
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-ionic self-assembling amphiphilic polyester dendrimers as new drug delivery excipients†
Dhiraj R. Sikwal,
Rahul S. Kalhapure *,
Mahantesh Jadhav,
Sanjeev Rambharose,
Chunderika Mocktar and
Thirumala Govender*
*,
Mahantesh Jadhav,
Sanjeev Rambharose,
Chunderika Mocktar and
Thirumala Govender*
Discipline of Pharmaceutical Sciences, University of KwaZulu-Natal, Private Bag X54001, Durban, 4000, KwaZulu-Natal, South Africa. E-mail: govenderth@ukzn.ac.za; kalhapure@ukzn.ac.za; rahul.kalhapure@rediffmail.com; Fax: +27 31 260 7792; Tel: +27 31 260 7358
First published on 2nd March 2017
Abstract
Solubility enhancement of poorly soluble antibiotics via self-assembling nano systems could be a promising approach to effectively treat bacterial infections in the current scenario of evolving resistant species. The study in this paper reports the synthesis of novel biocompatible G2 and G3 polyester amphiphilic dendrimers (ADs) (GMOA-G2-OH, GMOA-G3-OH, GMS-G2-OH and GMS-G3-OH) and their application as: (i) solubility enhancers for fusidic acid (FSD) as a model antibiotic with poor aqueous solubility and (ii) as stearic stabilizers in the preparation of solid lipid nanoparticles (SLNs). Two different series of ADs from glycerol monostearate (GMS) and glycerol monooleate (GMOA) were synthesized and their structures were confirmed employing FT-IR, NMR (1H and 13C) and HR-MS. The MTT assay confirmed their non-toxicity to mammalian cells. The critical aggregation concentration value order for ADs was GMS-G3-OH (5 × 10−6 mol l−1) < GMOA-G3-OH (7 × 10−6 mol l−1) < GMOA-G2-OH = GMS-G2-OH (5 × 10−5 mol l−1). All ADs formed micelles in the size range of 6.48 ± 0.04 nm to 12.38 ± 0.36 nm. At 1% w/w concentration FSD solubility enhancement in GMOA-G2-OH, GMOA-G3-OH, GMS-G2-OH and GMS-G3-OH was 43, 11, 9.1 and 6.8-fold respectively compared to water. As GMOA-G2-OH enabled the highest solubility of FSD, it was further evaluated for its antibacterial activity against Staphylococcus aureus and methicillin-resistant S. aureus (MRSA). The minimum inhibitory concentration values for FSD with and without GMOA-G2-OH against S. aureus were 0.23 μg ml−1 and 0.53 μg ml−1 respectively whereas the values were 0.23 μg ml−1 and 0.39 μg ml−1 against MRSA respectively. These results suggested that GM-OA-G2 not only enhanced the solubility but also enhanced antibacterial potency of FSD. Furthermore, these ADs showed their potential as promising pharmaceutical excipients as they acted as stearic stabilizers in the preparation of SLNs. Using these ADs stable SLNs with zeta potential value in the range of −15.30 ± 1.44 to −38.46 ± 3.04 were formed.
1. Introduction
Solubility, a phenomenon of dissolution of solute in solvent, is considered as one of the most important parameters to achieve a required concentration of drug in the systemic circulation for a desired pharmacological action.1 Poor aqueous solubility of many active pharmaceutical ingredients (APIs) limits their bioavailability and absorption and thus has become a major obstacle in the development of highly potent drugs.1–4 More than 40% of newly developed APIs are practically insoluble in water and are rejected by the pharmaceutical industry due to inadequate or variable bioavailability and will never benefit patients. Another 17% of available drugs in the market also suffer from suboptimal performance due to their limited or no aqueous solubility.1–4 Among various classes of APIs, antimicrobial drugs are one of the classes in which many antibiotics have poor or practically no aqueous solubility. Antibiotics with low aqueous solubility include polyene antibiotics [amphotericin B (0.0819 mg ml−1),5 nystatin (3.6 mg ml−1),6 pimaricin (0.39 mg ml−1)6], macrolide antibiotics [erythromycin (2 mg ml−1),6 azithromycin (0.6 mg ml−1)7], quinolone antibiotics [nadifloxacin and prulifloxacin (sparingly soluble)8] and steroidal antibiotics [fusidic acid, helvolic acid and cephalosporin P (sparingly soluble)9]. In the current scenario of development of resistance to antibiotics by various bacteria, solubility enhancement via self-assembling nano systems is one of the promising approaches to improve the efficacy of poorly soluble antibiotics.10Numerous techniques have been reported in the literature for solubilization of poorly water soluble antibiotics which includes (i) physical modification by particle size reduction11 (ii) chemical modification such as salt formation,12 and (iii) nanocarrier based strategies including formulation of nanosuspension,13 nanoemulsion,14 polymeric nanocarriers,15 micellization,16 nanoplexes17 and use of dendrimers.18
Fusidic acid (FSD) a steroidal antibiotic, isolated from fungus Fusidium coccineum. It is primarily active against Gram-positive bacteria such as Staphylococcus, Streptococcus, and Corynebacterium species.12 FSD has also been reported as an effective antibiotic against methicillin resistant Staphylococcus aureus (MRSA),19–21 a bacteria resistant to many antibiotics. However, its applicability is limited by the fact that it is sparingly soluble in water and tends to partition into oil in in vitro oil/water partition experiments.12 It is therefore prepared as the diethanolamine salt for intravenous administration and as sodium salt for oral administration.22 Different nanocarrier systems such as microemulsion,23 liposomes,24,25 polymeric micropheres26 and polymeric nanofibers27 were reported for the delivery of FSD alone or in combination with other antibiotics. However, all these formulations need tedious preparation procedures and use of organic solvents. On the contrary, amphiphilic dendrimers (ADs) can easily self-assemble into ultra-small micelles by simple mixing with drug without the use of any organic solvent28 thus leading to easy and green formulations. The ultra-small size features of these micelles will further provide a larger surface area to interact with the bacterial cell membrane thereby possibly, improving the antibacterial potency of an antibiotic. Thus, strategies to enhance solubility of poorly soluble antibiotics using ADs would be beneficial to improve their pharmacokinetic properties and subsequently antibacterial activity to control emergence of new resistant bacteria.
The concept of nano drug delivery is of great interest as it provides improved pharmacokinetic properties such as enhanced solubility, increased half-life and improved bioavailability to existing drugs.29 The use of non-ionic amphiphilic polymeric materials to form self-assembled nanostructures have gained importance in the field of nanomedicine and nanobiotechnology due to the formation of various morphological structures including micelles, vesicles and complex hierarchical structures such as fibers, ribbons and helices.30 Various poly(ethylene glycol) based amphiphiles for example Cremophore, Pluronics, and Tween have been extensively used as solubilizing agents for hydrophobic drugs.31 However, it is reported that nonionic surfactants leads to the formation of unstable, diverse and ill-defined micellar structure with high polydispersity properties.32 Therefore, there is a need to develop nonionic amphiphiles which can generate structurally uniform micelles that can act as stable nanocarriers for solubility enhancement of hydrophobic drugs. Recently much attention has been focused on a novel class of amphiphilic systems known as amphiphilic dendrimers (ADs). Multiple functional groups at hydrophilic end and single aliphatic hydrocarbon chain on hydrophobic side makes these structures align at the interface of conventional surfactants and amphiphilic polymers. Owing to their unique structural arrangement, the AD micelles can harbor a large void space within inner cores for high drug loading.33 The field of ADs is comparatively new and there are very few reports on their applicability as nanocarriers for hydrophobic drug molecules,28 carriers for gene therapy,34 photo-responsive delivery,35 and solubility enhancement.36 The limited number of biocompatible ADs for pharmaceutical and biomedical applications necessitates the search for new AD scaffolds. ADs have only been explored as materials for micellar delivery and solubility enhancement despite of their other potential applications for various purposes in pharmaceutical and biomedical sciences. To the best of our knowledge, there are no literature reports on use of ADs as steric stabilizers for colloidal drug delivery systems (DDS). Therefore, investigating the applicability of these ADs as stearic stabilizers will provide new insights that will assist in the development of ADs as colloidal drug delivery excipients. Therefore, the synthesis of new ADs will widen the pool of available structures for application as new pharmaceutical excipients with enhanced performance.
