
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functional organic click-materials: application in phosphorescent organic light emitting diodes†
Paul Kautny *a,
Chenyang Zhaob,
Thomas Kadera,
Berthold Stögerc,
Ernst Horkela,
Jiangshan Chen*b,
Dongge Mab,
Johannes Fröhlicha and
Daniel Lumpia
*a,
Chenyang Zhaob,
Thomas Kadera,
Berthold Stögerc,
Ernst Horkela,
Jiangshan Chen*b,
Dongge Mab,
Johannes Fröhlicha and
Daniel Lumpia
aInstitute of Applied Synthetic Chemistry, TU Wien, Getreidemarkt 9/163, A-1060 Vienna, Austria. E-mail: paul.kautny@tuwien.ac.at
bState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, China. E-mail: jschen@ciac.ac.cn
cInstitute of Chemical Technologies and Analytics, TU Wien, Getreidemarkt 9/164, A-1060 Vienna, Austria
First published on 20th February 2017
Abstract
In the presented work click chemistry is utilized to introduce 1,2,3-triazoles as a functional linker in organic donor–acceptor materials. A systematic series of materials was prepared and characterized to investigate the effect of the linkage mode on the molecular properties. The 1,2,3-triazole linker allowed control of the degree of intramolecular charge transfer over a wide range depending on the substitution pattern of the triazole moiety. The prepared materials were successfully employed as host materials for green and red dopants in phosphorescent organic light emitting diodes. Thus, this work presents the first application of this novel linkage mode in the design and synthesis of functional π-conjugated organic donor–acceptor materials and their application in organic light emitting diodes.
Introduction
Controlling the conjugation in organic molecules is one of the major challenges in the design of new materials for the rapidly evolving field of organic materials sciences,1–7 as the molecular properties of such compounds are predominantly determined by the π-conjugated system.1,2,7,8 One of these new technologies, which is now entering our everyday life in small portable displays and also large area TV screens,9 is organic light emitting diodes (OLEDs).10 Tremendous efforts have been made to advance OLEDs and in particular the introduction of phosphorescent heavy transition metal complexes as emitters significantly boosted the device efficiency of OLEDs.11,12In contrast to purely fluorescent materials, phosphorescent emitters harvest singlet and triplet excitons for light emission simultaneously, thus allowing for theoretically 100% internal quantum efficiency.13,14 Those metal complexes are typically dispersed in an organic host material to avoid concentration quenching.15,16 The application of bipolar host materials proved particularly successful to provide balanced charge transfer properties and thus high device efficiency.1,3,17
Unfortunately, the combination of electron rich donor and electron poor acceptor subunits in one host molecule lowers the triplet band gap (ET) via intramolecular charge transfer, resulting in undesired energy transfer from the guest emitter to the host.1,3 This effect particularly hampers the applicability of bipolar host materials for large band gap blue emitters.18 An improved efficiency of blue emitting components is, however, a prerequisite for the application of OLEDs in energy efficient solid-state lighting.9,19,20
Thus, the focus of current research is on the control of this undesired interaction. In order to minimize these donor–acceptor exchange via the conjugated π-system and to retain high ETs various strategies have been proposed. Among those are (i) the introduction of saturated sp3 hybridized bridges;21,22 (ii) induction of large twist angles by applying sterically demanding groups23,24 or ortho linkage of molecular subunits;25–27 (iii) decreased π-conjugation as result of meta linkage.25,26,28 Nonetheless, the challenge to design and prepare large band gap host materials remains.29
In particular, complex synthetic efforts are often required to join donor and acceptor units within one molecule, realizing a specific linkage mode at the same time. In contrast, copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) represents the most successful example of click chemistry30 and provides a convenient methodology to join two molecular building blocks by 1,2,3-triazole formation.31 Whereas widely applied in many fields of organic synthesis32,33 the application of this specific linkage mode and 1,2,3-triazoles as integral π-conjugated moiety in functional organic materials34–43 and in particular in host materials for PhOLEDs44 are relatively scarce. In contrast, 1,2,4-triazoles have been frequently employed as electron accepting units in the design of bipolar host materials.25,45–48 Furthermore, triazoles have been used as ligands for phosphorescent iridium complexes.49–51
In our previous work on small molecules we showed that the application of 1,2,3-triazole linkers allows for the subtle control of the photophysical properties of donor–acceptor materials.52 Thus, we envisioned to employ this strategy in the design of wide band gap host materials.
Results and discussion
Molecular design
The aim of this work was to incorporate the 1,2,3-triazole as functional linker in a large π-conjugated system in order to decrease the overall conjugation and utilize the CuAAC as key step for the assembly of the molecular subunits. This strategy allows for a building block approach enabling the convenient synthesis of a large set of molecules.The CuAAC was applied to connect electron rich triphenylamine (TPA) or phenylcarbazole (PCz) units to a central electron poor core system (Scheme 1). To explore the effect of the triazole linker on the overall conjugation and intramolecular charge transfer phenomena an electron accepting pyridine as well as benzene were chosen as core units. Furthermore, different substitution patterns of the central core units as well as of the triazole linker were investigated.
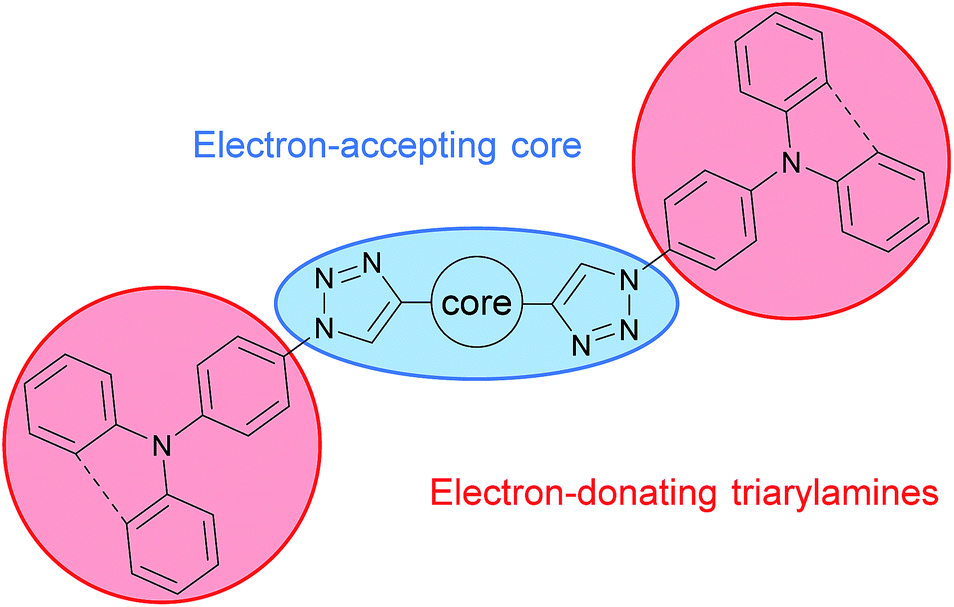 | ||
| Scheme 1 Molecular design of 1,2,3-triazole linked host materials. | ||
Beside the mere function as linkage unit the intrinsically weak electron accepting properties of the 1,2,3-triazole unit38,44 have to be considered in the molecular design of the materials. Thus, the triazole units will enhance the electron accepting properties of the core unit in case of pyridine acceptors or establish an electron accepting subunit itself in case of the benzene core.
Synthesis
TPA was chosen as primary electron rich subunit, owing to its superior electron donation properties. The TPA-substituted materials 3a, 3b, 3d and 3e were synthesized starting from azide 1i and the corresponding dialkynes (Scheme 2). Standard condition using CuSO4·5H2O and sodium ascorbate in a mixture of H2O and t-BuOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were employed and the application of a microwave reactor allowed for high reaction temperatures of 150 °C and convenient reaction times (30–60 min) yielding the target materials in reasonable to excellent yields (52–91%). In case of 3d single crystals were obtained by crystallization from EtOH, permitting the unambiguous determination of the triazole substitution pattern (Fig. 1 left). The five central aromatic units deviate only slightly from coplanarity with torsion angles of the least square planes of 10.03(10)° and 8.74(10)° for the pyridine–triazole and triazole–benzene fragments, respectively. Only one angle is listed in each case because the molecule is symmetric by twofold rotation with an axis passing through the pyridine fragment.
:1) were employed and the application of a microwave reactor allowed for high reaction temperatures of 150 °C and convenient reaction times (30–60 min) yielding the target materials in reasonable to excellent yields (52–91%). In case of 3d single crystals were obtained by crystallization from EtOH, permitting the unambiguous determination of the triazole substitution pattern (Fig. 1 left). The five central aromatic units deviate only slightly from coplanarity with torsion angles of the least square planes of 10.03(10)° and 8.74(10)° for the pyridine–triazole and triazole–benzene fragments, respectively. Only one angle is listed in each case because the molecule is symmetric by twofold rotation with an axis passing through the pyridine fragment.
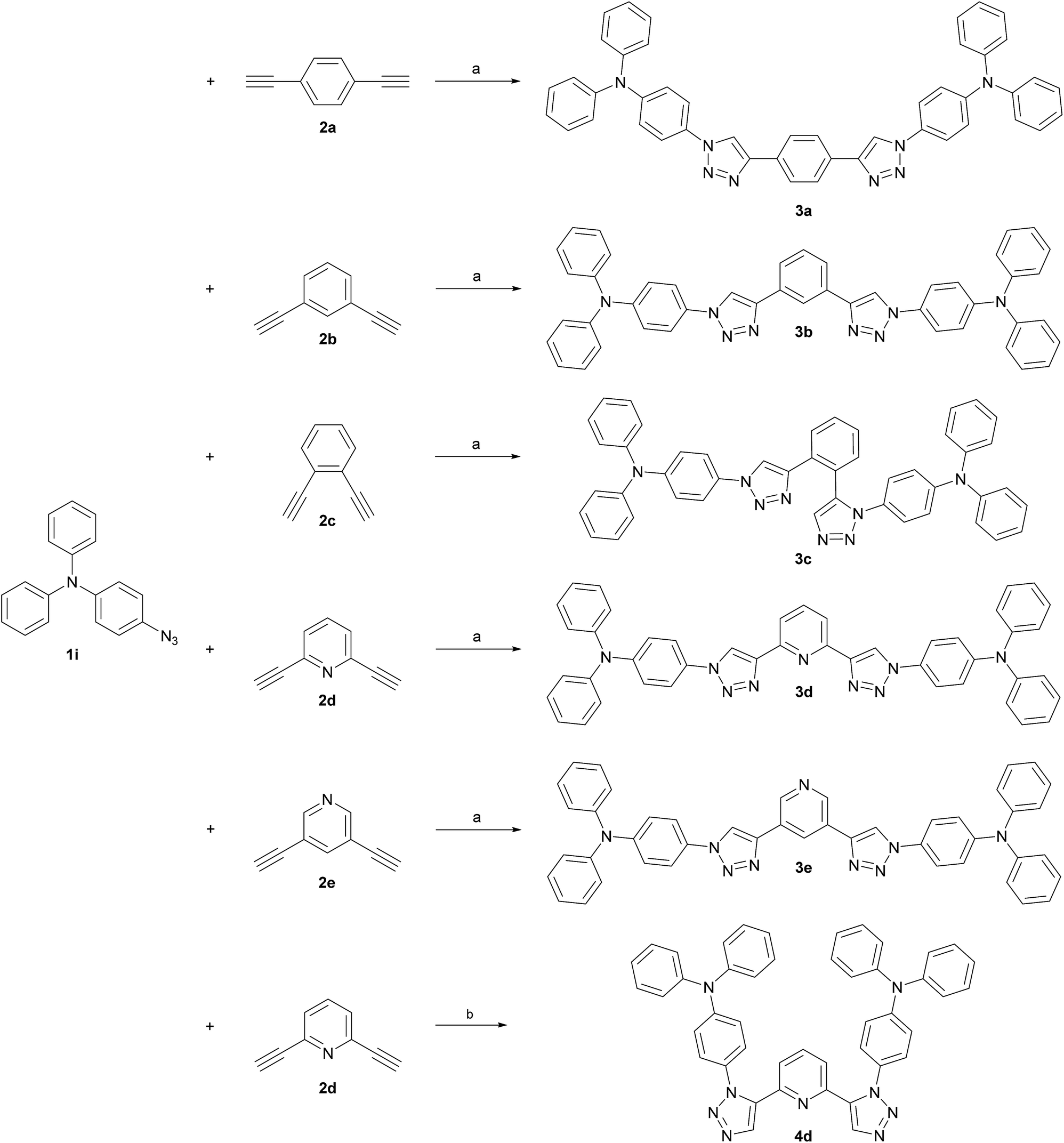 | ||
| Scheme 2 Synthesis of TPA-substituted host materials. (a) t-BuOH/H2O (1:1, 0.4 M), CuSO4·5H2O (0.20 eq.), sodium ascorbate (0.40 eq.), 150 °C microwave irradiation; (b) DMSO (0.25 M), tetramethylammonium hydroxide (0.1 eq., 25 wt% in H2O), 80 °C. | ||
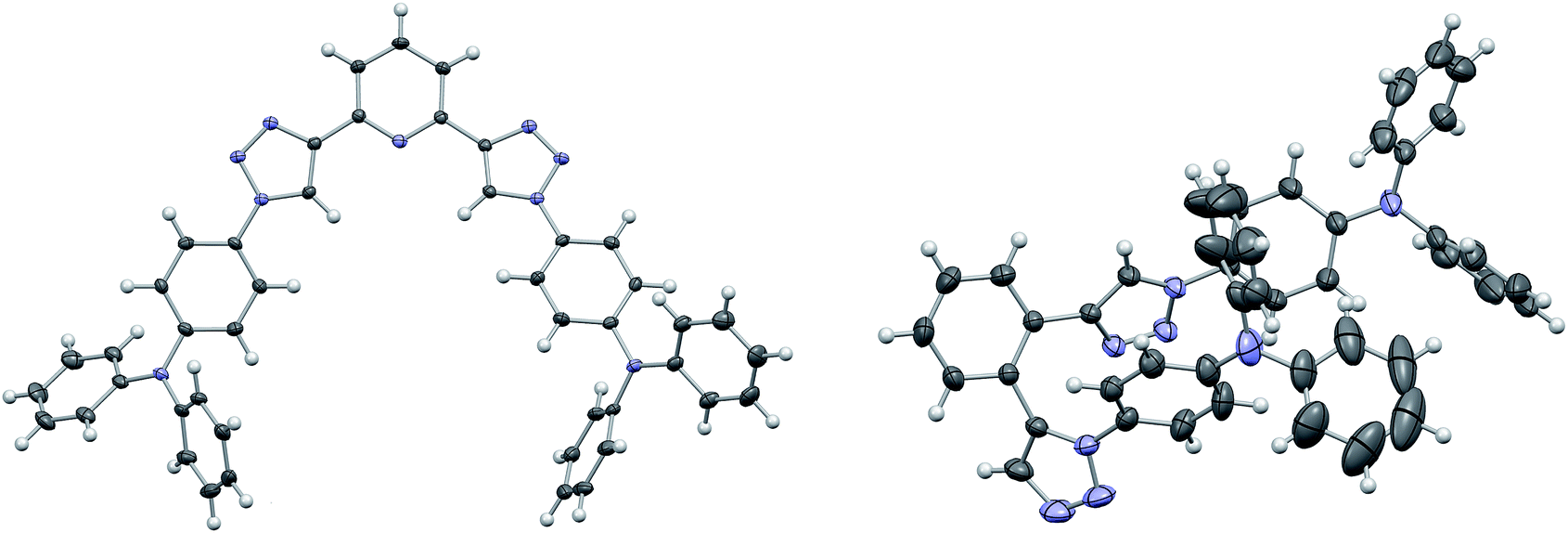 | ||
| Fig. 1 Molecular structures of 3d (left) and 3c (right). C and N atoms are represented by grey and blue ellipsoids drawn at 50% probability levels, H atoms by white spheres of arbitrary radius. The atoms of one diphenylamine fragment of 3c feature enlarged displacement parameters owing to disorder, which could not be resolved. | ||
Whereas the synthesis of the para- and meta-substituted benzene derivatives 3a and 3b proceeded smoothly, the corresponding ortho-substituted product could not be isolated. Instead a significant amount of a byproduct, that exhibited twice the number of signals in the carbon NMR spectrum, was obtained. Single crystals grown from acetonitrile enabled the assignment of structure 3c (Scheme 2). Surprisingly, 3c featured a mixed triazole substitution pattern (Fig. 1 right). Apparently, the primary installed bulky TPA-substituted 1,4-triazole unit inhibits the formation of the second 1,4-substituted triazole moiety, but favours the 1,5-substituted cycloaddition product. Nonetheless, the isolated product allows for an interesting comparison of the molecular properties of 3c and the purely 1,4-substituted derivatives.
Accordingly, a twofold 1,5-substituted material was prepared.
To maximize the donor–acceptor interaction electron poor pyridine was chosen as core and TPA as electron donating unit. A transition metal free methodology was utilized in the conversion of 2d and 1i applying tetramethylammonium hydroxide as alkaline promotor for cycloaddition.53 In such a manner 4d could be isolated with a low yield (18%).
Finally, two phenylcarbazole (PCz)-substituted derivatives were prepared to investigate the influence of weaker electron donors. Dialkynes 2b and 2d were chosen as templates for the central core (Scheme 3), since the meta relationship between the alkyne groups allows for a better comparability between the benzene and pyridine congeners. Identical reaction conditions were applied as for the synthesis of the 1,4-substituted TPA derivatives and 5b and 5d were isolated in 65% and 68% yield, respectively.
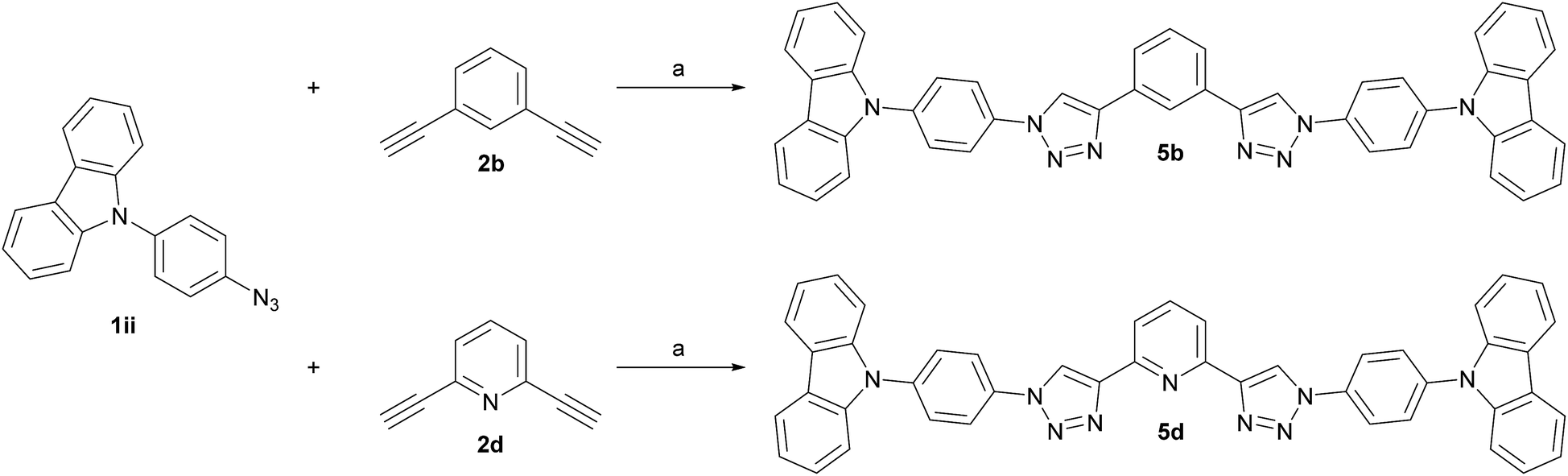 | ||
| Scheme 3 Synthesis of PCz-substituted host materials. (a) t-BuOH/H2O (1:1, 0.4 M), CuSO4·5H2O (0.20 eq.), sodium ascorbate (0.40 eq.), 150 °C microwave irradiation. | ||
Photophysical properties
UV/Vis absorption and photoluminescence (PL) spectra of all compounds as solutions in dichloromethane (5 μM) as well as low temperature phosphorescence spectra were recorded to investigate the impact of the conducted molecular variations on the photophysical properties of the materials. All TPA-substituted host materials exhibited two absorption maxima (Fig. 2). The high energy band is located around 300 nm and can be attributed to a π–π* transition centered at the TPA unit, while the peaks at longer wavelengths between 330 nm and 340 nm are due to charge transfer (CT) transitions between the electron rich TPA units and the electron withdrawing central cores. Notably, the intensity of this CT transition decreases for the 1,5-substituted derivatives. While for 1,4-substituted materials the CT transition is more intense compared to the π–π* transition, intensities are equal for 3c and inverted for 4d. The optical band gaps of all TPA materials are relatively similar and located between 3.31 and 3.37 eV.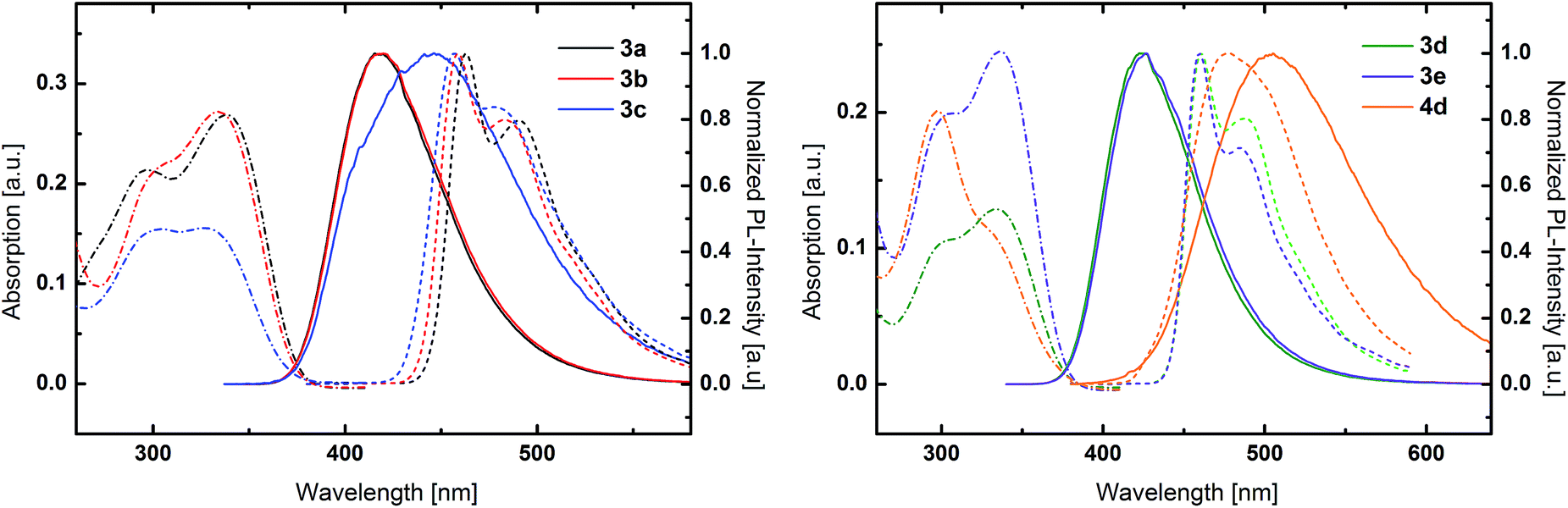 | ||
| Fig. 2 UV/Vis absorption (dashed and dotted lines), normalized fluorescence spectra at room temperature (solid lines) and normalized phosphorescence spectra at 77 K (dashed lines) of TPA-substituted materials 3a, 3b and 3c (left) and 3d, 3e and 4d (right). | ||
Inspecting the PL spectra of benzene based 3a and 3b, nearly identical emission spectra were observed (Fig. 2 left) with peak maxima at 418 nm. In contrast, the emission of 3c, featuring mixed 1,4 and 1,5 substitution pattern, is much broader. Remarkably, the emission onset is the same compared to the purely 1,4-substituted derivatives, however the emission extends to longer wavelength regions.
In analogy to the materials with benzene core, nearly identical emission features of 3d and 3e were observed in case of the pyridine based host materials, albeit with peak maxima at somewhat higher wavelengths of 424 nm and 425 nm, respectively. In contrast, the emission of 4d is distinctly red shifted compared to the 1,4-substituted derivatives. However, in the case of purely 1,5-substituted 4d also the onset of the emission is shifted compared to 3c with mixed substitution pattern.
Most strikingly, though, the absorption and emission properties of both meta-substituted derivative 3b and 3d are nearly identical. Additionally, no influence of the substitution pattern of the central aromatic ring on the photophysical properties was observed for 3a and 3b as well as 3d and 3e. Thus, the constitution and nature of the central aromatic core seems to be electronically insignificant due to the decreased conjugation as result of the triazole linkage.52 The negligible differences in the emission of the benzene and pyridine derivatives indicate that CT takes place between the peripheral TPA donors and an electron accepting core, which is established by the triazole moiety. In contrast, the central aromatic unit only plays a minor role, due to the restricted overall conjugation, which is unlike the behavior of molecules with less extended π-systems.52 Notably, the decreased overall conjugation can be solely attributed to the triazole linkage effect and is independent of any steric effect as conjugation is feasible in 3d due to the favorable planar configuration of the central aromatic units (Fig. 1).
PCz based host materials exhibited a sharp absorption peak at 293 nm due to the π–π* transition centered at the PCz unit (Fig. 3). The fine-structured absorption features at longer wavelengths resemble the absorption spectra of plain phenylcarbazole.54 No distinct CT band can be observed as result of the decreased donor strength of PCz compared to TPA. In relation to the TPA based derivatives 5b and 5d display distinctly blue shifted emission with maxima at 387 nm and 390 nm. Again an insignificant difference between the benzene and pyridine was observed.
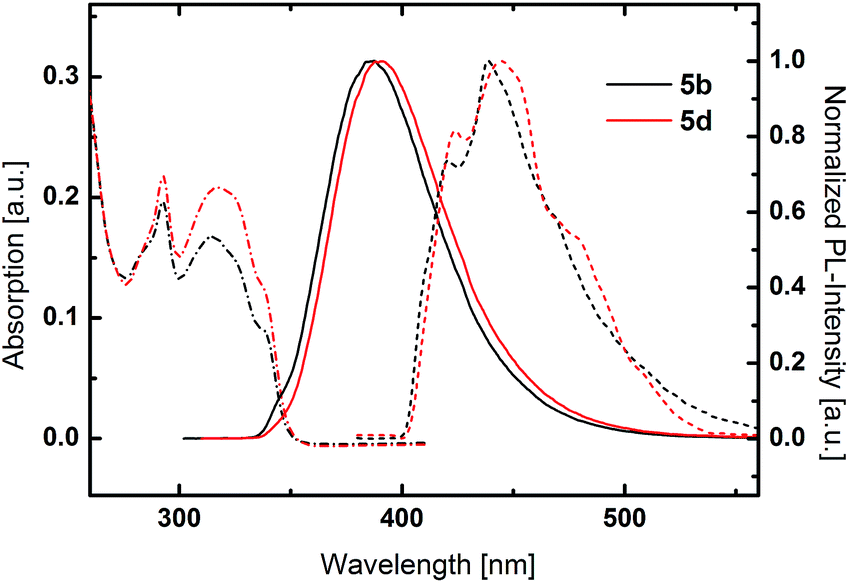 | ||
| Fig. 3 UV/Vis absorption (dashed and dotted lines), normalized fluorescence spectra at room temperature (solid lines) and normalized phosphorescence spectra at 77 K (dashed lines) of PCz-substituted materials 5b and 5d. | ||
Low temperature phosphorescence spectra were recorded at 77 K, to determine the triplet energies (ETs) of the potential host materials. Unlike the room temperature PL, low temperature phosphorescence featured vibronically resolved emission spectra (Fig. 2 and 3). Within the benzene series 3a and 3b exhibited similar ETs of 2.68 eV and 2.70 eV. Surprisingly, 3c displayed identical emission features indicating that the relevant triplet states are of similar nature. Analogously, pyridine based materials 3d and 3e featured ETs of 2.69 eV and 2.70 eV, respectively. However, 1,5-substituted 4d displayed diverse behavior with a broad phosphorescence emission without vibronic resolution and a lower ET of 2.59 eV. Notably, the red shift of the phosphorescence emission is distinctly lower compared to the room temperature PL, which exhibits a large Stokes-shift in medium polar dichloromethane. This particular effect indicates that the red shifted emission is only partly due to the intramolecular charge transfer but also induced by a strong structural relaxation that is inhibited in the solid phase and/or specific interaction with the solvent. In analogy to room temperature PL also the triplet emission of the PCz-substituted host materials are blue shifted compared to the TPA derivatives and are located at very high values of 2.95 eV and 2.92 eV, respectively.
Thermal and electrochemical properties
The thermal properties of the materials were investigated by TGA and DSC. DSC analysis (Fig. S17 and S18 left†) revealed the formation of stable films with glass transition temperatures (Tgs; determined from the second heating cycle of the DSC experiments) between 112 °C and 130 °C for the TPA-substituted derivatives. The introduction of PCz donors resulted in significantly increased Tgs of 182 °C and 171 °C for 5b and 5d, respectively.Unlike the satisfying film properties, the thermal durability of the compounds was found to be average (Fig. S18 right and S19†). In contrast to the Tgs the decomposition temperatures Tds (corresponding to the temperature of 5% mass loss) are not determined by the electron donor but the core ring system. Within the benzene series the Tds span a narrow range between 332 °C and 338 °C, whereas the range of the Tds of pyridine based materials is wider with 3d (293 °C) and 1,5-substituted 4d (355 °C) featuring the lowest and highest thermal stability, respectively.
Cyclic voltammetry (CV) was used to investigate the electrochemical properties of the materials (Fig. S20†). All TPA containing derivatives exhibited separated oxidation peaks, which are attributed to the two TPA units as exemplarily shown for 3b in Fig. 4. Whereas the first oxidation is reversible, a further increase of voltage led to the formation of reactive species. The HOMO levels of all TPA based compounds are located in a narrow range from −5.35 eV to −5.39 eV. The sole exception is 3a with a slightly higher lying HOMO at −5.27 eV. In contrast to the TPA compounds, PCz based host materials displayed irreversible oxidation, as typically observed for carbazole derivatives due to the instability of the formed cations.55 Owing to the less electron rich PCz moieties, the HOMO levels of 5b and 5d are located at lower energies of −5.66 eV and −5.71 eV, respectively. The suitable located HOMO energies indicate no significant barrier for charge injection from adjacent layers in PhOLED devices.
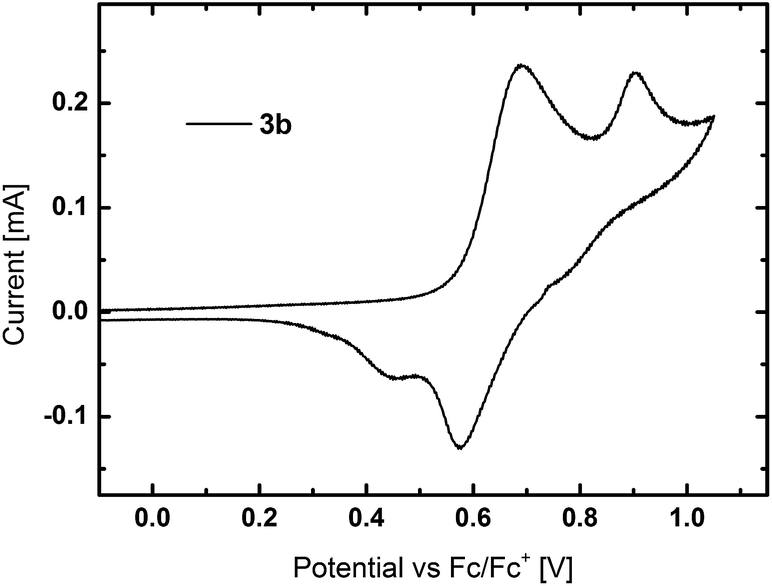 | ||
| Fig. 4 CV curve of 3b. | ||
The LUMO levels were calculated from the location of the HOMO and the optical bandgap and are located between −1.95 eV and −2.15 eV. Surprisingly, the incorporation of pyridine into the central core unit only negligibly influenced the energetic location of the LUMOs and the LUMO levels of pyridine based derivatives are only slightly lower compared to the corresponding benzene based congeners.
Computational investigations
Density functional theory (DFT) and time-dependent DFT (TDDFT) calculations were applied to obtain further insight into the electronic layout of the developed materials. In general, the calculated HOMO and LUMO energy levels as well as ETs are in good agreement with the experimental values (Table 1).| Tg/Tm/Tda [°C] | Opt. BGb,c [eV] | λmax,PLc [nm] | HOMO/LUMO [eV] | ET [eV] | |||
|---|---|---|---|---|---|---|---|
| Exp.d | Cal.e | Exp.f | Cal.g | ||||
| a Determined from TGA and DSC analysis; Tm – melting point, determined from the first cycle of the DSC experiments.b Estimated from the absorption onset.c Measured in DCM (5 μM) at room temperature.d Calculated from the onset of the oxidation peak and the optical bandgap.e Calculated applying the density functional theory level (B3LYP/6-311+G*).f Estimated from the highest energy vibronic transition in solid solutions (1 mg ml−1; toluene:EtOH = 9:1) at 77 K.g Calculated applying the time-dependent density functional theory level (B3LYP/6-311+G*).h Not observed. |
|||||||
| 3a | 130/312/336 | 3.32 | 418 | −5.27/−1.95 | −5.46/−1.58 | 2.68 | 2.74 |
| 3b | 121/n.o.h/338 | 3.34 | 418 | −5.38/−2.04 | −5.46/−1.53 | 2.70 | 2.79 |
| 3c | 112/n.o.h/338 | 3.37 | 446 | −5.37/−2.00 | −5.42/−1.58 | 2.71 | 2.80 |
| 3d | 122/220/293 | 3.31 | 424 | −5.36/−2.06 | −5.53/−1.64 | 2.69 | 2.79 |
| 3e | 125/n.o.h/335 | 3.31 | 425 | −5.35/−2.04 | −5.52/−1.63 | 2.70 | 2.78 |
| 4d | 112/n.o.h/355 | 3.33 | 505 | −5.39/−2.06 | −5.66/−1.91 | 2.59 | 2.87 |
| 5b | 182/317/332 | 3.56 | 387 | −5.66/−2.10 | −5.83/−1.85 | 2.95 | 2.94 |
| 5d | 171/323/340 | 3.56 | 390 | −5.71/−2.15 | −5.89/−2.01 | 2.92 | 3.01 |
As expected the HOMO levels of the materials are mainly located at the electron rich triarylamine groups with the exception of 3a (Fig. S21–S25†), as exemplarily depicted for 3b, 5b and 5d (Fig. 5). This localization can be explained by the prevention of full conjugation resulting from the triazole linker, as already observed for smaller asymmetric chromophores.52
 | ||
| Fig. 5 Spatial distribution of HOMOs and LUMOs of 3b, 5b and 5d. | ||
The spatial extension of the HOMO levels of 5b and 5d is even smaller compared to the TPA derivatives, due to the decreased electron donating character of the PCz moiety. In case of 3a the increased expansion of the HOMO onto the benzene core is due to the para substitution of the central aromatic moiety, which allows for better overall conjugation. Notably, this extension of the MO leads to a higher experimental determined HOMO energy compared to the corresponding meta and ortho derivatives. Furthermore, there are no significant differences between the extension of the HOMO levels between the benzene and pyridine based materials, which is again in agreement with the experimental results.
In analogy to the HOMOs, the LUMOs are mainly located on the three central aromatic rings and extend to the first phenyl rings of the triarylamine moieties. In case of the materials with benzene cores the strong localization of the LUMO levels indicates the establishment of an electron accepting subunit caused by the triazole linkers without the presence of an additional electron withdrawing group.
For the 1,5-linked compound 4d, a stronger localization of the MOs is found as result of the strongly twisted conformation and thus decreased conjugation, leading to slightly decreased energy levels. Furthermore, in 3c with mixed substitution pattern the HOMO is exclusively located on the TPA unit connected to the 1,5-substituted triazole and the corresponding triazole itself, due to the asymmetric architecture of the molecule.
Electroluminescent properties
Prototype PhOLED devices were fabricated to explore the practical applicability of the developed materials. Compounds 3b and 3d were chosen as representative examples for device fabrication. Their common substitution pattern allows for a direct comparability. Furthermore, the good solubility of the two materials permits device fabrication from solution. As the ETs of 3b and 3d are higher than those of green and red dopants, green emitting Ir(ppy)2(acac) and red emitting Ir(MDQ)2(acac) were chosen as emitters. Accordingly, prototype PhOLEDs were fabricated with the device architecture of ITO/PEDOT:PSS (45 nm)/EML (35 nm)/TPBi (GI–II: 50 nm; RI–II: 60 nm)/LiF/Al. PEDOT:PSS was employed as a hole injection and hole transporting layer, while TPBi was utilized as electron transporting and hole blocking layer. The emissive layers were made up of host materials 3b (GI and RI) and 3d (GII and RII) with emitters Ir(ppy)2(acac) (G) and Ir(MDQ)2(acac) (R) at a doping concentration of 8%.Luminance–voltage–current density curves are depicted in Fig. 6 and current efficiency–luminance and power efficiency–luminance curves are depicted in Fig. 7. The key electroluminescent properties of the devices are summarized in Table 2.
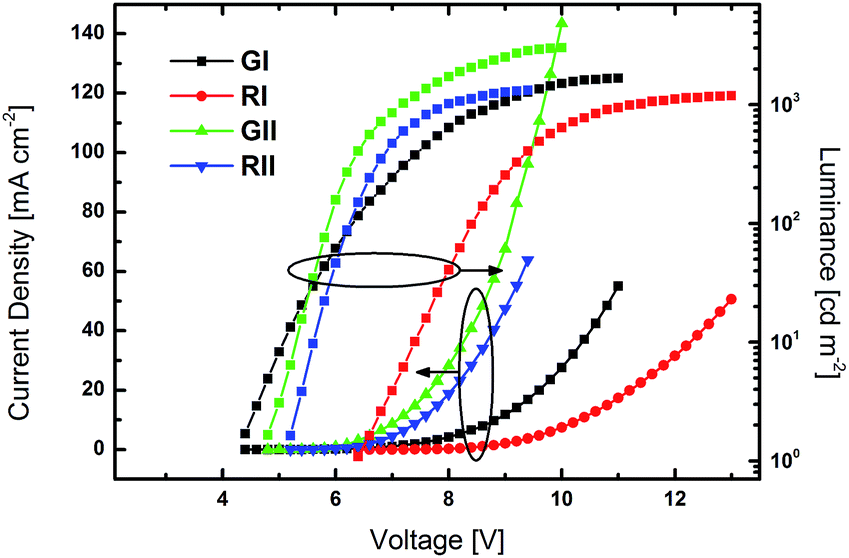 | ||
| Fig. 6 Luminance–voltage–current density curves of devices GI, RI, GII and RII. | ||
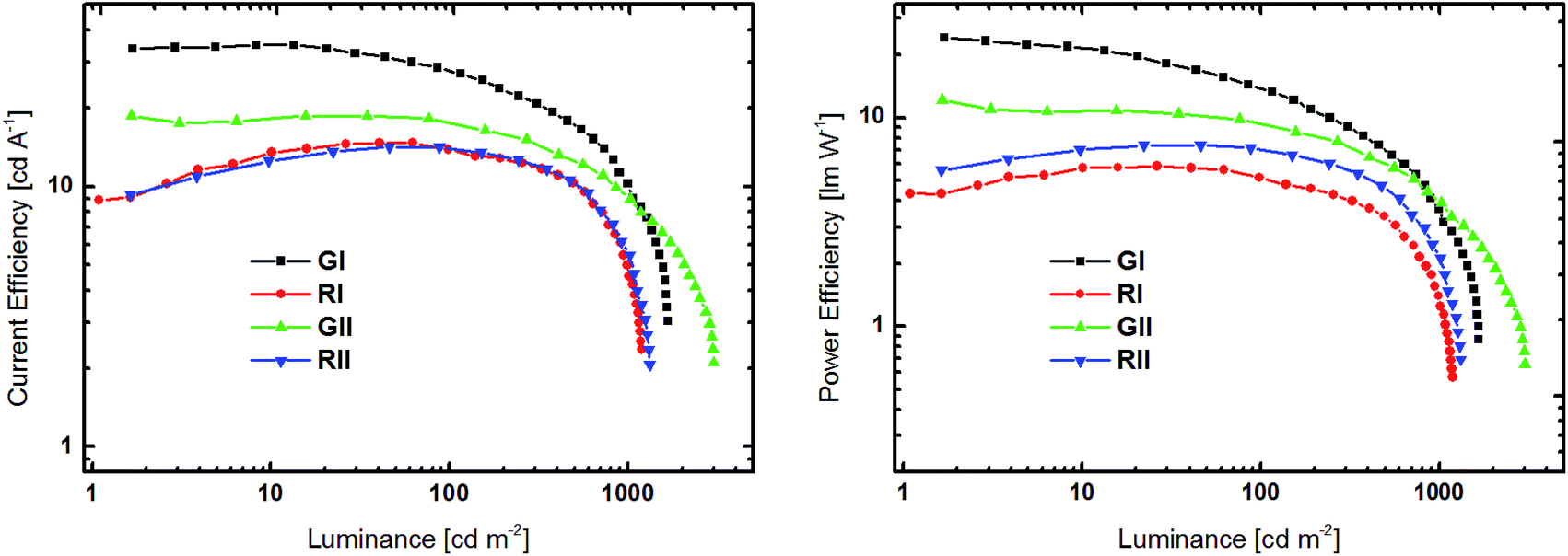 | ||
| Fig. 7 Current efficiency–luminance (left) and power efficiency–luminance (right) curves devices GI, RI, GII and RII. | ||
All devices exclusively exhibited emission from the phosphorescent emitters (Fig. S26†), indicating that energy transfer from the host to the dopant was effective and that excitons were successfully confined on the emitters. Green devices GI and GII exhibited turn-on voltages (Von) of 4.4 and 4.0 V, respectively, while the Vons of RI and RII were slightly higher at 6.4 and 5.4 V. Notably, the current density in GII and RII, utilizing host material 3d with a pyridine core, were significantly higher at lower voltage than in devices based on 3b (Fig. 6). Among the green devices GI displayed the better performance with a maximum current efficiency (CEmax) of 35.0 cd A−1 and a maximum power efficiency (PEmax) of 24.1 lm W−1, while the values were lower for GII. In contrast RI and RII featured basically the same CE characteristics with CEmax of 14.7 and 14.1 cd A−1, respectively. However, the PE of RII was slightly higher compared to RI, due to the lower driving voltage of RII.
Thin films of the four EMLs were investigated using AFM to explore their morphology (Fig. 8). In the case of all four EMLs films with a comparable low roughness (root mean square = 0.38, 0.35, 0.46 and 0.36 nm for GI, GII, RI and RII) were obtained. However, films based on 3b and in particular GI are distributed more uniformly compared to films based on 3d. This behaviour probably reduces the leak current, thus resulting in the superior device efficiency of GI compared to GII.
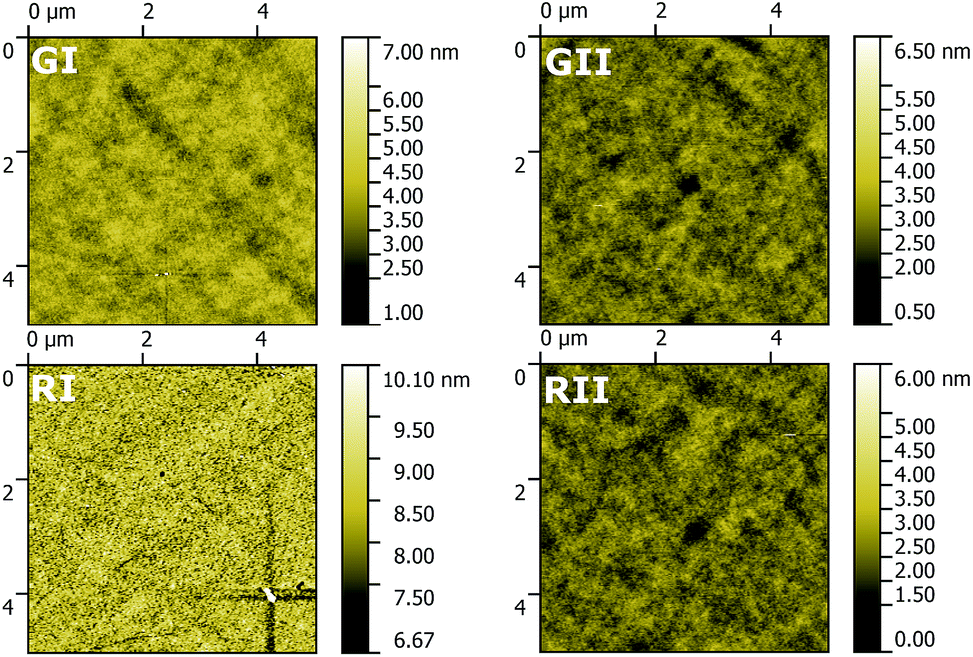 | ||
| Fig. 8 Top-view AFM images of the EMLs of GI, GII, RI and RII. | ||
Conclusions
We have successfully demonstrated the application of a novel click linkage mode for the preparation of large band gap conjugated donor–acceptor materials. Beside the convenient synthetic approach this strategy allowed the modulation of the molecular properties, which solely depend on the substitution pattern of the employed 1,2,3-triazole linker but are independent of the nature and substitution pattern of the core units. The prepared materials were successfully employed as host materials in red and green prototype PhOLED devices to demonstrate a first practical application of this class of materials. The intriguingly simple preparation in combination with the significant impact on the molecular properties predestines the presented approach as a novel tool to tune the molecular properties of π-conjugated materials. Therefore, we present a useful methodology for the field of organic materials chemistry, also beyond optoelectronics.Experimental section
General information
All reagents and solvents were purchased from commercial suppliers and used without further purification. Anhydrous solvents were prepared by filtration through drying columns. Experiments under microwave irradiation were performed in a Biotage Initiator Sixty microwave reactor. Column chromatography was performed on silica 60 (Merck, 40–63 μm). NMR spectra were recorded on a Bruker Avance DRX-400 Spectrometer. A Thermo Scientific LTQ Orbitrap XL hybrid FTMS (Fourier Transform Mass Spectrometer) equipped with an APCI source was used for high resolution mass spectrometry. Thermogravimetric (TG) and differential scanning calorimetry (DSC) measurements were carried out with a heating rate of 10 K min−1 in a flowing argon atmosphere (25 ml min−1). For the TG measurements, a Netzsch TG 209 F9 Tarsus system with open aluminium oxide crucibles was used. For the DSC measurements, a Netzsch DSC 200 F3 Maia, working with aluminium pans with pierced lids was employed. UV/Vis absorption and fluorescence emission spectra were recorded in DCM solutions (5 μM) with a Perkin Elmer Lambda 750 spectrometer and an Edinburgh FLS920, respectively. Time resolved low temperature phosphorescence spectra were recorded in solid solutions (1 mg ml−1; toluene:EtOH = 9:1) at 77 K with a Perkin Elmer Instruments LS 50B luminance spectrometer. Cyclic voltammetry was performed using a three electrode configuration consisting of a Pt working electrode, a Pt counter electrode and an Ag/AgCl reference electrode and a PGSTAT128N, ADC164, DAC164, External, DI048 potentiostat provided by Metrohm Autolab B.V. Measurements were carried out in a 0.5 mM solution in anhydrous DCM with Bu4NBF4 (0.1 M) as supporting electrolyte. The solutions were purged with nitrogen for 15 minutes prior to measurement. HOMO energy levels were calculated from the onset of the oxidation peaks. The onset potential was determined by the intersection of two tangents drawn at the background and the rising of the oxidation peaks.
Synthetic details
4-Azido-N,N-diphenylbenzenamine 1i56 and 9-(4-azidophenyl)-9H-carbazole 1ii57 have been prepared from the corresponding boronic acid esters58 and physical data acquired for the synthesized materials were identical with literature values. 1,4-Diethynylbenzene 2a, 1,2-diethynylbenzene 2c, 2,6-diethynlypyridine 2d and 3,5-diethynylpyridine 2e have been prepared by Sonogashira coupling starting from the corresponding dibromoaryls and trimethylsilyacetylene followed by desilylation.General procedure for the CuAAC (“click”) reaction
A microwave reaction vessel was charged with dialkyne (1.00 eq.) and azide (2.50 eq.) in t-BuOH/H2O (1:1, 0.4 M). Immediately before the start of the reaction CuSO4·5H2O (0.20 eq.) and sodium ascorbate (0.40 eq.) were added, the vial was sealed with a septum and heated to 150 °C under microwave irradiation. After complete conversion (TLC; 30–60 min) the reaction mixture was poured on water and extracted with dichloromethane. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. Purification of the crude products was accomplished by column chromatography.
4,4′-(1,4-Phenylendi-1H-1,2,3-triazol-4,1-diyl)bis[N,N′-diphenylbenzenamine] (3a)
Starting from 1i (573 mg, 2.00 mmol), 2a (101 mg, 0.80 mmol), CuSO4·5H2O (40 mg, 0.16 mmol) and sodium ascorbate (63 mg, 0.32 mmol) 3a (346 mg, 0.50 mmol, 62%) was isolated after column chromatography (dichloromethane/Et2O = 99:1 → 94:6). 1H-NMR (400 MHz, CD2Cl2): δ = 8.23 (s, 2H), 8.01 (s, 4H), 7.63 (d, J = 8.8 Hz, 4H), 7.32 (t, J = 7.9 Hz, 8H), 7.20–7.09 (m, 16H) ppm. 13C-NMR (100 MHz, CD2Cl2): δ = 149.1 (s), 148.1 (s), 147.7 (s), 131.5 (s), 131.0 (s), 130.1 (d), 126.7 (d), 125.6 (d), 124.4 (d), 123.7 (d), 122.1 (d), 118.5 (d) ppm. HRMS (APCI): m/z calculated for C46H34N8: 698.29009 [M]+, 699.29792 [M + H]+; found: 698.28987 [M]+, 699.29685 [M + H]+.
4,4′-(1,3-Phenylendi-1H-1,2,3-triazol-4,1-diyl)bis[N,N′-diphenylbenzenamine] (3b)
Starting from 1i (143 mg, 0.50 mmol), 2b (25 mg, 0.20 mmol), CuSO4·5H2O (10 mg, 0.04 mmol) and sodium ascorbate (16 mg, 0.08 mmol) 3b (128 mg, 0.18 mmol, 91%) was isolated after column chromatography (dichloromethane/Et2O = 97:3 → 93:7). 1H-NMR (400 MHz, CD2Cl2): δ = 8.45 (s, 1H), 8.30 (s, 2H), 7.92 (dd, J1 = 7.7 Hz, J2 = 1.8 Hz, 2H), 7.63 (d, J = 8.8 Hz, 4H), 7.56 (t, J = 7.7 Hz, 1H), 7.32 (t, J = 7.9 Hz, 8H), 7.19–7.09 (m, 16H) ppm. 13C-NMR (100 MHz, CD2Cl2): δ = 149.1 (s), 148.1 (s), 147.7 (s), 131.8 (s), 131.5 (s), 130.1 (d), 130.1 (d), 126.0 (d), 125.6 (d), 124.3 (d), 123.6 (d), 123.4 (d), 122.0 (d), 118.7 (d) ppm. HRMS (APCI): m/z calculated for C46H34N8: 698.29009 [M]+, 699.29792 [M + H]+, found: 698.28976 [M]+, 699.29685 [M + H]+.
4-[4-[2-[1-[4-(Diphenylamino)phenyl]-1H-1,2,3-triazol-5-yl]phenyl]-1H-1,2,3-triazol-1-yl]-N,N-diphenylbenzenamine (3c)
Starting from 1i (573 mg, 2.00 mmol), 2c (101 mg, 0.80 mmol), CuSO4·5H2O (40 mg, 0.16 mmol) and sodium ascorbate (63 mg, 0.32 mmol) 3c (158 mg, 0.23 mmol, 28%) was isolated after column chromatography (dichloromethane/Et2O = 97:3 → 91:9). 1H-NMR (400 MHz, CD2Cl2): δ = 7.92 (d, J = 7.9 Hz, 1H), 7.85 (s, 1H), 7.66–7.56 (m, 1H), 7.52–7.47 (m, 2H), 7.37 (d, J = 8.9 Hz, 2H), 7.33–7.23 (m, 8H), 7.13–7.03 (m, 15H), 6.80–6.78 (m, 4H) ppm. 13C-NMR (100 MHz, CD2Cl2): δ = 149.2 (s), 148.8 (s), 147.7 (s), 147.6 (s), 146.3 (s), 137.3 (s), 134.8 (d), 131.9 (d), 131.2 (s), 131.1 (s), 130.8 (d), 130.4 (s), 130.1 (d), 130.0 (d), 129.8 (d), 129.2 (d), 125.8 (s), 125.6 (d), 125.5 (d), 124.7 (d), 124.4 (d), 124.2 (d), 123.5 (d), 122.8 (d), 122.2 (d), 120.0 (d). HRMS (APCI): m/z calculated for C46H34N8: 698.29009 [M]+, 698.28980 [M + H]+, found: 698.28987 [M]+, 699.29675 [M + H]+.
4,4′-(2,6-Pyridindi-1H-1,2,3-triazol-4,1-diyl)bis[N,N′-diphenylbenzenamine] (3d)
Starting from 1i (143 mg, 0.50 mmol), 2d (25 mg, 0.20 mmol), CuSO4·5H2O (10 mg, 0.04 mmol) and sodium ascorbate (16 mg, 0.08 mmol) 3d (116 mg, 0.17 mmol, 83%) was isolated after column chromatography (dichloromethane/Et2O = 96:4). 1H-NMR (400 MHz, CD2Cl2): δ = 8.62 (s, 2H), 8.16 (d, J = 8.0 Hz, 2H), 7.94 (t, J = 7.9 Hz, 1H) 7.67 (d, J = 8.8 Hz, 4H), 7.32 (t, J = 7.9 Hz, 8H), 7.20–7.09 (m, 16H) ppm. 13C-NMR (100 MHz, CD2Cl2): δ = 150.6 (s), 149.2 (s), 149.2 (s), 147.7 (s), 138.4 (d), 131.5 (s), 130.1 (d), 125.6 (d), 124.4 (d), 123.6 (d), 122.1 (d), 120.7 (d), 119.9 (d) ppm. HRMS (APCI): m/z calculated for C45H33N9: 700.29317 [M + H]+, found: 700.29304 [M + H]+.
4,4′-(3,5-Pyridindi-1H-1,2,3-triazol-4,1-diyl)bis[N,N′-diphenylbenzenamine] (3e)
Starting from 1i (129 mg, 0.45 mmol), 2e (23 mg, 0.18 mmol), CuSO4·5H2O (9 mg, 0.04 mmol) and sodium ascorbate (14 mg, 0.07 mmol) 3e (65 mg, 0.09 mmol, 52%) was isolated after column chromatography (dichloromethane/Et2O = 95:5 → 82:18). 1H-NMR (400 MHz, CD2Cl2): δ = 9.09 (s, 2H), 8.74 (t, J = 1.9 Hz, 1H), 8.36 (s, 2H), 7.63 (d, J = 8.8 Hz, 4H), 7.32 (t, J = 7.9 Hz, 8H), 7.20–7.09 (m, 16H) ppm. 13C-NMR (100 MHz, CD2Cl2): δ = 149.3 (s), 147.7 (s), 147.0 (d), 145.3 (s), 131.2 (s), 130.1 (d), 130.0 (d), 127.2 (s), 125.6 (d), 124.4 (d), 123.5 (d), 122.1 (d), 119.2 (d) ppm. HRMS (APCI): m/z calculated for C45H33N9: 699.28534 [M]+, 700.29317 [M + H]+. Found: 699.28400 [M]+, 700.29201 [M + H]+.
4,4′-(2,6-Pyridindi-1H-1,2,3-triazol-5,1-diyl)bis[N,N′-diphenylbenzenamine] (4d)
The synthesis of 4d was accomplished following a previously published procedure.53 1i (515 mg, 1.80 mmol, 2.40 eq.) and 2d (95 mg, 0.75 mmol, 1 eq.) were dissolved in 3 ml degassed DMSO in a reaction vial. Subsequently, tetramethylammonium hydroxide (27 mg, 0.075 mmol, 0.1 eq., 25 wt% in H2O) was added an the reaction mixture was heated to 80 °C in a heating block for 27 h. The reaction mixture was poured on H2O and repeatedly extracted with dichloromethane. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. 4d (95 mg, 0.14 mmol, 18%) was isolated after column chromatography (dichloromethane/Et2O = 98:1 → 91:9). 1H-NMR (400 MHz, CD2Cl2): δ = 7.88 (s, 2H), 7.74 (t, J = 8.1 Hz, 1H), 7.34–7.29 (m, 10H), 7.17–7.05 (m, 20H) ppm. 13C-NMR (100 MHz, CD2Cl2): δ = 149.6 (s), 147.6 (s), 147.2 (s), 138.3 (d), 137.0 (s), 134.7 (d), 130.9 (s), 130.1 (d), 127.0 (d), 125.8 (d), 124.5 (d), 122.8 (d), 122.6 (d) ppm. HRMS (APCI): m/z calculated for C45H33N9: 699.28534 [M]+, 700.29317 [M + H]+, found: 699.28455 [M]+, 700.29232 [M + H]+.
9,9′-[1,3-Phenylendiylbis(1H-1,2,3-triazol-4,1-diyl-4,1-phenylen)]bis[9H-carbazole] (5b)
Starting from 1ii (745 mg, 2.62 mmol), 2b (134 mg, 1.06 mmol), CuSO4·5H2O (53 mg, 0.21 mmol) and sodium ascorbate (83 mg, 0.42 mmol) 5b (533 mg, 0.77 mmol, 72%) was isolated after column chromatography (dichloromethane/Et2O = 99:1 → 97:3). 1H-NMR (400 MHz, CD2Cl2): δ = 8.59 (t, J = 1.6 Hz, 1H), 8.52 (s, 2H), 8.18 (d, J = 7.8 Hz, 4H), 8.11 (d, J = 8.9 Hz, 4H), 8.02 (dd, J1 = 7.7, J2 = 1.9 Hz, 2H), 7.83 (d, J = 8.8 Hz, 4H), 7.65 (t, J = 7.8 Hz, 1H), 7.52–7.45 (m, 8H), 7.34 (td, J1 = 7.3, J2 = 1.4 Hz, 4H) ppm. 13C-NMR (100 MHz, CD2Cl2): δ = 148.6 (s), 141.2 (s), 138.7 (s), 136.3 (s), 131.6 (s), 130.3 (d), 128.9 (d), 126.8 (d), 126.3 (d), 124.1 (s), 123.6 (d), 122.5 (d), 120.9 (d), 120.9 (d), 118.8 (d), 110.2 (d) ppm. HRMS (APCI): m/z calculated for C46H30N8: 695.26662 [M + H]+, found: 695.26575 [M + H]+.
9,9′-[2,6-Pyridindiylbis(1H-1,2,3-triazol-4,1-diyl-4,1-phenylen)]bis-[9H-carbazole] (5d)
Starting from 1ii (142 mg, 0.50 mmol), 2d (25 mg, 0.20 mmol), CuSO4·5H2O (10 mg, 0.04 mmol) and sodium ascorbate (16 mg, 0.08 mmol) 5d (94 mg, 0.13 mmol, 68%) was isolated after column chromatography (dichloromethane/Et2O = 99:1 → 95:5). 1H-NMR (400 MHz, CD2Cl2): δ = 8.86 (s, 2H), 8.26 (d, J = 8.1 Hz, 2H), 8.19–8.13 (m, 8H), 8.02 (t, J = 7.9 Hz, 1H), 7.84 (d, J = 8.4 Hz, 4H), 7.52–7.44 (m, 8H), 7.33 (t, J = 7.3 Hz, 4H) ppm. 13C-NMR (100 MHz, CD2Cl2): δ = 150.5 (s), 149.7 (s), 141.2 (s), 138.8 (s), 138.6 (d), 136.3 (s), 128.9 (d), 126.8 (d), 124.1 (s), 122.6 (d), 121.0 (d), 120.9 (d), 120.9 (d), 120.3 (d), 110.2 (d) ppm. HRMS (APCI): m/z calculated for C45H29N9: 695.25404 [M]+, 696.26187 [M + H]+, found: 695.25171 [M]+, 696.26094 [M + H]+.
Computational details
All (TD)DFT computations were performed using the Gaussian 09 package, revision D.01.59 Density functional theory (DFT) and time-dependent (TD)DFT calculations were performed using the Becke three parameters hybrid functional with Lee–Yang–Perdew correlation (B3LYP),60,61 in combination with Pople basis sets (6-31G*, 6-311+G*).62 Geometry optimizations were performed in gas phase and without symmetry constraints. For the calculation of HOMO/LUMO levels, ground state (S0) geometries were optimized applying the 6-311+G* basis set. The determination of triplet energy (ET) was achieved by the calculation of the T1 excitation energy applying TDDFT level and the 6-311+G* basis to a S0 geometry optimized at DFT level using the 6-31G* basis set. Orbital plots were generated using GaussView.63Single crystal diffraction
Crystals suitable for single crystal diffraction were selected under a polarizing microscope, embedded in perfluorinated oil and attached to Kapton® micromounts. Intensity data were collected in a dry stream of nitrogen at 100 K on a Bruker KAPPA APEX II diffractometer system. Data were reduced using SAINT-Plus64 and an empirical absorption correction using the multi-scan approach implemented in SADABS64 was applied. The crystal structures were solved by charge-flipping implemented in SUPERFLIP65 and refined against F with the JANA2006 (ref. 66) software package. The non-H atoms were refined with anisotropic displacement parameters. The H atoms were placed at calculated positions and refined as riding on the parent C atoms.Device fabrication and measurement
All devices were fabricated on glass substrates pre-coated with a 180 nm thick layer of indium tin oxide (ITO) having a sheet resistance of 10 Ω per square. The ITO substrates were ultrasonically cleaned with detergent, deionized water, acetone and isopropanol, and then dried by blowing nitrogen over them. A layer of 45 nm thick PEDOT:PSS was spin-coated onto the pre-cleaned ITO substrates, and then baked at 120 °C in a vacuum oven for 30 min to extract residual water. Afterwards, the samples were moved into a glove box under a nitrogen-protected environment (oxygen and water contents less than 1 ppm), and the emissive layers (EMLs) were spin-coated on top of PEDOT:PSS from chlorobenzene and then annealed at 100 °C in a vacuum oven for 10 min to remove the residual solvent; the resulting thickness of EMLs is about 35 nm. Following that, the samples were transferred to a thermal evaporator chamber (pressure less than 5 × 10−4 Pa) connected to the glove box without exposure to the atmosphere. TBPi, LiF and Al were deposited sequentially by thermal evaporation. The overlap between ITO and Al electrodes was 16 mm2 as the active emissive area of the devices. The luminance–voltage–current density characteristics were measured by a Keithley source measurement unit (Keithley 2400 and Keithley 2000) with a calibrated silicon photodiode. The EL spectra were measured by SpectraScan PR650 spectrophotometer. The film thickness was determined by Dektak 6M Profiler (Veeco Metrology Inc.). All the measurements were carried out in ambient atmosphere.Acknowledgements
This work was supported in part by the TU Wien “Innovative Projects” research funds and the Austrian Federal Ministry of Science, Research and Economy. The X-ray centre of the Vienna University of Technology is acknowledged for providing access to the single-crystal diffractometer. The authors acknowledge P. Gauss, E. Schönegger and D. Liu for contributing to the synthesis of the materials and C. Hametner for assistance during NMR characterization. P. K., T. K. and J. F. gratefully acknowledge financial support by the Austrian Science Fund (FWF) (grant No. I 2589-N34). J. C. and D. M. gratefully acknowledge financial support by the National Natural Science Foundation of China (grant No. 11661131001).Notes and references
- Y. Tao, C. Yang and J. Qin, Chem. Soc. Rev., 2011, 40, 2943–2970 RSC.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- A. Chaskar, H.-F. Chen and K.-T. Wong, Adv. Mater., 2011, 23, 3876–3895 CrossRef CAS PubMed.
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed.
- M. J. Cho, D. H. Choi, P. A. Sullivan, A. J. P. Akelaitis and L. R. Dalton, Prog. Polym. Sci., 2008, 33, 1013–1058 CrossRef CAS.
- S.-i. Kato and F. Diederich, Chem. Commun., 2010, 46, 1994–2006 RSC.
- G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245–1330 CrossRef CAS PubMed.
- Y. Shirota, J. Mater. Chem., 2000, 10, 1–25 RSC.
- S. Reineke, Nat. Mater., 2015, 14, 459–462 CrossRef CAS PubMed.
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- M. A. Baldo, S. Lamansky, P. E. Burrows, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 1999, 75, 4–6 CrossRef CAS.
- M. A. Baldo, D. F. O'Brien, M. E. Thompson and S. R. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 14422–14428 CrossRef CAS.
- C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2001, 90, 5048–5051 CrossRef CAS.
- M. A. Baldo, C. Adachi and S. R. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 10967–10977 CrossRef CAS.
- C. Murawski, K. Leo and M. C. Gather, Adv. Mater., 2013, 25, 6801–6827 CrossRef CAS PubMed.
- S. O. Jeon and J. Y. Lee, J. Mater. Chem., 2012, 22, 4233–4243 RSC.
- K. S. Yook and J. Y. Lee, Adv. Mater., 2012, 24, 3169–3190 CrossRef CAS PubMed.
- S. Reineke, M. Thomschke, B. Lüssem and K. Leo, Rev. Mod. Phys., 2013, 85, 1245–1293 CrossRef CAS.
- W.-C. Chen, C.-S. Lee and Q.-X. Tong, J. Mater. Chem. C, 2015, 3, 10957–10963 RSC.
- M. Guan, Z. Q. Chen, Z. Q. Bian, Z. W. Liu, Z. L. Gong, W. Baik, H. J. Lee and C. H. Huang, Org. Electron., 2006, 7, 330–336 CrossRef CAS.
- L. Zeng, T. Y.-H. Lee, P. B. Merkel and S. H. Chen, J. Mater. Chem., 2009, 19, 8772–8781 RSC.
- Y. Zheng, A. S. Batsanov, V. Jankus, F. B. Dias, M. R. Bryce and A. P. Monkman, J. Org. Chem., 2011, 76, 8300–8310 CrossRef CAS PubMed.
- P. Kautny, Z. Wu, B. Stöger, A. Tissot, E. Horkel, J. Chen, D. Ma, H. Hagemann, J. Fröhlich and D. Lumpi, Org. Electron., 2015, 17, 216–228 CrossRef CAS.
- Y. Tao, Q. Wang, L. Ao, C. Zhong, C. Yang, J. Qin and D. Ma, J. Phys. Chem. C, 2010, 114, 601–609 CAS.
- Y. Tao, Q. Wang, C. Yang, C. Zhong, K. Zhang, J. Qin and D. Ma, Adv. Funct. Mater., 2010, 20, 304–311 CrossRef CAS.
- P. Kautny, D. Lumpi, Y. Wang, A. Tissot, J. Bintinger, E. Horkel, B. Stöger, C. Hametner, H. Hagemann, D. Ma and J. Fröhlich, J. Mater. Chem. C, 2014, 2, 2069–2081 RSC.
- Z. Ge, T. Hayakawa, S. Ando, M. Ueda, T. Akiike, H. Miyamoto, T. Kajita and M.-a. Kakimoto, Org. Lett., 2008, 10, 421–424 CrossRef CAS PubMed.
- C.-L. Ho and W.-Y. Wong, New J. Chem., 2013, 37, 1665–1683 RSC.
- A. Qin, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2010, 39, 2522–2544 RSC.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- F. Amblard, J. H. Cho and R. F. Schinazi, Chem. Rev., 2009, 109, 4207–4220 CrossRef CAS PubMed.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- I. Stengel, A. Mishra, N. Pootrakulchote, S. J. Moon, S. M. Zakeeruddin, M. Grätzel and P. Bäuerle, J. Mater. Chem., 2011, 21, 3726–3734 RSC.
- M. Juricek, M. Felici, P. Contreras-Carballada, J. Lauko, S. R. Bou, P. H. J. Kouwer, A. M. Brouwer and A. E. Rowan, J. Mater. Chem., 2011, 21, 2104–2111 RSC.
- R. M. Meudtner, M. Ostermeier, R. Goddard, C. Limberg and S. Hecht, Chem.–Eur. J., 2007, 13, 9834–9840 CrossRef CAS PubMed.
- Y. H. Lau, P. J. Rutledge, M. Watkinson and M. H. Todd, Chem. Soc. Rev., 2011, 40, 2848–2866 RSC.
- M. Parent, O. Mongin, K. Kamada, C. Katan and M. Blanchard-Desce, Chem. Commun., 2005, 2029–2031 RSC.
- D. Lumpi, F. Glöcklhofer, B. Holzer, B. Stöger, C. Hametner, G. A. Reider and J. Fröhlich, Cryst. Growth Des., 2014, 14, 1018–1031 CAS.
- S. Ast, T. Fischer, H. Müller, W. Mickler, M. Schwichtenberg, K. Rurack and H. J. Holdt, Chem.–Eur. J., 2013, 19, 2990–3005 CrossRef CAS PubMed.
- S. S. Bag and R. Kundu, J. Org. Chem., 2011, 76, 3348–3356 CrossRef CAS PubMed.
- A.-S. Cornec, C. Baudequin, C. Fiol-Petit, N. Plé, G. Dupas and Y. Ramondenc, Eur. J. Org. Chem., 2013, 2013, 1908–1915 CrossRef CAS.
- D. J. V. C. van Steenis, O. R. P. David, G. P. F. van Strijdonck, J. H. van Maarseveen and J. N. H. Reek, Chem. Commun., 2005, 4333–4335 RSC.
- M. K. Kim, J. Kwon, T.-H. Kwon and J. I. Hong, New J. Chem., 2010, 34, 1317–1322 RSC.
- D. Liu, D. Li, M. Wang and W. Li, J. Mater. Chem. C, 2016, 4, 7260–7268 RSC.
- M.-k. Leung, Y.-H. Hsieh, T.-Y. Kuo, P.-T. Chou, J.-H. Lee, T.-L. Chiu and H.-J. Chen, Org. Lett., 2013, 15, 4694–4697 CrossRef CAS PubMed.
- J. Zhuang, W. Su, W. Li, Y. Zhou, Q. Shen and M. Zhou, Org. Electron., 2012, 13, 2210–2219 CrossRef CAS.
- J. Zhuang, W. Li, W. Su, M. Zhou and Z. Cui, New J. Chem., 2014, 38, 650–656 RSC.
- Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2010, 39, 638–655 RSC.
- Y. Zhao, J. Tang, H. Zhang and Y. Ma, Eur. J. Inorg. Chem., 2014, 2014, 4843–4851 CrossRef CAS.
- J. Feldman, G. D. Vo, C. D. McLaren, T. C. Gehret, K.-H. Park, J. S. Meth, W. J. Marshall, J. Buriak, L. M. Bryman, K. D. Dobbs, T. H. Scholz and S. G. Zane, Organometallics, 2015, 34, 3665–3669 CrossRef CAS.
- P. Kautny, D. Bader, B. Stöger, G. A. Reider, J. Fröhlich and D. Lumpi, Chem.–Eur. J., 2016, 22, 18887–18898 CrossRef CAS PubMed.
- S. W. Kwok, J. R. Fotsing, R. J. Fraser, V. O. Rodionov and V. V. Fokin, Org. Lett., 2010, 12, 4217–4219 CrossRef CAS PubMed.
- H. Li, Y. Wang, K. Yuan, Y. Tao, R. Chen, C. Zheng, X. Zhou, J. Li and W. Huang, Chem. Commun., 2014, 50, 15760–15763 RSC.
- J. F. Ambrose and R. F. Nelson, J. Electrochem. Soc., 1968, 115, 1159–1164 CrossRef CAS.
- Q. Zhang, Z. Ning and H. Tian, Dyes Pigm., 2009, 81, 80–84 CrossRef CAS.
- A. Hörner, D. Volz, T. Hagendorn, D. Fürniss, L. Greb, F. Rönicke, M. Nieger, U. Schepers and S. Bräse, RSC Adv., 2014, 4, 11528–11534 RSC.
- K. D. Grimes, A. Gupte and C. C. Aldrich, Synthesis, 2010, 1441–1448 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- R. Dennington, T. Keith and J. Millam, GaussView, Version 5, Semichem, Inc., Shawnee Mission, KS, 2009 Search PubMed.
- Bruker computer programs: APEX2, SAINT and SADABS, Bruker AXS Inc., Madison, WI, 2015 Search PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- V. Petříček, M. Dušek and L. Palatinus, Z. Kristallogr.–Cryst. Mater., 2014, 229, 345–352 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, DSC and TGA analysis, cyclic voltammetry, theoretical calculations and crystallographic information. CCDC 1497478–1497479. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra28212a |
| This journal is © The Royal Society of Chemistry 2017 |