
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A stronger reversal effect of the combination of dasatinib and menadione on P-gp-mediated multidrug resistance in human leukemia K562/Adr cell line
Changyuan
Wang†
 a,
Zhihao
Liu†
a,
Yaoting
Sun
a,
Ting
Chen
a,
Xiaokui
Huo
a,
Qiang
Meng
a,
Qi
Liu
a,
Huijun
Sun
a,
Pengyuan
Sun
a,
Jinyong
Peng
a,
Xiaodong
Ma
a and
Kexin
Liu
*ab
a,
Zhihao
Liu†
a,
Yaoting
Sun
a,
Ting
Chen
a,
Xiaokui
Huo
a,
Qiang
Meng
a,
Qi
Liu
a,
Huijun
Sun
a,
Pengyuan
Sun
a,
Jinyong
Peng
a,
Xiaodong
Ma
a and
Kexin
Liu
*ab
aDepartment of Clinical Pharmacology, College of Pharmacy, Dalian Medical University, 9 West Section, Lvshun South Road, Lvshunkou District, Dalian 116044, China. E-mail: kexinliu@dlmedu.edu.cn; Fax: +86 411 86110407; Tel: +86 411 86110407
bProvincial Key Laboratory for Pharmacokinetics and Transport, Dalian Medical University, Dalian 116044, Liaoning, China
First published on 21st March 2017
Abstract
Multidrug resistance (MDR) leads to poor efficiency of chemotherapy. Overexpression of membrane transporters including P-gp could result in MDR. In this study, we investigated the effect of the combination of dasatinib (DA) and menadione (MQ) on P-gp-mediated MDR in the human leukemia K562/Adr cell line. The combination of DA and MQ had a stronger effect on potentiating the sensitivity of K562/Adr cells to DOX and reducing the formation of cell colonies, further increasing the accumulation of DOX and rhodamine 123 in P-gp-overexpressing K562/Adr cells. The increasing cellular accumulation of DOX led to the enhanced increase of DOX-induced and caspase-3-mediated cell apoptosis via ROS production. The stronger reversal effect of the combination of DA and MQ might partly be due to the increased downregulation of mRNA and protein levels of P-gp as opposed to direct inhibition of the P-gp efflux ability. However, the ERK and AKT/mTOR signal pathways were not involved. The combination of DA and MQ shows potential advantages in overcoming MDR in cancer chemotherapy.
Introduction
Chemotherapy is one of the most pivotal treatments for cancers. However, exposing cancer patients to one kind of antineoplastic agent for too long may lead to multidrug resistance (MDR), which may in turn result in resistance to many other antitumor drugs of different structures and different mechanisms of action. MDR is one of the major reasons for the failure of chemotherapy.1 Mechanisms of drug resistance vary, including alterations of targeted enzymes and receptors, enhanced repairs of antidrug-induced defects, decreased amount of active drugs and so forth.2The ATP-binding cassette (ABC) transporter family contains several main transporters involved in MDR, such as P-glycoprotein (P-gp; MDR1), breast cancer resistance protein (BCRP), and multidrug resistance associated proteins (MRP). These transporters energy-dependently pumped various molecules, including anti-cancer drugs and toxic agents. P-gp, a 170 kDa membrane protein, is coded by the ABCB1 gene. Among all the transporters involved in MDR, P-gp was first figured out to be overexpressed in resistant Chinese hamster ovary cells.3 Since then, P-gp was reported to be overexpressed in various multidrug resistant cancer cells,4–6 and thus it plays a key role in the development of MDR.
P-gp is capable of recognizing and excreting a large range of compounds, unrelated to the cellular target or chemical structure, tyrosine kinase inhibitors (TKIs) included. Dasatinib (DA, BMS-354825), a dual Src/Abl kinase inhibitor, has been granted FDA approval for the treatment of imatinib resistant chronic myelogenous leukemia (CML) and (Ph+) acute lymphoblastic leukemia (ALL).7 A previous study has already demonstrated that DA could reverse P-gp-mediated MDR via the MAPK/ERK signal pathway.8 Nevertheless, since DA is a substrate of P-gp,9 it may be less potent to overcome P-gp-mediated MDR alone.
Menadione (MQ, 2-methyl-1,4-naphthoquinone, vitamin K3) is a synthetic vitamin K. It functions as a cofactor for the post-translational modification of proteins like blood clotting factors and osteocalcin, and thus it is necessary for the regular function of blood coagulation and bone metabolism.10,11 Also, MQ can be effective against various cancer cell lines,12–14 and suppresses growth of transplanted tumors.15 As a substrate of P-gp,16 MQ could down-regulate P-gp via ROS production.17
