
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural stability and thermoelectric property optimization of Ca2Si
Rui Xionga,
Baisheng Sa *ab,
Naihua Miaob,
Yan-Ling Lic,
Jian Zhoub,
Yuanchun Panb,
Cuilian Wen*a,
Bo Wua and
Zhimei Sun*b
*ab,
Naihua Miaob,
Yan-Ling Lic,
Jian Zhoub,
Yuanchun Panb,
Cuilian Wen*a,
Bo Wua and
Zhimei Sun*b
aCollege of Materials Science and Engineering, Fuzhou University, and Key Laboratory of Eco-materials Advanced Technology (Fuzhou University), Fujian Province University, Fuzhou 350100, P. R. China. E-mail: bssa@fzu.edu.cn; clwen@fzu.edu.cn
bSchool of Materials Science and Engineering, and Center for Integrated Computational Materials Engineering, International Research Institute for Multidisciplinary Science, Beihang University, 100191 Beijing, P. R. China. E-mail: zmsun@buaa.edu.cn
cSchool of Physics and Electronic Engineering, Jiangsu Normal University, 221116, Xuzhou, People's Republic of China
First published on 31st January 2017
Abstract
By using an ab initio evolutionary algorithm structure search, low enthalpy criterion as well as stability analysis, we have found that cubic Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m Ca2Si can be achieved under a negative external pressure, and the cubic phase is dynamically and mechanically stable at ambient conditions and high pressure. From first-principle hybrid functional calculations, we have unraveled the direct bandgap nature and bandgap variation of cubic Fmm Ca2Si with respective to pressure. Moreover, by combining with Boltzmann transport theory and the phonon Boltzmann transport equation, we have predicted that the figure of merit ZT for the cubic Fmm Ca2Si reaches the maximum value of 0.52 by p-type doping. Our results provide an interesting insight and feasible guidelines for the potential applications of cubic Fmm Ca2Si and related alkaline-earth metals silicides as the thermoelectric materials for heat-electricity energy convertors.
m Ca2Si can be achieved under a negative external pressure, and the cubic phase is dynamically and mechanically stable at ambient conditions and high pressure. From first-principle hybrid functional calculations, we have unraveled the direct bandgap nature and bandgap variation of cubic Fmm Ca2Si with respective to pressure. Moreover, by combining with Boltzmann transport theory and the phonon Boltzmann transport equation, we have predicted that the figure of merit ZT for the cubic Fmm Ca2Si reaches the maximum value of 0.52 by p-type doping. Our results provide an interesting insight and feasible guidelines for the potential applications of cubic Fmm Ca2Si and related alkaline-earth metals silicides as the thermoelectric materials for heat-electricity energy convertors.
1. Introduction
Thermoelectric materials have received global attention because of their potential application in heat-electricity energy conversion, power generation and cooling thermoelectric devices.1,2 Many specialized applications have been realized in the thermoelectric devices due to high reliability, good scalability and simple device structure. The efficiency of thermoelectric devices can be determined by a dimensionless figure of merit ZT = TS2σ/κ, where T, S, σ and κ (κ = κl + κe) are the absolute temperature, the Seebeck coefficient, electrical conductivity and thermal conductivity (as the sum of the lattice and electronic contributions), respectively. The product of S2σ is the thermoelectric power factor (PF) which should be maximized and the thermal conductivity κ should be minimized to achieve the expected high thermoelectric performance. The typical ways to optimize ZT are alloying,3,4 pressure & strain engineering,5,6 nanostructuring7 and low dimensioning.8,9 Besides, the investigations of novel thermoelectric materials with intrinsic high ZT value are still anticipated.10Since 1950, the Bi2Te3-based chalcogenides have been developed and proposed as the best materials for room-temperature thermoelectric applications.11–13 The α-MgAgSb-based materials with the high ZT ∼ 1 near room-temperature has been recently reported to be the candidates of the next generation thermoelectric energy production materials.14–16 Unfortunately, the high-cost and scarcity of Te and Ag elements hinder the commercial applications of these thermoelectric materials in large scale. Recently, the semiconducting alkaline-earth metals (AEMs) silicides have gained great attention for their potential applications in the thermoelectric power generator utilizing waste heat sources.17,18 Ca2Si is one of the environmental friendly and inexpensive AEMs, which can be synthesized by the heat treatment from the Mg2Si powders under Ca vapor.19,20 It is interesting to see that the Seebeck coefficient of ∼300 μV K−1 with p-type conduction nature of Ca2Si was experimentally measured.21 However, in ambient condition Ca2Si is stabilized in the orthorhombic phase with a space group of Pnma.22 Considering the fact that the high symmetry cubic phase present larger number of equivalent degenerated valleys of the electronic structure than the low symmetry orthorhombic phase, better performance of the thermoelectric properties of cubic Ca2Si is anticipated. And the low symmetry of the orthorhombic phase leads to the anisotropic mechanical and optical properties,23,24 which is not desired associated with the Cannikin law. From the self-consistent scalar-relativistic full potential linearized augmented plane wave (FPLAPW) calculation, cubic Ca2Si shows smaller bandgap value then the orthorhombic phase.25,26 The authors also assumed that suitable cubic substrate like diamond can help the growth of the cubic Ca2Si.25 It is worth noting that as the silicon source and crystal template, Mg2Si is crystallized in the cubic phase with the space group of Fmm, which is isotropic.27,28 But till now, the cubic Ca2Si is not experimentally observed. Hence theoretically, a more complete picture is required to elucidate the existence condition of cubic Ca2Si.
Applying external pressure is an effective way to stabilize the metastable or unstable phases of materials.29–31 In this work, by means of the ab initio evolutionary algorithm calculations, we have revealed the stable conditions for the Ca2Si isomers under a wide (including positive and negative) pressure range. We found that the cubic phase of Ca2Si is a metastable phase under negative pressure and can be stabilized to the ambient condition, such negative-pressure condition could be probably achieved by increasing the synthetic temperature. Based on further density functional theory calculations combined with Boltzmann transport theory, we have systematically analyzed the electronic structure, lattice dynamical properties and the thermoelectric properties for cubic Ca2Si.
2. Computational details
We studied Ca2Si based on density functional theory (DFT) using the Vienna ab initio simulation package32 (VASP) in conjunction with projector augmented wave (PAW) pseudopotentials within the generalized gradient approximations33 (GGA) of Perdew–Burke–Ernzerhof34 (PBE). Structure searches for the Ca2Si isomers under positive and negative pressures were carried out through the ab initio evolutionary algorithm (EA) using the USPEX code.35,36 The valence electron configuration for Ca and Si were 3s23p64s2 and 3s23p2 respectively. The cutoff energy for the plane-wave was set to 600 eV. The first Brillouin zone (1BZ) was sampled with a grid of 11 × 11 × 11 for the cubic phase and equivalent dense for the other phases in the structure optimization and energy calculation processes. The YPHON37 code was applied to obtain the phonon frequencies through the density functional perturbation theory (DFPT) methods38 with 2 × 2 × 2 supercells and 3 × 3 × 3 K-points sampling. The thermodynamic properties were gained by using the quasi-harmonic approximation (QHA) method.39 The relaxation convergence for both ions and electrons were 1 × 10−6 eV. The crystal structures were visualized using the VESTA40 tool. It is well known that PBE calculations normally underestimate the electronic band gap, and the hybrid function with the mixing of the Hartree–Fock and DFT exchange terms is thought to be a practical solution to solve the band gap problem.41,42 Herein, we introduced the Heyd–Scuseria–Ernzerhof (HSE06) hybrid functional43 to precisely evaluate the bandgap of Ca2Si. The thermoelectric properties were computed from the semi-classical Boltzmann transport theory within the constant relaxation time approximation (τ = 10−15 s, which is a normal magnitude for relaxation time in semiconductors44) and a rigid band approach using the BoltzTraP code.45 For the prediction of the thermoelectric properties, denser k-point meshes (twice as large as for the geometry optimization) were adopted for Brillouin zone integration to ensure the convergence and accuracy of eigenvalues. The second harmonic force constants from the Phonopy46 package and the third order anharmonic interatomic force constants created by the ShengBTE47 code with 4 × 4 × 4 supercells and gamma K-point sampling were introduced to evaluate the lattice thermal conductivity.3. Results and discussions
We have found four possible structures for Ca2Si, which belong to the space groups of Pnma (Fig. 1(a)), Fmm (Fig. 1(b)), R3m (Fig. 1(c)) and Pnm (Fig. 1(d)), respectively. The equilibrium lattice parameters and atomic Wyckoff positions for the predicted phases at the ambient condition are listed in Tables 1 and 2. Herein the Pnma phase refers to the orthorhombic phase which has been experimentally synthesized. At the ambient condition, our GGA-PBE relaxed lattice parameters for orthorhombic Pnma Ca2Si are a = 7.605, b = 4.821, c = 9.038, agree well with the experimental results within less than 1% mismatch.48 The Fmm and Pnm are two cubic phases. It is obvious that the Fmm phase is more close-packing than the Pnm phase, indicating that the Fmm phase is more stable. In fact, the Fmm phase shows the same crystal structure as stable Mg2Si. Our calculated lattice constant a is 7.165 Å for the cubic Fmm Ca2Si at the ambient condition, which is in good agreement with the previous theoretical prediction.25 Fig. 1(e) illustrates the enthalpies vs. pressure curves of Ca2Si in difference space groups. As we can see from the figure that Pnma and Fmm phases hold very close enthalpy, which locate at obviously lower level than the R3m and Pnm cases. Hence in the full pressure range, the Pnma and Fmm phases are favorable. Moreover, one can see that the Pnma phase is the most stable structure at the ambient condition and the full positive pressure range. The Fmm phase become the most stable one only under a negative external pressure of −0.82 GPa. To achieve such negative pressure condition, we can experimentally enhance the synthesis temperature. As seen in Fig. 1(f), the equilibrium volume of cubic Fmm Ca2Si gradually increases with the increase of the temperature. The volume of cubic Fmm Ca2Si at −0.82 GPa (the transition pressure) is 94.22 Å3, corresponding to the environmental temperature around 480 K. Hence we assume that the synthetic temperature higher than 480 K is necessary to achieve the cubic Fmm Ca2Si. The insert figures in Fig. 1(e) show the pressure dependence of the lattice constants of Pnma and Fmm Ca2Si, we found that the lattice constants of both the phases decrease gradually with the increase of the external pressure. Considering the fact that the enthalpy difference between the Pnma and Fmm phases is as small as 44 meV per u.c. (unit cell), it is natural to expect that the cubic Fmm phase might be stabilized at the ambient condition as well as under finite positive external pressure.
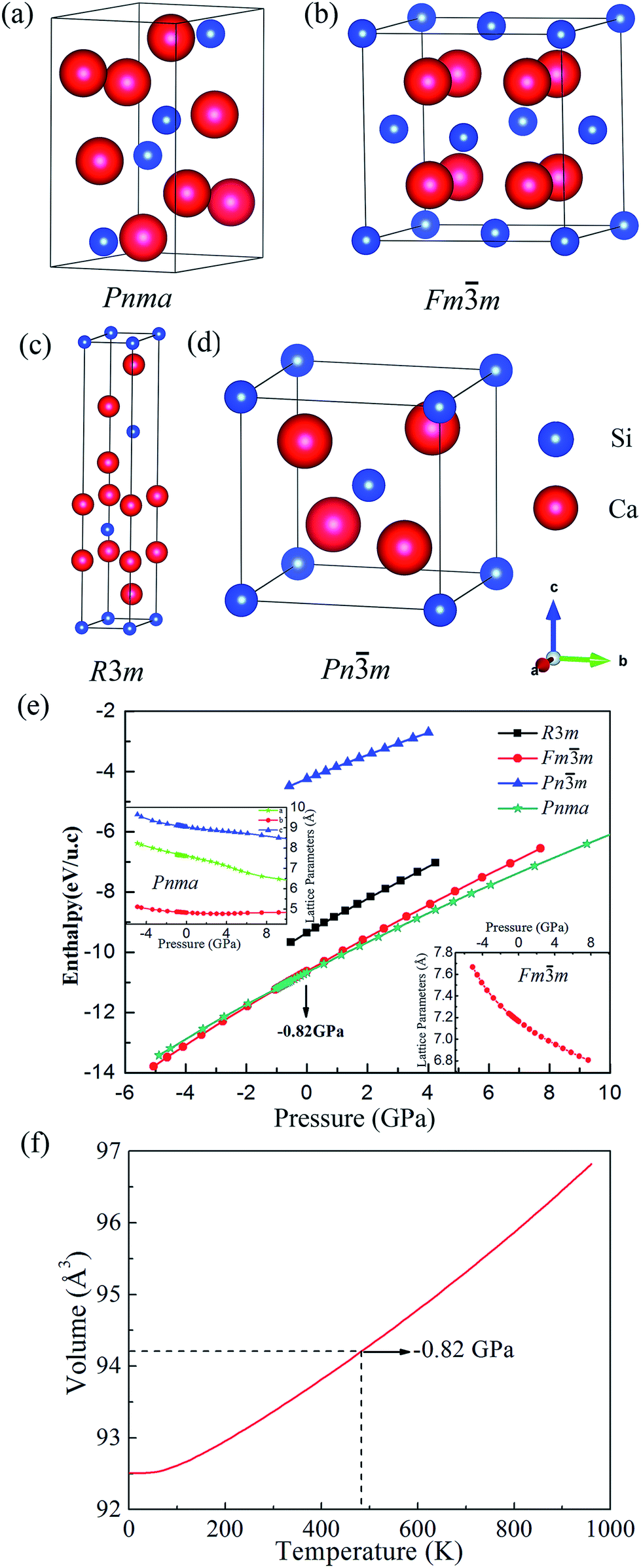 | ||
| Fig. 1 The crystal structures of (a) Pnma, (b) Fmm, (c) R3m and (d) Pnm Ca2Si, respectively. (e) The enthalpy as a function of pressure for the predicted phases of Ca2Si, the insert figures show the pressure dependence of the lattice constants of Pnma and Fmm Ca2Si and (f) the volume as a function of temperature for Fmm Ca2Si. | ||
| Phase | E0 (eV per u.c.) | a0 (Å) | b0 (Å) | c0 (Å) | α (°) | β (°) | γ (°) |
|---|---|---|---|---|---|---|---|
| Pnma | −10.667 | 7.605 | 4.821 | 9.038 | 90 | 90 | 90 |
| Pnma25 | −10.773 | 7.618 | 4.793 | 9.001 | 90 | 90 | 90 |
| Pnma47 | Expt. | 7.691 | 4.816 | 9.035 | 90 | 90 | 90 |
| Fmm |
−10.623 | 7.165 | 7.165 | 7.165 | 90 | 90 | 90 |
| Fmm25 |
−10.740 | 7.148 | 7.148 | 7.148 | 90 | 90 | 90 |
| R3m | −9.352 | 3.819 | 3.819 | 22.748 | 90 | 90 | 120 |
| Pnm |
−8.473 | 6.435 | 6.435 | 6.435 | 90 | 90 | 90 |
| Phase | Atom | Site | x | y | z |
|---|---|---|---|---|---|
| Pnma | Ca1 | 4c | 0.3459 | 0.2500 | 0.0742 |
| Ca2 | 4c | 0.9802 | 0.2500 | 0.8221 | |
| Si1 | 4a | 0.7476 | 0.2500 | 0.1052 | |
| Fmm |
Ca1 | 8c | 0.2500 | 0.2500 | 0.2500 |
| Ca2 | 8c | 0.7500 | 0.7500 | 0.7500 | |
| Si1 | 4a | 0.0000 | 0.0000 | 0.0000 | |
| R3m | Ca1 | 3a | 0.0000 | 0.0000 | 0.2500 |
| Ca2 | 3a | 0.0000 | 0.0000 | 0.7500 | |
| Si1 | 3a | 0.0000 | 0.0000 | 0.0000 | |
| Pnm |
Ca1 | 4b | 0.7500 | 0.7500 | 0.2500 |
| Ca2 | 4b | 0.2500 | 0.2500 | 0.2500 | |
| Si1 | 2a | 0.5000 | 0.5000 | 0.5000 |
To investigate the stability of cubic Fmm Ca2Si under different pressures, we calculated the phonon dispersion curves both without and with LO–TO splitting in the first Brillouin zone (1BZ), which are illustrated in Fig. 2. As we can see, no negative or imaginary frequency was found in cubic Fmm Ca2Si for all the cases. The results suggest that cubic Fmm Ca2Si shows very good lattice dynamical stability at the pressure range we have studied. Hence the Fmm structure is a metastable phase of Ca2Si under pressure higher than −0.82 GPa. With the increase of pressure, the Ca–Si bonds were compressed, resulting in the increase of the maximum dispersion frequencies of the optical modes. Without LO–TO splitting, the calculated maximum frequencies of the twofold degenerated optical modes at the Γ point are 6.2 THz, 6.5 THz and 8.1 THz under −0.82 GPa negative pressure, at the ambient condition and under a positive pressure of 7.8 GPa, respectively. The LO–TO splitting is accounted by collecting the Born effective charge tensor and dielectric constant tensor. Under the influence of LO–TO splitting, we found that the twofold degenerated optical modes at the Γ point split into a higher frequency discontinue LO branch and a lower frequency TO branch. The TO branch holds the frequency of the optical branches without LO–TO splitting. The discontinuity of the LO branch is due to the difference in the magnitude of LO–TO splitting along the L–Γ and Γ–X directions.49 It is well known that the soft mode of the acoustic branch is the critical feature of a material with low lattice thermal conductivity, which is expected to achieve high ZT.50 We have found that cubic Fmm Ca2Si represents very similar soft mode characteristics to cubic Fmm Mg2Si.51 It is worth noting that the low frequency optical branches overlap with the acoustic branches along the X–W–K path are softer than Mg2Si. Hence lower lattice thermal conductivity in cubic Fmm Ca2Si is anticipated. On the other hand, similar to Mg2Si, the increase of external pressure leads to the frequency enhancement of the soft modes, which is not expected for thermoelectric applications.
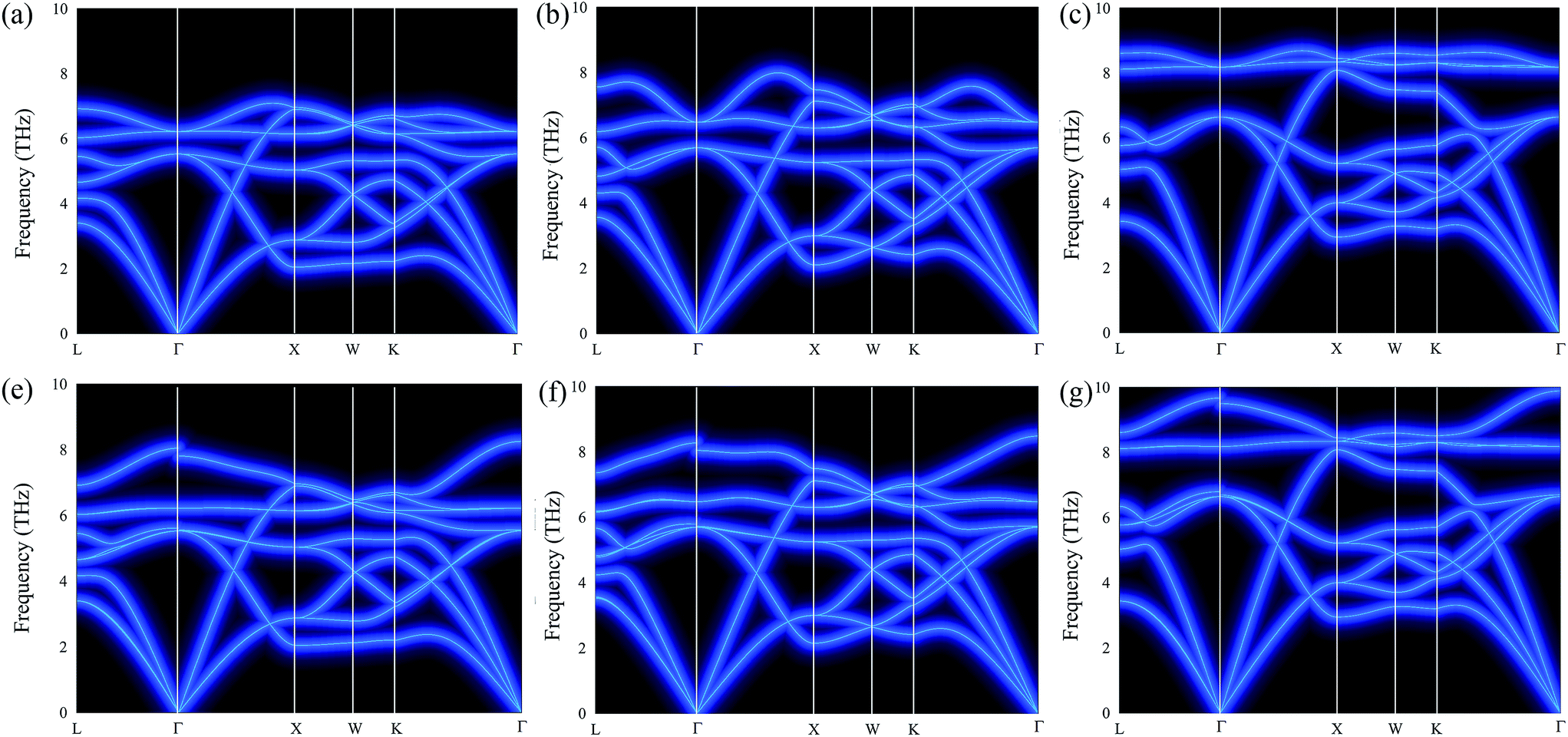 | ||
| Fig. 2 The calculated phonon dispersion curves of cubic Fmm Ca2Si (a–c) without and (e–g) with LO–TO splitting, where (a) and (e) at −0.82 GPa external pressure, (b) and (f) at the ambient condition, (c) and (g) at 7.8 GPa external pressure. | ||
In additions, to further confirm the stability of cubic Fmm Ca2Si, we predicted the mechanical stability via the elastic constants criterion. For a cubic crystal, there are three independent elastic constants C11, C12 and C44. The mechanical stability can be judged by C11 > 0, C44 > 0, C11 > |C12| and (C11 + 2C12) > 0. Fig. 3(a) shows the calculated pressure dependence of elastic constants by a step by step stress–strain method42,52 for cubic Fmm Ca2Si. It is obviously that the elastic constants satisfy all the stability conditions shown above, indicating that cubic Fmm Ca2Si is mechanical stable in the pressure range from −2 to 8 GPa. The positive correlation between the elastic constants and external pressure can be found, which is due to the enhanced interaction between atoms under external pressure.53 Fig. 3(b) shows the calculated bulk modulus B, Young's modulus E, and shear modulus G from the elastic constants as a function of pressure. As we can see, with the increase of pressure, the mechanical properties of B, E, G increase gradually, which are similar to the trend of elastic constants. Moreover, according to the calculated Poisson's ratio presented in Fig. 3(c), we found that the Poisson's ratio is very small, although it is increasing with the growth of pressure, indicating cubic Fmm Ca2Si is relatively stable against the shear strain.54
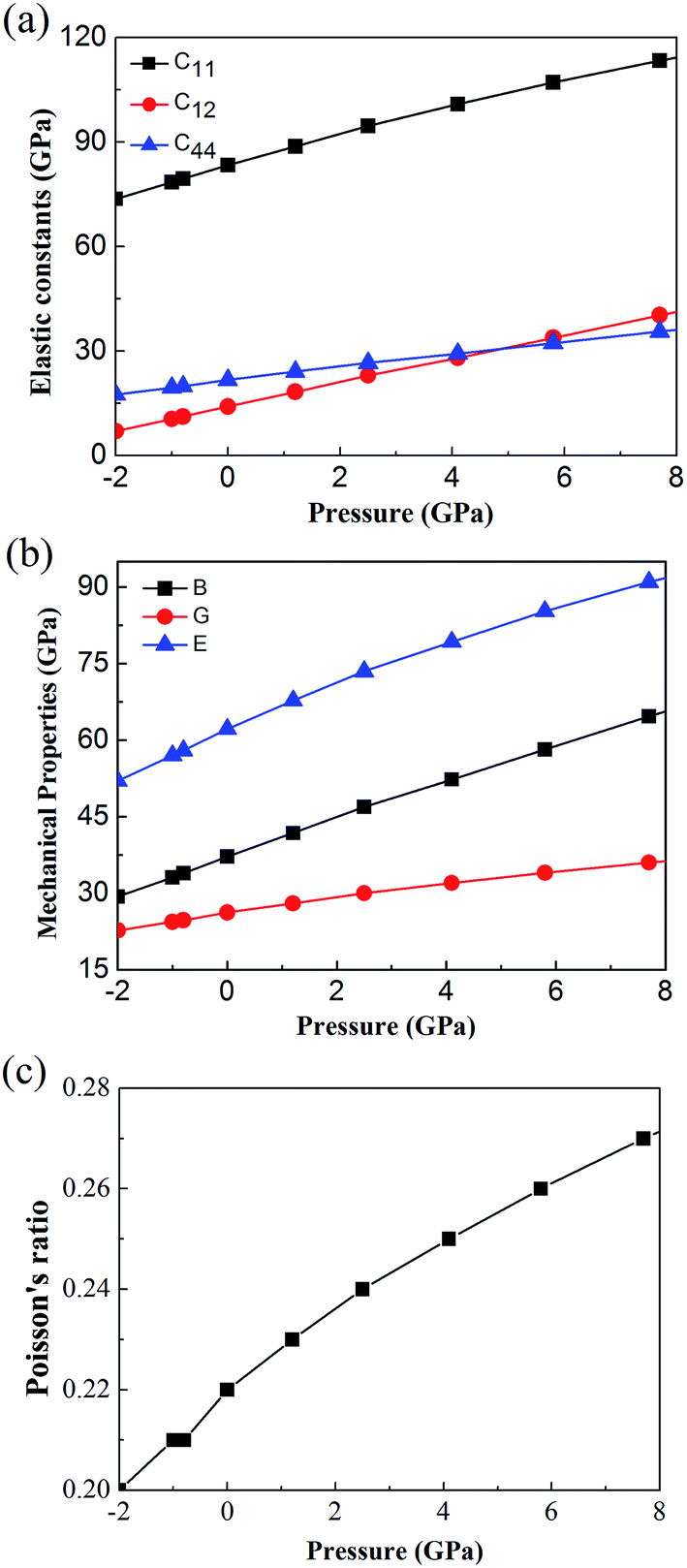 | ||
| Fig. 3 Calculated (a) elastic constants (b) bulk modulus B, Young's modulus E, shear modulus G and (c) Poisson's ratio of cubic Fmm Ca2Si as a function of pressure. | ||
In order to get better understanding, we introduced both the standard GGA-PBE functional as well as the HSE06 hybrid functional to calculate the electronic structures. Fig. 4(a) illustrates the total density of states (DOS) for cubic Fmm Ca2Si. As we can see from the figure, we have obtained 0.59 eV band gap using GGA-PBE functional, agrees well with the previous FPLAPW values of 0.56 eV.25 It is noted that HSE06 functional enlarges the bandgap to 1.13 eV. The band gap value is obviously larger than the narrow gap Bi2Te3-based alloys, which is beneficial for the high temperature thermoelectric applications due to the decrease of the high temperature bipolar conduction effect.55 By the analysis of the bandgap at various conditions in Fig. 4(b), we found that both GGA-PBE and HSE06 bandgaps reduce linearly with the increase of the pressure. The fitting slope for the GGA-PBE and HSE06 results are −0.0326 and −0.0283 eV GPa−1, respectively. We have found that GGA-PBE and HSE06 methods present very similar band gap changing tendency of cubic Fmm Ca2Si under pressure, where the HSE06 gaps are about 0.6 eV larger than the PBE gaps. According to the projected band structure at the ambient condition in Fig. 4(c) and (e), we found that cubic Fmm Ca2Si has been theoretically predicted to be a direct band gap semiconductor using PBE, where both the valance band maximum (VBM) conduction band minimum (CBM) are located at the X (0.5, 0, 0.5) point of 1BZ. However, for the HSE06 hybrid functional calculation, we found that the energy level of VBM at the L (0.5, 0.5, 0.5) point is slightly (∼0.04 eV) higher than that at the X point. Similar to black phosphorene,56 we can still briefly consider cubic Fmm Ca2Si as a direct gap semiconductor using HSE06 as well. For the both cases, the double generated VBM at the X point is mainly occupied by the Si p electrons and the CBM is characterized as the Ca-d electrons. It is worth noting that the Si-p valence band states at the K point and in the middle of the W–K high symmetry tie line show very close energy level to the VBM, agrees well with the previous FPLAPW prediction.25 These energy states give rise to the flat valence band states and high DOS peaks below the Fermi level and indicate good thermoelectric properties of cubic Fmm Ca2Si. To verify our hypothesis, we calculated the effective masses of the hole and electron at the X point by fitting the states around the VBM and CBM, respectively. The effective masses of the electron along the X–Γ direction using PBE 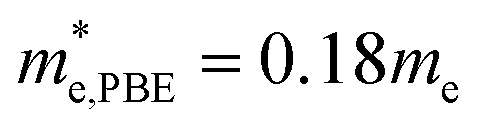 and using HSE06
and using HSE06 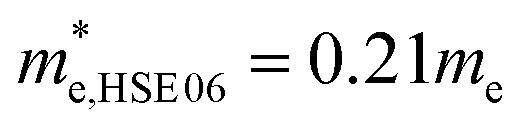 . The effective masses of the hole along the X–Γ direction using PBE
. The effective masses of the hole along the X–Γ direction using PBE 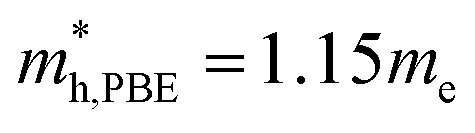 and using HSE06
and using HSE06 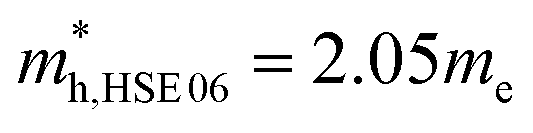 . The outstanding effective masses of the hole indicating the potential high thermoelectric properties with p type doping of cubic Fmm Ca2Si. According to the projected band structure under 7.8 GPa external pressure in Fig. 4(d) and (f), we found that the effects of the external pressure pull up the energy level of VBM and push down the energy level of CBM at the X point for both PBE and HSE06. As a result, the X to X direct gap nature of cubic Fmm Ca2Si can be protected. The Ca-s dominated conduction band state at the Γ point and the Si-s dominated conduction band state at the L point move upwards to higher energy levels under certain pressure. Besides, the other valence and conduction states are not sensitive to the external pressure. Herein, the stability of the electronic structure can protect the structure stability of cubic Fmm Ca2Si. Although the effective masses of the electron are insensitive to the external, however, with the increasing of the pressure up to 7.8 GPa, the effective masses of the hole along the X–Γ direction reduce to
. The outstanding effective masses of the hole indicating the potential high thermoelectric properties with p type doping of cubic Fmm Ca2Si. According to the projected band structure under 7.8 GPa external pressure in Fig. 4(d) and (f), we found that the effects of the external pressure pull up the energy level of VBM and push down the energy level of CBM at the X point for both PBE and HSE06. As a result, the X to X direct gap nature of cubic Fmm Ca2Si can be protected. The Ca-s dominated conduction band state at the Γ point and the Si-s dominated conduction band state at the L point move upwards to higher energy levels under certain pressure. Besides, the other valence and conduction states are not sensitive to the external pressure. Herein, the stability of the electronic structure can protect the structure stability of cubic Fmm Ca2Si. Although the effective masses of the electron are insensitive to the external, however, with the increasing of the pressure up to 7.8 GPa, the effective masses of the hole along the X–Γ direction reduce to 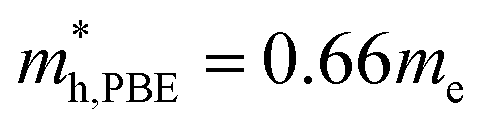 and
and 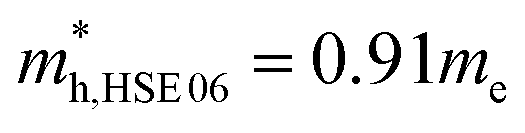 . The decreasing of the hole effective mass indicating the increasing of the external pressure cannot improve the thermoelectric properties of cubic Fmm Ca2Si. Hence combining with the lattice dynamic analysis, our further thermoelectric property study is focused on cubic Fmm Ca2Si at the ambient conduction.
. The decreasing of the hole effective mass indicating the increasing of the external pressure cannot improve the thermoelectric properties of cubic Fmm Ca2Si. Hence combining with the lattice dynamic analysis, our further thermoelectric property study is focused on cubic Fmm Ca2Si at the ambient conduction.
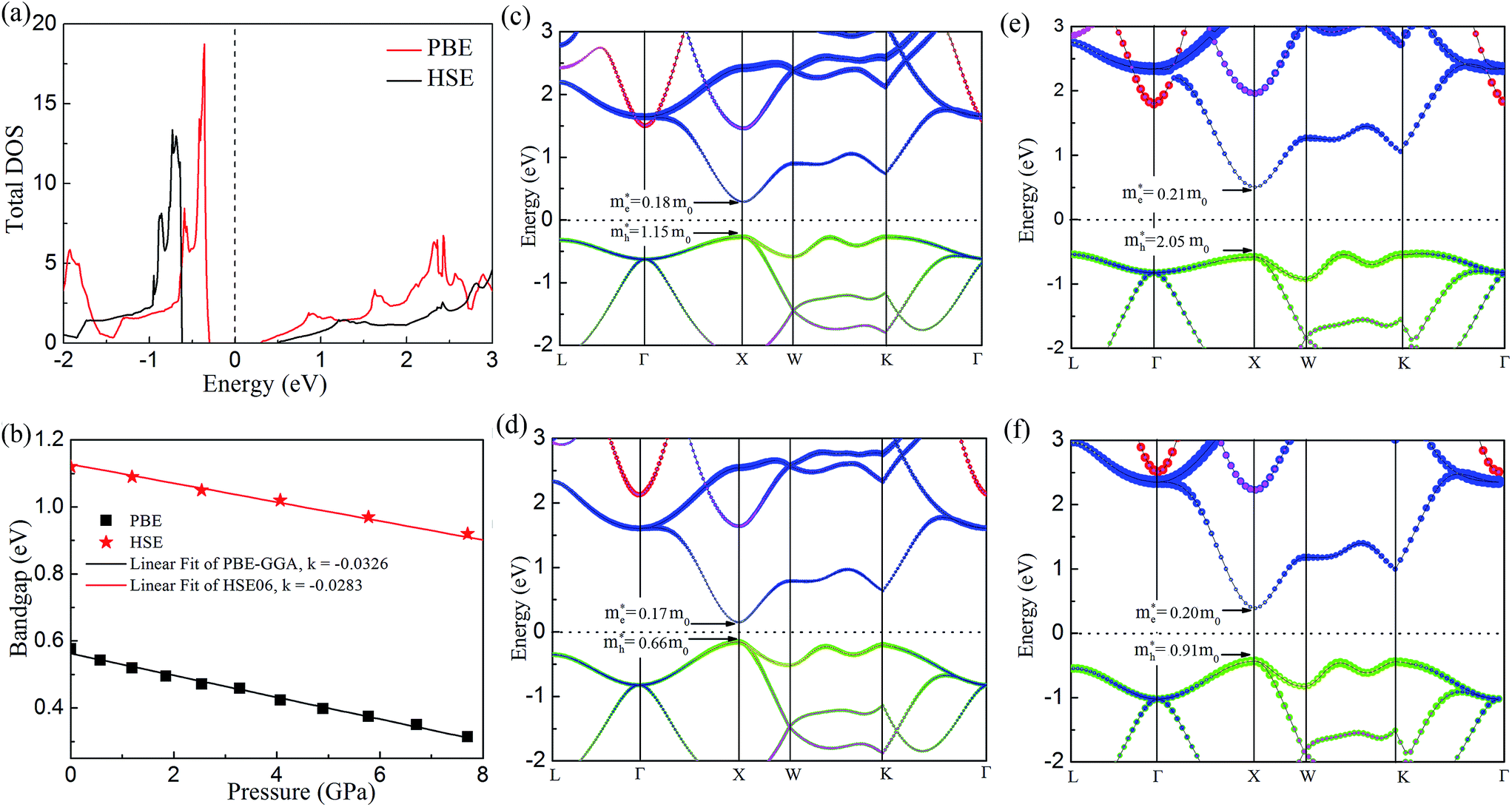 | ||
| Fig. 4 (a) The total density of states (TDOS) of cubic Fmm Ca2Si at the ambient condition using PBE and HSE06 functionals. (b) The calculated band gap of cubic Fmm Ca2Si as a function of pressure. The projected PBE band structure (c) at the ambient condition and (d) at 7.8 GPa external pressure of cubic Fmm Ca2Si. The projected HSE06 band structure (e) at the ambient condition and (f) at 7.8 GPa external pressure of cubic Fmm Ca2Si. The Fermi energy is set to 0 eV. The size of red, blue, pink and green circles illustrates the project weight of Ca-s, Ca-d and Si-s Si-p electrons, respectively. | ||
By solving the Boltzmann transport equation based on the HSE06 electronic eigenvalues, we first analyzed the thermoelectric power factor (PF) S2σ to examine the thermoelectric properties of cubic Fmm Ca2Si. The Seebeck coefficients S as a functional of the chemical potential at difference temperature are illustrated in Fig. 5(a). We found that the Seebeck coefficient is very sensitive to the chemical potential. Hence the efficient thermoelectric properties can be tuned via the carrier concentration. Generally, we found that the Seebeck coefficient of cubic Fmm Ca2Si decreases with the increase of temperature. Nevertheless, it is higher than the experimental measured peak value 300 μV K−1 for the p-type doped orthorhombic Ca2Si in most of the chemical potential and temperature range, which indicates that cubic Fmm Ca2Si is expected to show better thermoelectric performances than the orthorhombic phase. The calculated electrical conductivity σ within the constant relaxation time approximation is plotted in Fig. 5(b). It is easily to understand that there shows a positive correlation between the electrical conductivity and chemical potential, since more positive (negative) chemical potential corresponds to the higher electron (hole) concentration. Hence large chemical potential is expected to achieve high electrical conductivity. However, the thermoelectric PF S2σ does not follow this rule due to the fact that the Seebeck coefficient peaks at chemical potential very close to 0, indicating the finite carrier concentration is needed for the optimized thermoelectric PF. As seen from the calculated thermoelectric PF in Fig. 5(c), the thermoelectric PF reaches a peak around −0.5 to −0.6 eV for the p-type doping and 0.4–0.6 eV for the n-type doping at different temperatures. We found that the thermoelectric PF increases with the increase of temperature, which is contrary to the Seebeck coefficient. It is because the electrical conductivity is gradually increased when the temperature is increasing at the chemical potential larger than −0.64 eV. Moreover, we found that the p-type doping is preferred for the thermoelectric applications of cubic Fmm Ca2Si with larger optimized thermoelectric PF S2σ.
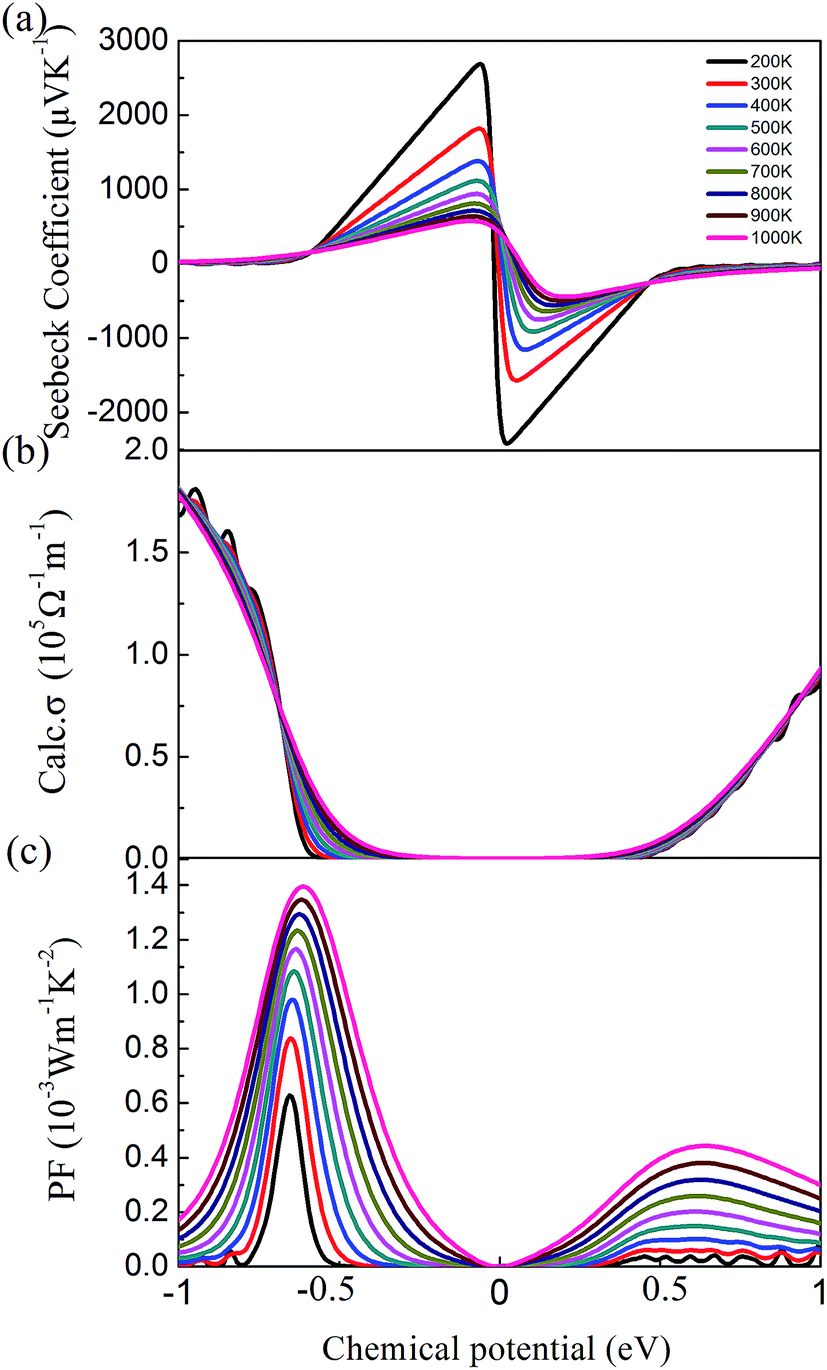 | ||
| Fig. 5 (a) Calculated Seebeck coefficient, (b) electrical conductivity and (c) power factor of cubic Fmm Ca2Si as a function of chemical potential. | ||
In the denominator of the figure of merit ZT, the thermal conductivity can be divided to the electronic (κe) and lattice (κl) contribution terms. The electronic thermal conductivity in Fig. 6(a) can also be solved from the well-known Wiedemann–Franz law 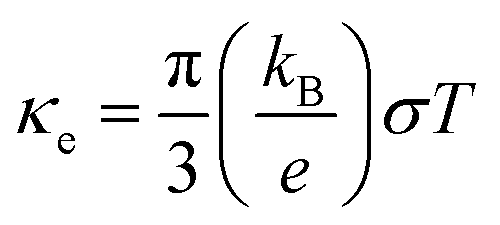 . Hence as a function of the chemical potential, the electronic thermal conductivity curves present very similar trend to the electrical conductivity. Meanwhile, in the whole chemical potential range, positive correlation can be found between the electronic thermal conductivity and temperature. The lattice thermal conductivity in Fig. 6(b) is estimated by the phonon Boltzmann transport equation. We found that the lattice thermal conductivity of cubic Fmm Ca2Si show the same order of magnitude to the Pnma Ca2Si and Bi2Te3-based chalcogenides around serval W m−1 K−1, which is lower than Mg2Si as we have anticipated. Considering the large differences between the covalent radii of Ca (1.76 Å) and Si (1.11 Å)57 as well as the soft mode nature of the phonon dispersion curves, the low lattice thermal conductivities can be understood. As seen in the insert figure of Fig. 6(b), the lattice thermal conductivity can be linearly fitted to the reciprocal of temperature, indicating the fact that the phonon scattering is dominated by the anharmonic phonon–phonon interactions. To the better understanding of the low thermal conductivity, we analyze the Grüneisen parameter to quantify the anharmonicity. The average Grüneisen parameter is 1.97 for cubic Fmm Ca2Si, which is comparable with typical thermoelectric materials PbTe of 1.96 with low thermal conductivity.58
. Hence as a function of the chemical potential, the electronic thermal conductivity curves present very similar trend to the electrical conductivity. Meanwhile, in the whole chemical potential range, positive correlation can be found between the electronic thermal conductivity and temperature. The lattice thermal conductivity in Fig. 6(b) is estimated by the phonon Boltzmann transport equation. We found that the lattice thermal conductivity of cubic Fmm Ca2Si show the same order of magnitude to the Pnma Ca2Si and Bi2Te3-based chalcogenides around serval W m−1 K−1, which is lower than Mg2Si as we have anticipated. Considering the large differences between the covalent radii of Ca (1.76 Å) and Si (1.11 Å)57 as well as the soft mode nature of the phonon dispersion curves, the low lattice thermal conductivities can be understood. As seen in the insert figure of Fig. 6(b), the lattice thermal conductivity can be linearly fitted to the reciprocal of temperature, indicating the fact that the phonon scattering is dominated by the anharmonic phonon–phonon interactions. To the better understanding of the low thermal conductivity, we analyze the Grüneisen parameter to quantify the anharmonicity. The average Grüneisen parameter is 1.97 for cubic Fmm Ca2Si, which is comparable with typical thermoelectric materials PbTe of 1.96 with low thermal conductivity.58
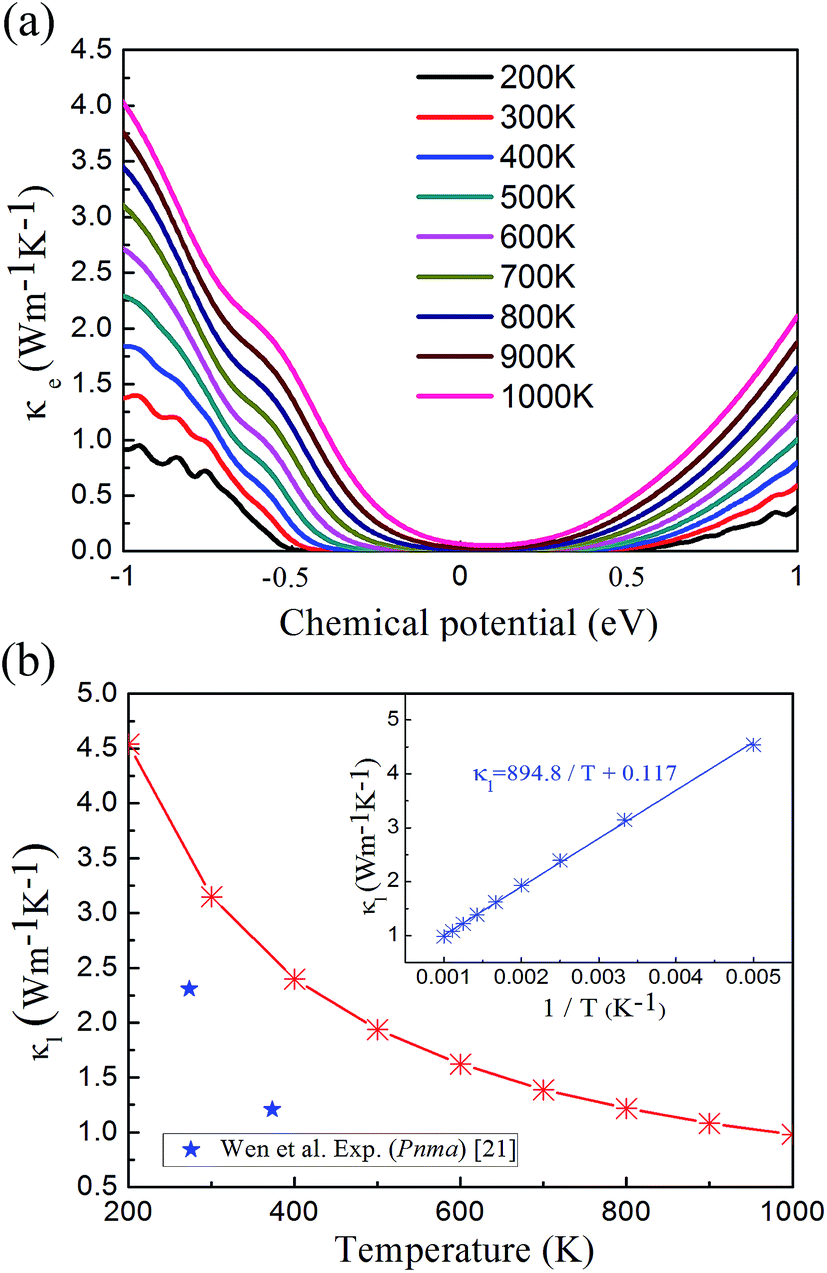 | ||
| Fig. 6 (a) The electronic thermal conductivity and (b) lattice thermal conductivity of cubic Fmm Ca2Si as a function of temperature. The star flags are experimental lattice thermal conductivity of Pnma Ca2Si in ref. 21. | ||
After all, we can evaluate the thermoelectric figure of merit ZT based on the above results. Fig. 7(a) illustrates the calculated ZT as a function of the chemical potential. We found that the plot of ZT shows very similar feature to the plot of PF, and it is interesting that ZT increases with the increase of temperature up to 1000 K. The peak values of ZT at different temperatures are plotted in Fig. 7(b). The maximal ZT value of 0.07 and 0.52 are observed with the chemical potential of −0.628 eV and −0.533 eV at 300 K and 1000 K, corresponding to the hole concentration of 2.45 × 1018 cm−3 and 2.83 × 1018 cm−3, respectively. The ZT value of the p-type doped Pnma Ca2Si is 2 × 10−5 around 350 K (ref. 21) (marked as the violet star in Fig. 7(b)), which is much smaller than our predicted values for the cubic Fmm Ca2Si. As seen the ZT of intrinsic Mg2Si in Fig. 7(b), we found that the thermoelectric properties of cubic Fmm Ca2Si are comparable to intrinsic Mg2Si.59,60 Since the solid solution treatment and doping can effectively enhance the figure of merit ZT of Mg2Si to as large as 1.1 (ref. 61), further optimizing of the thermoelectric properties of the cubic Fmm Ca2Si is anticipated.
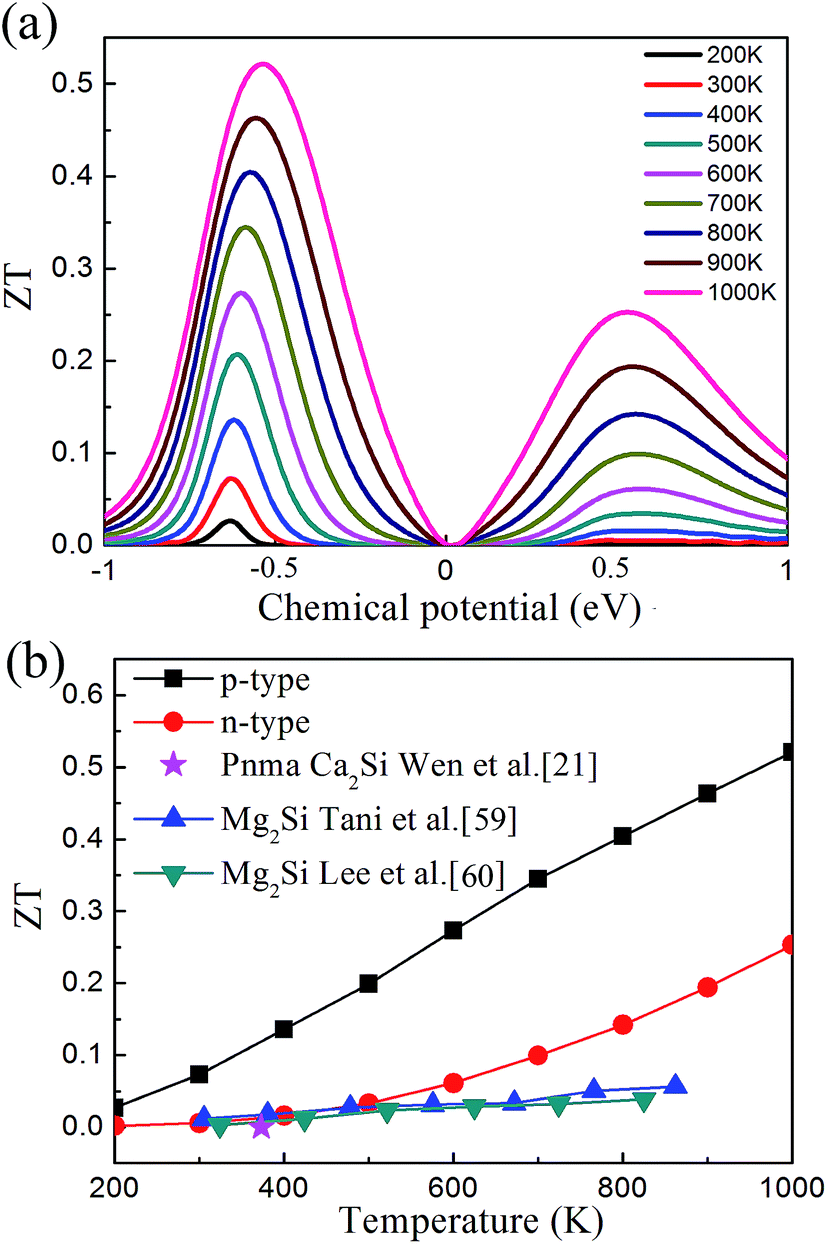 | ||
| Fig. 7 (a) Thermoelectric figure of merit ZT of cubic Fmm Ca2Si as a function of chemical potential. (b) The maximum ZT of cubic Fmm Ca2Si as a function of temperature. | ||
4. Conclusion
In summary, based on the ab initio evolutionary algorithm structure searching and density functional theory calculations, we have found that the orthorhombic Pnma phase is the most stable structure at pressure higher than −0.82 GPa, while the cubic Fmm phase is a meta-stable one in certain conditions. The stability of the cubic Fmm Ca2Si has been confirmed by the lattice dynamical phonon dispersion calculations and the mechanic criterion studies. The direct bandgap nature of cubic Fmm Ca2Si at various conditions was explored. Combining the HSE06 hybrid functional calculations, Boltzmann transport equation and the phonon Boltzmann transport equation, we have theoretically predicted the thermoelectric properties of cubic Fmm Ca2Si, which present larger Seebeck coefficient than orthorhombic Ca2Si. The optimized thermoelectric figure of merit ZT value of 0.52 is achieved for the p-type doping with the hole concentration 2.83 × 1018 cm−3 at 1000 K. We expect our findings will facilitate the experimental synthesis of Ca2Si isomers as well as their practical applications in thermoelectric devices.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 61504028, 61274005, 51301039, 11347007 and 11674131), the National Natural Science Foundation for Distinguished Young Scientists of China (Grant No. 51225205), the Natural Science Foundation of Fujian Province (No. 2016J01214 and 2016J01216), the Qing Lan Project, and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).References
- M. Beekman, D. T. Morelli and G. S. Nolas, Nat. Mater., 2015, 14, 1182–1185 CrossRef CAS PubMed.
- J. Yang, L. Xi, W. Qiu, L. Wu, X. Shi, L. Chen, J. Yang, W. Zhang, C. Uher and D. J. Singh, npj Computational Materials, 2016, 2, 15015 CrossRef.
- J. Zhou, B. Sa and Z. Sun, Intermetallics, 2010, 18, 2394–2398 CrossRef CAS.
- J. Zhou, Z. Sun, X. Cheng and Y. Zhang, Intermetallics, 2009, 17, 995–999 CrossRef CAS.
- N. Miao, J. Zhou, B. Sa, B. Xu and Z. Sun, Appl. Phys. Lett., 2016, 108, 213902 CrossRef.
- H. Zhu, W. Sun, R. Armiento, P. Lazic and G. Ceder, Appl. Phys. Lett., 2014, 104, 082107 CrossRef.
- I. Gonzalez-Valls and M. Lira-Cantu, Energy Environ. Sci., 2009, 2, 19–34 CAS.
- D. Parker, X. Chen and D. J. Singh, Phys. Rev. Lett., 2013, 110, 146601 CrossRef PubMed.
- D. I. Bilc, G. Hautier, D. Waroquiers, G. M. Rignanese and P. Ghosez, Phys. Rev. Lett., 2015, 114, 136601 CrossRef PubMed.
- S. Xun and L. Chen, Nat. Mater., 2016, 15, 691–692 CrossRef PubMed.
- B. Poudel, Q. Hao, Y. Ma, Y. Lan, A. Minnich, B. Yu, X. Yan, D. Wang, A. Muto, D. Vashaee, X. Chen, J. Liu, M. S. Dresselhaus, G. Chen and Z. Ren, Science, 2008, 320, 634–638 CrossRef CAS PubMed.
- R. Venkatasubramanian, E. Siivola, T. Colpitts and B. O'Quinn, Nature, 2001, 413, 597–602 CrossRef CAS PubMed.
- F. Q. Wang, S. Zhang, J. Yu and Q. Wang, Nanoscale, 2015, 7, 15962–15970 RSC.
- H. Zhao, J. Sui, Z. Tang, Y. Lan, Q. Jie, D. Kraemer, K. Mcenaney, A. Guloy, G. Chen and Z. Ren, Nano Energy, 2014, 7, 97–103 CrossRef CAS.
- N. Miao and P. Ghosez, J. Phys. Chem. C, 2015, 119, 14017–14022 CAS.
- P. Ying, X. Liu, C. Fu, X. Yue, H. Xie, X. Zhao, W. Zhang and T. Zhu, Chem. Mater., 2015, 27, 909–913 CrossRef CAS.
- T. D. Huan, V. N. Tuoc, N. B. Le, N. V. Minh and L. M. Woods, Phys. Rev. B: Condens. Matter Mater. Phys., 2016, 93, 094109 CrossRef.
- C. Wen, T. Nonomura, Y. Warashina, Y. Kubota, T. Nakamura, Y. Hayakawa, M. Tanaka, K. Isobe and H. Tatsuoka, Int. J. Mater. Res., 2011, 102, 401–405 CrossRef CAS.
- C. Wen, A. Kato, T. Nonomura and H. Tatsuoka, J. Alloys Compd., 2011, 509, 4583–4587 CrossRef CAS.
- C. Wen, T. Nonomura, K. Isobe, Y. Kubota, T. Nakamura, Y. Hayakawa, A. Kato and H. Tatsuoka, IOP Conf. Ser.: Mater. Sci. Eng., 2011, 18, 142014 CrossRef.
- C. Wen, T. Nonomura, A. Kato, Y. Kenichi, H. Udono, K. Isobe, M. Otake, Y. Kubota, T. Nakamura, Y. Hayakawa and H. Tatsuoka, Phys. Procedia, 2011, 11, 106–109 CrossRef CAS.
- P. Eckerlin, E. Leicht and E. Wölfel, Z. Anorg. Allg. Chem., 1961, 307, 145–156 CrossRef.
- Z. Yang, D. Shi, B. Wen, R. Melnik, S. Yao and T. Li, J. Solid State Chem., 2010, 183, 136–143 CrossRef CAS.
- J.-I. Tani and H. Kido, Comput. Mater. Sci., 2015, 97, 36–41 CrossRef CAS.
- D. B. Migas, L. Miglio, V. L. Shaposhnikov and V. E. Borisenko, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 205203 CrossRef.
- S. Lebègue, B. Arnaud and M. Alouani, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 085103 CrossRef.
- T. Sakamoto, T. Iida, A. Matsumoto, Y. Honda, T. Nemoto, J. Sato, T. Nakajima, H. Taguchi and Y. Takanashi, J. Electron. Mater., 2010, 39, 1708–1713 CrossRef CAS.
- J. D. Boor, C. Compere, T. Dasgupta, C. Stiewe, H. Kolb, A. Schmitz and E. Mueller, J. Mater. Sci., 2014, 49, 3196–3204 CrossRef.
- Y. L. Li, W. Luo, Z. Zeng, H. Q. Lin, H. K. Mao and R. Ahuja, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9289–9294 CrossRef CAS PubMed.
- B. Sa, J. Zhou, Z. Song, Z. Sun and R. Ahuja, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 085130 CrossRef.
- Z. Sun, J. Zhou, Y. Pan, Z. Song, H. K. Mao and R. Ahuja, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10410–10414 CrossRef CAS PubMed.
- J. Hafner, J. Comput. Chem., 2008, 29, 2044–2078 CrossRef CAS PubMed.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter, 1992, 45, 13244 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- C. W. Glass, A. R. Oganov and N. Hansen, Comput. Phys. Commun., 2006, 175, 713–720 CrossRef CAS.
- A. R. Oganov and C. W. Glass, J. Chem. Phys., 2006, 124, 244704 CrossRef PubMed.
- Y. Wang, L.-Q. Chen and Z.-K. Liu, Comput. Phys. Commun., 2014, 185, 2950–2968 CrossRef CAS.
- S. Baroni, S. De Gironcoli, A. Dal Corso and P. Giannozzi, Rev. Mod. Phys., 2001, 73, 515–562 CrossRef CAS.
- M. B. Taylor, G. D. Barrera, N. L. Allan and T. H. K. Barron, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 14380 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- J. Muscat, A. Wander and N. M. Harrison, Chem. Phys. Lett., 2001, 342, 397–401 CrossRef CAS.
- B. Sa, J. Zhou, R. Ahuja and Z. Sun, Comput. Mater. Sci., 2014, 82, 66–69 CrossRef CAS.
- J. Paier, M. Marsman, K. Hummer, G. Kresse, I. C. Gerber and J. G. Angyán, J. Chem. Phys., 2006, 124, 154709 CrossRef CAS PubMed.
- N. Miao, B. Xu, N. C. Bristowe, D. I. Bilc, M. J. Verstraete and P. Ghosez, J. Phys. Chem. C, 2016, 120, 9112–9121 CAS.
- G. K. H. Madsen and D. J. Singh, Comput. Phys. Commun., 2006, 175, 67–71 CrossRef CAS.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- W. Li, J. Carrete, N. A. Katcho and N. Mingo, Comput. Phys. Commun., 2014, 185, 1747–1758 CrossRef CAS.
- P. Manfrinetti and A. Palenzona, Intermetallics, 2000, 8, 223–228 CrossRef CAS.
- A. Khare, B. Himmetoglu, M. Johnson, D. J. Norris, M. Cococcioni and E. S. Aydil, Appl. Phys. Lett., 2012, 111, 083707 Search PubMed.
- W. Schweika, R. P. Hermann, M. Prager, J. Persson and V. Keppens, Phys. Rev. Lett., 2007, 99, 125501 CrossRef CAS PubMed.
- W. Li, L. Lindsay, D. A. Broido, D. A. Stewart and N. Mingo, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 174307 CrossRef.
- Z. Sun, R. Ahuja and J. E. Lowther, Solid State Commun., 2010, 150, 697–700 CrossRef CAS.
- B. Sa, J. Zhou and Z. Sun, Intermetallics, 2012, 22, 92–98 CrossRef CAS.
- N. Miao, B. Sa, J. Zhou and Z. Sun, Comput. Mater. Sci., 2010, 50, 1559–1566 CrossRef.
- H. Shi, D. Parker, M. H. Du and D. J. Singh, Phys. Rev. Appl., 2015, 3, 014004 CrossRef.
- B. Sa, Y. L. Li, J. Qi, R. Ahuja and Z. Sun, J. Phys. Chem. C, 2014, 118, 26560–26568 CAS.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, C. E. F. Barragán and S. Alvarez, Dalton Trans., 2008, 21, 2832–2838 RSC.
- Y. Zhang, X. Ke, C. Chen, J. Yang and P. Kent, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 024304 CrossRef.
- J.-I. Tani and H. Kido, Intermetallics, 2007, 15, 1202–1207 CrossRef CAS.
- H. J. Lee, Y. R. Cho and I.-H. Kim, J. Ceram. Process. Res., 2011, 12, 16–20 Search PubMed.
- G. Jiang, J. He, T. Zhu, C. Fu, X. Liu, L. Hu and X. Zhao, Adv. Funct. Mater., 2014, 24, 3776–3781 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |