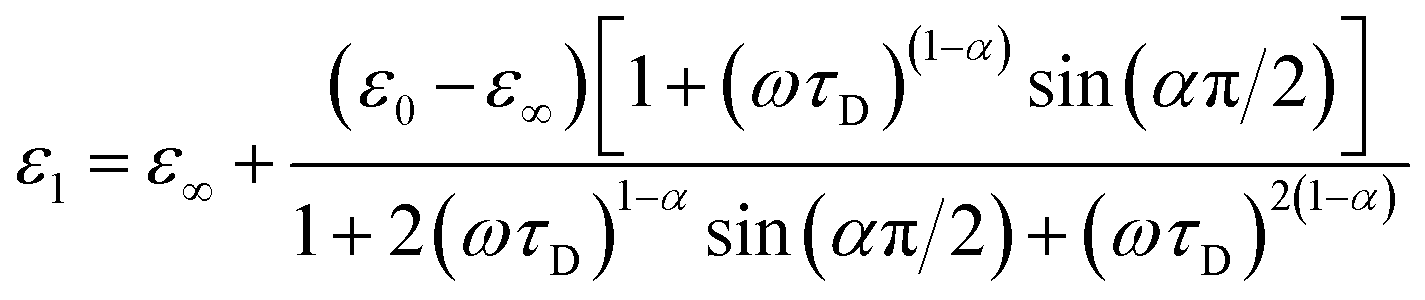 | (1) |
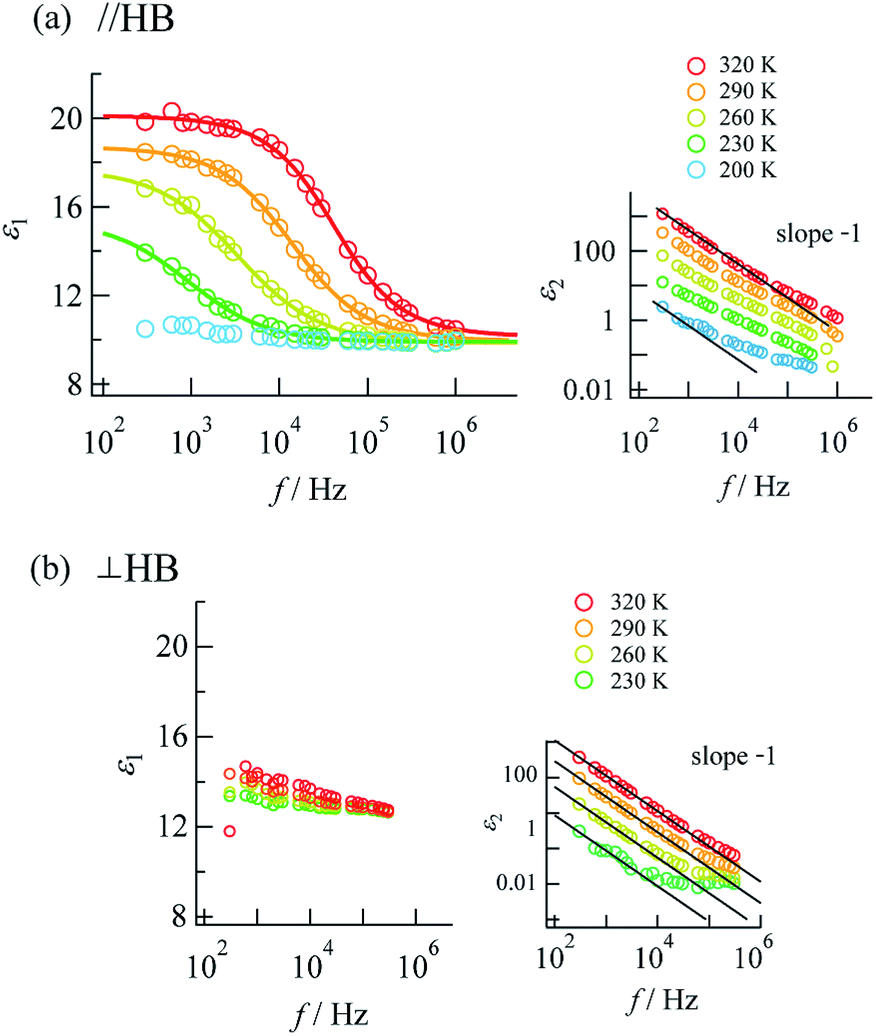 | ||
Fig. 4 Frequency dispersions of dielectric complex permittivity measured along (a) the a + c direction (parallel to the N⋯H⋯N hydrogen bonds) and (b) the b axis (perpendicular to the N⋯H⋯N hydrogen bonds). The inset graphs show the imaginary parts with fitting lines for log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε2 ∝ −logf(slope − 1). Fitting to eqn (1) was performed for the data at 230–320 K. ε2 ∝ −logf(slope − 1). Fitting to eqn (1) was performed for the data at 230–320 K. | ||
The frequency dependence of ε1 measured along the direction of the hydrogen bonds was well fitted by this model (shown by the solid lines in Fig. 4a; the parameters are summarized in the ESI†), which indicates dipole relaxation caused by the fluctuation of protons. The relaxation time is given by τD = τ0exp(−Ea,τ/kBT) (where Ea,τ is the activation energy and kB is the Boltzmann constant). Arrhenius analysis of τD performed by fitting to eqn (1) determined the activation energy to be 0.27 eV (ESI†), which was a comparable value to those determined by DC and AC conductivity analysis of 0.33 eV. It was suggested that a long-range charge transport process (DC conduction) can occur together with a local proton transfer process. However, further details are not discussed here. Investigations using deuterium instead of protons are desired.
Theoretical DFT calculations
Theoretical DFT calculations were also performed to estimate the dipole moment and electronic structure of TTFPy molecules for a process of proton fluctuation. Fig. 5a–d shows plots of the energy near the frontier orbitals, the molecular dipole, and the potential energy difference estimated for the hydrogen-bonded TTFPy dimer by DFT calculations based on SC-XRD data (at 93 K), in which the displacement of a proton from the midpoint between two N atoms is represented by d. The protonated form N–H+ corresponded to d = ±0.4 (N–H distance of ∼0.9 Å). A shallow double-minimum potential was estimated for local proton transfer with an activation energy of ∼0.05 eV. This low activation energy reflected a structure of N⋯H+⋯N-type hydrogen bonds where protons thermally fluctuated between two N atoms. Indeed, differential Fourier maps (|Fo| − |Fc|) originating from SC-XRD data show that the electron density at the midpoint of two N atoms had an ellipsoidal-shaped distribution (ESI†). The theoretical activation energy value (0.05 eV) for proton fluctuation was almost one order of magnitude lower than the activation energy determined by dielectric measurements (0.22 eV).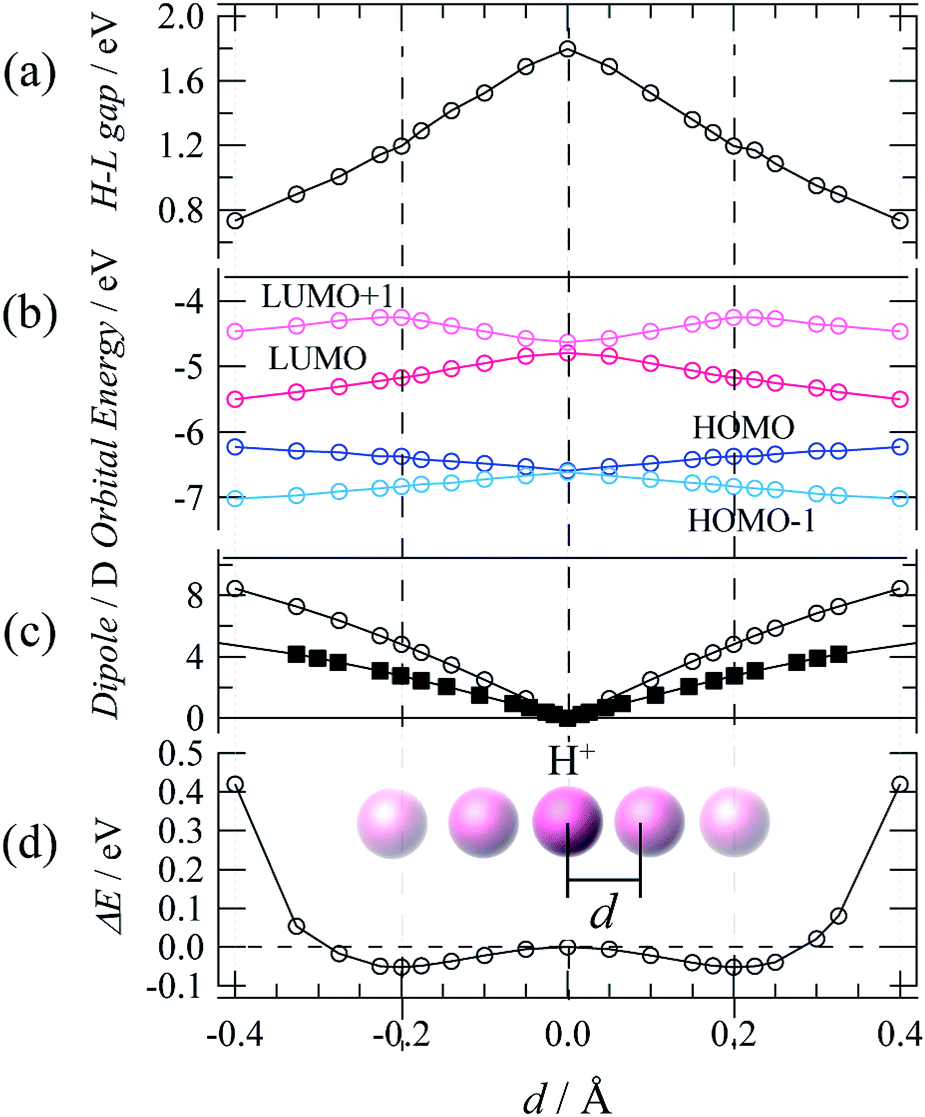 | ||
| Fig. 5 DFT results for the (a) HOMO–LUMO gap, (b) orbital energy, (c) dipole, and (d) potential energy difference estimated for the hydrogen-bonded TTFPy dimer (open circles) and Py dimer (squares) by DFT calculations based on SC-XRD data (at 93 K) in which the displacement of a proton from the midpoint between two N atoms is represented by d. | ||
Finally, dipole moments were also calculated as a function of d for the cases of both the TTFPy dimer and a pyridine-only dimer with the same structural conformation as 1. The displacement of a proton from the midpoint breaks the symmetry of the hydrogen bond and generates a dipole moment. Here, it is important to mention that the dipole moment in 1 is larger by a factor of 2 than that estimated for the pyridyl-only moiety. This is because the dipole moment originated from the TTF moiety owing to its rich π-electron system. We emphasize that sophisticated molecular design can be extended from our results shown here to amplify fluctuations in a small dipole moment by coupling and π-electron systems.
Conclusions
Here, we prepared single crystals of a TTFPy-based hydrogen-bonded dimer and details of the hydrogen bonding and electronic structure present in the closed-shell TTFPy dimer system were investigated via structural analysis by SC-XRD analysis and FTIR. The existence of thermally fluctuating protons between two TTFPy molecules was confirmed and it was concluded that 1 displayed isotropic long-range DC conduction with electron/hole carriers, whereas local proton transfer was involved along the a + c direction. Importantly, both processes had similar activation energies. Dielectric measurements using single crystals agreed well with a model of Debye-type dipole relaxation, in which DFT calculations suggested that the dipole moment caused by proton fluctuations was amplified owing to coupling with the π-electron systems of TTF moieties. Further developments based on the design of our materials are obviously interesting not only for proton-coupled electron transfer systems, but also for multifunctional systems with dielectric, optical, and/or magnetic characteristics.Experimental
Synthesis
TTFPy was synthesized according to the literature method.14 Commercially available copper nitrate trihydrate and potassium hexafluorophosphate were used without further purification. Single crystals of 1 were prepared by a slow evaporation method. A hot solution in methanol of TTFPy (0.020 mmol, 10 mL) was mixed with copper nitrate (5 μmol). After two days, a solution in methanol of potassium hexafluorophosphate (0.013 mmol, 1 mL) was added to the mixed solution. Black crystals of 1 were obtained after slow evaporation for a few days (yield of ∼5% as single crystals). A control experiment without copper nitrate was not successful in obtaining crystals of 1. The effect of copper nitrate will be investigated in more detail. FTIR (KBr disk): 1614(m), 1550(w), 1533(w), 1512(w), 839(s), 794(w), 779(w), 682(w), 661(w), and 559(m) cm−1.X-ray crystallography
SC-XRD analysis of 1 was performed using a Rigaku R-AXIS RAPID diffractometer using filtered Cu Kα radiation. Data were collected at 93 and 323 K. The structures were solved by direct methods and expanded using Fourier techniques. Crystal data: 1-LT, C22H15F6N2PS8, M = 708.82, monoclinic, a = 28.6106(6) Å, b = 6.0020(2) Å, c = 16.6897(3) Å, β = 113.9002(8)°, V = 2620.22(9) Å3, T = 93 K, space group C2/c, Z = 4, μ(Cu Kα) = 74.713 cm−1, 13911 reflections measured, 2401 independent reflections (Rint = 0.0321). The final R1 value (I > 2σ(I)) was 0.0314. The final wR(F2) value (all data) was 0.0882. The final R1 value (all data) was 0.0349. The goodness of fit on F2 was 1.137. CCDC number 1519984. Crystal data: 1-HT, C22H15F6N2PS8, M = 708.82, monoclinic, a = 29.2197(6) Å, b = 6.0807(1) Å, c = 16.8443(3) Å, β = 114.9637(7)°, V = 2713.21(9) Å3, T = 323 K, space group C2/c, Z = 4, μ(Cu Kα) = 72.152 cm−1, 14411 reflections measured, 2480 independent reflections (Rint = 0.0410). The final R1 value (I > 2σ(I)) was 0.0377. The final wR(F2) value (all data) was 0.1310. The final R1 value (all data) was 0.0439. The goodness of fit on F2 was 0.842. CCDC number 1519983.
Electrical measurements
Measurements of electrical conductivity were performed by a DC two-probe technique using a single crystal of 1. Gold paste was utilized for electrodes and electrical contacts were formed using gold wires. Temperature dependence and I–V characteristics were measured under vacuum using a commercially available cryostat with a temperature control system over the temperature range indicated in the main text. The current was monitored with a Keithley 6517 electrometer with a constant bias. Similar experiments for measurements of AC impedance (Keysight E4980AL) were performed using the same single crystal.DFT
The dependences of the N⋯H distance on the potential energy difference (ΔE), dipole moment, and molecular orbitals were calculated using the B3LYP/6-31G(d,p) level of theory. The structure of the dimer was derived from the crystal structure by removing the anion.Measurements
IR spectra were recorded using a Thermo Fisher Scientific Avatar 370 spectrometer (KBr pellets). The spectrum at 110 K was recorded under vacuum using a commercially available cryostat with a temperature control system. The spectrum at 393 K was recorded in a heating chamber.Acknowledgements
This work was supported by Opto-Energy Research Center of Yamaguchi University, Grant-in-Aid for Science Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan and the cooperative research program of the Network Joint Research Centre for Materials and Devices of Japan.Notes and references
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- G. R. Desiraju, Angew. Chem., Int. Ed., 1995, 34, 2311–2327 CrossRef CAS.
- K. D. Kreuer, Chem. Mater., 1996, 8, 610–641 CrossRef CAS.
- S. Horiuchi, R. Kumai and Y. Tokura, Adv. Mater., 2011, 23, 2098–2103 CrossRef CAS PubMed.
- J. Fujioka, S. Horiuchi, F. Kagawa and Y. Tokura, Phys. Rev. Lett., 2009, 102, 197601 CrossRef CAS PubMed.
- S. Horiuchi, R. Kumai and Y. Tokura, Chem. Commun., 2007, 2321–2329 RSC.
- W. Zhang and R. G. Xiong, Chem. Rev., 2012, 112, 1163–1195 CrossRef CAS PubMed.
- T. Norby, Nature, 2001, 410, 877–878 CrossRef CAS PubMed; T. Akutagawa, S. Takeda, T. Hasegawa and T. Nakamura, J. Am. Chem. Soc., 2004, 126, 291–294 CrossRef PubMed.
- J. S. Park, E. Karnas, K. Ohkubo, P. Chen, K. M. Kadish, S. Fukuzumi, C. W. Bielawski, T. W. Hudnall, V. M. Lynch and J. L. Sessler, Science, 2010, 329, 1324–1327 CrossRef CAS PubMed.
- Y. Kobayashi, M. Yoshioka, K. Saigo, D. Hashizume and T. Ogura, J. Am. Chem. Soc., 2009, 131, 9995–10002 CrossRef CAS PubMed.
- M. Fourmigue and P. Batail, Chem. Rev., 2004, 104, 5379–5418 CrossRef CAS PubMed; M. H. V. Huynh and T. J. Meyer, Chem. Rev., 2007, 107, 5004–5064 CrossRef PubMed.
- S. C. Lee, A. Ueda, H. Kamo, K. Takahashi, M. Uruichi, K. Yamamoto, K. Yakushi, A. Nakao, R. Kumai, K. Kobayashi, H. Nakao, Y. Murakami and H. Mori, Chem. Commun., 2012, 48, 8673–8675 RSC.
- A. Ueda, A. Hatakeyama, R. Kumai, Y. Murakami and H. Mori, Chem.–Eur. J., 2015, 21, 15020–15028 CrossRef CAS PubMed; T. Isono, H. Kamo, A. Ueda, K. Takahashi, A. Nakao, R. Kumai, H. Nakao, K. Kobayashi, Y. Murakami and H. Mori, Nat. Commun., 2013, 4, 1344 CrossRef PubMed.
- L. Wang, B. Zhang and J. Zhang, Inorg. Chem., 2006, 45, 6860–6863 CrossRef CAS PubMed.
- Y. F. Han, J. S. Zhang, Y. J. Lin, J. Dai and G. X. Jin, J. Organomet. Chem., 2007, 692, 4545–4550 CrossRef CAS.
- R. Tsunashima, Y. Iwamoto, Y. Baba, C. Kato, K. Ichihashi, S. Nishihara, K. Inoue, K. Ishiguro, Y. F. Song and T. Akutagawa, Angew. Chem., Int. Ed., 2012, 42, 11228–11231 Search PubMed.
- S. Lee, A. Ueda, A. Nakao, R. Kumai, H. Nakao, Y. Murakami and H. Mori, Chem.–Eur. J., 2014, 20, 1909–1917 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- E. P. Parry, J. Catal., 1963, 2, 371–379 CrossRef CAS.
- G. Saito and Y. Yoshida, Bull. Chem. Soc. Jpn., 2007, 80, 1–137 CrossRef CAS.
- K. S. Cole and R. H. Cole, J. Chem. Phys., 1941, 9, 341–351 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Differential Fourier maps, electrical properties, and crystal structure. CCDC 1519983 and 1519984. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra27814k |
| This journal is © The Royal Society of Chemistry 2017 |