In contrast to imatinib or nilotinib, DA shows an adverse reaction of haemorrhaging, which may be attributed to the inhibition of platelet-derived growth factor receptor (PDGFR).18 Besides, according to Abdelmohsen K. et al., phosphorylation of PDGFR could be induced by MQ.19 At this point, the combined application of DA and MQ in clinical use may help alleviate the haemorrhage adverse effect of DA in CML. In this study, to enhance the reversal effect of DA alone on P-gp-mediated MDR, we conducted a series of experiments to investigate the combined effect of DA and MQ on the reversal of P-gp-mediated MDR.
Materials and methods
Reagents and antibodies
DA, doxorubicin (DOX), verapamil (VRP) and cisplatin were obtained from Dalian Meilun Biotech Co., Ltd. (China). MQ sodium bisulfite and rhodamine 123 (Rho 123) were purchased from Sigma-Aldrich (St. Louis, MO). Mitogen-activated kinase (MEK) inhibitor U0126 and phosphatidylinositol 3-kinase (PI3K)/Akt/mTOR inhibitor LY294002 were from Selleck Chemicals (Houston, TX, USA). Rabbit polyclonal antibody against P-gp was from Abcam Company (UK). Phosphorylated extracellular signal-regulated kinase (P-ERK), extracellular signal-regulated kinase (ERK), Akt and phosphorylated Akt antibodies were purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA). Cleaved caspase-3 antibody and secondary antibodies were from Proteintech group (Chicago, USA). RPMI Medium 1640 and fetal bovine serum FBS were from GIBCO® (Grand Island, NY, USA). All the other chemicals were of analytic grade or purer.Cell line culture
Human leukemia K562 cells and DOX-selected P-gp-overexpressing K562/Adr were purchased from Nanjing KeyGen Biotech Co. Ltd. (China). Cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 1% non-essential amino acid solution, 100 units per mL penicillin and 50 units per mL streptomycin, at 37 °C in 5% CO2. K562/Adr cells were maintained in medium with 2 mg mL−1 DOX and were grown in DOX-free medium for more than 2 weeks before being used in experiments.Cell viability assay
Cell proliferation was determined using the MTT assay. Cells were harvested, seeded, incubated with drugs and finally the absorbance was read using a microplate reader (Bio-Rad, San Diego, CA, USA). The fold-reversal of MDR was calculated by dividing IC50 values of cells with DOX/cisplatin in the absence of inhibitors (DA, MQ or VRP) by IC50 values obtained with inhibitors. Resistance index was calculated by dividing the IC50 of the MDR cells by that of the sensitive cells.Soft agar colony formation assay
Cells were suspended in a medium containing 20% FBS with an equal amount of 0.3% agar with drugs, and plated in plates over a solidified medium containing 20% FBS with an equal amount of 0.5% agar. Plates were maintained at 37 °C in a 5% CO2 incubator for three weeks, and the number of colonies with no less than 50 cells was counted under an inverted microscope.DOX and rhodamine 123 (Rho 123) accumulation
Cells were seeded in 6-well plates with various treatments of drugs for 24 h. Then 8 µM DOX or 5 µM Rho 123 was added to wells for another 1 h or 0.5 h incubation, respectively. Cells were washed and resuspended in ice-cold phosphate buffered saline (PBS). Intracellular DOX or Rho 123 was evaluated by FACS flow cytometry (FACScalibar, BD, USA).Cell apoptosis assay
Apoptosis was explored by staining K562/Adr cells with FITC-Annexin V/SYTOX Red according to the manufacturer’s recommendations (Thermo Scientific, USA). Briefly, after pre-treatment with DA, MQ, or a combination for 24 h, cells were treated with DOX for another 48 h. Then, they were collected and stained with FITC-Annexin V (5 µL)/SYTOX Red (5 nM), and analyzed using a flow cytometry assay (FACScalibar, BD, USA).Caspase-3 activity assay
The activity of caspase-3 was determined using the caspase-3 activity kit from Promega following the manufacturers’ protocol. Briefly, after pre-treatment with DA, MQ, a combination of the two, or VRP for 24 h, cells were treated with DOX for another 48 h. Cells were collected at a concentration of 5 × 105 cells per mL and suspended in PBS. 100 µL of cells were added in a 96-well plate and mixed with an equal volume of Apo-ONE homogeneous caspase-3 reagent. The mixture was incubated for 5 h at room temperature. Fluorescence at an excitation wavelength of 492 nm and an emission wavelength of 521 nm was measured using a multimode plate reader (Perkin-Elmer Life Sciences).Detection of intracellular reactive oxygen species (ROS) level
The detection of the intracellular ROS level was conducted using 2′,7′-dichlorofluorescein diacetate (DCFH-DA) (Beyotime Biotechnology Inc., China). Cells were given DA, MQ, a combination of the two, or VRP for 24 h pre-incubation. Then, cells were harvested, washed twice with PBS and incubated in an FBS-free medium with DOX for another 48 h.20 After the incubation, cells were washed twice with an FBS-free medium, and incubated in an FBS-free medium containing 10 µM DCFH-DA for 30 min at 37 °C in the dark. Intracellular fluorescence was measured using FACS flow cytometry (FACScalibar, BD, USA).Molecular docking
The binding modes of DA and MQ with P-gp were recorded using molecular docking analysis according to the computational docking model CDOCKER protocol of Discovery Studio 3.0. Mouse and human P-gp shows 87% overall sequence identity and nearly 100% identity of the binding cavity except for mSer725/hAla729,21 thus in this study, the crystal structure of mouse P-gp (PDB: 3G60) was chosen as the receptor for molecular docking analysis.22 DA or MQ were docked into the receptor using default options after refining with the CHARMm force field. It would be considered as a failure if a docking was without any output pose. The most probable binding conformation regarded was with the lowest CDOCKER energy.23P-gp ATPase activity assay
A P-gp ATPase activity assay was performed using the Pgp-Glo™ assay system (Promega) according to the protocol of the manufacturer. VRP was a P-gp ATPase stimulator, and sodium orthovanadate (Na3VO4) was a P-gp ATPase inhibitor. Decreased luminescence of the drug-free sample compared to the Na3VO4-treated sample was considered as basal P-gp ATPase activity (ΔRLUbasal). Decreased luminescence of drug-treated samples compared to the Na3VO4-treated sample reflected the effect of drugs on P-gp ATPase activity (ΔRLUdrug). If ΔRLUdrug > ΔRLUbasal, the drug is a stimulator of P-gp ATPase activity. If ΔRLUdrug < ΔRLUbasal, the drug is an inhibitor of P-gp ATPase activity. If ΔRLUdrug = ΔRLUbasal, the drug shows no effect on P-gp ATPase activity.Quantitative real-time PCR
Cells were collected on ice and washed twice with ice-cold PBS. Total RNA was extracted using an RNA isoPlus® Reagent Kit (Takara Biotechnology) following the instructions of the manufacturer. 1 µg of total RNA was used to generate cDNA using PrimeScript® RT Reagent Kit with gDNA Eraser (Takara Biotechnology). cDNA was then amplified using a SYBR® Premix Ex Taq™ Kit (Takara Biotechnology) by ABI PRISM® 7500 Real-Time PCR System (Applied Biosystems). β-Actin was performed as the normalization control. Data obtained was analyzed by the comparative ΔΔCt method. Primers were as follows: MDR1 (forward: 5′-GGAGCCTACTTGGTGGCACATAA-3′, reverse: 5′-TGGCATAGTCAGGAGCAAATGAAC-3′); β-actin (forward: 5′-ATTGAACACGGCATTGTCAC-3′, reverse: 5′-CATCGGAACCGCTCATTG-3′).Western blot analysis
Cell proteins were extracted and determined using a BCA assay kit. Proteins were separated by SDS-PAGE, transferred to PVDF membrane, blocked with 5% fat-free dry milk or 5% BSA, and immunoblotted with primary antibodies at 4 °C overnight. Membranes were washed and incubated with corresponding secondary antibodies for 1 h at room temperature. Immunoreactivity was detected using an ECL kit by Bio-Spectrum Gel Imaging System (UVP, USA). Quantification of protein expression was analyzed using a Gel-Pro Analyzer 3.0 software.Statistical analysis
Differences between groups were analyzed using Student’s t-test (GraphPad PRISMR 4 software, GraphPad Software, San Diego, CA). All experiments were repeated at least three times and data obtained are given as the mean ± SD. Values of p < 0.05 were considered to be statistically significant.Results
The combination of DA and MQ had a stronger effect on potentiating sensitivity of K562/Adr cells to DOX and reducing cell colony formation