The study in this paper reports the synthesis of novel biocompatible G2 and G3 polyester ADs and their application as solubility enhancers for poorly soluble antibiotics and as a stearic stabilizer for colloidal DDS. Two different series of ADs from glycerol monostearate (GMS) and glycerol monooleate (GMOA) were synthesized. The hydrophobic tail in GMS and GMOA series consisted of saturated (stearic acid) and unsaturated (oleic acid) fatty acid respectively. Whilst the hydrophilic head was made up of 2,2-bis(hydroxymethyl)propionic acid (Bis-MPA) based polyester dendrons in both types. Bis-MPA was selected as the hydrophilic scaffold because of their characteristic features such as (i) in vitro and in vivo biocompatibility, (ii) solubility in biological media, (iii) biodegradability by enzymatic and non-enzymatic hydrolysis and (iv) ease of functionalization.37 The other structural components, stearic acid and oleic acid, are dietary fatty acids and are generally regarded as safe materials38 for pharmaceutical and cosmetic preparations.39 The aim of this study was therefore to synthesize and characterize novel biocompatible GMS and GMOA derived ADs for pharmaceutical applications by evaluating their ability to (i) enhance solubility and antibacterial activity of FSD (ii) form self-assembled nanoaggregates and (iii) act as stearic stabilizers for vancomycin, a glycopeptide antibiotic, loaded solid lipid nanoparticles (SLNs). The originality and novelty of this study can be specified as (i) most of the literature on ADs reports on synthesis of ADs with polyglycerol dendrons and single or double saturated C11, C16 and C18 aliphatic chains with aromatic linkers. ADs in this study are synthesized using simple esterification reaction between polyester dendrons and C18 saturated/unsaturated aliphatic chains, (ii) unlike other reports in the literature, we have demonstrated the biosafety of the synthesized ADs using three different cell lines, (iii) besides their use as solubility enhancer and micellar carrier, we have extended their applicability by proving the concept that ADs could be used as promising stearic stabilizers for colloidal drug delivery systems by preparing stable SLNs.
In terms of significance and impact, the findings presented in this study can be applied to facilitate the design of new biocompatible ADs for multiple biological applications. This study will also serve as a directive for the design and synthesis of novel ADs as substitutes for conventional non-ionic surfactant and gemini surfactants for application in various fields such as catalysis,40 pharmaceuticals,41 environmental sciences,42 detergents, cosmetics, textile and dyeing.43
2. Material and methods
2.1. Materials
GMS, purified powder was purchased from Alfa-Aesar (Germany). Dowex®50WX2 hydrogen form (50–100 mesh), Bis-MPA, 4-(dimethylamino)pyridine (DMAP) and oleic acid (99%) were purchased from Sigma-Aldrich (USA). The 2,2-dimethoxypropane was obtained from Sigma-Aldrich (Germany), DL-1,2-isopropylideneglycerol from Sigma-Aldrich (Spain), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide·HCl (EDC) from Carbosynth (UK), p-toluenesulfonic acid monohydrate (PTSA) from Sigma-Aldrich (France) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Merck Chemicals (Germany). Pluronic F-68 (PF-68) from Sigma-Aldrich (USA) and fusidic acid was purchased from Aopharm Co. (China). Compritol 888 ATO was obtained as a generous gift from Gattefossé (France) and vancomycin (VCM) was purchased from Sinobright Import and Export Co. Ltd. (China). For thin layer chromatography (TLC), Merck precoated silica gel 60F254 plates (Germany) were used. Purified water used during the study was obtained with a Milli-Q purification system (Millipore corp., USA) in the laboratory. Nutrient Broth, Mueller–Hinton Broth (MHB) and Mueller–Hinton Agar (MHA) were obtained from Biolab (South Africa). The bacterial cultures used Staphylococcus aureus (S. aureus) ATCC 25923 and MRSA (Rosenbach ATCC BAA 1683). Cell lines A549 (ATCC® CCL-185™), Hep G2 (ATCC® HB-8065™), MCF7 (ATCC® HTB-22™) were originally purchased from ATCC and donated to us as generous gift by Dr S Naidoo (Department of Therapeutics and Medicinal Management, University of KwaZulu-Natal). All other reagents and chemicals were of analytical grade purchased from Merck Chemicals (Germany).2.2. Synthesis of GMS and GMOA ADs
G1, G2 and G3 GMS/GMOA ADs were synthesized from GMS and GMOA through iterative generation growth by acetonide protected Bis-MPA and subsequent deprotection by Dowex® acidic resin as depicted in Schemes 1 and 2.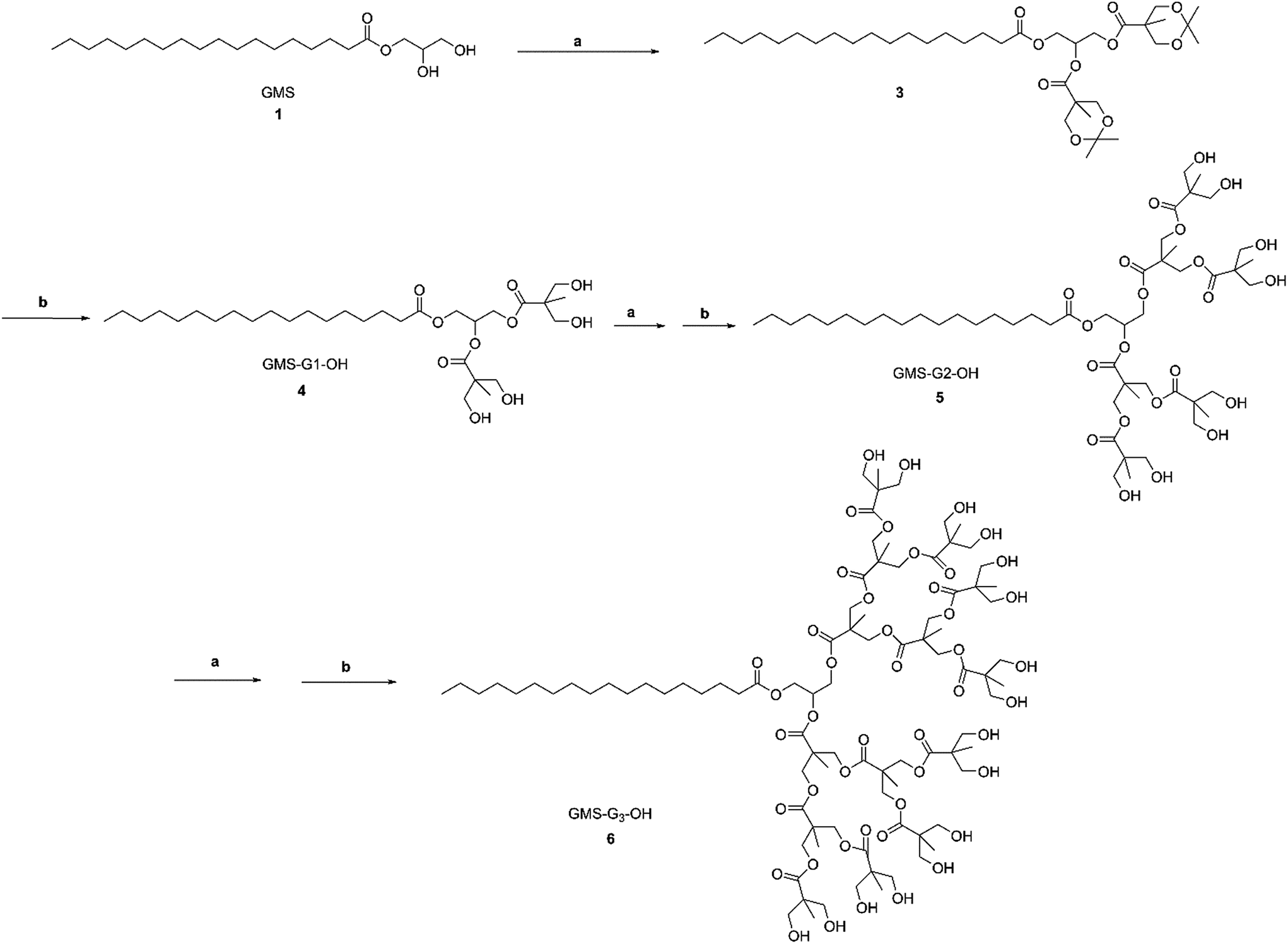 | ||
| Scheme 1 Synthesis of GMS-G1, G2 and G3 ADs. Reagents and conditions: (a) Bis-MPA 2, DMAP, EDC, MDC, room temperature, 12 h; (b) Dowex® H+ resin (10% w/w), MeOH, room temperature, 20 min–3 h. | ||
 | ||
| Scheme 2 Synthesis of GMOA ADs. Reagents and conditions: (a) DMAP, EDC, MDC, room temperature, 12 h; (b) Bis-MPA 2, DMAP, EDC, MDC, room temperature, 12 h; (c) Dowex® H+ resin (10% w/w), MeOH, room temperature, 20 min–3 h. | ||
2.2.1.1. General procedure for esterification (I). Synthesis of acetonide protected Bis-MPA was undertaken using a previously reported procedure.44 The esterification reaction between acetonide protected Bis-MPA and terminal hydroxyl group was performed using Steglich esterification.45 The hydroxyl terminated dendrimer, acetonide protected Bis-MPA and DMAP were dissolved in dried methylene dichloride (MDC) and stirred under nitrogen for 30 minutes. EDC was then added and the reaction mixture was further stirred under nitrogen and at room temperature for 12 h. The reaction was monitored by TLC, and the crude product obtained was purified by column chromatography using silica gel #60-200.
2.2.1.2. General procedure for acetonide deprotection (II). Acidic Dowex® H+ resin (10% w/w) was added to a methanolic solution of acetonide protected dendrimer and stirred at room temperature until completion of the reaction. The resin was removed by filtration and MeOH was evaporated under reduced pressure to obtain final product.
2.2.1.3. GMS-G1-OH 4. GMS 1 (8.00 g, 0.022 mol), Bis-MPA 2 (9.71 g, 0.056 mol), DMAP (1.36 g, 0.011 mol) were dissolved in dry MDC (40 ml) and EDC (10.69 g, 0.056 mol) was added to the reaction mixture under stirring. The reaction was further continued according to the general procedure I, and the crude product was purified by column chromatography (hexane/ethyl acetate, 80
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20) to obtain GMS-G1-Me 3 as a colorless viscous oil (13.88 g, 92.78%). GMS-G1-Me, 3 (13.00 g, 0.019 mol) was dissolved in MeOH (80 ml) and subjected to acetonide deprotection following the general procedure II to obtain GMS-G1-OH 4 as a white solid (11.05 g, 96.50%).
:20) to obtain GMS-G1-Me 3 as a colorless viscous oil (13.88 g, 92.78%). GMS-G1-Me, 3 (13.00 g, 0.019 mol) was dissolved in MeOH (80 ml) and subjected to acetonide deprotection following the general procedure II to obtain GMS-G1-OH 4 as a white solid (11.05 g, 96.50%).
2.2.1.4. GMS-G2-OH 5. Compound 4 (11.00 g, 0.019 mol), 2 (17.84 g, 0.102 mol), DMAP (1.14 g, 0.009 mol) and EDC (19.63 g, 0.102 mol) were reacted in dry MDC (50 ml) as per the general esterification procedure I to obtain GMS-G2-Me as an intermediate product after column chromatographic purification (hexane/ethyl acetate, 60
:40) (20.08 g, 88.73%). GMS-G2-Me (20.00 g, 0.016 mol) was then deprotected as per the general procedure II using MeOH (100 ml) to give GMS-G2-OH 5 as a white solid (17.00 g, 97.92%).
2.2.1.5. GMS-G3-OH 6. To a solution of compound 5 (6.00 g, 0.006 mol), 2 (15.85 g, 0.09 mol) and DMAP (1.04 g, 0.09 mol) in dry MDC (40 ml), EDC (17.44 g, 0.09 mol) was added and the reaction was continued as per the general procedure I. After completion of the reaction, the crude product was purified by column chromatography (hexane/ethyl acetate, 50
:50) to get GMS-G3-Me as a colorless oil (11.20 g, 85.5%). GMS-G3-Me (11.00 g, 0.004 mol) was dissolved in MeOH (80 ml) and deprotected as per the general procedure II to give compound 6 as a white solid (8.30 g, 87.7%).
2.2.2.1. GMOA (10). EDC (11.19 g, 0.06 mol) was added to a solution of oleic acid (OA) 7 (15.00 g, 0.05 mol), DL-1,2-isopropylideneglycerol 8 (7.70 g, 0.06 mol) and DMAP (6.48 g, 0.05 mol) in dry MDC (50 ml) and the reaction was continued following the general procedure I. Intermediate crude product, GMOA-Me 9, was purified by chromatography (hexane/ethyl acetate, 80
:20) (20.80 g, 98.85%). GMOA-Me 9 (20.50 g, 0.05 mol) was dissolved in MeOH (250 ml) and deprotected as per the general procedure II to obtain GMOA 10, a colorless oil (18.00 g, 96.2%).
2.2.2.2. GMOA-G1-OH 12. Compound 9 (10.00 g, 0.028 mol), 2 (12.22 g, 0.07 mol), DMAP (1.71 g, 0.014 mol) were dissolved in dry MDC (50 ml) and EDC (13.45 g, 0.07 mol) was added. The reaction mixture was stirred and continued as described in the general procedure I. Crude product was purified by column chromatography (hexane/ethyl acetate, 80
:20) to give GMOA-G1-Me 11 as colorless oil (17.55 g, 93.55%). Compound 11 (17.00 g, 0.025) was deprotected following the general procedure II using MeOH (100 ml) as a reaction medium to obtain 12 as white sticky solid (14.00 g, 93.58%).
2.2.2.3. GMOA-G2-OH 13. To the solution of compound 11 (13.00 g, 0.022 mol), 2 (19.23 g, 0.11 mol) and DMAP (2.70 g, 0.022 mol) in dry MDC (80 ml), EDC (21.16 g, 0.110 mol) was added and the reaction proceeded as described in the general procedure I. Crude intermediate product (GMOA-G2-Me) was purified with column chromatography (hexane/ethyl acetate, 60
:40) to obtain GMOA-G2-Me as a thick oil (25.90 g, 96.68%). GMOA-G2-Me (25.00 g, 0.020 mol) was subjected to deprotection in MeOH (250 ml) according to the general procedure II to obtain GMOA-G2-OH 13 as a white solid (20.00 g, 92.25%).
2.2.2.4. GMOA-G3-OH 14. GMOA-G2-OH, 12 (10.00 g, 0.01 mol), 2 (26.46 g, 0.152 mol), EDC (29.12 g, 0.152 mol) and DMAP (1.16 g, 0.009 mol) were dissolved in dry MDC (80 ml) and reacted as per the general procedure I. GMOA-G3-Me was purified by column chromatography (hexane/ethyl acetate, 50
:50) to oily product (19.30 g, 88.30%). GMOA-G3-Me (19.00 g, 0.008 mol) was dissolved in MeOH (200 ml) and deprotected as per the general procedure II to obtain GMOA-G3-OH 14 as a white solid (13.00 g, 79.51%).
2.3. Structural characterization
FT-IR spectra of all the compounds were recorded on a Bruker Alpha-p spectrometer with diamond ATR (Germany) as per standard protocols. 1H NMR and 13C NMR measurements were performed using a Bruker 400/600 Ultrashield™ (United Kingdom) NMR spectrometer. HRMS was performed on a Waters Micromass LCT Premier TOF-MS (United Kingdom).2.4. In vitro cytotoxicity
MCF 7, Hep G2 and A549 human carcinoma cells were selected to investigate the in vitro cytotoxicity using MTT assay.46 The details of the MTT assay are provided in the ESI.†2.5. Determination of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) Poctanol/water and HLB values
Poctanol/water and HLB values
The logPoctanol/water value of all ADs were calculated using ACD/ChemSketch version 2016, Advanced Chemistry Development, Inc., Toronto, ON (Canada), whereas HLB values were calculated by method described by Davies and Rideal47 using the following formula:| HLB = Ʃ (hydrophilic group numbers) − Ʃ (lipophilic group numbers) + 7 |
2.6. Determination of critical aggregation concentration (CAC)
The CAC of samples were determined by dynamic light scattering (DLS),46 using a zeta sizer nano ZS (Malvern, UK) equipped with 4 mm helium–neon laser operating at wavelength of 633 nm at an angle of 173° for AD concentration ranging from 1 × 10−6 mol l−1 to 1 × 10−3 mol l−1 prepared from aqueous stock solution (1% w/v) for each derivative. Intensity measurements in kilo cycles per second (kcps) were performed in polystyrene cuvettes at 25 °C. A graph of kcps versus concentration was plotted and an intersection point of two straight lines drawn was recorded as CAC.2.7. Self-assembly and characterization
Self-assembly of GMS-G2-OH, GMS-G3-OH, GMOA-G2-OH, GMOA-G3-OH and PF-68 was studied by dissolving the respective amphiphiles in Milli-Q water at a concentration of 1% w/v. The formulated micelles were characterized in terms of size, poly dispersity index (PDI) and zeta potential (ZP) by DLS (Nano ZS, Malvern, UK).2.8. Solubility studies
Solubilization of FSD in GMS-G2-OH, GMS-G3-OH, GMOA-G2-OH and GMOA-G3-OH ADs and PF-68, a commercialized solubilizing agents, was studied by a simple shaking method.36 Excess (ca. 10 mg) of FSD was added in screw-top glass vials containing 2 ml of amphiphiles dispersion (0.2 and 1% w/w) in Milli-Q water. The obtained suspensions were incubated at 37 °C in a shaking incubator (100 rpm) for 24 h. After incubation, undissolved FSD was removed by filtering through syringe filters (cellulose acetate membrane, 0.2 μm, GVS filter technology, USA). FSD content was determined by a reported High Performance Liquid Chromatography (HPLC) method48 using an Agilent HPLC (Shimadzu Prominence DGU-20A3) (HPLC procedure details are given in ESI†).2.9. In vitro antibacterial activity
In vitro antibacterial study was performed by using broth dilution method against S. aureus and MRSA. Both the bacterial cultures were grown in Mueller–Hinton Broth (MHB) and further diluted to 5 × 105 colony forming units per ml (CFU ml−1). To determine the minimum inhibitory concentration (MIC) values for FSD in the presence and absence of GMOA-G2-OH (test substances) against both cultures, serial dilutions of drug and micelles were prepared in MHB broth and incubated with bacterial cultures for 18 h in a shaking incubator at 37 °C and 100 rpm. Dilutions (10 μl) were spotted on Mueller–Hinton Agar (MHA) plates and incubated further for 18 h. The minimum concentration at which no visible bacterial growth was observed was considered as the MIC.2.10. Bacterial growth kinetics
To perform bacterial growth kinetics study, 50 ml of bacterial suspension of S. aureus and MRSA (5 × 105 CFU ml−1) in Luriya–Bertani (LB) media was incubated with bare FSD and FSD with GMOA-G2-OH at MIC in sterilized bottles at 37 °C. Plain bacterial cultures were incubated as the untreated control, 3 ml of samples were withdrawn from the bottles at different time points (0, 2, 4, 6, 8, 12, 18 and 24 h) and absorbance was recorded at 600 nm by UV spectrophotometer (Shimadzu UV 1601, Japan).2.11. Formulation of SLNs using ADs as stearic stabilizer
Application of synthesized ADs as stearic stabilizer was evaluated by formulating SLNs using VCM as model drug, Compritol as lipid and ADs as surfactants. SLNs were prepared by reported ultrasound dispersion technique49 with slight modifications. Briefly, Compritol 888 ATO (0.3%) and VCM (0.1%) were heated in water bath at 80 °C and surfactant solution was prepared by adding ADs (0.3%) in 15 ml of Milli-Q water and heated at the same temperature as that of lipid phase. Hot surfactant solution was added to the melted lipid drug mixture and homogenized for 5 min at 6000 rpm with an Ultra Turrax T-25 homogenizer (IKA Labortechnik, Germany). The resultant emulsion was immediately subjected to high intensity probe sonication at 30% amplitude for 5 min under same temperature and then cooled to 20 °C to obtain SLNs. Volume of formulation was adjusted to 10 ml if necessary. Blank formulations were prepared using similar method without VCM. For comparison purpose, SLNs were also prepared using PF-68 as a standard non-ionic surfactant.2.12. Characterization of SLNs
The particle size, PDI and ZP were measured by DLS technique using zeta sizer (Nano ZS, Malvern, UK). All measurements were performed in triplicate by diluting 300 μl of SLNs dispersion in to 10 ml of Milli-Q water. The percentage entrapment efficiency (%EE) was determined by a previously reported method50 using following formula.| %EE = (weight of VCM in SLN/weight of VCM added) × 100% |
The morphology of formulated SLNs was determined by using scanning electron microscopy (SEM). Briefly, samples were prepared by placing a few drops of appropriately diluted SLNs on glass coverslip placed on carbon tape. The SLNs were allowed to air dry, sputter coated with gold and images were captured by field-emission gun SEM (ZEISS FEGSEM Ultra Plus, Germany) at an accelerated voltage of 10 kV.
2.13. Statistical analysis
GraphPad Prism® 5 (GraphPad Software Inc., USA) was used to statistically analyze the data. The data is representative of at least three replicates and reported as mean ± standard deviation (SD). The results were analyzed using one-way ANOVA and unpaired t-test followed by Welch's correction and the difference was considered significant when p < 0.05.3. Results and discussion
3.1. Characterization
The synthetic methods involved simple protection and deprotection sequences, where the hydrophilic dendron of ADs were grown divergently with acetonide protected Bis-MPA. Activation of the focal point was achieved through deprotection of the acetonide group by using acidic Dowex® H+ resin. Synthesis of these amphiphiles is simple and cost effective as overall yield of final compounds ranged from 52.13% to 77.79% even after multiple synthetic steps and therefore can be easily manufactured on a commercial scale.As the synthesis of these ADs involved formation of esters and aliphatic alcohols, it was easy to monitor the progress of the reaction using TLC, FT-IR and NMR. The formation of an ester bond between acetonide protected Bis-MPA acid and hydroxyl terminated dendron was confirmed by disappearance of the peak at 3300 cm−1 of –OH and appearance of the characteristic ester peak at 1730 cm−1. Deprotection of acetonide group of Bis-MPA could be identified by disappearance of acetonide (C–O stretch) peak at 1077 cm−1 and appearance of peaks at 1036 cm−1 (O–H bending) and 3326 cm−1 (O–H stretching) of the free hydroxyl group (Fig. S2 of ESI†). Deprotection of acetonide could also be confirmed by 1H NMR spectroscopy as shown in Fig. 1. The two peaks at 1.37 and 1.41 δ ppm corresponding to terminal CH3 of acetonide disappeared after deprotection. To test the chemical identity of ADs, elemental combustion analysis was performed on a ThermoScientific Flash2000 Elemental Analyser. For GMS-G3-OH the observed C/H values were consistent with the theoretical values. However, for GMOA-G2-OH, GMOA-G3-OH and GMS-G2-OH the observed %C value were lower than theoretical values (Table S1 and Fig. S3–S6 of ESI†). This could be due to the incomplete adsorption and combustion of gases and moisture of the samples.51 It has been reported in the literature that elemental analysis for polymers could give wrong results such as low carbon content.51,52 Further, High Resolution Mass spectrometry (HRMS) analysis was performed on a Waters Micromass LCT Premier TOF-MS (United Kingdom). The accurate mass recorded for all ADs matched with the theoretical mass which gave confirmation of their structural purity (Fig. S23, S31, S51 and S59 of ESI†).
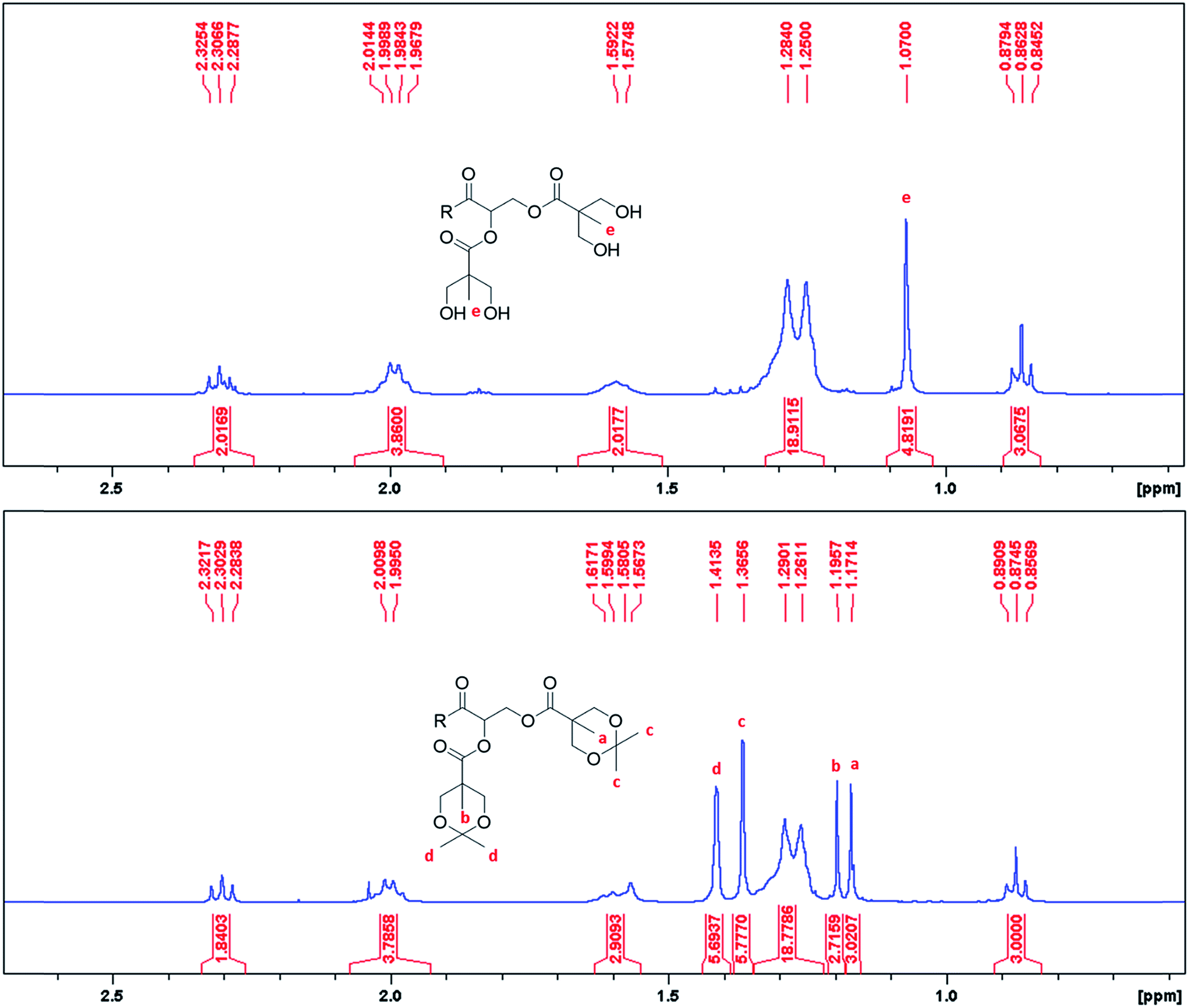 | ||
| Fig. 1 1H NMR data confirming deprotection of acetonide group of GMOA-G1-Me to GMOA-G1-OH. | ||
3.2. In vitro cytotoxicity
The MTT results indicated a high percentage of cell viability for all derivatives tested (>78%) across the concentration range (Fig. 2). No concentration dependent trend was observed across all cell lines studied. The results obtained in this study are in line with previous studies which report on materials that exhibit a toxicity profile that are independent of the concentration.53,54 The test material displaying percentage cell viability >70% can be considered as biologically safe and nontoxic to mammalian cells.55 The biosafety of all ADs can be attributed to selection of non-toxic and biodegradable building blocks used for their synthesis. | ||
| Fig. 2 Results of MTT assay showing % cell viability vs. concentration of GMOA (A, B, C) and (D, E, F) ADs against MCF 7, A549 and Hep G2 cell lines respectively. | ||
3.3. Determination of logPoctanol/water and HLB values
Calculated logPoctanol/water and HLB values for all GMS and GMOA ADs are presented in Table 1. Calculations showed that logPoctanol/water values decreased with increasing generation of polyester dendrimeric head group of ADs making G2 and G3 derivatives more hydrophilic than G1. GMOA ADs exhibited lower logPoctanol/water values than their respective GMS counter parts. The order of logPoctanol/water was GMOA-G3-OH < GMS-G3-OH < GMOA-G2-OH < GMS-G2-OH < GMOA-G1-OH < GMS-G1-OH. HLB calculations showed that the values increased from 8.5 to 34.9 for G1 to G3 respectively with an increase in hydrophilicity. G2 and G3 ADs showed HLB values more than 15 indicating they can used as solubilizing agents.49
P and HLB values for synthesized ADs
| ADs | LogPoctanol/water |
HLB |
|---|---|---|
| GMS-G1-OH | 5.42 ± 0.71 | 8.5 |
| GMOA-G1-OH | 4.90 ± 0.71 | 8.5 |
| GMS-G2-OH | 4.19 ± 0.93 | 15.7 |
| GMOA-G2-OH | 3.67 ± 0.93 | 15.7 |
| GMS-G3-OH | 0.99 ± 1.07 | 34.9 |
| GMOA-G3-OH | 0.47 ± 1.07 | 34.9 |
3.4. Determination of CAC
Avoidance of the influence of an external probe makes non-invasive methods as the most advantageous methods for determination of CAC.46,56 DLS, one of the non-invasive techniques, was successfully used for the determination CAC of GMS-G2-OH, GMS-G3-OH, GMOA-G2-OH and GMOA-G3-OH ADs. DLS measures time-dependent change in intensity of scattered light from particles in the Brownian motion within the colloidal system.46,57 Intensity of scattered light for concentration below CAC is low and similar to that of Milli-Q water. Once concentration reaches the CAC, amphiphiles self-assemble to form micelles which results in an increased intensity of scattered light.46,57 First generation derivatives (GMS-G1-OH and GMOA-G1-OH) were very slightly soluble, and were therefore not used in the study. In the case of G2 and G3 ADs, there was a decrease in CAC value with an increase in the generation. This decrease in CAC could be attributed to an increase in the number of hydrophilic polyester functions with an increase in the generation. The CAC order was GMS-G3-OH (5 × 10−6 mol l−1) < GMOA-G3-OH (7 × 10−6 mol l−1) < GMOA-G2-OH = GMS-G2-OH (5 × 10−5 mol l−1) (Table 2). The results obtained are in line with previous findings where a similar trend of a decrease in CAC with an increase in generation number was observed for polyglycerol dendrimers.28 When comparing GMS and GMOA ADs, dendrimers of same generation had no significant difference in their CAC values. This suggests that unsaturation in the aliphatic chain had no effect on CAC value.| ADs | CAC (mol l−1) | Size (nm) | PDI | ZP (mV) |
|---|---|---|---|---|
| GMS-G2-OH | 5 × 10−5 | 7.60 ± 0.13 | 0.263 ± 0.053 | −2.61 ± 0.121 |
| GMS-G3-OH | 5 × 10−6 | 6.48 ± 0.04 | 0.620 ± 0.060 | −0.011 ± 0.053 |
| GMOA-G2-OH | 5 × 10−5 | 8.62 ± 0.48 | 0.272 ± 0.028 | −5.51 ± 0.760 |
| GMOA-G3-OH | 7 × 10−6 | 12.38 ± 0.36 | 0.339 ± 0.019 | −7.89 ± 0.763 |
3.5. Self-assembly and characterization
Aggregation behavior of all synthesized ADs was evaluated by dissolving 1% w/w in Milli-Q water (concentration well above CAC). DLS study results showed the existence of smaller aggregates for G2 and G3 derivatives of GMS and GMOA with average size in the range of 6.48 to 12.38 nm and narrow PDI (Table 2). In the case of GMOA ADs, the G2 derivative formed significantly smaller aggregates (p = 0.0016) than G3 with low PDI, whilst with GMS ADs, no significant (p = 0.0835) difference was observed for average size in between G2 and G3 derivative. Amongst all ADs, GMOA derivatives formed larger aggregates compared to GMS. The average particle size for GMOA G2 and GMOA G3 was 8.62 ± 0.48 nm and 12.38 ± 0.36 nm respectively whereas for GMS G2 and G3 the size was 7.6 ± 0.13 nm and 6.48 ± 0.04 nm respectively. ZP for G2 and G3 GMS and GMOA ADs were −2.61, −0.011, −5.51 and −7.87 respectively. Furthermore, stability of all micelles were carried out for 30 days at 4 °C by measuring particle size, PDI and ZP. There was a significant (p < 0.05) change observed in PDI and ZP of GMS ADs whereas no significant difference was observed in these values in the case of GMOA ADs after 1 month storage (Table 3). These results indicated that, though particle sizes for GMOA G2 and G3 ADs were high, they were more stable as compared to their respective GMS derivatives after 30 days.| ADs | Size (nm) | PDI | ZP (mV) |
|---|---|---|---|
| GMS-G2-OH | 8.93 ± 0.08 | 0.400 ± 0.011 | −1.43 ± 0.483 |
| GMS-G3-OH | 12.45 ± 0.86 | 0.685 ± 0.018 | −0.000 ± 0.005 |
| GMOA-G2-OH | 8.16 ± 0.61 | 0.291 ± 0.011 | −6.62 ± 0.868 |
| GMOA-G3-OH | 13.32 ± 0.39 | 0.381 ± 0.015 | −6.86 ± 0.337 |
3.6. Solubility studies
Solubilization results of FSD in G2 and G3 ADs and in PF-68 are presented in Fig. 3. The results were expressed as factor of solubilization (F solubilization, where F is x fold increase in solubility of FSD compared to water). PF-68 was selected in the study as a commercial non-ionic surfactant for comparison purposes. FSD showed 0.0003% w/v solubility in water, whilst all ADs at 0.2% w/w concentration showed higher solubilization of FSD [GMS-G2-OH (1.7-fold), GMOA-G2-OH (5.6-fold), GMS-G3-OH (1-fold) and GMOA-G3-OH (2.9-fold) increase in solubility compared to water]. After increasing the amphiphile concentration to 1% w/w, there was an enhancement in the solubility of FSD [GMOA-G2-OH (43-fold), GMOA-G3-OH (11-fold), GMS-G2-OH (9.1-fold) and GMS-G3-OH (6.8-fold)]. However, at both concentrations FSD showed significantly lower solubility (p = 0.0001) in the standard solubilizing excipient PF-68 (0.8-fold and 3.5-fold at 0.2 and 1% respectively). These results confirmed the superiority of the synthesized ADs over the marketed non-ionic surfactant. The results of the solubilization of FSD by PF-68 are in line with previous reports where PF-68 showed low solubilization enhancement compared to non-ionic glycerol based dendritic amphiphiles studied for poorly soluble anticancer drug sagopilone.36 The order of solubilization of FSD in all amphiphiles was GMOA-G2-OH > GMOA-G3-OH > GMS-G2-OH > GMS-G3-OH > PF-68 (Fig. 3). The higher solubility of FSD in GMOA-G2-OH ADs can be attributed to the chain kink due to cis confirmation of unsaturated bond in oleic acid thereby leading to lower curvatures.58 This spatial arrangement could have assisted in accommodating more FSD molecules leading to its higher entrapment.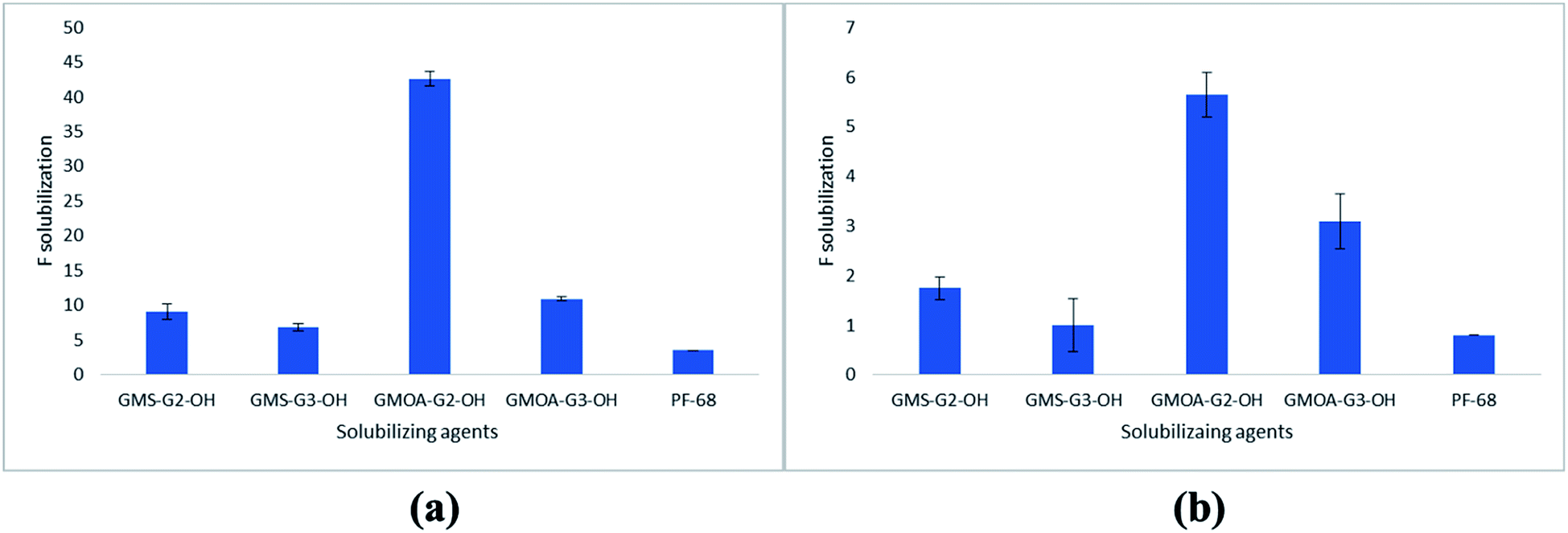 | ||
| Fig. 3 Solubilization of FSD by ADs and PF-68 at 0.2% w/w (A) and at 1% w/w (B). | ||
Effect of FSD on size, PDI and ZP of ADs and PF-68 aggregates were also studied. The size, PDI and ZP values for FSD loaded aggregates are presented in Table S2 of ESI.† The results showed that FSD loading significantly increases size of aggregates except in the case of GMOA-G2-OH where the size slightly decreased from 8.62 ± 0.48 to 7.20 ± 0.09. There was a slight increase in PDI and decrease in ZP values of aggregates after FSD solubilization. In case of PF-68 aggregates, size increased form 28.67 ± 4.87 to 142.6 ± 9.86 nm with a bimodal distribution curve indicating its instability after FSD loading. After noting the differences in ZP values of plain and FSD loaded micelles, we hypothesized that there could be a presence of unencapsulated FSD in filtered solution. The drug's positive or negative charge can affect the zeta potential value.59 To support this statement, we determined the encapsulation efficiency (%EE) and drug loading (%DL) of FSD in all amphiphiles using an ultrafiltration method following a literature reported procedure60 with detailed procedure provided in the ESI.† It was observed that %EE and %DL for FSD was in the range of 4.5 ± 0.9 to 25.76 ± 0.1% and 0.009 ± 0.001 to 0.35 ± 0.001 (Table S3 of ESI†). The unencapsulated percentages (74.24 to 95.5%) of FSD could have been solubilized by one or combination of one or more mechanisms such as (i) reduction in surface tension,1 (ii) wetting and disintegration of solids forming fine particles61 and; (iii) incorporation of FSD onto the micellar surface.62
3.7. In vitro antibacterial activity
As FSD exhibited highest solubilization in GMOA-G2-OH, it was selected for further evaluation to assess the effect of these ADs on antibacterial activity of FSD. GMOA-G2-OH exhibited no activity against S. aureus and MRSA. The minimum inhibitory concentration (MIC) values for FSD with and without GMOA-G2-OH against S. aureus were 0.23 μg ml−1 and 0.53 μg ml−1 respectively whereas the values were 0.23 μg ml−1 and 0.39 μg ml−1 against MRSA respectively (Table 4). These results suggested that GM-OA-G2 not only enhanced the solubility but also enhanced the antibacterial potency of FSD. There was a significant difference between MIC of FSD with GMOA-G2-OH and bare FSD (p < 0.05). This enhanced activity could be ascribed to increased solubility of FSD which enabled its better diffusion. The results were in good agreement with a previous reported finding where self-assembled β-cyclodextrin enhanced the antimicrobial potency of chlorhexidine.63| Formulation | MIC (μg ml−1) | |
|---|---|---|
| S. aureus | MRSA | |
| a NA = no activity, * = results statistically significant (p < 0.05) compared to control (FSD). | ||
| FSD | 0.53 | 0.39 |
| GMOA-G2-OH | NA | NA |
| FSD with GMOA-G2-OH | 0.23* | 0.23* |
3.8. Bacterial growth kinetics
Investigation of bacterial growth kinetics in LB media quantitatively evaluated the antibacterial potency of FSD GMOA-G2-OH micelles against S. aureus and MRSA. Fig. 4 shows the optical density at 600 nm for different incubation times. The control group achieved a cell concentration of 0.120 OD and 0.103 OD for S. aureus and MRSA respectively at 4 h of incubation time. After 18 h of incubation, the control group growth curve in both strains achieved exponential growth with maximum bacterial concentration of 1.388 OD and 1.308 OD. In bare FSD treated samples, bacterial cells started growing at incubation of 12 h with concentration of 0.410 OD and 0.414 OD attending maximum concentration of 1.225 OD and 1.231 OD for S. aureus and MRSA respectively at 24 h of incubation time. It was observed that GMOA-G2-OH treated samples effectively controlled the bacterial growth for both the strains throughout the study period. At the end of 24 h, the OD values for FSD GMOA-G2-OH treated samples were 0.388 and 0.130 against S. aureus and MRSA respectively. These growth curve kinetic studies demonstrated that the growth of both strains was significantly (p < 0.05) inhibited by FSD ADs, thus proving its superiority over bare FSD.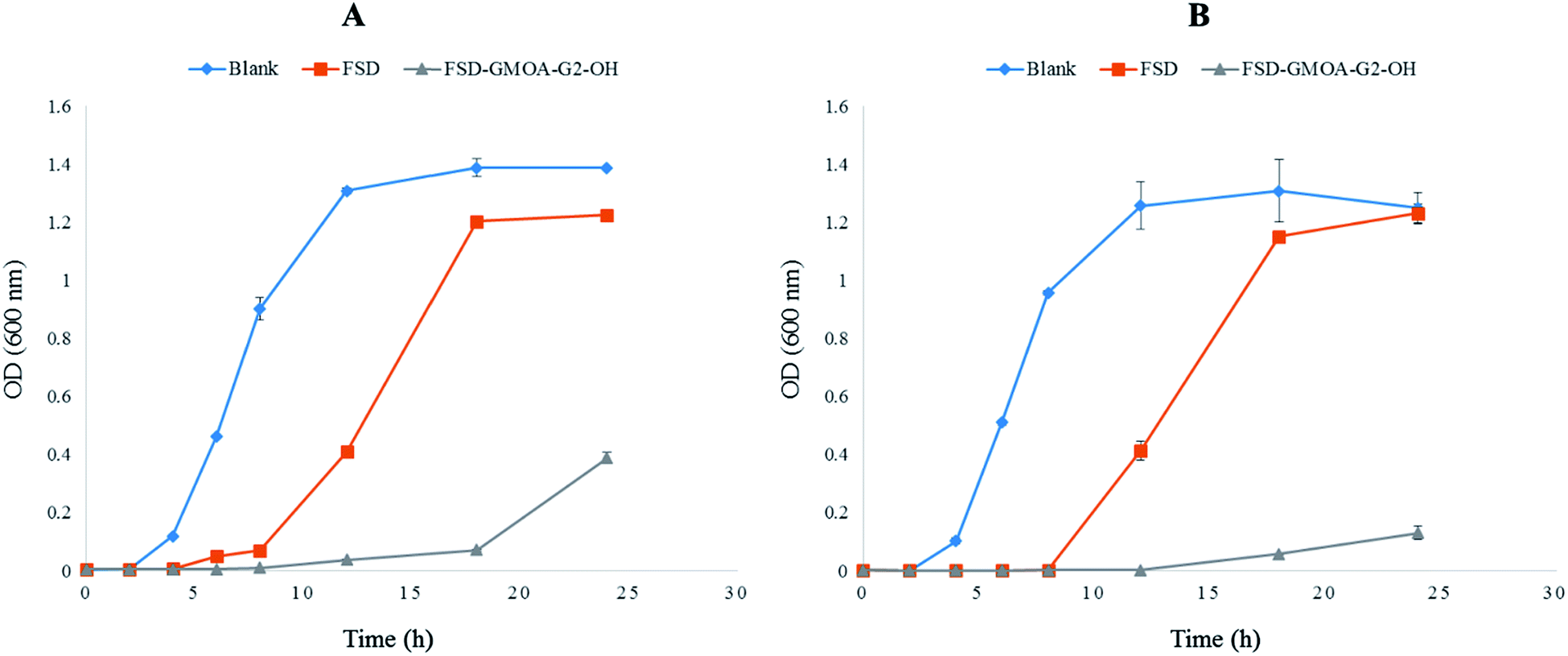 | ||
| Fig. 4 Bacterial growth kinetics curve for (A) S. aureus (B) MRSA (n = 3). | ||
3.9. Formulation and characterization of SLNs
SLNs were successfully formulated by using Compritol 888 ATO as a solid lipid ADs as surfactants using ultrasound dispersion technique. For comparative evaluation PF-68 was chosen as commercial non-ionic surfactant. All results of particle size, PDI and ZP for blank formulation are summarized in Table 5. The PDI values were <0.3 and ZP values were between −15.3 ± 1.44 mV to −38.46 ± 3.04 mV for SLNs prepared using ADs whereas PDI was >0.3 and ZP was 4.16 ± 0.14 mV for SLNs prepared using PF-68 (Table 5). The results showed that SLNs formulated from GMOA ADs were more stable than those prepared using PF-68 though their particle sizes were higher.| Formulation | Size (nm) | PDI | ZP (mV) |
|---|---|---|---|
| DF-GMOA-G2-OH SLNs | 252.56 ± 12.53 | 0.154 ± 0.017 | −26.06 ± 2.04 |
| DF-GMS-G2-OH SLNs | 250.6 ± 5.41 | 0.160 ± 0.011 | −15.3 ± 1.44 |
| DF-GMOA-G3-OH SLNs | 365.53 ± 3.86 | 0.233 ± 0.011 | −38.46 ± 3.04 |
| DF-GMS-G3-OH SLNs | 273.1 ± 2.81 | 0.204 ± 0.029 | −31.23 ± 1.98 |
| DF-PF-68 SLNs | 161.76 ± 3.60 | 0.325 ± 0.035 | 4.16 ± 0.14 |
VCM loading produced SLNs (containing ADs as stabilizers) with size, PDI and ZP values in the range of 133.93 ± 3.44 to 291.17 ± 22.02 nm, 0.296 ± 0.008 to 0.485 ± 0.055 and −22.33 ± 1.02 mV to −34.10 ± 1.83 mV respectively (Table 6). The %EE of VCM in all the formulations was in between 8.34 ± 1.04 to 12.81 ± 1.02%. This low %EE could be ascribed to the hydrophilicity of VCM. The results are consistent with previous findings where low %EE (16.81 ± 3.64) was observed for VCM.50
| Formulation | Size (nm) | PDI | ZP (mV) | %EE |
|---|---|---|---|---|
| DL-GMOA-G2-OH SLNs | 133.93 ± 3.44 | 0.296 ± 0.008 | −25.08 ± 0.65 | 12.81 ± 1.02 |
| DL-GMS-G2-OH SLNs | 192.80 ± 4.44 | 0.338 ± 0.042 | −22.33 ± 1.02 | 10.46 ± 0.57 |
| DL-GMOA-G3-OH SLNs | 291.17 ± 22.02 | 0.379 ± 0.018 | −34.10 ± 1.83 | 10.68 ± 1.72 |
| DL-GMS-G3-OH SLNs | 278.97 ± 8.26 | 0.485 ± 0.055 | −26.13 ± 3.16 | 8.34 ± 1.04 |
Morphological investigation showed that SLNs formulated from ADs were homogenous and spherical in shape (Fig. S3 and S4 of ESI†). Overall results showed that ADs can formulate nano-sized particulate systems with lower PDI and higher ZP making them attractive stearic stabilizers for SLNs and other colloidal drug delivery systems.
4. Conclusion
In this paper, new G2 and G3 ADs were synthesized by conjugation of polyester based dendrons to aliphatic hydrocarbon chains. These ADs which were found to be non-toxic to mammalian cells were further characterized for their CAC to determine their micelle forming ability. All the G2 and G3 ADs exhibited lower CAC values and were able to form self-assembled nano structures as confirmed by DLS. These properties of ADs suggested their potential to be new and attractive biocompatible pharmaceutical excipients. Considering the poor solubility of antibiotics, one of the reasons limiting their effectiveness against bacteria and subsequently resulting in antibiotic resistance, these new ADs were evaluated for their solubility enhancement of FSD, a poorly soluble antibiotic effective against MRSA. The solubility results suggested that all ADs were able to significantly improve solubility of FSD compared to water and the commercial PF-68. In addition, it was observed that during the solubilization process, ADs self-assembled to form nano micelles with % FSD encapsulation in the range of 1.1 to 3%. This confirmed their applicability in the formulation of nano drug delivery systems. Amongst all ADs, FSD exhibited highest solubility in GMOA-G2-OH with formation of nano micelles of 8.62 ± 0.48 nm particle size. In vitro antibacterial activity and cell kill kinetic studies confirmed that ADs enhanced the antibacterial activity of FSD against S. aureus and MRSA. The ADs were further evaluated for their application as stearic stabilizers for SLNs by using them as surfactants. The results from formulation studies proved that ADs are capable of providing sufficient stability to SLNs, as confirmed by higher zeta potential values.The overall results of the study direct that ADs reported in this study could be promising excipients for pharmaceutical applications. The findings reported in this paper will also be a guide towards the use of simple and efficient synthetic methods to obtain biocompatible amphiphilic ADs which can self-assemble to ultra-small aggregates and can acts as stearic stabilizer for colloidal systems for various applications in pharmaceutical and biomedical sciences.
Acknowledgements
The authors acknowledge College of Health Sciences (CHS), University of KwaZulu-Natal (UKZN), UKZN Nanotechnology Platform and the National Research Foundation (NRF) of South Africa for financial support (NRF Grant No. 87790 and 88453).References
- K. T. Savjani, A. K. Gajjar and J. K. Savjani, ISRN Pharm., 2012, 2012, 195727 Search PubMed.
- H. Chen, C. Khemtong, X. Yang, X. Chang and J. Gao, Drug Discovery Today, 2011, 16, 354–360 CrossRef CAS PubMed.
- P. Khadka, J. Ro, H. Kim, I. Kim, J. T. Kim, H. Kim, J. M. Cho, G. Yun and J. Lee, Asian J. Pharm. Sci., 2014, 9, 304–316 CrossRef.
- S. Svenson and A. S. Chauhan, Nanomedicine, 2008, 3, 679–702 CrossRef CAS PubMed.
- S. A. Adediran, T. P. Day, D. Sil, M. R. Kimbrell, H. J. Warshakoon, S. S. Malladi and S. A. David, Mol. Pharm., 2009, 6, 1582–1590 CrossRef CAS PubMed.
- P. Weiss, M. Andrew and W. Wright, Antibiot. Chemother., 1957, 7, 374–377 CAS.
- M. Aucamp, R. Odendaal, W. Liebenberg and J. Hamman, Drug Dev. Ind. Pharm., 2015, 41, 1100–1108 CrossRef CAS PubMed.
- Y. Cheng, H. Qu, M. Ma, Z. Xu, P. Xu, Y. Fang and T. Xu, Eur. J. Med. Chem., 2007, 42, 1032–1038 CrossRef CAS PubMed.
- M. C. Carey, J. C. Montet and D. M. Small, Biochemistry, 1975, 14, 4896–4905 CrossRef CAS PubMed.
- S. Ranghar, P. Sirohi, P. Verma and V. Agarwal, Braz. Arch. Biol. Technol., 2014, 57, 209–222 CrossRef CAS.
- A. Chaudhary, U. Nagaich, N. Gulati, V. Sharma, R. Khosa and M. U. Partapur, J. Adv. Pharm. Educ. Res., 2012, 2, 32–67 Search PubMed.
- J. Turnidge, Int. J. Antimicrob. Agents, 1999, 12, S23–S34 CrossRef CAS PubMed.
- D. Zhang, T. Tan, L. Gao, W. Zhao and P. Wang, Drug Dev. Ind. Pharm., 2007, 33, 569–575 CrossRef CAS PubMed.
- S. Vatsraj, K. Chauhan and H. Pathak, J. Nanosci., 2014, 2014 Search PubMed.
- G. Mohammadi, H. Valizadeh, M. Barzegar-Jalali, F. Lotfipour, K. Adibkia, M. Milani, M. Azhdarzadeh, F. Kiafar and A. Nokhodchi, Colloids Surf., B, 2010, 80, 34–39 CrossRef CAS PubMed.
- P. A. Bhat, A. A. Dar and G. M. Rather, J. Chem. Eng. Data, 2008, 53, 1271–1277 CrossRef CAS.
- W. S. Cheow and K. Hadinoto, J. Colloid Interface Sci., 2012, 367, 518–526 CrossRef CAS PubMed.
- K. Winnicka, M. Wroblewska, P. Wieczorek, P. T. Sacha and E. A. Tryniszewska, Molecules, 2013, 18, 8607–8617 CrossRef CAS PubMed.
- P. Grohs, M.-D. Kitzis and L. Gutmann, Antimicrob. Agents Chemother., 2003, 47, 418–420 CrossRef CAS PubMed.
- R. C. Moellering, G. R. Corey and M. L. Grayson, Clin. Infect. Dis., 2011, 52, S467–S468 CrossRef PubMed.
- M. Whitby, Int. J. Antimicrob. Agents, 1999, 12(2), S67–S71 CrossRef CAS PubMed.
- D. Reeves, J. Antimicrob. Chemother., 1987, 20, 467–476 CrossRef CAS PubMed.
- T. Chhibber, S. Wadhwa, P. Chadha, G. Sharma and O. P. Katare, J. Drug Targeting, 2015, 23, 943–952 CrossRef CAS PubMed.
- D. Nicolosi, S. Cupri, C. Genovese, G. Tempera, R. Mattina and R. Pignatello, Int. J. Antimicrob. Agents, 2015, 45, 622–626 CrossRef CAS PubMed.
- S. Wadhwa, B. Singh, G. Sharma, K. Raza and O. P. Katare, Drug Delivery, 2016, 23, 1204–1213 CrossRef CAS PubMed.
- C. Yang, D. Plackett, D. Needham and H. M. Burt, Pharm. Res., 2009, 26, 1644–1656 CrossRef CAS PubMed.
- S. E. Gilchrist, D. Lange, K. Letchford, H. Bach, L. Fazli and H. M. Burt, J. Controlled Release, 2013, 170, 64–73 CrossRef CAS PubMed.
- B. Trappmann, K. Ludwig, M. R. Radowski, A. Shukla, A. Mohr, H. Rehage, C. Böttcher and R. Haag, J. Am. Chem. Soc., 2010, 132, 11119–11124 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS PubMed.
- A. Sorrenti, O. Illa and R. M. Ortuno, Chem. Soc. Rev., 2013, 42, 8200–8219 RSC.
- E. V. Batrakova and A. V. Kabanov, J. Controlled Release, 2008, 130, 98–106 CrossRef CAS PubMed.
- M. J. Schick, Nonionic surfactants: physical chemistry, Marcel Dekker, New York, 1987 Search PubMed.
- T. Wei, C. Chen, J. Liu, C. Liu, P. Posocco, X. Liu, Q. Cheng, S. Huo, Z. Liang and M. Fermeglia, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2978–2983 CrossRef CAS PubMed.
- S. Malhotra, H. Bauer, A. Tschiche, A. M. Staedtler, A. Mohr, M. Calderón, V. S. Parmar, L. Hoeke, S. Sharbati, R. Einspanier and R. Haag, Biomacromolecules, 2012, 13, 3087–3098 CrossRef CAS PubMed.
- C. Kordel, C. S. Popeney and R. Haag, Chem. Commun., 2011, 47, 6584–6586 RSC.
- A. Richter, A. Wiedekind, M. Krause, T. Kissel, R. Haag and C. Olbrich, Eur. J. Pharm. Sci., 2010, 40, 48–55 CrossRef CAS PubMed.
- A. Carlmark, E. Malmström and M. Malkoch, Chem. Soc. Rev., 2013, 42, 5858–5879 RSC.
- A. C. Rustan and C. A. Drevon, in eLS, John Wiley & Sons, Ltd, 2001, DOI:10.1038/npg.els.0003894.
- F.-Q. Hu, S.-P. Jiang, Y.-Z. Du, H. Yuan, Y.-Q. Ye and S. Zeng, Colloids Surf., B, 2005, 45, 167–173 CrossRef CAS PubMed.
- H. Y. Rhyoo, H.-J. Park, W. H. Suh and Y. K. Chung, Tetrahedron Lett., 2002, 43, 269–272 CrossRef CAS.
- J. Eastoe and R. F. Tabor, in Colloidal Foundations of Nanoscience, ed. G. Palazzo, Elsevier, Amsterdam, 2014, pp. 135–157, DOI:10.1016/B978-0-444-59541-6.00006-0.
- F. Volkering, A. M. Breure and W. H. Rulkens, Biodegradation, 1997, 8, 401–417 CrossRef CAS PubMed.
- N. Kumar and R. Tyagi, J. Dispersion Sci. Technol., 2014, 35, 205–214 CrossRef CAS.
- H. Ihre, A. Hult, J. M. J. Fréchet and I. Gitsov, Macromolecules, 1998, 31, 4061–4068 CrossRef CAS.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed. Engl., 1978, 17, 522–524 CrossRef.
- Surfactant micelle characterization using dynamic light scattering, http://www.malvern.com/en/pdf/secure/AN101104SurfactantMicelleCharacterization.pdf, accessed 23 January 2016.
- J. T. Davies and E. K. Rideal, Interfacial Phenomenon, Academic Press, New York, 1963, pp. 343–450 Search PubMed.
- A. H. Hikal, A. Shibl and S. El-Hoofy, J. Pharm. Sci., 1982, 71, 1297–1298 CrossRef CAS PubMed.
- R. S. Kalhapure and K. G. Akamanchi, Colloids Surf., B, 2013, 105, 215–222 CrossRef CAS PubMed.
- R. S. Kalhapure, C. Mocktar, D. R. Sikwal, S. J. Sonawane, M. K. Kathiravan, A. Skelton and T. Govender, Colloids Surf., B, 2014, 117, 303–311 CrossRef CAS PubMed.
- Y. Yang, Q. Zhang, S. Zhang and S. Li, RSC Adv., 2014, 4, 5568–5574 RSC.
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- S. Di Gioia, C. Sardo, G. Belgiovine, D. Triolo, M. d'Apolito, S. Castellani, A. Carbone, I. Giardino, G. Giammona, G. Cavallaro and M. Conese, Int. J. Pharm., 2015, 491, 359–366 CrossRef PubMed.
- M. D. Romic, M. S. Klaric, J. Lovric, I. Pepic, B. Cetina-Cizmek, J. Filipovic-Grcic and A. Hafner, Eur. J. Pharm. Biopharm., 2016, 107, 67–79 CrossRef CAS PubMed.
- S. Rambharose, R. S. Kalhapure, K. G. Akamanchi and T. Govender, J. Mater. Chem. B, 2015, 3, 6662–6675 RSC.
- U. Anand, C. Jash and S. Mukherjee, J. Colloid Interface Sci., 2011, 364, 400–406 CrossRef CAS PubMed.
- Ö. Topel, B. A. Çakır, L. Budama and N. Hoda, J. Mol. Liq., 2013, 177, 40–43 CrossRef.
- S. M. Sagnella, C. E. Conn, I. Krodkiewska, M. Moghaddam, J. M. Seddon and C. J. Drummond, Langmuir, 2010, 26, 3084–3094 CrossRef CAS PubMed.
- S. Hassani, A. Laouini, H. Fessi and C. Charcosset, Colloids Surf., A, 2015, 482, 34–43 CrossRef CAS.
- L. L. Cai, P. Liu, X. Li, X. Huang, Y. Q. Ye, F. Y. Chen, H. Yuan, F. Q. Hu and Y. Z. Du, Int. J. Nanomed., 2011, 6, 3499–3508 CAS.
- L. Di and E. H. Kerns, Drug-like properties: concepts, structure design and methods from ADME to toxicity optimization, Academic Press, 2015 Search PubMed.
- D. Everett, Pure Appl. Chem., 1972, 31, 577–638 CrossRef.
- Â. M. L. Denadai, K. I. Teixeira, M. M. Santoro, A. M. C. Pimenta, M. E. Cortés and R. D. Sinisterra, Carbohydr. Res., 2007, 342, 2286–2296 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28100a |
| This journal is © The Royal Society of Chemistry 2017 |