
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomimetic adsorption of zwitterionic–xyloglucan block copolymers to CNF: towards tailored super-absorbing cellulose materials†
F. L. Hatton‡
 a,
J. Engströmab,
J. Forslinga,
E. Malmströma and
A. Carlmark*a
a,
J. Engströmab,
J. Forslinga,
E. Malmströma and
A. Carlmark*a
aKTH Royal Institute of Technology, School of Chemical Science and Engineering, Department of Fibre and Polymer Technology, Teknikringen 56, SE-100 44 Stockholm, Sweden. E-mail: annac@kth.se
bKTH Royal Institute of Technology, School of Chemical Science and Engineering, Wallenberg Wood Science Centre, Teknikringen 56, SE-100 44 Stockholm, Sweden
First published on 7th March 2017
Abstract
A biomimetic, facile approach to cellulose modification is the utilisation of self-adsorbing, naturally occurring biopolymers, such as the hemicellulose xyloglucan (XG). Herein, XG-block-poly(sulfobetaine methacrylate) (XG-b-PSBMA) zwitterionic block copolymers have been prepared and assessed for their ability to adsorb to cellulose, specifically cellulose nanofibrils (CNF). The polymers were synthesised using reversible addition–fragmentation chain-transfer (RAFT) polymerisation, employing an XG macromolecular RAFT agent (XG-RAFT), polymerising a sulfobetaine methacrylate (SBMA) under aqueous conditions. The incorporation of the XG block shifted the upper critical solution temperature (UCST) values to higher temperatures (20 and 30 °C) compared with the PSBMA homopolymers (17 and 22 °C) and the transition was also broadened. The adsorption of the polymers to a CNF surface was monitored using quartz crystal microbalance with dissipation monitoring (QCM-D), showing that the XG block enhanced the adsorption of the zwitterionic polymer. The formation of CNF-composite films was achieved utilising a facile vacuum filtration methodology, and the targeted compositions were confirmed by FT-IR and TGA analyses. The films exhibited high degrees of swelling in water, which were investigated at two different temperatures, 5 and 60 °C (below and above the polymer USCT values). These results highlight the advantage of using an XG block for the biomimetic modification of cellulose to form new cellulose-composite materials such as super-absorbing films.
1. Introduction
The desire to prepare sustainable, renewable materials has motivated significant research in utilising naturally occurring polymers such as cellulose. Cellulose, as well as being abundant and renewable, has incredibly interesting mechanical properties, which is also true for its nanocellulosic counterparts: bacterial nanocellulose (BNC), cellulose nanocrystals (CNC) and cellulose nanofibrils (CNF).1 In particular, CNF is foreseen as a component in several interesting applications due to its high aspect ratio.2,3 CNF is liberated from cellulose fibres through chemical modification, followed by mechanical treatment, and dimensions reported are usually within 5–20 nm in diameter and up to several micrometres in length.1 CNF is considered hydrophilic in character and has been used for several 3-D structured materials such as water or oil absorbing aerogels4,5 and more moisture stable films by forming a stated core–shell structure of CNF coated with xyloglucan.6 CNF is commonly used to produce transparent films by either casting a CNF water dispersion in a Petri dish, or by vacuum filtration to give strong and stiff networks, as shown previously by Isogai et al.7 and Berglund et al.8Modification of cellulose can be necessary to impart specific properties to the cellulose surface or cellulose bulk materials. Specifically, modification of cellulose with polymers has been achieved through covalent and non-covalent, or physical attachments. Covalent grafting of cellulose with polymers is an efficient method to modify cellulose; however, it usually requires inert atmospheres and organic solvents.9–12 Physical adsorption of polymers to cellulose13 can be performed under benign aqueous conditions and has been utilising in the pulp and paper industry for decades. Most commonly electrostatic interactions are exploited, owing to the slight overall negative charge of native cellulose, which can easily be enhanced to a higher negative charge through simple chemical oxidation. In paper making, for example, cationic polyelectrolytes are used to modify the cellulose, to improve properties such as wet-strength, for example. Examples of CNF modification through physical adsorption include a recent work by Isogai and coworkers, where the anionic carboxylate groups, imparted to the CNF during their preparation, were utilised to ionically bind to a quaternised amine at the chain end of a poly(ethylene glycol) (PEG).14 The PEG-grafted CNF were evaluated as fillers in a poly(L-lactide) (PLLA) matrix and the PEG modification improved the dispersion of the CNF in the matrix, which subsequently increased the stiffness, strength and toughness of the nanocomposites. Recently, several research groups have investigated the adsorption of non-polyelectrolytes to cellulose, specifically hemicelluloses, which have a natural affinity for cellulose and bind through van der Waals and hydrogen bonding interactions.13
Xyloglucan (XG) is a branched polysaccharide (see Fig. S1† for the chemical structure) which has been utilised in the biomimetic modification of cellulose in its natural state,15,16 with chemical modifications,17 and as a component of block copolymers18,19 as well as XG-functional latex nanoparticles.20 This has resulted in an interest to study the adsorption of XG to nanocelluloses. Cathala and coworkers have performed several investigations of the adsorption behaviour of XG to CNC in concentrated and dilute regimes,15,21 and have prepared XG-CNC multi-layer films.22,23 Likewise, the adsorption of XG to CNF has also been studied by Österberg and colleagues, showing that XG binds efficiently and has a positive influence on the dispersibility of the CNF.24–26 Our group has recently reported the synthesis of XG-decorated PMMA latex particles prepared via emulsion reversible addition–fragmentation chain-transfer (RAFT) polymerisation, for the biomimetic modification of cellulose substrates; filter paper and model cellulose surfaces.20
Zwitterionic polymers, such as poly(sulfobetaines), are an interesting class of materials, composed of positive and negative charges resulting in an overall neutral net charge.27 They exhibit the “anti-polyelectrolyte” effect, whereby in an aqueous solution increasing the ionic strength, through the addition of salts, which leads to an improvement in aqueous solubility.28 Poly(sulfobetaines) are well-known for their biocompatible properties, including anti-biofouling properties.15,28 N,N-Dimethyl(methacryloylethyl)ammonium propanesulfonate (SBMA) is a well-studied sulfobetaine methacrylate zwitterionic monomer, which can be polymerised via aqueous RAFT polymerisation yielding the corresponding polymer, PSBMA, with controlled molecular weights and narrow dispersities.29 The upper critical solution temperature (UCST) of this polymer has been shown to vary with the polymer molecular weight, concentration and presence of salt.30 Recently Willcock et al. have studied the effect of polymer architecture on the UCST of poly(sulfobetaines) which have been copolymerised with a divinyl monomer to yield branched poly(sulfobetaines), highlighting that polymer architecture also affects the UCST.31 Several publications report the covalent grafting of cellulosic substrates with zwitterionic polymers, most commonly PSBMA, to give hemocompatibility,32–34 applications in sensors35 and membranes,36 and generally impart anti-biofouling properties.37,38 However, all of these reports use polymerisation techniques which require organic solvents, inert atmospheres and the subsequent removal of the catalyst, therefore, it is desirable to find methods that can be performed under benign aqueous conditions.
In the present work we describe the synthesis of xyloglucan-block-poly(sulfobetaine methacrylate) (XG-b-PSBMAn) copolymers synthesised via aqueous RAFT polymerisation for the subsequent modification of CNF through biomimetic adsorption. The combination of this thermoresponsive, antibacterial polymer with CNF is foreseen to result in exciting new functional materials that could find applications such as packaging, membranes, for controlled release and wound dressings.39 Herein, the adsorption of the polymers to cellulose was investigated in detail through QCM-D experiments using CNF surfaces prepared in situ. The polymers were further utilised to prepare CNF-polymer composite films which showed effective swelling properties at both 5 and 60 °C, compared to a reference CNF film. This route allows a facile biomimetic modification of cellulose towards super-absorbing, anti-biofouling CNF-based materials.
2. Results and discussion
The preparation of XG-b-PSBMA block copolymers was performed utilising RAFT polymerisation. The XG block was introduced through the use of an XG-functional macromolecular RAFT chain transfer agent, denoted here as XG-RAFT, see Fig. 1. The XG-RAFT was synthesised according to previous reports,20 and used to prepare XG-b-PSBMA block copolymers, whilst 4-cyano-4(phenylcarbothioylthio) pentanoic acid (CTP) was used to prepare the reference PSBMA homopolymers.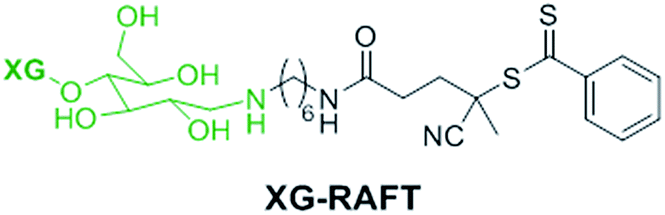 | ||
| Fig. 1 Chemical structure of the XG-RAFT agent used in this study. | ||
2.1 Preparation of XG-b-PSBMA block copolymers
The RAFT polymerisation of the SBMA monomer was conducted in 0.5 M NaCl aqueous solution employing XG-RAFT and the radical initiator 4,4′-azobis(4-cyanovalenic acid) (ACVA), see Scheme 1, with similar reaction conditions to literature procedures.29,31 Two targeted degrees of polymerisation (DPn) were chosen; 266 and 710 (corresponding to 75 and 200 kg mol−1), to observe differences in behaviour between shorter and longer polymer chains. Reference PSBMAn homopolymers were prepared utilising 4-cyano-4(phenylcarbonothioylthio)pentanoic acid (CTP) as the RAFT agent and the same polymerisation conditions as those used to prepare the XG block copolymers.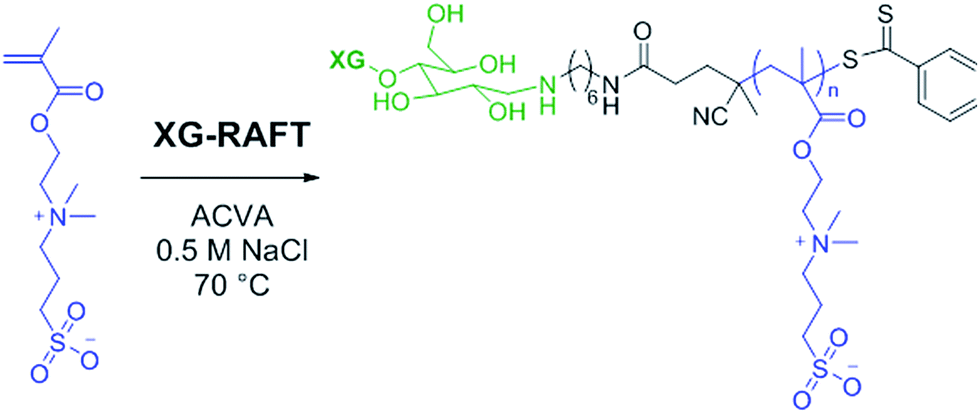 | ||
| Scheme 1 Reaction conditions for the RAFT-mediated polymerisation of SBMA utilising the XG-RAFT agent. | ||
Due to the broad dispersity of the XG-RAFT, the resulting block copolymers also had similar dispersities, see Table 1. However, the kinetic evaluation of the block copolymers showed similar results to reference polymerisations, with the polymerisations following first order kinetics as expected for a controlled radical polymerisation, see Fig. S2.† The polymers were purified by dialysis at elevated temperatures (45–50 °C) with a dialysis membrane with a molecular weight cut off of 50 kg mol−1. This was to ensure that any unreacted XG-RAFT was removed and an elevated temperature was used to ensure the polymers were above their upper critical solution temperature (UCST), in an extended conformation, to allow full removal of any NaCl present.
| Targeted polymer composition | Conversiona (%) | Mn, theoretical (kg mol−1) | Mnb (kg mol−1) | Mwb (kg mol−1) | Đb | UCSTc (°C) |
|---|---|---|---|---|---|---|
| a Calculated by 1H NMR.b Determined using 10 mM NaOH aqueous SEC, using conventional calibration with pullulan standards.c Determined by UV-Vis turbidity measurements.d Determined by SEC with DMSO as the eluent, using conventional calibration with pullulan standards. | ||||||
| PSBMA266 | 87 | 65.7 | 27.0 | 32.6 | 1.21 | 16.6 |
| PSBMA710 | 83 | 166 | 50.3 | 64.4 | 1.28 | 22.2 |
| XG-b-PSBMA266 | 92 | 68.7 | 44.0 | 94.4 | 2.15 | 20.1 |
| XG-b-PSBMA710 | 89 | 177 | 46.4 | 126 | 2.71 | 29.8 |
| XG-RAFT | — | — | 11.4d | 32.5d | 2.85d | — |
The 1H NMR spectra of the polymers are shown in Fig. 2A, where the PSBMA polymer peaks in PBSMA266 and PBSMA710 are observed as expected, and in the XG-b-PSBMAn polymers the presence of the XG block is most clearly seen with singlet peaks at 5.1 and 4.9 ppm, corresponding to the hydrogens bonded to the anomeric carbon centres in the XG backbone. The FT-IR spectra of these samples; Fig. 2B, shows sharp peaks at 1730 cm−1 corresponding to the carbonyl group in all the samples containing PSBMA. In the FT-IR spectrum of XG-b-PSBMA266 and XG-b-PSBMA710 a larger contribution for the broad hydroxyl peak between 3600–3200 cm−1 gives an indication that XG is present. Thermogravimetric analysis of the samples show a slightly different thermal stability between the PSBMA homopolymers and XG-b-PSBMAn block copolymers, indicating that the presence of the XG block causes a reduction in the thermal stability of the block copolymers (Fig. S3†).
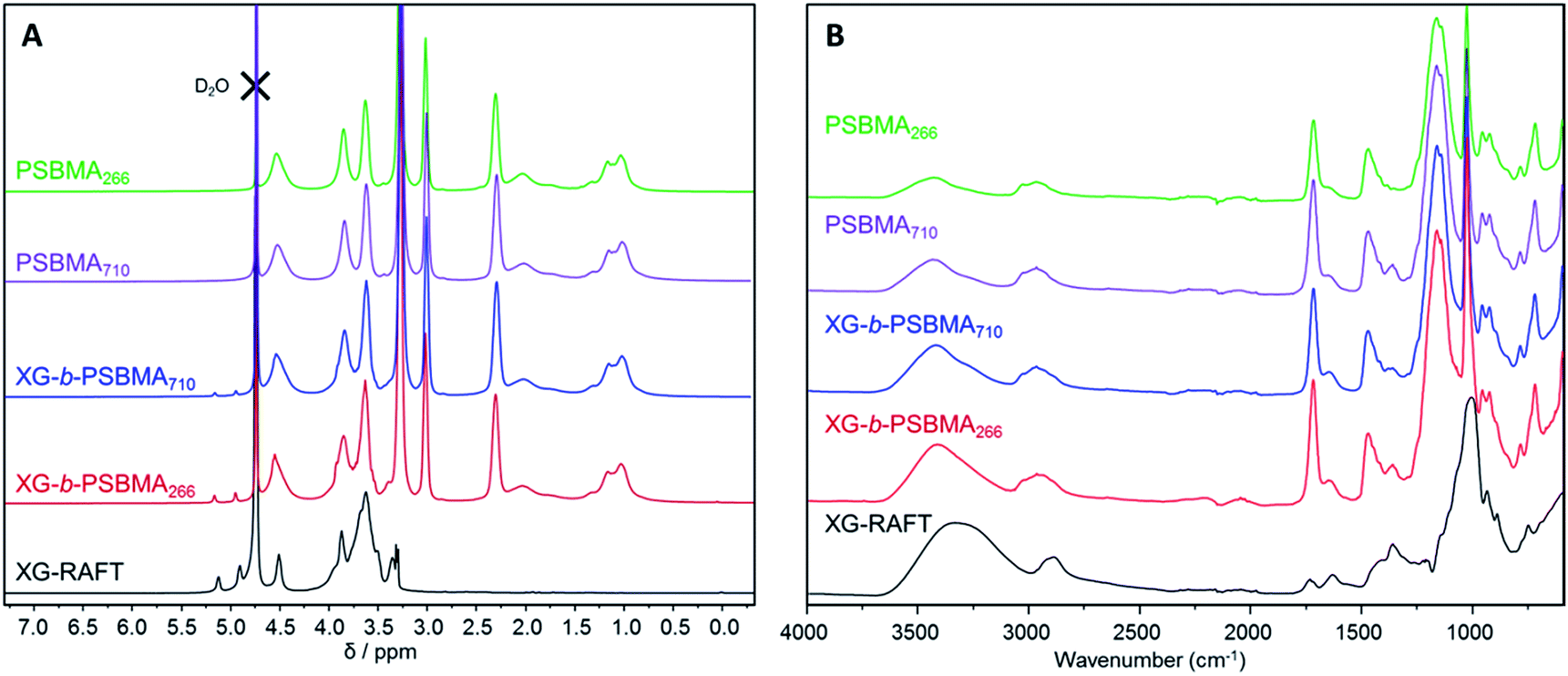 | ||
| Fig. 2 Characterisation of XG-RAFT, PSBMAn and XG-b-PSBMAn samples; (A) 1H NMR (0.5 M NaCl in D2O, 400 MHz) and (B) FT-IR spectroscopy. | ||
PSBMA is well-known to be thermoresponsive, exhibiting a UCST dependent on the molar mass, architecture of the polymer, polymer concentration and electrolyte concentration.30,31 Here, we observed an increase in UCST with increasing molar mass for both the XG-b-PSBMA samples and the reference PSBMA samples, Fig. 3, with polymer concentrations of 5 mg mL−1. This increase in UCST with increased PSBMA molar mass is well-known, and can be attributed to the inter- and/or intra-molecular interactions between the ammonium cation and sulfo-anion present in the SBMA monomer.30 Therefore, the increase in UCST values for the block copolymers containing XG was considered to be due to the presence of higher molar mass polymer chains present, as observed by increased Mw by SEC (Table 1). The two XG-block copolymers also showed a broader transition, due to the broader dispersity of polymer molar mass in these samples. The UCST measurements were also conducted at a lower concentration, i.e. 2.5 mg mL−1 (Fig. S4†), where the UCST showed a clear shift to a lower temperature, as expected, and the UCST was not detectable for the lowest molecular weight sample; PSBMA266.
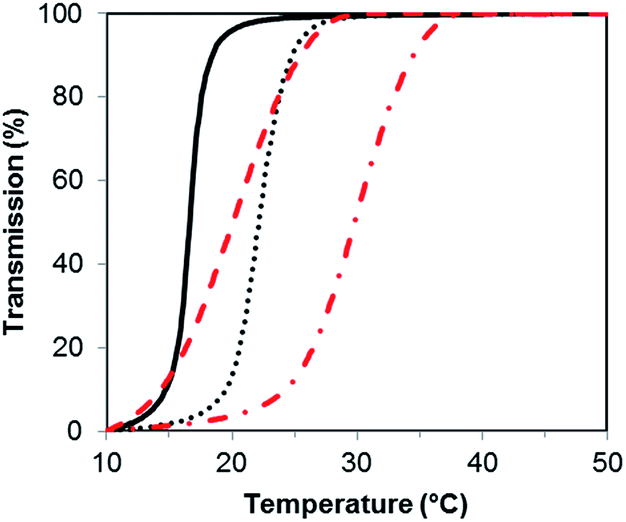 | ||
| Fig. 3 Turbidity curves for samples; PSBMA266 (solid black line), PSBMA710 (dotted black line), XG-b-PSBMA266 (dashed red line) and XG-b-PSBMA710 (dot-dash red line), for the cooling step (1 °C min−1) at a concentration of 5 mg mL−1 in MilliQ water. | ||
2.2 In situ QCM-D adsorption
To evaluate the XG-b-PSBMAn zwitterionic polymers adsorption to cellulose nanofibrils (CNF), quartz crystal microbalance with dissipation monitoring (QCM-D) was utilised. QCM-D allows the observation of a build-up of polymer layers in situ as an increase in mass adsorbed is detected as a decrease in frequency. The change in dissipation provides information about the viscoelastic properties of the adsorbed layer. The block copolymers were studied for their adsorption to a CNF surface in the QCM-D (Fig. 4), as the ultimate aim is to use them for CNF modification. The CNF surfaces were prepared in situ during the QCM-D experiments as previously reported.40,41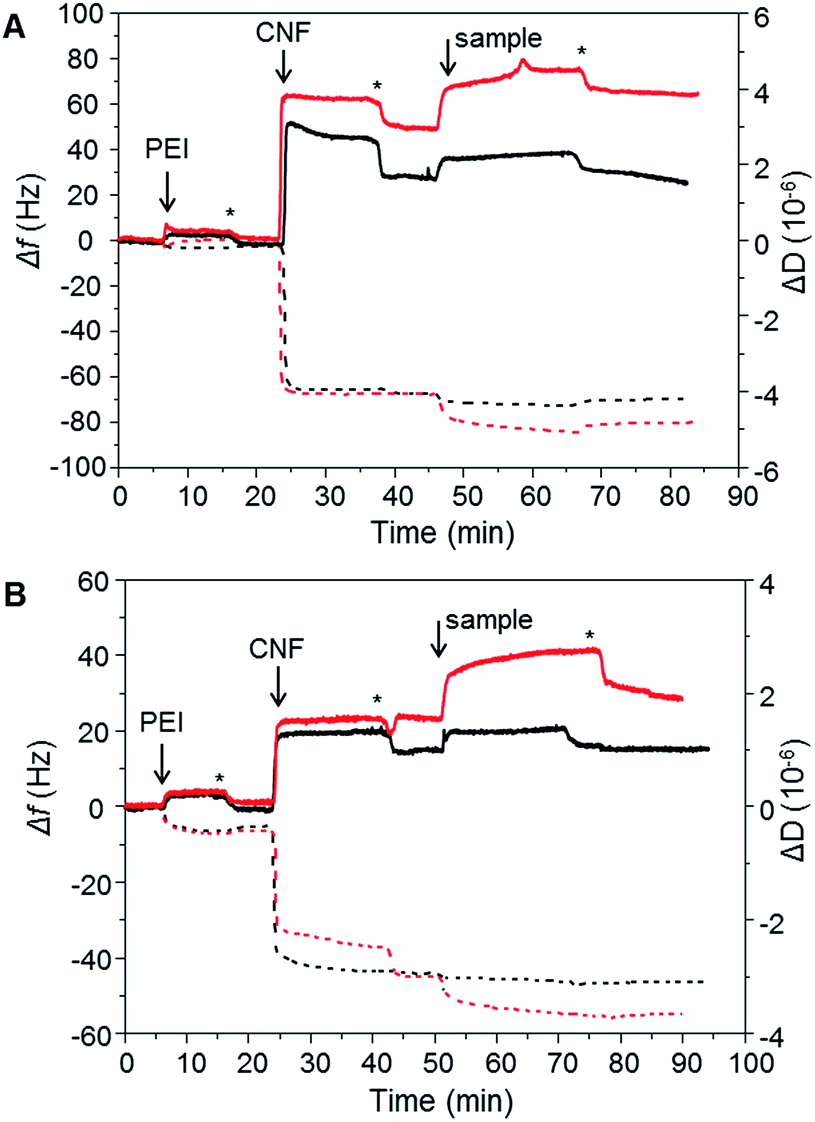 | ||
| Fig. 4 In situ QCM-D experiment showing adsorption of PEI, CNF and the polymer samples. (A) PSBMA266 (black lines) and XG-b-PSBMA266 (red lines), (B) PSBMA710 (black lines) and XG-b-PSBMA710 (red lines). Change in frequency, Δf, (dashed lines) and change in dissipation, ΔD, (solid lines). Rinsing steps with MilliQ water are denoted with an asterisk (*). | ||
The synthesised polymers were introduced to the QCM-D flow chamber after the CNF surface had been treated to a rinsing step. Each QCM-D experiment was performed in duplicate. The change in frequency (Δf) and change in dissipation (ΔD) for each of the experiments are shown in Fig. 4. When introducing the XG-b-PSBMAn polymers to the CNF layer, adsorption was observed in both cases. After a rinsing step the frequency did not increase for either sample, indicating that the XG-b-PSBMAn copolymers adsorbed to the CNF irreversibly. More specifically, the XG-b-PSBMA266 (Fig. 4A) showed a larger decrease in frequency with adsorption compared with XG-b-PSBMA710 (Fig. 4B), indicating a larger mass adsorbed for the shorter block copolymer. This result was not expected as it was hypothesised that the longer block copolymer; XG-b-PSBMA710, would adsorb a higher mass to the CNF surface than the shorter block copolymer; XG-b-PSBMA266, as has been shown in our previous work on block copolymers42,43 and latex nanoparticles.20,44 It may be that this polymer, which is water soluble and does not form micelles in aqueous solution, shields the adsorbing block rather than exposes it, as is the case with previously studied amphiphilic block copolymers,42 which then would cause the unexpected low adsorption. Furthermore, in these QCM-D experiments the initial mass of CNF adsorbed to the surface was different between the two samples. To understand how the mass of CNF adsorbed impacted the adsorbed masses of polymer, the mass adsorbed after each step was calculated using the Sauerbrey model,45 see Table 2. The ratio of CNF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :polymer adsorbed to the surface was calculated from the mass of adsorbed CNF and the mass of adsorbed polymer. These calculations showed that the block-copolymers (XG-b-PSBMA266 and XG-b-PSMBA710) gave CNF:polymer mass ratios of 1:0.18 and 1:0.23 respectively, whilst the homopolymers (PSBMA266 and PSBMA710) gave values of 1:0.03 and 1:0.05, respectively, showing a clear advantage of using the XG block as a biomimetic anchoring block to the CNF with a zwitterionic polymer as the functionalising component. Furthermore, the dissipation increased with adsorption of both XG-b-PSBMAn copolymers, indicating that the adsorbed layer was softer and more hydrated than the CNF surface before adsorption had occurred.
:polymer adsorbed to the surface was calculated from the mass of adsorbed CNF and the mass of adsorbed polymer. These calculations showed that the block-copolymers (XG-b-PSBMA266 and XG-b-PSMBA710) gave CNF:polymer mass ratios of 1:0.18 and 1:0.23 respectively, whilst the homopolymers (PSBMA266 and PSBMA710) gave values of 1:0.03 and 1:0.05, respectively, showing a clear advantage of using the XG block as a biomimetic anchoring block to the CNF with a zwitterionic polymer as the functionalising component. Furthermore, the dissipation increased with adsorption of both XG-b-PSBMAn copolymers, indicating that the adsorbed layer was softer and more hydrated than the CNF surface before adsorption had occurred.
| Sample | Δfplat CNF (Hz) | Δfplat poly (Hz) | Mass adsorbed CNFa (mg m−2) | Mass adsorbed polymera (mg m−2) | Mass ratio CNF:poly |
|---|---|---|---|---|---|
| a Mass adsorbed calculated using the Sauerbrey model. | |||||
| PSBMA266 | −67.9 | −70.2 | 12.0 | 0.4 | 1:0.03 |
| PSBMA710 | −44.2 | −46.5 | 7.8 | 0.4 | 1:0.05 |
| XG-b-PSBMA266 | −67.9 | −80.4 | 12.0 | 2.2 | 1:0.18 |
| XG-b-PSBMA710 | −45.0 | −54.9 | 8.0 | 1.8 | 1:0.23 |
The PSBMA266 homopolymer exhibited a low amount of adsorption to the CNF layer, observed as a small decrease in frequency, however, with rinsing the frequency increased to a similar value as before the adsorption (Fig. 4A). This indicated that whilst the homopolymer did adsorb to the CNF, it was a weak and reversible binding which caused the homopolymer to be washed away with the rinsing step. The dissipation value also decreased to the same value as observed before the polymer was introduced to the CNF surface, corroborating that the PSBMA266 was indeed rinsed away from the surface due to a weak and reversible binding. The PSBMA710 homopolymer showed no adsorption to the CNF surface by QCM-D as no decrease in frequency was observed, see Fig. 4B, although the dissipation did increase slightly which suggests that a small amount was adsorbed. As the PSBMA polymer is easily hydrated, a small amount of adsorbed polymer can be enough to increase the absorbed water and therefore increase the dissipation. As expected, the QCM-D experiments show that the XG block is necessary for an irreversible binding to the cellulose, which is not observed for the zwitterionic homopolymer.
After adsorption of each polymer sample to CNF surfaces prepared in situ, the surfaces were dried and analysed by atomic force microscopy (AFM), see Fig. S5.† The surfaces with XG-b-PSBMAn adsorbed showed a slight difference in surface topography, where the CNF are more difficult to individually distinguish after block copolymer adsorption, compared to the PSBMAn homopolymer surfaces, indicating the presence of the block copolymers on the CNF surface.
2.3 CNF and composite film formation
To produce composite films of CNF and the homo- and block-copolymers, a facile vacuum filtration technique was applied. The XG-b-PSBMA710 sample was chosen for preparing CNF-composite films, as this polymer adsorbed the highest mass ratio in the QCM-D experiments, vide supra, and exhibited an UCST above room temperature (29.8 °C). The corresponding homopolymer, PSBMA710, was also utilised to prepare a CNF-composite film in order to assess any differences in film properties due to the XG-adsorbing block. The CNF was added to polymer solutions (1.25 mg mL−1) and mixed at 40 °C to ensure adsorption above the UCST of the polymers, with a targeted solids content ratio of CNF:polymer of 75:25 wt%. The suspensions were filtered under vacuum and subsequently dried in a fume cupboard, resulting in three different composite films; CNF-reference (CNF-ref), i.e. no polymer present, CNF/PSBMA710 and CNF/XG-b-PSBMA710. The CNF-ref and CNF-composite films all formed translucent films after drying, with thicknesses between 62–76 μm, see Table 3.
| Sample name | Thickness (μm) | Densitya (kg m−3) | Porosityb (%) |
|---|---|---|---|
| a Density calculated from at least three data points.b Porosity calculated from equation, porosity = 1 − (densityfilm/densitycellulose), where the density for cellulose is assumed to be 1500 kg m−3. | |||
| CNF-ref | 76 | 1487 ± 20 | 1 |
| CNF/XG-b-PSBMA710 | 67 | 1477 ± 3 | 2 |
| CNF/PSBMA710 | 62 | 1406 ± 30 | 6 |
The calculated density was highest for the CNF-ref and the CNF/XG-b-PSBMA710 films; 1487 ± 20 and 1477 ± 3 kg m−3 respectively, and decreased for the CNF/PSBMA710 composite film to 1406 ± 30 kg m−3. Accordingly, the calculated porosity of the films was lowest for the CNF-ref and CNF/XG-b-PSBMA710 (1–2%), whilst the CNF/PSBMA710 composite film had the highest porosity of 6%. This suggests a poorer compatibility of the PSBMA710 homopolymer with the CNF compared with the XG-b-PSBMA710 block copolymer. The CNF-ref and composite films were analysed by FT-IR and TGA analyses to confirm the presence of both CNF and polymer in the samples, see Fig. 5. The FT-IR spectra of the composite films (Fig. 5A) show carbonyl peaks (1730 cm−1) corresponding to the PSBMA polymer: as expected, the peak is higher in intensity for the CNF/XG-b-PSBMA710 composite film. This suggested that in the case of the homopolymer, some polymer was lost during film formation as the PSBMA710 does not adsorb as well to the CNF as the XG-b-PSBMA710. This is corroborated by the QCM-D adsorption experiment results (Fig. 4) which highlighted that the XG-b-PSBMA710 copolymer showed irreversible binding to CNF whereas the PSBMA710 homopolymer did not.
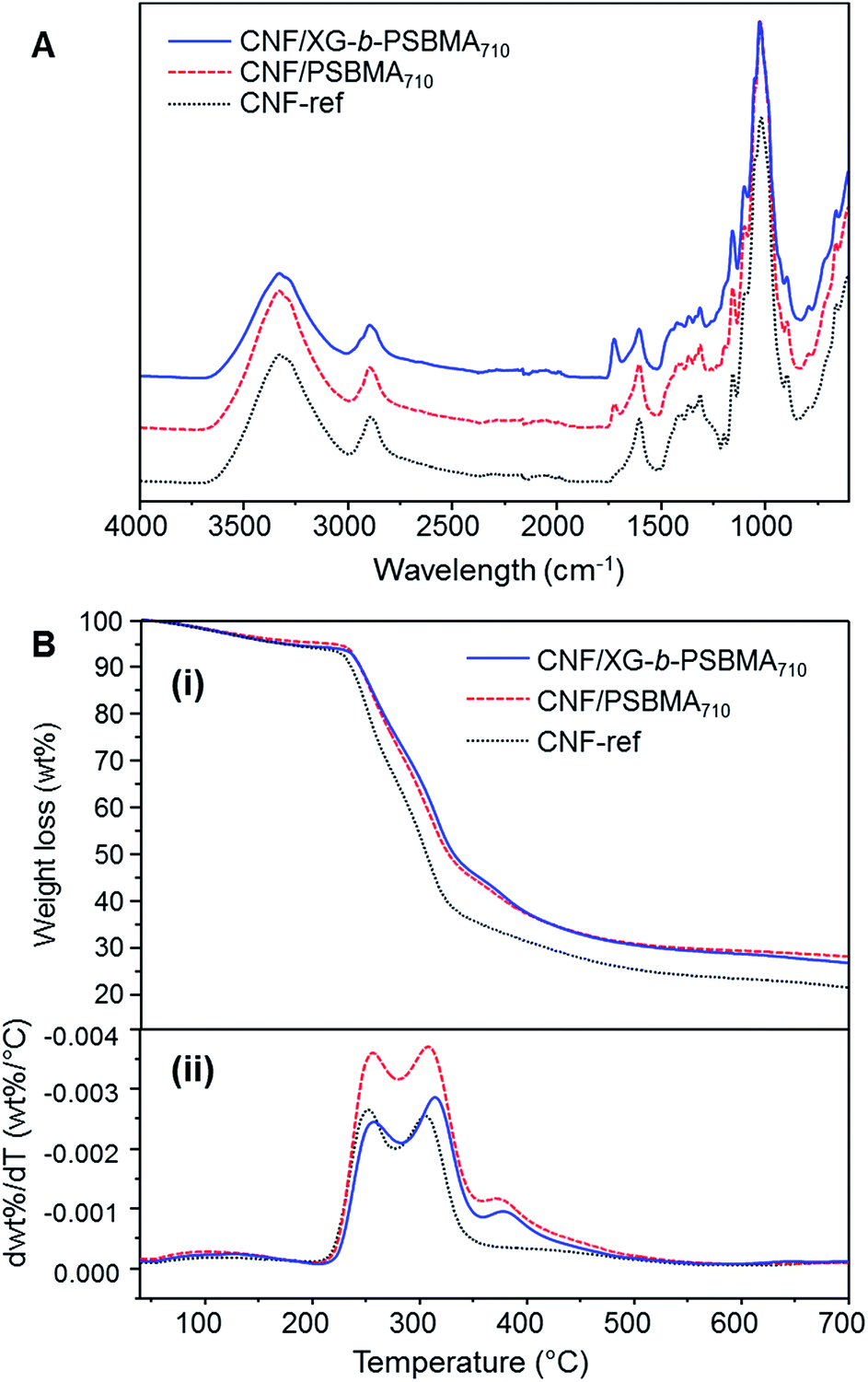 | ||
| Fig. 5 Characterisation of CNF composite films; CNF-ref, CNF/PSBMA710 and CNF/XG-b-PSBMA710. (A) FT-IR spectra, (B) (i) thermogravimetric weight loss curves and (ii) thermogravimetric derivative curves. | ||
The lower thickness of the CNF/PSBMA710 film may also indicate that some material was lost during film formation, whereas when using the XG-b-PSBMA710 the XG anchoring block minimised loss of material. After film formation the filtrate was collected and freeze-dried in order to analyse any residual polymer by 1H NMR analysis. PSBMA710 was clearly observed in the filtrate from the CNF/PSBMA710 film formation, whilst a very low amount of residual polymer could be observed from the filtrate of the CNF/XG-b-PSBMA710 film formation. This further indicated that the XG block was necessary to improve polymer retention in the CNF/polymer composite films, and also agrees with the assumption that the CNF/PSBMA710 film was thinner than the CNF/XG-b-PSBMA710 film due to loss of polymer during film formation. TGA analyses (Fig. 5B) confirmed the presence of polymer due to the difference in thermal degradation patterns observed, which is clearly shown through the first derivative curves (Fig. 5B(ii)). For the CNF/PSBMA710 and the XG-b-PSBMA710 composite films, the first derivatives show a third peak or inflection point at 375 °C, which is not present for the CNF-ref sample, indicating that this peak is due to the presence of the added polymers in these CNF composite films.
The composite films were imaged by SEM in order to make structural comparisons, see Fig. 6. The dried films were prepared so that the morphology through the cross-section of a cryo-fracture could be imaged. It was apparent that the dried films varied in morphology in the cross-section; the CNF-ref shows a more commonly reported CNF film cross-section structure,8,43 while the two CNF/polymer composites appear slightly more compact in structure. This seems counterintuitive as the calculated density was lower for the CNF/PSBMA710 composite film compared with the CNF-ref. Furthermore, the CNF/PSBMA710 film exhibited a more ordered layered structure to some extent, possibly due to phase separation.
 | ||
| Fig. 6 Scanning electron micrographs of cryo-fracture cross section surfaces of each film; (A) CNF-ref, (B) CNF/XG-b-PSBMA710 and (C) CNF/PSBMA710. | ||
2.4 Swelling of CNF composite films
It was noted that during the filtration step the CNF composite films exhibited high swelling, especially for the polymer containing films, shown by a slow dewatering process. Therefore, the swelling of the films without further treatment (except for drying in a fume cupboard at room temperature) was systematically studied. Interestingly, we could find no previous reports on the swelling of TEMPO-oxidised CNF films. However, a recently reported study of the swelling of quaternised-CNF nanopapers, at room temperature, indicated swelling in the region of 100 g of water per gram of cellulose, for the CNF nanopaper with the lowest degree of quaternisation.46 The CNF-ref film exhibited maximal swelling after 120 h at 60 °C, of 2540%, corresponding to 25.4 g of water per gram of cellulose. However, due to the difference in the chemical functionality of the CNF and the processing of the film after formation, through heating at 93 °C under 70 mBar vacuum pressure, it is unsurprising that we observed different degrees of swelling. Here, we investigated the films swelling properties at two different temperatures: 5 and 60 °C (Fig. 7), corresponding to temperatures either above or below the polymers UCSTs.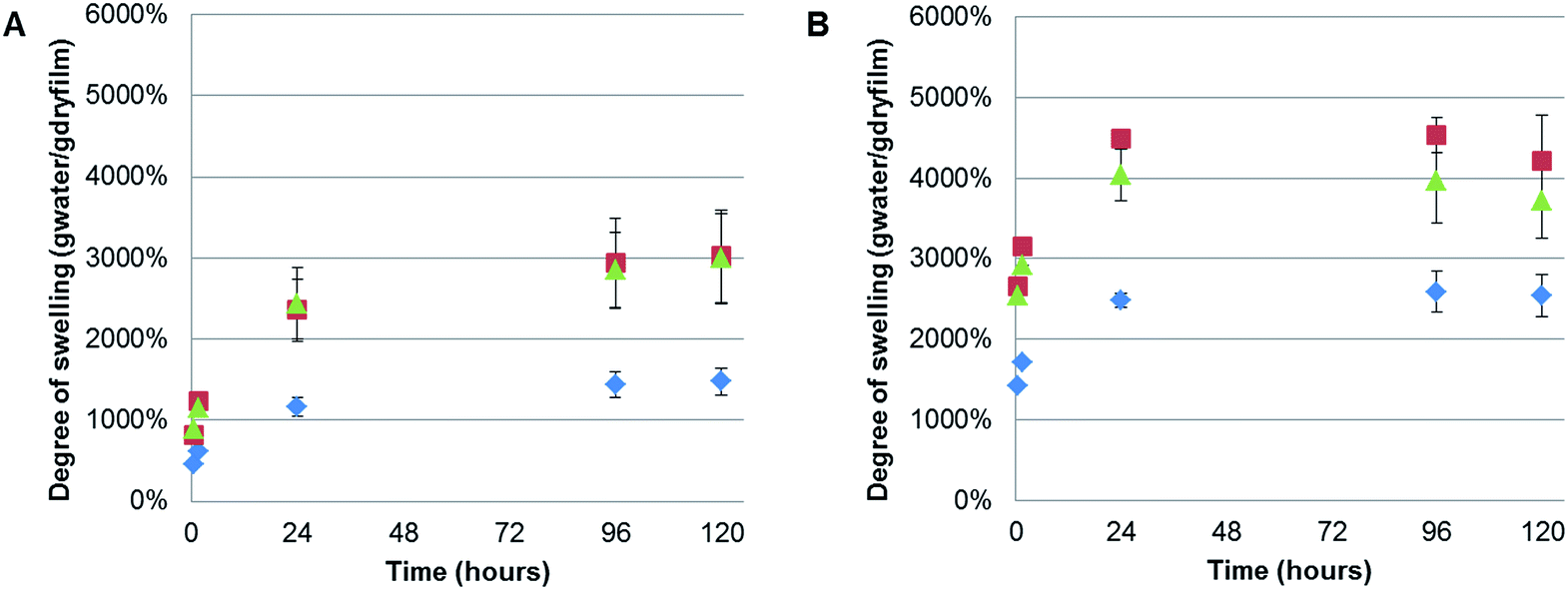 | ||
| Fig. 7 Swelling test in deionised water at two different temperatures; (A) 5 °C and (B) 60 °C for 0.5, 1, 24, 96 and 120 h, for CNF-ref (blue diamonds), CNF/PSBMA710 (green triangles) and CNF/XG-b-PSBMA710 films (red squares). | ||
All of the films exhibited greater swelling (approximately 1000% more) at 60 °C than at 5 °C, including the CNF-ref, which was initially considered to have no thermo-responsive properties. However, previous reports have shown that the swelling of wood occurred at faster rates with elevated temperatures in water,47 and the same result has also been shown for various compressed cellulose fibre pellets.48 Moreover, this strong swelling dependence on temperature closely obeyed the classical Arrhenius equation, indicating that the swelling of wood was an activated process similar to a classical chemical reaction.47 Therefore, the observed difference in swelling at 5 and 60 °C with the currently described CNF films is to be expected. Consequently, no significant difference due to the thermo-responsive properties, UCST, of the polymers incorporated in the composite films could be concluded, due to the temperature dependence of the CNF swelling.
Nonetheless, it was clear that incorporating either PSBMA710 or XG-b-PSBMA710 in the films resulted in a significant increase of the degree of swelling at both temperatures. The maximal degree of swelling, measured in gram of absorbed water per gram of dry film, was observed at 60 °C after 120 h for all samples. The CNF/PSBMA710 film resulted in a higher degree of swelling (3120%) which is an increase of 45% compared with the CNF-ref film. The CNF/XG-b-PSBMA710 film exhibited the highest degree of swelling of 4200%, an increase of 70% compared to the CNF-ref. An additional reference film was prepared, comprising XG-RAFT and CNF, with the amount of added XG-RAFT corresponding to the amount of XG present in the XG-b-PSBMA/CNF composite film. It was shown that the CNF/XG-RAFT film swelled 24% more than the CNF-ref, highlighting that the XG itself also attributes to the swelling.
This increase in the swelling of the CNF/XG-b-PSBMA710 film is a further indication that the XG resulted in an improved compatibility between the PSBMA and the CNF. The final degree of swelling, at 5 °C after 120 h, was 3020% for CNF/XG-PSBMA710 and 2990% for CNF/PSBMA710 while the CNF-ref swelled to merely 1480%. It was hypothesised that the porosity would affect both the rate at which, and how much, the films could swell in water. However, as the porosity was similar for the CNF-ref and CNF/XG-b-PSBMA710, but the swelling varied dramatically, it was concluded that the polymer functionality had a greater influence over the swelling of the films. Additionally, temperature affected the degree of swelling for all samples. This could be rationalised by entropically favourable swelling at higher temperatures, as discussed above. Another contributing factor is that the pH of water at 5 °C is lower than the pH at 60 °C, therefore the carboxylate groups present on the CNF surface are protonated to a larger extent at lower temperature, resulting in decreased water solubility and decreased swelling.
The kinetics of swelling appeared slightly different between the samples swelled at 5 and 60 °C. At 5 °C the swelling increased more slowly at the beginning of the experiment and reach a plateau after 96 hours, whilst at 60 °C a plateau was reached after 24 hours with a slight decrease in swelling over time. This could be attributed to the slower diffusion at lower temperatures, resulting in longer times needed to reach a plateau and therefore equilibrium. Whilst the reduction in the degree of swelling after 120 h at 60 °C is within error, and may be due to a slight loss of water during measurements.
Photographs of the films before and after swelling at 60 °C, Fig. 8A, highlight the large difference between the relative thicknesses. Due to the similarity of all films before swelling only the CNF-ref is shown for the illustration of swelling direction, Fig. 8A(i). The swollen films Fig. 8A(ii)–(iv) were all similar in appearance. The geometries of the samples were measured before and after swelling, which showed that all samples swelled significantly (from 72 μm to over 7 mm) in the z-direction, i.e. the thickness/height, and not in the width.
 | ||
| Fig. 8 (A) Photographs showing films before swelling; (i) CNF-ref, and after swelling; (ii) CNF-ref, (iii) CNF/XG-b-PSBMA710 and (iv) CNF/PSBMA710, for 24 h at 60 °C. Scanning electron micrographs show cross-sections of the cryo-fractured swollen films; (B) CNF-ref and (C) CNF/XG-b-PSBMA710. | ||
In order to elucidate the possible swelling mechanism of the CNF composite films, SEM was used to investigate the structural properties of the hydrogels before and after swelling. The hydrogel samples were swollen at 5 or 60 °C for at least 120 h and were subsequently were frozen with liquid nitrogen in the swollen state and cryo-fractured after freeze drying to give cross-sections. The porous structure of the swollen CNF-ref and CNF/XG-b-PSBMA710 can be seen in Fig. 8B and C respectively, showing the large pores which are able to entrap a large amount of water inside the network. It was considered that the swelling kinetics would be dependent on any pores and channels present in the samples specifically in the z-direction, or thickness of the films.
The swollen films were also assessed by compression testing to ascertain the mechanical properties of the swollen hydrogels, see Table S1 and Fig. S6.† The compressive stress reduced from 0.386 ± 0.050 MPa for the CNF-ref, to 0.060 ± 0.003 MPa and 0.093 ± 0.011 MPa for the CNF/XG-b-PSBMA710 and CNF/PSBMA710 respectively. The compressive strain of the CNF-ref (58%) was higher than that of the CNF/XG-b-PSBMA710 film (38%) and the CNF/PSBMA710 film (42%). These results suggest that the incorporation of polymer reduces then number of contact points between fibrils, ultimately weakening the hydrogel, which may explain the improved swelling of the networks containing the XG-b-PSBMA710 and PSBMA710 polymers.
3. Conclusions
A biomimetic approach towards the modification of CNF has been presented through the preparation of zwitterionic block copolymers containing a xyloglucan (XG) block. The zwitterionic polymer poly(sulfobetaine methacrylate) (PSBMA) was prepared utilising aqueous RAFT polymerisation, with an XG-functional macromolecular RAFT chain transfer agent (XG-RAFT), targeting two different degrees of polymerisation. The resulting polymers exhibited UCST behaviour which was dependent on molecular weight and the homopolymer or block copolymer architecture. Adsorption of the polymers to a CNF surface prepared through a quartz crystal microbalance with dissipation monitoring (QCM-D) in situ experiment showed that the XG block was necessary to achieve irreversible binding of the zwitterionic polymer to the CNF surface. A facile vacuum filtration methodology was utilised to prepare neat CNF, CNF/PSBMA710 and CNF/XG-b-PSBMA710 films, where the successful formation of the films was confirm by FT-IR and TGA analyses. The XG block enabled a higher amount of the XG-b-PSBMA710 block copolymer to be retained within the CNF-composite film than the PSBMA710 homopolymer. The density and porosities of the resultant dry films suggested an improved compatibility between the CNF and the zwitterionic polymer in the presence of the XG anchoring block. Whilst the swelling properties of the CNF-composite films seemed to be dominated by the presence of polymer, the kinetics of swelling and overall degree of swelling were strongly affected by the temperature of the swelling experiment. The composite films showed super-absorbing properties of up to 4200% (for CNF/XG-b-PSBMA710) when placed in water at 60 °C. Whilst there was no significant thermo-response which could be attributed to the PSBMA block, for example due to the UCST, it was clear that for an improved compatibility between the CNF and zwitterionic block, the XG block was crucial. These results demonstrate a biomimetic approach for preparing cellulose-based smart materials, with potential in applications such as super-absorbent, anti-biofouling materials.4. Experimental
4.1 Materials
The xyloglucan macromolecular RAFT agent (XG-RAFT), bearing a dithiobenzoate moiety, was prepared as previously described,20 the synthesis of which relies on the reductive amination of the XG reducing chain end,49 followed by coupling the RAFT chain transfer agent using EDC chemistry, and had an average molecular weight of 17.4 kg mol−1. [2-(Methacryloyloxy)ethyl] dimethyl-(3-sulfopropyl)ammonium hydroxide (SBMA, 97%), 4-cyano-4(phenylcarbonothioylthio)pentanoic acid (CTP, 97%), 4,4′-azobis(4-cyanovaleric acid) (ACVA, 98%), polyethyleneimine (PEI, 50 wt% solution, Mn = 60 kg mol−1 by GPC, Mw = 750 kg mol−1 by LS) were purchased from Aldrich and used as received. Dialysis membrane tubing (MWCO 50 kDa) was received from Spectrumlabs. The QCM crystals (AT-cut quartz crystals, 5 mHz resonance frequency, SiO2 surface with 50 nm thickness) were purchased from Q-sense AB and rinsed with MilliQ water then ethanol, dried under N2 and plasma cleaned for 2 min before use.4.2 Characterisation
1H NMR spectra were recorded at room temperature with a Bruker Avance 400 MHz spectrometer, using D2O or a 0.5 M NaCl D2O solution as the deuterated solvent.FT-IR spectra were recorded on a Perkin-Elmer Spectrum 2000 FT-IR equipped with a MKII Golden Gate, single reflection ATR System from Specac Ltd, (London, UK). The ATR-crystal used was a MKII heated diamond 45° ATR top plate. For each spectrum 16 scans were recorded. The spectra were normalised to the region between 4000–3100 cm−1 unless stated otherwise.
Molecular weights Mn and Mw and dispersities, Đ, of the polymers were determined by SEC; a HPLC-Aqueous SEC system with a Dionex Ultimate-3000 HPLC system (Dionex, Sunnyvale, CA, USA) was used, containing three PSS suprema columns in series (300 × 8.00 mm, 10.0 μm particle size) with 30.0 Å, 1000 Å and 1000 Å pore sizes, together with a guard column (50.0 × 8.00 mm, 10.0 μm particle size) with 10.0 mM NaOH as the mobile phase and a flow rate of 1.0 mL min−1 at 40 °C. The columns were calibrated with using narrow linear pullulan standards, nominal molecular weight ranging from 342 kDa to 708 kDa (Polymer Standard Services, Germany). The device was fitted with a LPG-3400SD gradient pump, a WPS-3000SL autosampler and a DAD-3000 UV-Vis detector (Dionex, Sunnyvale, CA, USA) in addition to a Waters-410 refractive index (RI) detector (Waters, Milford, MA, USA). SEC measurements in DMSO were performed on a SECcurity 1260 (PSS, Mainz, Germany) equipped with refractive index (RI) detector (40 °C) and three columns (PSS GRAM; precolumn, 10 μm 100 Å and 10 μm 10000 Å), with DMSO + 0.5 w/w% LiBr as the mobile phase at 60 °C and a flow rate of 0.5 mL min−1. A conventional calibration method was employed using pullulan standards.
UV-visible spectroscopy was used to determine the UCST of the polymer samples in deionised water. The method was designed with a heating and cooling rate of 1 °C min−1. The UCST values were calculated from the cooling measurement at 50% transmittance, using a wavelength of 450 nm. The measurements were performed with a Shimadzu UV-2550 UV-VIS Spectrophotometer (Kyoto, Japan), software UVProbe 2.0 and temperature controller: S-1700 Thermoelectric Single Cell Holder.
Thermogravimetric analysis was performed with a TA Instruments Hi-Res TGA 2950 analyser, operating in a N2 flow of 30 mL min−1, a heating rate of 10 °C min−1, heating the samples from 40 to 700 °C.
Quartz crystal microbalance with dissipation monitoring (QCM-D) was used to measure the adsorption of the samples to model CNF surfaces. Each sample was diluted with MilliQ water (0.1 g L−1) for the adsorption experiments, and the experiments were performed at 25 °C with a continuous flow of 0.15 mL min−1. This instrument measures the change in resonance frequency of the crystal, corresponding to a change in mass attached to the surface. To convert the change in frequency to its corresponding change in adsorbed mass per area unit, the Sauerbrey model45 was used, eqn (1), where C is a sensitivity constant, −0.177 (mg (m2 Hz)−1), Δf the change in resonance frequency (Hz), and n the overtone number.
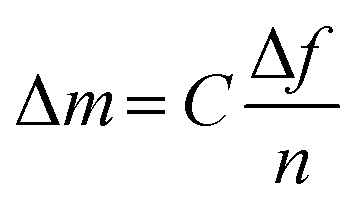 | (1) |
The dissipation can be related to the viscoelastic properties of the adsorbed layer. A thin, rigid attached film is expected to yield a low change in dissipation. A more water-rich and mobile film is expected to yield a larger change in dissipation. The dissipation factor, D, is defined in eqn (2), where Edissipated is the energy dissipated during one oscillation period, and Estored, the energy stored in the oscillating system.
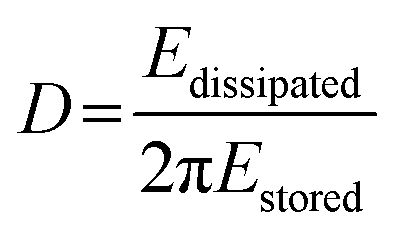 | (2) |
This Sauerbrey model assumes rigidly attached layers, and the attached amount determined contains both polymer and other compounds coupled to the surface. Earlier work has shown that this model is also valid for layers with higher dissipations and comparable to more advanced models.41
Atomic force microscopy (AFM) was used to image the QCM-D crystal surface topographies. A Multimode 8 (Bruker, USA) was used with the ScanAsyst in Air mode, using a cantilever with 70 kHz resonance frequency, spring constant 0.4 N m−1, and tip radius 2 nm (ScanAsyst-Air, Bruker, USA).
Contact angle (CA) measurements were performed at 50% RH and 23 °C on a KSV instrument CAM 200 equipped with a Basler A602f camera, using 5 μL droplets of MilliQ water. A Young–Laplace fitting model was used to process the images. Contact angle values reported were those observed after 30 s of measurement.
Conductometric titration of the TEMPO-oxidised fibres was performed to confirm the target charge density of around 600 μeq g−1.50 The pulp fibres were mechanically disintegrated using a high pressure microfluidiser (M-110EH, Microfluidics Corp, US). Ultra-sonication of CNF was performed using a Vibracell VCX750 (Sonics & Materials, Inc., US) with a microtip probe (6 mm in diameter). Centrifugation of CNF was conducted with a Rotina 420 bench top centrifuge (Andreas Hettich GmbH & Co., KG, Germany). Polyelectrolyte titration of diluted CNF dispersions in MilliQ was carried out with a Stabino Particle Charge Mapping system (Particle Matrix, Microtrac Europe GmbH, Germany) using polyelectrolyte titration with poly(diallyl dimethyl ammonium chloride) (PDADMAC) solution, as reported previously.51
An Ultra-Turrax T25 (IKA Germany) was used to disintegrate CNF prior to nanocomposite formation, at 10000 rpm for 20 min.
Compression testing was performed on the composite films swollen in water at 60 °C for 120 h using an Instron 5566. The method for compression was 10% min−1 strain rate using a 500 N load cell, performed at 23 °C and 50% RH.
4.3 Experimental procedure
For the CNF employed in the QCM-D experiments further treatments were implemented in order to remove aggregated fibrils.40,53 The aqueous CNF dispersion (50 mL, 2 wt%) was sonicated for 10 min at 27% amplitude. The dispersions were centrifuged for 1 h at 4500 rpm in order to remove any aggregated fibrils. The supernatant was collected and a CNF stock dispersion (1 g L−1) was prepared by dilution with MilliQ water. The surface charge after fibrillation and preparation of the dispersion was determined by polyelectrolyte titration and assessed to be approximately 800 μeq g−1.
The degree of swelling was calculated using eqn (3). Where masswet film is the mass of the film after swelling in water and massdry film is the mass before swelling in water, the % will be the mass increase of water.
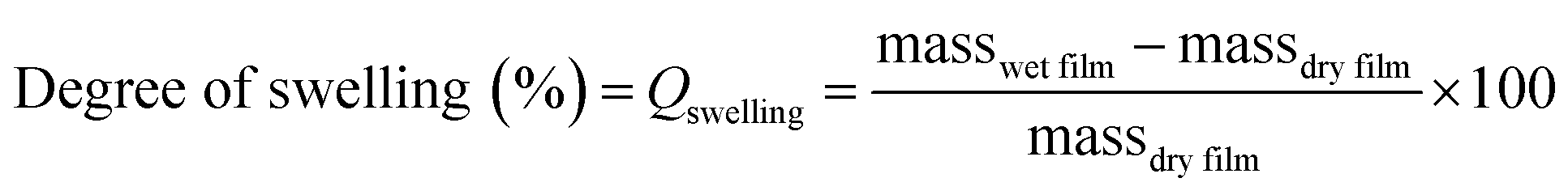 | (3) |
The water phase from swelling was freeze-dried and analysed with 1H-NMR in D2O. It was shown that only miniscule amounts of polymer (PSBMA homopolymer or PSBMA-b-XG) was leaked out of the gels during the swelling tests along with trace amounts of CNF.
Acknowledgements
The authors wish to thank Tahani Kaldéus for preparing the CNF used in the QCM-D experiments. FH is grateful to the Tunholmen Foundation for funding. JE gratefully acknowledges the Wallenberg Wood Science Center (WWSC) for funding.References
- D. Klemm, F. Kramer, S. Moritz, T. Lindström, M. Ankerfors, D. Gray and A. Dorris, Angew. Chem., Int. Ed., 2011, 50, 5438–5466 CrossRef CAS PubMed
.
- H. P. S. A. Khalil, A. H. Bhat and A. F. I. Yusra, Carbohydr. Polym., 2012, 87, 963–979 CrossRef
- S. J. Eichhorn, A. Dufresne, M. Aranguren, N. E. Marcovich, J. R. Capadona, S. J. Rowan, C. Weder, W. Thielemans, M. Roman, S. Renneckar, W. Gindl, S. Veigel, J. Keckes, H. Yano, K. Abe, M. Nogi, A. N. Nakagaito, A. Mangalam, J. Simonsen, A. S. Benight, A. Bismarck, L. A. Berglund and T. Peijs, J. Mater. Sci., 2010, 45, 1–33 CrossRef CAS
- N. T. Cervin, C. Aulin, P. T. Larsson and L. Wågberg, Cellulose, 2012, 19, 401–410 CrossRef CAS
- F. Jiang and Y.-L. Hsieh, J. Mater. Chem. A, 2014, 2, 350–359 CAS
- K. Prakobna, C. Terenzi, Q. Zhou, I. Furo and L. A. Berglund, Carbohydr. Polym., 2015, 125, 92–102 CrossRef CAS PubMed
- H. Fukuzumi, T. Saito, T. Iwata, Y. Kumamoto and A. Isogai, Biomacromolecules, 2008, 10, 162–165 CrossRef PubMed
- M. Henriksson, L. A. Berglund, P. Isaksson, T. Lindström and T. Nishino, Biomacromolecules, 2008, 9, 1579–1585 CrossRef CAS PubMed
- D. Roy, M. Semsarilar, J. T. Guthrie and S. Perrier, Chem. Soc. Rev., 2009, 38, 2046–2064 RSC
- E. Malmström and A. Carlmark, Polym. Chem., 2012, 3, 1702–1713 RSC
- A. Carlmark, E. Larsson and E. Malmström, Eur. Polym. J., 2012, 48, 1646–1659 CrossRef CAS
- A. Carlmark, Macromol. Chem. Phys., 2013, 214, 1539–1544 CrossRef CAS
- F. L. Hatton, E. Malmström and A. Carlmark, Eur. Polym. J., 2015, 65, 325–339 CrossRef CAS
- S. Fujisawa, T. Saito, S. Kimura, T. Iwata and A. Isogai, Biomacromolecules, 2013, 14, 1541–1546 CrossRef CAS PubMed
- A. Villares, C. Moreau, A. Dammak, I. Capron and B. Cathala, Soft Matter, 2015, 11, 6472–6481 RSC
- T. Benselfelt, E. D. Cranston, S. Ondaral, E. Johansson, H. Brumer, M. W. Rutland and L. Wågberg, Biomacromolecules, 2016, 17, 2801–2811 CrossRef CAS PubMed
- Q. Zhou, M. W. Rutland, T. T. Teeri and H. Brumer, Cellulose, 2007, 14, 625–641 CrossRef CAS
- Q. Zhou, L. Greffe, M. J. Baumann, E. Malmström, T. T. Teeri and H. Brumer, Macromolecules, 2005, 38, 3547–3549 CrossRef CAS
- Q. Zhou, H. Brumer and T. T. Teeri, Macromolecules, 2009, 42, 5430–5432 CrossRef CAS
- F. L. Hatton, M. Ruda, M. Lansalot, F. D'Agosto, E. Malmström and A. Carlmark, Biomacromolecules, 2016, 17, 1414–1424 CrossRef CAS PubMed
- A. Dammak, B. Quémener, E. Bonnin, C. Alvarado, B. Bouchet, A. Villares, C. Moreau and B. Cathala, Biomacromolecules, 2015, 16, 589–596 CrossRef CAS PubMed
- A. Dammak, C. Moreau, F. Azzam, B. Jean, F. Cousin and B. Cathala, J. Colloid Interface Sci., 2015, 460, 214–220 CrossRef CAS PubMed
- C. V. Cerclier, A. Guyomard-Lack, F. Cousin, B. Jean, E. Bonnin, B. Cathala and C. Moreau, Biomacromolecules, 2013, 14, 3599–3609 CrossRef CAS PubMed
- S. Ahola, P. Myllytie, M. Österberg, T. Teerinen and J. Laine, BioResources, 2008, 3, 1315–1328 Search PubMed
- P. Eronen, K. Junka, J. Laine and M. Österberg, BioResources, 2011, 6, 4200–4217 CAS
- J. Lucenius, K. Parikka and M. Österberg, React. Funct. Polym., 2014, 85, 167–174 CrossRef CAS
- A. Laschewsky, Polymers, 2014, 6, 1544 CrossRef CAS
- G. S. Georgiev, E. B. Kamenska, E. D. Vassileva, I. P. Kamenova, V. T. Georgieva, S. B. Iliev and I. A. Ivanov, Biomacromolecules, 2006, 7, 1329–1334 CrossRef CAS PubMed
- M. S. Donovan, B. S. Sumerlin, A. B. Lowe and C. L. McCormick, Macromolecules, 2002, 35, 8663–8666 CrossRef CAS
- D. N. Schulz, D. G. Peiffer, P. K. Agarwal, J. Larabee, J. J. Kaladas, L. Soni, B. Handwerker and R. T. Garner, Polymer, 1986, 27, 1734–1742 CrossRef CAS
- H. Willcock, A. Lu, C. F. Hansell, E. Chapman, I. R. Collins and R. K. O'Reilly, Polym. Chem., 2014, 5, 1023–1030 RSC
- J. Zhang, J. Yuan, Y. L. Yuan, J. Shen and S. C. Lin, Colloids Surf., B, 2003, 30, 249–257 CrossRef CAS
- P.-S. Liu, Q. Chen, X. Liu, B. Yuan, S.-S. Wu, J. Shen and S.-C. Lin, Biomacromolecules, 2009, 10, 2809–2816 CrossRef CAS PubMed
- M. Wang, J. Yuan, X. Huang, X. Cai, L. Li and J. Shen, Colloids Surf., B, 2013, 103, 52–58 CrossRef CAS PubMed
- Y. Zhu, X. Xu, N. D. Brault, A. J. Keefe, X. Han, Y. Deng, J. Xu, Q. Yu and S. Jiang, Anal. Chem., 2014, 86, 2871–2875 CrossRef CAS PubMed
- Y.-H. Zhao, K.-H. Wee and R. Bai, ACS Appl. Mater. Interfaces, 2010, 2, 203–211 CAS
- P. Liu, Q. Chen, L. Li, S. Lin and J. Shen, J. Mater. Chem. B, 2014, 2, 7222–7231 RSC
- P.-S. Liu, Q. Chen, S.-S. Wu, J. Shen and S.-C. Lin, J. Membr. Sci., 2010, 350, 387–394 CrossRef CAS
- Y. Zhang, T. Nypelo, C. Salas, J. Arboleda, I. C. Hoeger and O. J. Rojas, J. Renewable Mater., 2013, 1, 195–211 CrossRef CAS
- L. Wågberg, G. Decher, M. Norgren, T. Lindström, M. Ankerfors and K. Axnäs, Langmuir, 2008, 24, 784–795 CrossRef PubMed
- C. Aulin, I. Varga, P. M. Claesson, L. Wågberg and T. Lindström, Langmuir, 2008, 24, 2509–2518 CrossRef CAS PubMed
- S. Utsel, C. Bruce, T. Pettersson, L. Fogelström, A. Carlmark, E. Malmström and L. Wågberg, ACS Appl. Mater. Interfaces, 2012, 4, 6795–6806 Search PubMed
- E. Larsson, C. C. Sanchez, C. Porsch, E. Karabulut, L. Wågberg and A. Carlmark, Eur. Polym. J., 2013, 49, 2689–2696 CrossRef CAS
- L. Carlsson, A. Fall, I. Chaduc, L. Wågberg, B. Charleux, E. Malmström, F. D'Agosto, M. Lansalot and A. Carlmark, Polym. Chem., 2014, 5, 6076–6086 RSC
- G. Sauerbrey, Z. Phys., 1959, 155, 206–222 CrossRef CAS
- A. Pei, N. Butchosa, L. A. Berglund and Q. Zhou, Soft Matter, 2013, 9, 2047–2055 RSC
- G. I. Mantanis, R. A. Young and R. M. Rowell, Wood Sci. Technol., 1994, 28, 119–134 CAS
- G. I. Mantanis, R. A. Young and R. M. Rowell, Cellulose, 1995, 2, 1–22 CAS
- C. Schatz and S. Lecommandoux, Macromol. Rapid Commun., 2010, 31, 1664–1684 CrossRef CAS PubMed
- S. Katz, R. P. Beatson and A. M. Scallan, Sven. Papperstidn., 1984, 87, 48–53 Search PubMed
- I. Usov, G. Nyström, J. Adamcik, S. Handschin, C. Schutz, A. Fall, L. Bergström and R. Mezzenga, Nat. Commun., 2015, 6, 7564 CrossRef PubMed
- T. Saito, M. Hirota, N. Tamura, S. Kimura, H. Fukuzumi, L. Heux and A. Isogai, Biomacromolecules, 2009, 10, 1992–1996 CrossRef CAS PubMed
- A. B. Fall, S. B. Lindström, O. Sundman, L. Ödberg and L. Wågberg, Langmuir, 2011, 27, 11332–11338 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Polymerisation kinetic data, TGA analyses, AFM images and compression data. See DOI: 10.1039/c6ra28236a |
| ‡ Present address: Department of Chemistry, University of Sheffield, Dainton Building, Brook Hill, Sheffield, S3 7HF, UK. |
| This journal is © The Royal Society of Chemistry 2017 |