The cellular toxicity of DA and MQ alone in K562 and K562/Adr cells was investigated to determine the IC10 values (a concentration at which more than 90% of cells survived) of DA and MQ. The IC10 value was considered to be the reversal concentration. The IC10 values of DA and MQ were 0.02 µM and 20 µM, respectively. The IC50 values of DOX or cisplatin in K562 and K562/Adr cells with or without DA and MQ are shown in Table 1. The combination of DA and MQ decreased the IC10 values and resistance index of DOX to resistant K562/Adr cells more than when either was being used alone. However, neither the IC50 values and resistance index of DOX to sensitive K562 cells were altered nor change was presented when cells were treated with cisplatin in K562 and K562/Adr cells. Further, in the combined group of DA and MQ, the number of cell colonies was the least of all experimental groups (Fig. 1). Combining the results from Table 1 and Fig. 1, it is suggested that the combination of DA and MQ revealed a stronger effect on increasing the sensitivity of resistant cells to P-gp substrate DOX than when either was being used alone, while there was little effect on the sensitivity of resistant cells to cisplatin which is not a substrate of P-gp.| Compounds | IC50 ± SD (µM; fold-reversal) | Resistance index | |
|---|---|---|---|
| K562 | K562/Adr (P-gp) | ||
| a Independent experiments were performed at least three times. Data are expressed as the mean ± SD. *P < 0.05, versus the values obtained without DA, MQ or VRP. | |||
| DOX | 0.97 ± 0.149 (1.00) | 7.77 ± 0.624 (1.00) | 8.01 |
| +DA | 1.09 ± 0.132 (0.89) | 2.37 ± 0.277* (3.28) | 2.17 |
| +MQ | 1.21 ± 0.157 (0.80) | 5.71 ± 0.412* (1.49) | 4.72 |
| +DA + MQ | 0.94 ± 0.093 (1.03) | 1.93 ± 0.097* (4.03) | 2.05 |
| +VRP | 0.92 ± 0.079 (1.05) | 1.47 ± 0.036* (5.29) | 1.60 |
| Cisplatin | 7.02 ± 0.513 (1.00) | 7.79 ± 0.667 (1.00) | 1.11 |
| +DA | 7.44 ± 0.428 (0.94) | 7.11 ± 0.597 (1.10) | 0.96 |
| +MQ | 6.82 ± 0.346 (1.03) | 7.03 ± 0.538 (1.11) | 1.03 |
| +DA + MQ | 6.97 ± 0.471 (1.01) | 8.23 ± 0.512 (0.95) | 1.18 |
| +VRP | 7.21 ± 0.677 (1.03) | 7.92 ± 0.601 (1.02) | 1.10 |
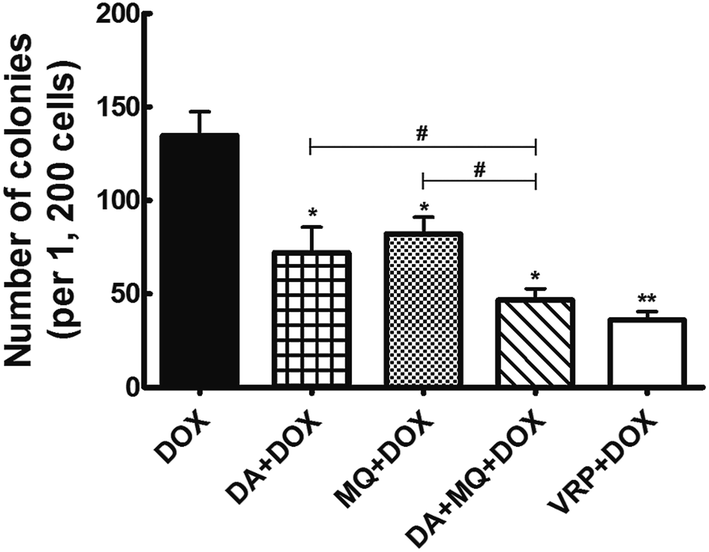 | ||
| Fig. 1 Effect of the combination of DA and MQ on colony formation in soft agar. Each column reflects the mean ± SD of at least three independent experiments. Bars represent SD values. *P < 0.05 and **P < 0.01, versus the control group of DOX. #P < 0.05, versus the single group. | ||
The combination of DA and MQ significantly increased the accumulation of DOX and Rho 123 in P-gp-overexpressing K562/Adr cells more than either of the single groups
To figure out the possible mechanism involved in the reversal effect of the combination of DA and MQ, we conducted experiments to test the cellular accumulation of DOX and Rho 123 both of which are classical P-gp substrates in K562 and K562/Adr cells (Fig. 2). We found that the cellular accumulation of DOX (Fig. 2A) and Rho 123 (Fig. 2B) in K562 cells was much higher than in K562/Adr cells. All of the groups couldn’t alter the cellular accumulation of DOX and Rho 123 in K562 cells. However, compared with a positive control group of VRP, the combination of DA and MQ significantly increased cellular DOX and Rho 123 accumulation 2.39 and 2.61-fold in K562/Adr cells, respectively. A single group of DA or MQ remarkably increased cellular DOX accumulation 1.82- or 1.45-fold, respectively, and increased cellular Rho 123 accumulation 1.97- or 1.63-fold, respectively. The combined group showed a stronger effect than the single groups, suggesting that the combination of DA and MQ had a stronger effect on inhibiting P-gp-mediated transport in resistant K562/Adr cells.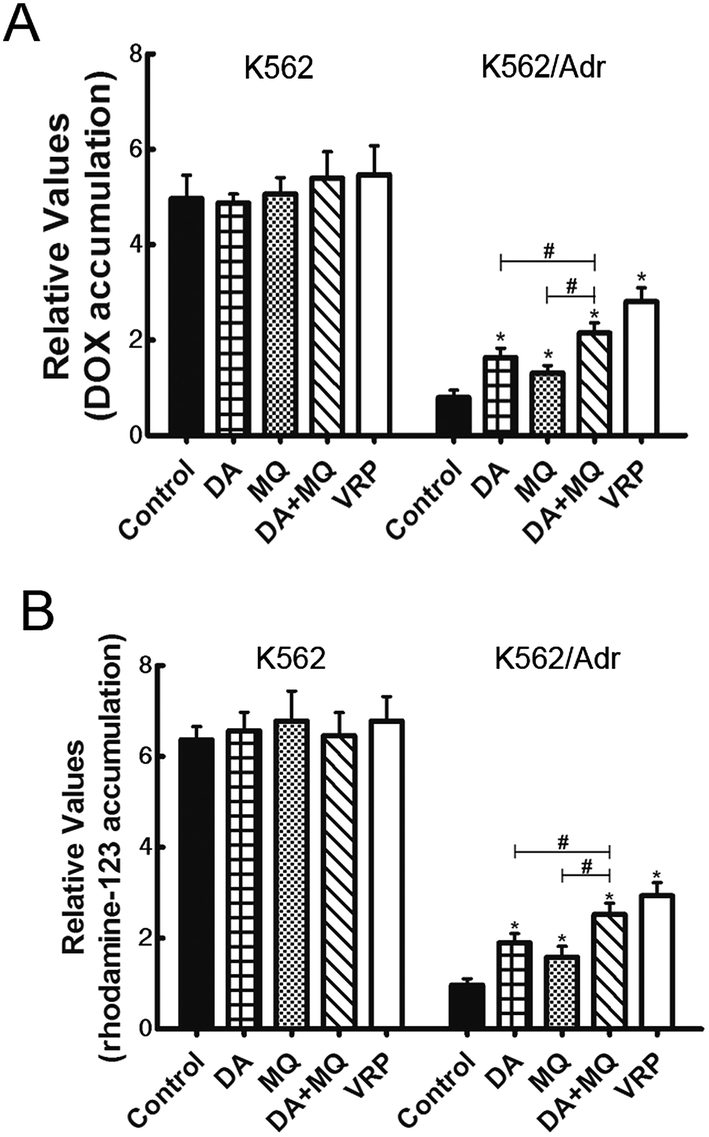 | ||
| Fig. 2 Effect of the combination of DA and MQ on the cellular accumulation of DOX (A) and Rho 123 (B). Results are shown as fold change of fluorescence intensity relative to the control group of K562/Adr cells. Each column shows the mean ± SD of at least three independent experiments. Bars represent SD values. *P < 0.05, versus the control group of K562/Adr cells. #P < 0.05, versus the single group. | ||
The combination of DA and MQ enhanced the increase of DOX-induced and caspase-3-mediated apoptosis in K562/Adr cells via ROS production
Since cellular DOX accumulation was increased, we further investigated cell apoptosis in the presence of the combination of DA and MQ. Results suggested that the combination of DA and MQ could facilitate DOX-induced apoptosis (Fig. 3). It was further revealed that expression of cleaved caspase-3 was increased the most when treated with the combination of DA and MQ compared to single groups (Fig. 4A and B). Caspase-3 activity was higher in the combined group than in single groups (Fig. 4C). Results suggested that the combination of DA and MQ led to a much higher increase of caspase-3-mediated apoptosis induced by DOX in K562/Adr cells than when being treated with either alone. Since ROS production shows some relationship with cell apoptosis, results in Fig. 4D showed that the ROS level in the combined group was generated higher than from either of the single groups, which was in accordance with the results of caspase-3-mediated apoptosis. Thus it could be speculated that the increase of DOX-induced caspase-3-mediated apoptosis by the combination of DA and MQ may occur due to ROS production.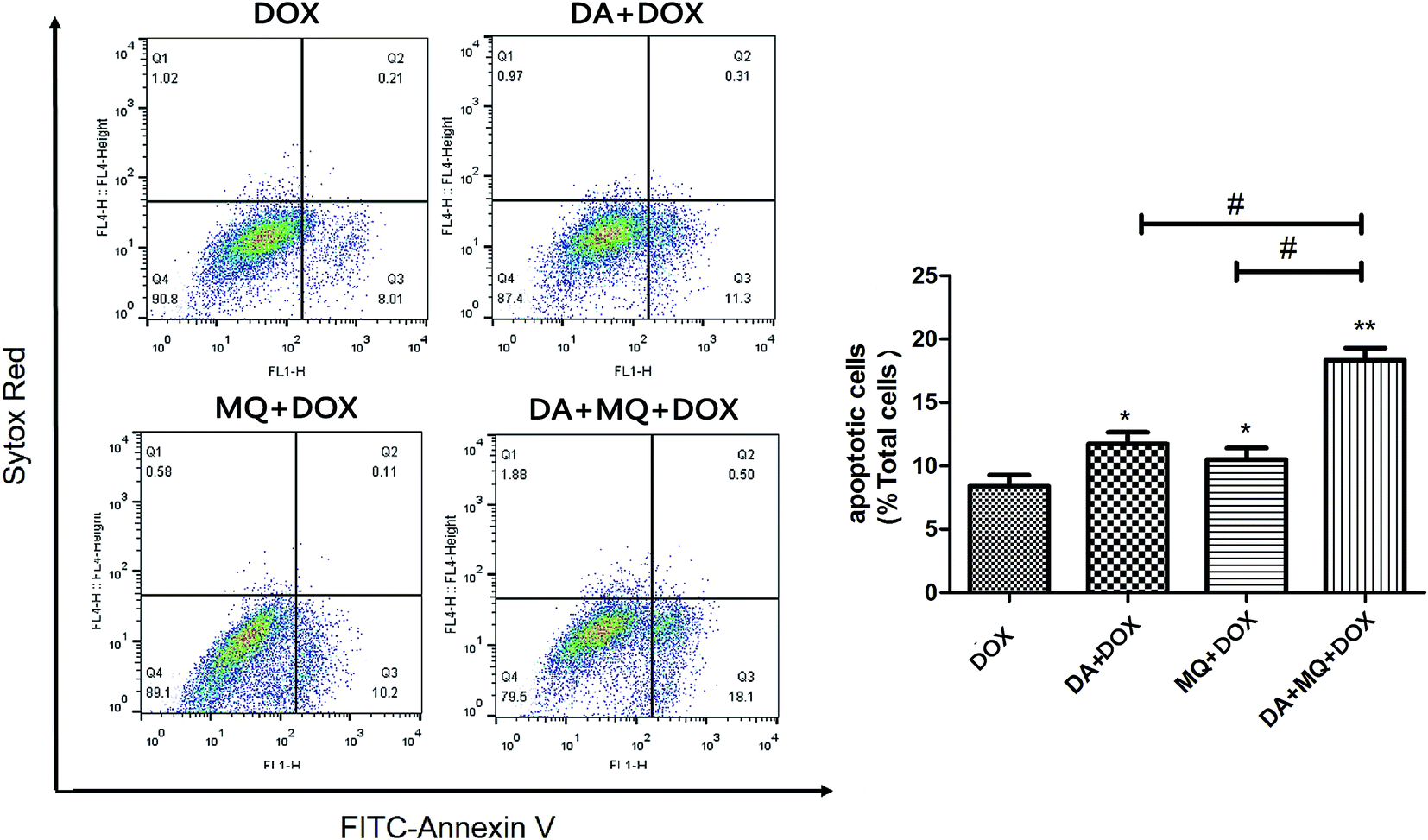 | ||
| Fig. 3 Effect of the combination of DA and MQ on DOX-induced cell apoptosis in K562/Adr cells. After pre-treatment with DA, MQ, or a combination of the two for 24 h, cells were then treated with DOX for another 48 h. Then cells were stained with Annexin V/SYTOX Red, followed by flow cytometry analysis. One representative experiment is shown. *P < 0.05 and **P < 0.01, versus the control group of DOX. #P < 0.05, versus the single group. | ||
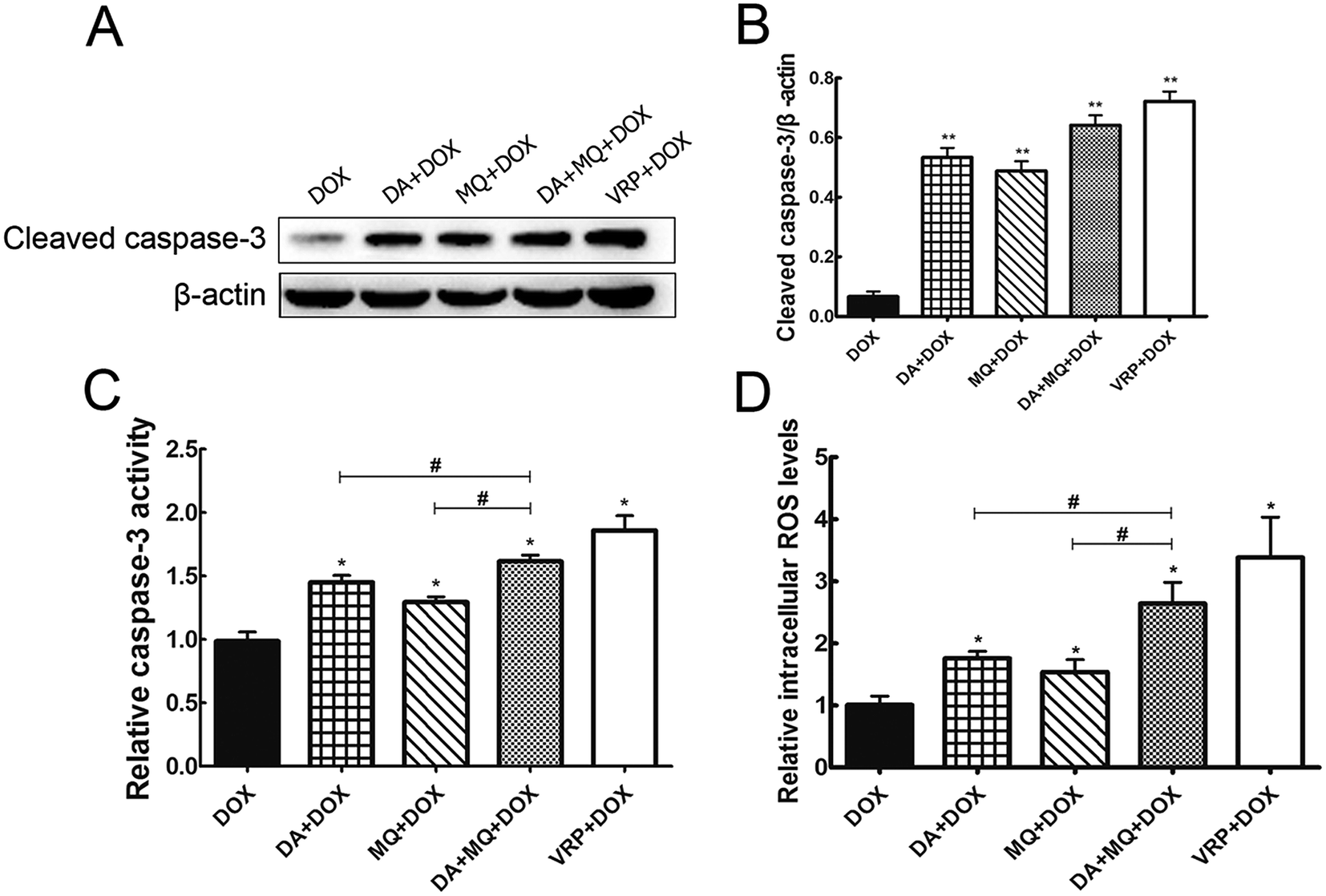 | ||
| Fig. 4 Effect of the combination of DA and MQ on the levels of caspase-3 and ROS in K562/Adr cells. Cleaved caspase-3 expression was performed (A) with quantification (B), caspase-3 activity (C) and ROS level (D) detections using kits. The quantification of caspase-3 expression was normalized to β-actin expression. Each column reflects the mean ± SD of at least three independent experiments. Bars represent SD values. *P < 0.05 and **P < 0.01, versus the control group of DOX. #P < 0.05, versus the single group. | ||
Molecular docking of DA and MQ to P-gp
Molecular binding modes of DA and MQ with P-gp were recorded in Fig. 5. Various amino acid residues lying in the internal cavity of P-gp were responsible for P-gp to bind with DA and MQ. DA not only had two potential pi interactions with Phe974 and one potential pi interaction with Phe728, but also formed a hydrogen bond with Gln721, which might take a critical role in the strong inhibition of P-gp (Fig. 5B). As for MQ, it had one potential pi interaction with Phe974, which suggested a weaker inhibition to P-gp (Fig. 5C).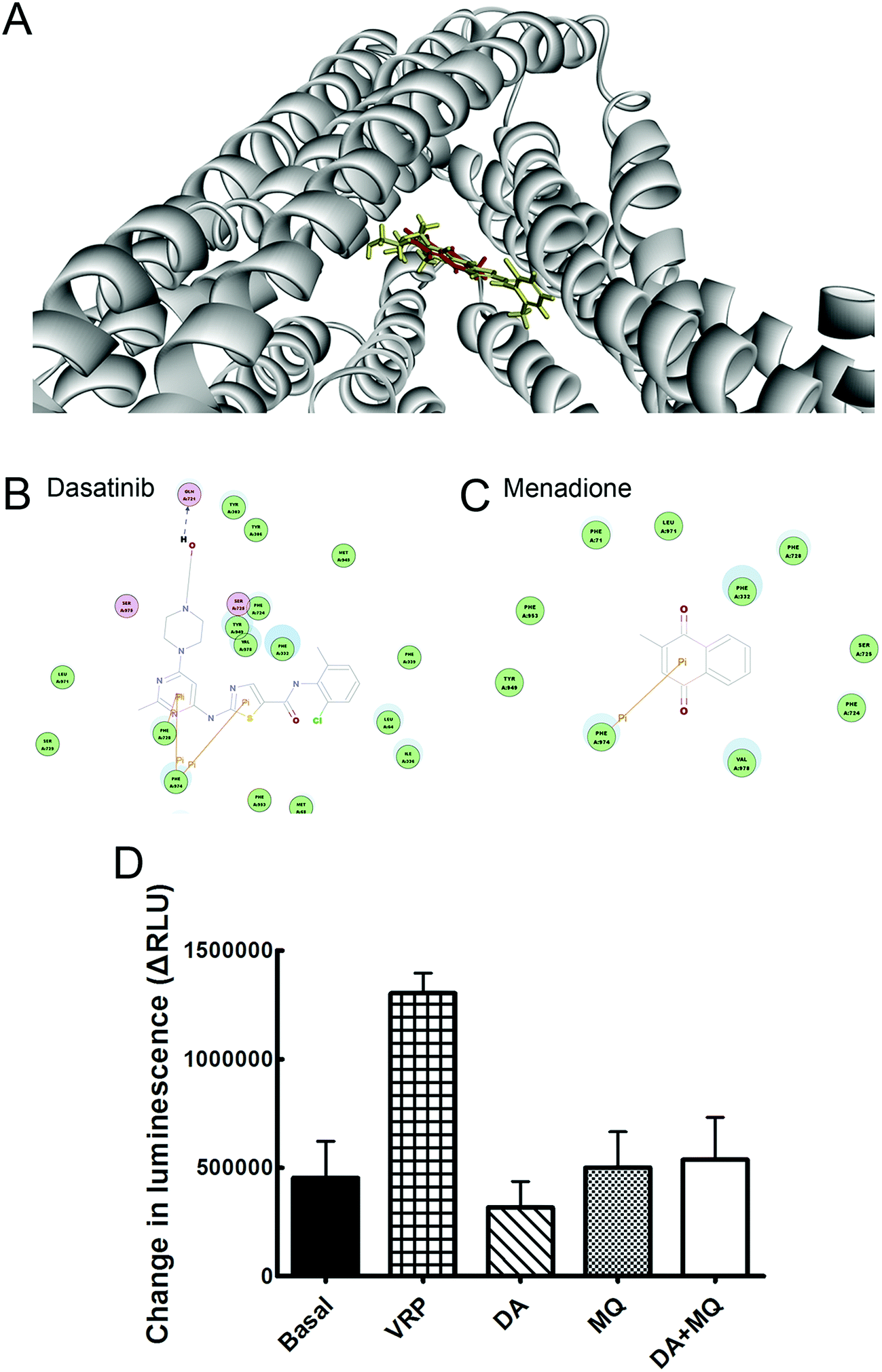 | ||
| Fig. 5 Effect of the combination of DA and MQ on P-gp efflux ability. (A) The conformation with the lowest CDOCKER energy was chosen as the most probable binding modes of DA or MQ. The three-dimensional diagrams present the binding conformation of DA (red) and MQ (yellow) with P-gp. (B and C) The two-dimensional diagrams show amino acid residues in the internal active site of P-gp, and they may potentially be involved in the binding of DA and MQ. The colors of residue represent the types of interaction: green (van der Waals), yellow (pi interaction), magenta (polar), blue arrows (hydrogen bond). (D) Changes in luminescence were compared to the changed luminescence of basal P-gp ATPase activity. All the experiment groups showed similar values with the basal P-gp ATPase activity, which indicated no effect on the P-gp ATPase activity. Each column reflects the mean ± SD of at least three independent experiments. Bars represent SD values. | ||
The combination of DA and MQ showed little effect on P-gp ATPase activity
To confirm whether the combination of DA and MQ could directly inhibit P-gp efflux ability, we conducted experiments to examine its effect on P-gp ATPase activity. ΔRLUDA, ΔRLUMQ and ΔRLUDA+MQ all had no significant difference compared with ΔRLUbasal (Fig. 5D), elucidating that all of the three groups showed no effect on the inhibition of P-gp efflux ability. Combining this with the molecular docking results, it is suggested that DA, MQ, and the combination of them, at the reversal concentrations, couldn’t inhibit P-gp efflux function.The combination of DA and MQ remarkably downregulated the mRNA and protein levels of P-gp more than either of the two being used alone
Since the possibility of the inhibition of the combined group to P-gp efflux ability has been excluded, we further investigated its effect on the regulation of P-gp expression. Compared to the single group of DA or MQ, the combined group showed much more downregulation of mRNA (Fig. 6A) and protein (Fig. 6B and C) expression of P-gp. Results suggested that the stronger reversal effect of the combination of DA and MQ might be attributed to its increased downregulation of P-gp expression both in mRNA and protein levels.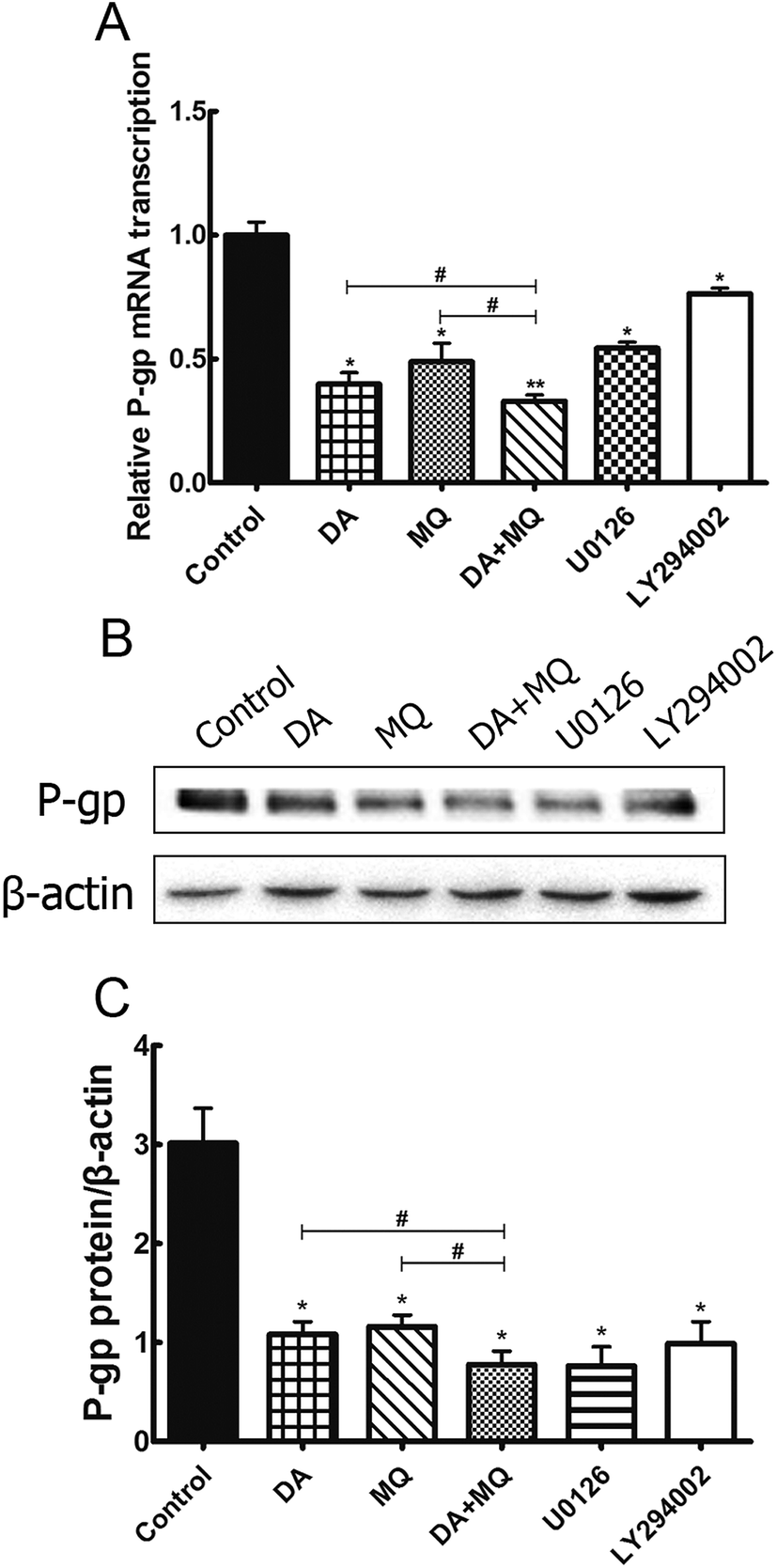 | ||
| Fig. 6 Effect of the combination of DA and MQ on the mRNA (A) and protein (B and C) levels of P-gp in K562/Adr cells. The quantification of P-gp expression was normalized to β-actin expression. Each column reflects the mean ± SD of at least three independent experiments. Bars represent SD values. *P < 0.05 and **P < 0.01, versus the control group. #P < 0.05, versus the single group. | ||
ERK and AKT/mTOR signal pathways were not involved in the stronger effect of the combination of DA and MQ on downregulating P-gp expression
It was shown in Fig. 6 that ERK pathway inhibitor U0126 and AKT/mTOR pathway inhibitor LY294002 downregulated P-gp expression significantly, thus in order to figure out if the stronger reversal effect of the combination of DA and MQ has something to do with the ERK or AKT/mTOR signal pathway, we examined the expressions of phosphorylated ERK, phosphorylated AKT and phosphorylated mTOR after being treated with DA and MQ together. We found that DA could significantly inhibit the expressions of phosphorylated forms of ERK, AKT, and mTOR, while MQ on the contrary activated their expressions. As a result, in the combined group, the inhibition of phosphorylated forms of ERK (Fig. 7A and B), AKT (Fig. 7C and D), and mTOR (Fig. 7E and F) are weaker than that in the single group of DA. Results suggested that the combination of DA and MQ had a weaker effect in blocking the ERK and AKT/mTOR signal pathways than DA alone, therefore, ERK and AKT/mTOR signal pathways were not involved in the stronger effect of the combination of DA and MQ on downregulating P-gp expression.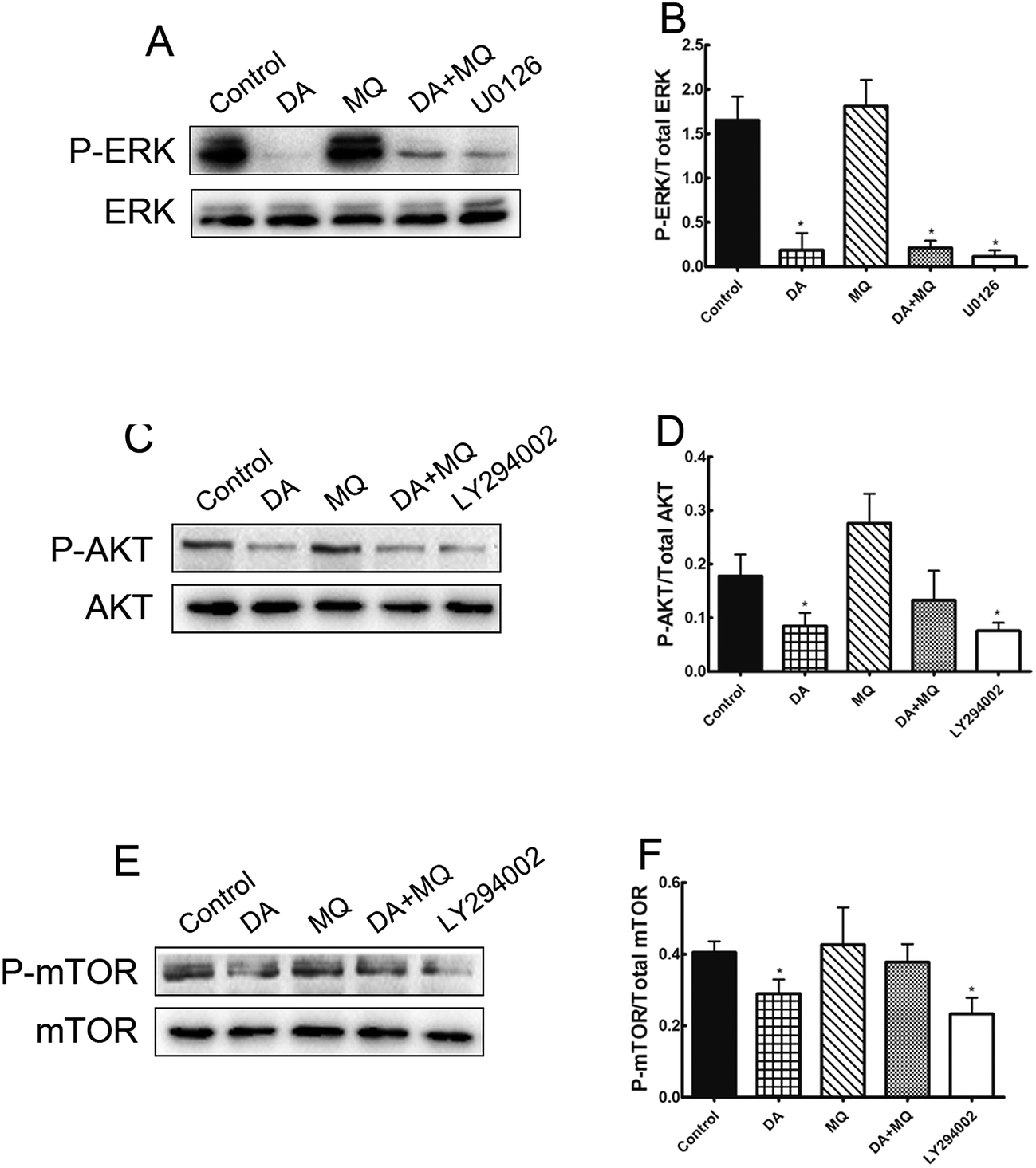 | ||
| Fig. 7 Effect of the combination of DA and MQ on the expressions of phospho-ERK (A and B), phospho-AKT (C and D) and phospho-mTOR (E and F). The quantifications of phospho-ERK, phospho-AKT and phospho-mTOR expression were normalized to the total content of ERK, AKT and mTOR, respectively. Each column reflects the mean ± SD of at least three independent experiments. Bars represent SD values. *P < 0.05, versus the control group. | ||
Discussion
Chronic myeloid leukemia (CML) was the first cancer discovered with molecular mechanisms (Philadelphia chromosome (Ph) and the related fusion gene BCR-ABL) leading to malignant transformation. One strategic therapy of CML is to develop novel drugs targeting inhibition of the BCR-ABL oncogene.Dasatinib (DA), a second generation BCR-ABL inhibitor, is approved for imatinib resistant CML and Ph+ acute lymphoblastic leukemia (ALL) in clinical use.24 Vitamin K is an important nutritional factor in human beings contributing to the blood clotting process. It mainly has three types, vitamin K1, K2 and K3. Menadione (MQ, vitamin K3) shows a potential effect of inhibiting cancer cell lines.12–14 It has been proven that a combination of MQ and ascorbate could kill cancer cells which express oncoprotein Bcr-Abl through inducing ROS.25 It has been reported that the imatinib resistant cell line shows overexpression of both Bcr-Abl and P-gp protein.26 The inhibition of DA and MQ to oncogenic protein Bcr-Abl has already been reported, and there is a possibility that the haemorrhage side effect of DA, which is partly due to the inhibition of PDGFR, may be improved by MQ through inducing phosphorylation of PDGFR,18,19 thus in this study, we aimed at investigating the combined effect of DA and MQ on P-gp-mediated MDR in P-gp-overexpressing K562/Adr cells.
In the present study, the combination of DA and MQ significantly enhanced the increase of cellular toxicity of DOX compared to either of the two drugs being used alone in K562/Adr cells. However, all of the experimental groups neither altered the toxicity of DOX to parental K562 cells nor changed the sensitivity of cisplatin to both K562 and K562/Adr cells (Table 1). In Fig. 1, the least number of cell colonies were produced in the combined group, which suggested that the combined group restored more of the sensitivity of K562/Adr cells to DOX than other groups. To confirm the reversal ability of the combination of DA and MQ, we investigated the effect of the combined group on cellular accumulation of DOX and Rho 123 in K562 and K562/Adr cells. Cellular accumulation of DOX and Rho 123 was increased more when treated with DA and MQ together compared with being treated with either of the two alone, whereas both the cellular accumulation of DOX and Rho 123 showed few changes in K562 cells (Fig. 2). The results above suggested that the combination of DA and MQ enhanced the sensitivity of MDR cells to DOX via increasing the cellular DOX accumulation.
Apoptosis is one of the fundamental components in the pathogenesis of cancer. At the molecular level, caspase activation and mitochondrial dysfunction are key elements for the description of apoptosis.27 Mitochondria and ROS play important roles in inducing apoptosis under physiologic and pathologic conditions. Interestingly, mitochondria are sources and targets of ROS.28 Therefore, in our study, we examined the expression of cleaved caspase-3 and caspase-3 activity to elucidate cell apoptosis induced by the chemotherapeutic drug DOX, and further detected the cellular ROS level. It is shown that the expression of cleaved caspase-3 and caspase-3 activity were both increased the most in the combined group, so did that of ROS production (Fig. 4). Besides, it is reported that ROS could reduce P-gp transport rate.29 Combined with the results of cellular accumulation of DOX, it could be suggested that the combination of DA and MQ could enhance the cellular accumulation of DOX, which led to increased cell apoptosis via ROS production in K562/Adr cells. Increased ROS production might further reduce P-gp transport rate.
Since DOX and Rho 123 are both classical substrates of P-gp,30 the enhanced accumulation of cellular DOX and Rho 123 indicates some relationships between drugs and P-gp transporters. Therefore, we wondered whether this reversal effect was achieved by downregulating P-gp expression or by inhibiting efflux function of P-gp. Molecular docking and P-gp ATPase activity assays were conducted to figure out whether drugs could inhibit efflux ability of P-gp. Molecular docking assay showed that DA and MQ both were substrates of P-gp (Fig. 5A), DA showed a stronger binding capacity with P-gp than MQ, and they shared one common binding site of amino acid residue Phe974 which might indicated the possibility of competitive inhibition (Fig. 5B and C). However, the results of the P-gp ATPase activity assay demonstrated that DA and MQ, both at reversal concentrations, had no effect on P-gp ATPase activity (Fig. 5D). These data suggested that the combination of DA and MQ, both at reversal doses, might have no effect on the efflux function of P-gp.
Since the possible relationship between drugs and efflux ability of P-gp could be excluded, we then investigated the effect of the combined group on the regulation of P-gp expression. Compared with drugs being given alone, the combined group downregulated more of the mRNA and protein levels of P-gp (Fig. 6). Previously, it has been reported that the main EGFR downstream pathways MAPK/ERK and PI3K/AKT/mTOR are constitutively activated in tumor cells and closely involved in the proliferation, invasion and migration processes of cancer cells.31,32 Therefore, we examined the effect of the combination of DA and MQ on ERK, AKT and mTOR phosphorylation. Results showed that DA significantly inhibited phosphorylated ERK, AKT and mTOR, while MQ on the contrary activated phosphorylated ERK, AKT and mTOR, which led to a non-enhanced inhibition effect of all the phosphorylated pathways in K562/Adr cells (Fig. 7). ERK and AKT signal pathways did not account for the enhanced reversal effect of the combination of DA and MQ.
In conclusion, this study provided evidences that the combination of DA and MQ significantly enhanced more of the efficacy of DOX in P-gp-overexpressing MDR K562/Adr cells, resulting in increased cell apoptosis via ROS production. The involved mechanism of the reversal effect might be the downregulation of the mRNA and protein levels of P-gp. Increased ROS production might further reduce P-gp transport rate. In addition, the reversal effect is independent of ERK and AKT/mTOR signal pathways. Our findings may support the potential advantages of combining DA with MQ in overcoming drug resistance in cancer chemotherapy, and lay a theoretical basis for the clinical combined use of DA and MQ.
Acknowledgements
This work was supported by grants from National Natural Sciences Foundation of China (NSFC) No. 81273580, 81473280.References
- E. Marthinet, G. Divita, J. Bernaud, D. Rigal and L. G. Baggetto, Gene Ther., 2000, 7, 1224–1233 CrossRef CAS PubMed.
- A. A. Stavrovskaya, Biochemistry, 2000, 65, 95–106 CAS.
- R. L. Juliano and V. Ling, Biochim. Biophys. Acta, Bioenerg., 1976, 455, 152–162 CrossRef CAS.
- F. Shen, S. Chu, A. K. Bence, B. Bailey, X. Xue, P. A. Erickson, M. H. Montrose, W. T. Beck and L. C. Erickson, J. Pharmacol. Exp. Ther., 2008, 324, 95–102 CrossRef CAS PubMed.
- A. Mayer, M. Takimoto, E. Fritz, G. Schellander, K. Kofler and H. Ludwig, Cancer, 1993, 71, 2454–2460 CrossRef CAS PubMed.
- J. Zhou, M. Liu, R. Aneja, R. Chandra, H. Lage and H. C. Joshi, Cancer Res., 2006, 66, 445–452 CrossRef CAS PubMed.
- E. Weisberg, P. W. Manley, S. W. Cowan-Jacob, A. Hochhaus and J. D. Griffin, Nat. Rev. Cancer, 2007, 7, 345–356 CrossRef CAS PubMed.
- T. Chen, C. Wang, Q. Liu, Q. Meng, H. Sun, X. Huo, P. Sun, J. Peng, Z. Liu, X. Yang and K. Liu, Cancer Biol. Ther., 2015, 16, 106–114 CrossRef CAS PubMed.
- Y. Chen, S. Agarwal, N. M. Shaik, C. Chen, Z. Yang and W. F. Elmquist, J. Pharmacol. Exp. Ther., 2009, 330, 956–963 CrossRef CAS PubMed.
- E. C. Cranenburg, L. J. Schurgers and C. Vermeer, Thromb. Haemostasis, 2007, 98, 120–125 CAS.
- C. Vermeer, M. J. Shearer, A. Zittermann, C. Bolton-Smith, P. Szulc, S. Hodges, P. Walter, W. Rambeck, E. Stocklin and P. Weber, Eur. J. Nutr., 2004, 43, 325–335 CrossRef CAS PubMed.
- S. A. Akman, M. Dietrich, R. Chlebowski, P. Limberg and J. B. Block, Cancer Res., 1985, 45, 5257–5262 CAS.
- L. M. Nutter, A. L. Cheng, H. L. Hung, R. K. Hsieh, E. O. Ngo and T. W. Liu, Biochem. Pharmacol., 1991, 41, 1283–1292 CrossRef CAS PubMed.
- D. W. Lamson and S. M. Plaza, Alternative Med. Rev., 2003, 8, 303–318 Search PubMed.
- M. Hitomi, F. Yokoyama, Y. Kita, T. Nonomura, T. Masaki, H. Yoshiji, H. Inoue, F. Kinekawa, K. Kurokohchi, N. Uchida, S. Watanabe and S. Kuriyama, Int. J. Oncol., 2005, 26, 713–720 CAS.
- S. J. Oh, H. K. Han, K. W. Kang, Y. J. Lee and M. Y. Lee, Arch. Pharmacal Res., 2013, 36, 509–516 CrossRef CAS PubMed.
- M. Wartenberg, K. Fischer, J. Hescheler and H. Sauer, Int. J. Cancer, 2000, 85, 267–274 CrossRef CAS PubMed.
- A. Quintas-Cardama, H. Kantarjian, F. Ravandi, S. O’Brien, D. Thomas, G. Vidal-Senmache, W. Wierda, S. Kornblau and J. Cortes, Cancer, 2009, 115, 2482–2490 CrossRef CAS PubMed.
- K. Abdelmohsen, P. Patak, C. Von Montfort, I. Melchheier, H. Sies and L. O. Klotz, Methods Enzymol., 2004, 378, 258–272 CAS.
- M. Hayakawa, H. Miyashita, I. Sakamoto, M. Kitagawa, H. Tanaka, H. Yasuda, M. Karin and K. Kikugawa, EMBO J., 2003, 22, 3356–3366 CrossRef CAS PubMed.
- E. Dolghih, C. Bryant, A. R. Renslo and M. P. Jacobson, PLoS Comput. Biol., 2011, 7, e1002083 CAS.
- W. Zha, G. Wang, W. Xu, X. Liu, Y. Wang, B. S. Zha, J. Shi, Q. Zhao, P. M. Gerk, E. Studer, P. B. Hylemon, W. M. Pandak Jr and H. Zhou, PLoS One, 2013, 8, e54349 CAS.
- M. H. Bohari and G. N. Sastry, J. Mol. Model., 2012, 18, 4263–4274 CrossRef CAS PubMed.
- J. Cortes, P. Rousselot, D. W. Kim, E. Ritchie, N. Hamerschlak, S. Coutre, A. Hochhaus, F. Guilhot, G. Saglio, J. Apperley, O. Ottmann, N. Shah, P. Erben, S. Branford, P. Agarwal, A. Gollerkeri and M. Baccarani, Blood, 2007, 109, 3207–3213 CrossRef CAS PubMed.
- R. Beck, R. C. Pedrosa, N. Dejeans, C. Glorieux, P. Leveque, B. Gallez, H. Taper, S. Eeckhoudt, L. Knoops, P. B. Calderon and J. Verrax, Invest. New Drugs, 2011, 29, 891–900 CrossRef CAS PubMed.
- F. X. Mahon, F. Belloc, V. Lagarde, C. Chollet, F. Moreau-Gaudry, J. Reiffers, J. M. Goldman and J. V. Melo, Blood, 2003, 101, 2368–2373 CrossRef CAS PubMed.
- A. M. Hunter, E. C. LaCasse and R. G. Korneluk, Apoptosis, 2007, 12, 1543–1568 CrossRef CAS PubMed.
- H. U. Simon, A. Haj-Yehia and F. Levi-Schaffer, Apoptosis, 2000, 5, 415–418 CrossRef CAS PubMed.
- P. Trach, N. Afahaene, M. Nowak and O. Thews, Adv. Exp. Med. Biol., 2012, 737, 161–167 CrossRef CAS PubMed.
- M. D. Perloff, L. L. Von Moltke, J. E. Marchand and D. J. Greenblatt, J. Pharm. Sci., 2001, 90, 1829–1837 CrossRef CAS PubMed.
- N. K. Saxena, D. Sharma, X. Ding, S. Lin, F. Marra, D. Merlin and F. A. Anania, Cancer Res., 2007, 67, 2497–2507 CrossRef CAS PubMed.
- N. Huynh, K. H. Liu, G. S. Baldwin and H. He, Biochim. Biophys. Acta, Bioenerg., 2010, 1803, 1106–1113 CrossRef CAS PubMed.
Footnote |
| † Changyuan Wang and Zhihao Liu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |