
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-biological reduction of Cr(VI) by reacting with humic acids composted from cattle manure†
Min Wu *a,
Gongxia Lia,
Xiaolin Jiangb,
Qianqian Xiaoa,
Mingxing Niua,
Zhiyuan Wanga and
Yayi Wanga
*a,
Gongxia Lia,
Xiaolin Jiangb,
Qianqian Xiaoa,
Mingxing Niua,
Zhiyuan Wanga and
Yayi Wanga
aState Key Laboratory of Pollution Control and Resources Reuse, Tongji University, 1239 Siping Road, Shanghai 200092, China. E-mail: minw@tongji.edu.cn; Fax: +86 021 65984275; Tel: +86 021 65984275
bShanghai Pudong Veolia Water Corporation Limited, China
First published on 23rd May 2017
Abstract
Previous studies on reduction of Cr(VI) by humic acids (HAs) have seldom used the extracts from composted animal manure. For greater yields of HAs and resource reclamation of animal manure, cattle manure was used as the composting material in our study. The capacity of humic acids extracted from composted cattle manure (HAscm) to reduce Cr(VI) was tested under the influence of environmental factors (pH, illumination with light and dissolved oxygen). And the non-biological detoxification mechanism was investigated by using three-dimensional fluorescence, Fourier transform infrared spectroscopy (FTIR) and X-ray absorption near-edge structure (XANES) spectroscopy. The results indicated that Cr(III) after the reduction of Cr(VI) formed an outer sphere complex with –OH and inner sphere complex with carboxyl groups in HAscm.
1. Introduction
Chromium is a commonly identified contaminant in soils and water due to its frequent industrial application. It has been considered one of the top 20 contaminants on the Superfund priority list of hazardous substances for the past 15 years.1 Chromium normally exists in two different oxidation states: hexavalent (Cr(VI)) and trivalent (Cr(III)). At physiological pH, Cr(VI) enters the human cell more easily comparing with Cr(III), which is thus more dangerous in causing some toxicological effects such as cancer, activation of apoptosis, and cell death.2 Cr(III) is nearly insoluble at neutral pH. It is widely recognized that Cr(VI) is more toxic than Cr(III).3 To reduce the toxicity of Cr in environments, it is desirable to convert Cr(VI) to Cr(III).Up to now, various processes have been proposed for Cr(VI) reduction. Chemical reduction is the most commonly used method of Cr(VI) detoxification, by which inorganic or organic reductants (electron donors) are used to reduce Cr(VI) to Cr(III), and the Cr(III) further reacts with OH− to form insoluble and stable Cr(III) hydroxides.4 These reductants include reduced sulfur compounds such as sodium sulfide (Na2S)5 and calcium polysulfide (CaSx),6 and iron-based materials such as zero-valent iron nanoparticles (nZVI),7–9 dissolved ferrous iron,10,11 and solids containing ferrous iron.12,13 The sulfur compounds reduce Cr(VI) to Cr(III) which usually form Cr(III) hydroxides with lower mobility, however most of them inevitably are toxic and hazardous byproducts could be produced.14 nZVI is well known to immobilize Cr(VI) effectively, but it is usually prepared by a relatively expensive method,15 and it may have a harmful effect on microorganisms, animal cells, plant cells, and human cells.16,17 Thus, an eco-friendly and efficient strategy for Cr(VI) detoxification is urgently desirable.
Humic acids (HAs) are a group of high molecular aromatic polymers. Their structures make them to bond easily with hydrophobic and hydrophilic material. Experimental studies on Cr(VI) reduction, have found that Fe(II) and organic matter such as HAs had similar effect on Cr(VI) reduction in terms of total reduction capacity.18–20 As in the literature most of studies on Cr(VI) reduction by HAs have used commercial HAs or HAs extracted from soil, coal and organic matter from water.19,21 But the cost of the commercial products and the relatively low yields (mg of HAs extracted per g of parent material dry matter) of HAs from natural sources limited their potential use. However, HAs also can be obtained from composted agro-industrial and municipal organic wastes.22 It has been known that manure humification can be promoted in the composting process in which microbial assimilation and dissimilation are conducted. Our previous study has shown that the content of total humic substances in cattle manure could be increased to 8.71% after composting.23 The potential for using composted animal manure is attractive, as animal manure are no-cost, are widely available, and the HAs yield may be greater than the yield from natural sources. Additionally, widespread applying composted animal manure to obtain HAs provides an eco-friendly way of resource reclamation.
The aim of this work is thus to provide a scientific basis for the remediation of Cr(VI) contaminated sites, including water, by HAs extracted from composted cattle manure (HAscm). The capacity of HAscm to reduce Cr(VI) under the influence of environmental factors was tested. Additionally, the detoxification mechanism of Cr(VI) reduction by HAs was investigated by using three-dimensional fluorescence, Fourier transform infrared spectroscopy (FTIR) and X-ray absorption near-edge structure (XANES) spectroscopy in this study.
2. Materials and methods
2.1 Materials
The anaerobically composted cattle manure used in this study was purchased from a farm in Chongming County (Shanghai). This organic fertilizer was produced according to national agricultural fertilizer industry standard NY525-2012. The extraction of HAs from composted catlle manure was performed according to the method recommended by International Humic Substance Society (IHSS).24 Summarizing, HAscm extraction of the sample was performed in 0.1 M NaOH under a nitrogen atmosphere, separated from solution by setting at pH 1; HAscm was redissolved in 0.1 M KOH under nitrogen, separated from solution at pH 1; then HAscm was purified by 0.1 M HCl and 0.3 M HF three times, dialysed against demineralised water and finally freeze-dried.2.2 Experiments
Bench scale batch tests using 250 mL flask as a reactor was conducted. The flask was placed in the shaking table (DKY-II, China) to maintain the temperature (25 °C) and agitation (150 rpm). A stock solution of HAscm (0.1 g L−1) used in the experiment was made by dissolving 50 mg of the HAscm in 495 mL Milli-Q water added with 5 mL of 1 M NaOH solution. In each reaction, the initial Cr(VI) concentration was adjusted to 4 mg L−1. Each reaction was prolonged for 96 h and repeated for three times.To explore the influence of light irradiation, two reaction vessels at pH 2.5 under anaerobic condition were employed for the experiment. One of them was wrapped, while the other one wasn't.
To study the influence of aeration conditions, three reactors operated in three oxygen levels (anaerobic, natural ventilation, and aerobic) were employed. In this experiment, the reaction vessels were wrapped and the initial pH of the reaction solution was controlled at 2.5. For anaerobic condition, all solutions were purged with nitrogen (>99.99%) for at least 30 min before use and the vessel was prepared in an anaerobic glove box (Thermo 1029 Forma) during the experiment. The vessels under natural ventilation and aeration conditions were ventilated and aerated (using an air pump) for 20 min every 24 h, respectively.
To determine the effect of pH conditions, the initial pH of each reaction solutions were adjusted according to the experimental set points (2.5, 4.0, and 6.0) using 0.1 M HCl and NaOH. All the flasks are wrapped completely with aluminum foil to avoid any light irradiation and carried out under anaerobic condition.
2.3 Analytical methods
All the samples used for the analysis were collected from the reactions with initial pH of 4 and 6. The sample from the reaction condition with initial pH of 2.5 was not utilized because there were some suspended solid in the solution at this condition.
The three dimensional fluorescence spectra of the samples were analyzed by spectrophotometer (F-4600 FL Rili). The test sample was first filtered using a 0.45 μm membrane filter, and then its pH was adjusted to 5–6 using 0.1 M HCl and NaOH. Next, the solution was added into 1 cm quartz colorimetric utensil and then put it in the sample tank for analyzing. After removing Raman scattering and Rayleigh scattering etc. by using Milli-Q water as a blank, the fluorescence intensity of each regional were added up, and the result was standardized according to each area to get the fluorescent area percentage Pi, n. The tested data were processed by Origin 9.1 and the Sigma plot 12.5.
After fully grinding, small amount of samples was applied onto glass slices through KBr pressed method.32 The slices were then put into infrared drying oven to remove any water in the sample. Nicolet iS5 FTIR spectrometer with a measurement range of 4000–400 cm−1 and a resolution of 4 cm−1 was used in this research.
The reaction solution samples were first acidified to pH less than 2 with 6 M HCl. After setting for 20 minutes, the acidified liquid was centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. Then the precipitates were collected and put into a freeze dryzer (DYYB-10 Shanghai). After the precipitate become powder, it was completely grinded and screened through a 400 mesh sieve to ensure the small particle size for XANES analysis. The X-ray absorption data at the Cr K-edge of the samples were recorded by a 4 channel silicon drift detector (SDD) (Bruker 5040) at beamline BL14W1 of the Shanghai Synchrotron Radiation Facility (SSRF), Shanghai, China.35 All spectra were taken at room temperature in the transmission geometry. The station was operated with a Si(III) double crystal monochromator. The synchrotron was operated at energy of 3.5 GeV and a current between 150 and 210 mA in the measurement.
000 rpm. Then the precipitates were collected and put into a freeze dryzer (DYYB-10 Shanghai). After the precipitate become powder, it was completely grinded and screened through a 400 mesh sieve to ensure the small particle size for XANES analysis. The X-ray absorption data at the Cr K-edge of the samples were recorded by a 4 channel silicon drift detector (SDD) (Bruker 5040) at beamline BL14W1 of the Shanghai Synchrotron Radiation Facility (SSRF), Shanghai, China.35 All spectra were taken at room temperature in the transmission geometry. The station was operated with a Si(III) double crystal monochromator. The synchrotron was operated at energy of 3.5 GeV and a current between 150 and 210 mA in the measurement.
3. Results and discussion
3.1 The structure and properties of HAscm
The basic structure and properties of the HAscm were studied through FTIR, 13C CP-MAS NMR and elemental analysis. Preliminary FTIR analysis showed that the HAs extracted from the composted cattle manure contained the same functional groups with those extracted from soil, peat, sediment previously (Fig. S2†).29 It was found that N, C, H, S, O, in the HAscm were 6.24%, 53.55%, 5.30%, 2.15%, 32.76% respectively, according to elemental analysis (Table S1†); and the analysis using 13C CP-MAS NMR showed that alkyl C, O-alkyl C, aromatic C, carbonyl C, carboxyl C accounted for 26.08%, 24.46%, 24.64%, 16.26%, 8.57% of total carbon, respectively (Fig. S3, Table S2†).According to the H/C calculated from the date obtained in the elemental analysis and those recorded in some literatures, the sequence of the degree of humification (defined as the magnitude of H/C) in the composted manure and other materials was: composted manure < peat < lignite < soil.19,36 Ohta et al. found that the lower the degree of humification, the stronger the reduction ability of Cr(VI).37 So HAscm should have stronger reducibility than those from soil, lignite and peat. In addition, it is widely known that the number of active functional groups also can influence the reduction capacity. Except quinone groups, other heteroatomic groups such as aldehydes, phenols, sulfonium can reduce Cr(VI) as well.38 Besides, some researchers showed that phenolic C and O-alkyl C such as oligosaccharide, monosaccharide contained in HAs are well reductive substances to Cr(VI).38–40 So according to the content of O-alkyl C and phenolic C, it can be determined that the HAscm have a good reduction ability to Cr(VI).
3.2 Reduction of Cr(VI) by HAscm and adsorption of Cr(III) to HAscm
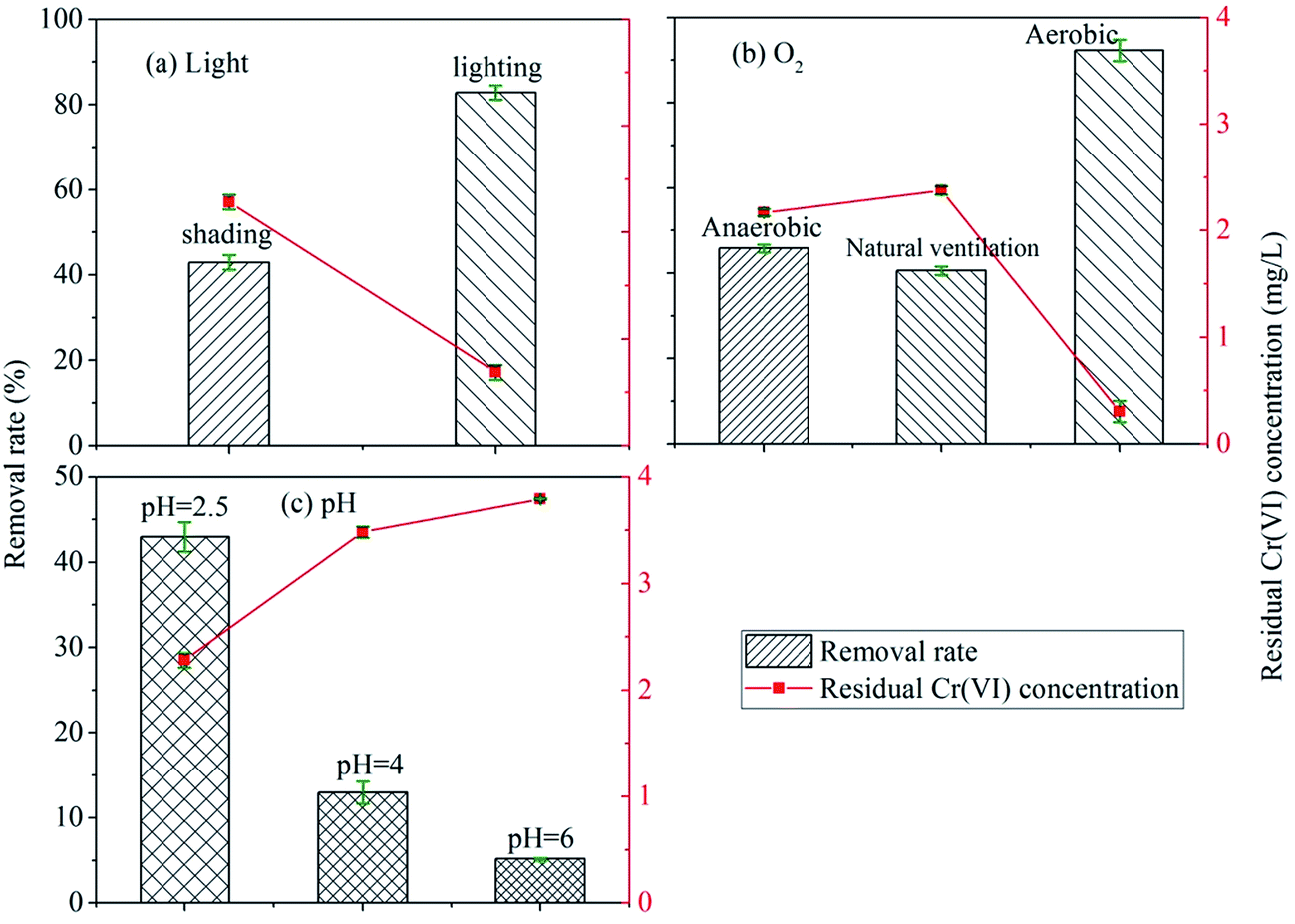 | ||
| Fig. 1 The influence of environmental factors on the reduction reaction of Cr(VI): (a) light; (b) O2; (c) pH. | ||
The effect of the illumination on the improvement of Cr(VI) reduction by HAscm can be explained as following: first, HAscm were a kind of organic matters on which illumination produced free radicals such as OH˙ or stimulated the electrons directly to promote the reduction of Cr(VI).41–43 Lipski et al. proposed that illumination will react with HAs to generate some active oxides such as O2˙ and H2O2.44 The mechanism of the illumination effect on the reduction of Cr(VI) by HAs through the intermediates such as free radicals or active oxides can be described by eqn (1)–(5).45,46
| HAscm + hν → O2HAscm+ + O2˙− | (1) |
| O2˙− + H2O → HO2˙ + OH− | (2) |
| O2˙− + HO2˙ + H+ → H2O2 + O2 | (3) |
| 2O2˙− + HCr2O7− + 9H+ → 5H2O + 2Cr3+ + 3O2 | (4) |
| 3H2O2 + HCr2O7− + 7H+ → 7H2O + 2Cr3+ + 3O2 | (5) |
Second, the first step that forms chromium–ester bond in Cr(VI) reduction was very rapid, and the electron transport between Cr(VI) and phenol or other functional groups was the rate-limiting step of Cr(VI) reduction. Illumination can improve the process of ligand-to-metal charge transfer (LMCT) in the related photochemical reactions.47,48
Oxygen is a strong oxidant with a redox potential E (O2/H2O) of 1.229 V. But the removal rate of Cr(VI) under natural ventilation condition was similar with that in anaerobic condition. This was probably because the adsorbed complex of Cr(III) on HAscm impeded the O2 to oxidize Cr(III) under natural ventilation condition.49
The removal rate in aerobic condition was enhanced significantly compared with both the anaerobic and natural ventilation conditions. The removal rate under aerobic condition has increased by 51.6% rather than fall. The reasons can be the followings: (1) under aeration condition using an air pump, the contact chance of O2 and HAscm increased; (2) O2 can react with HAs to form peroxide which has a stronger electron transfer capability with Cr(VI) than HAs,50 this means that O2 was an intermediate which improves the electron transfer of HAscm with Cr(VI).
| HCrO4− + 7H+ + 3e− ⇔ Cr3+ + 4H2O | (6) |
The lower pH value or higher concentration of H+ could enhance the redox potential of Cr(VI)/Cr(III), which made Cr(VI) to be reduced easily.
3.3 Reaction mechanism of Cr reacting with HAs
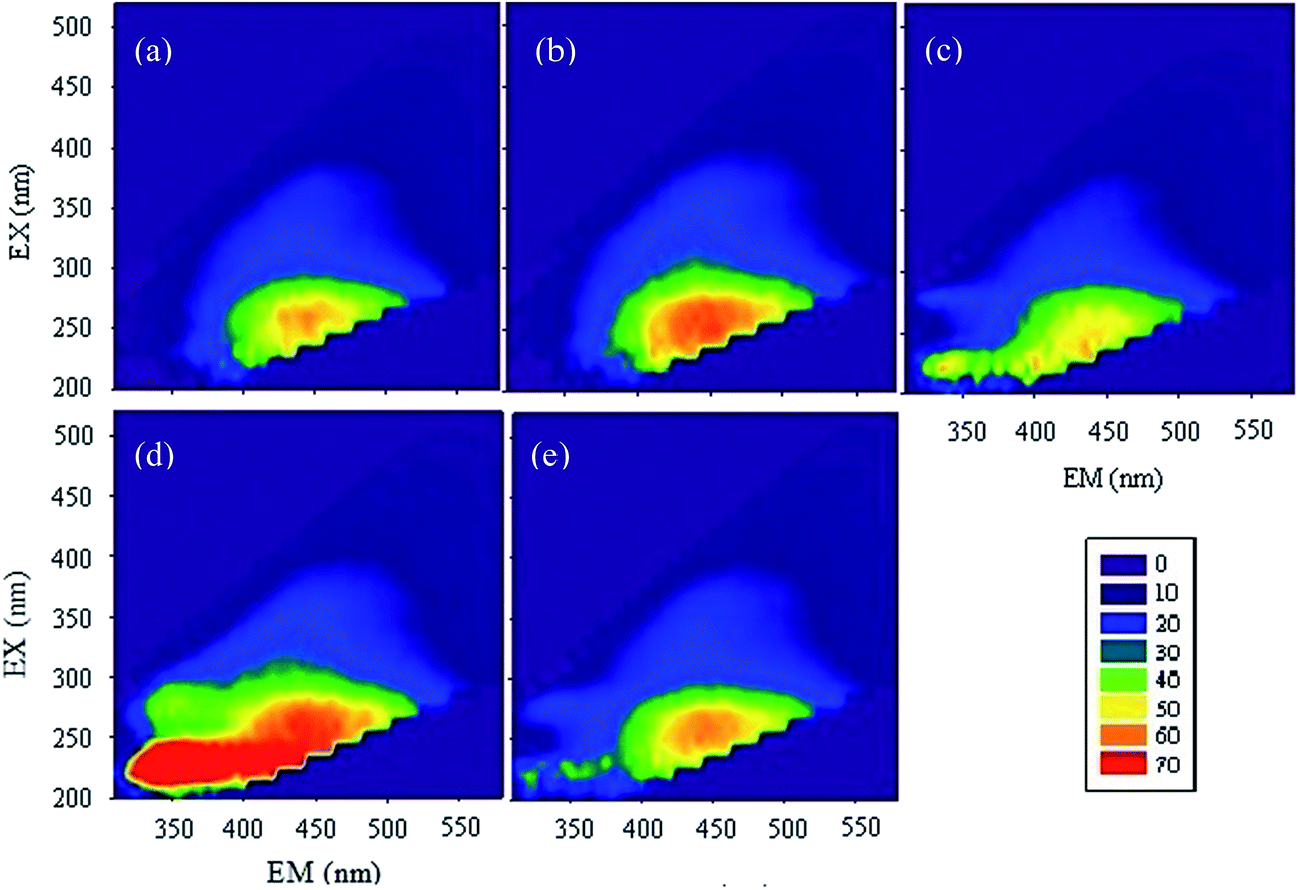 | ||
| Fig. 2 Three-dimensional fluorspar spectra under different reaction conditions: (a) pH = 4, time = 4 d, shading, anaerobic; (b) pH = 6, time = 4 d, shading, anaerobic; (c) pH = 4, time = 25 d, shading, anaerobic; (d) pH = 6, time = 25 d, shading, anaerobic; (e) HAscm: without reaction. The horizontal axis is fluorescence emission wavelength (EM), and the vertical axis is the fluorescence excitation wavelength (EX). To ensure the comparability of samples, we didn't choose the samples under pH = 2.5, for there are some suspended solid in the solution generated at this condition. | ||
| Samples | 310–330/200–250 (%) | 330–380/200–250 (%) | 380–580/200–250 (%) | 310–380/250–520 (%) | 380–580/250–520 (%) | Total intensity |
|---|---|---|---|---|---|---|
| a Initial: without reaction; pH = 4, 4 d: pH = 4, time = 4 d, shading, anaerobic; pH = 6, 4 d: pH = 6, time = 4 d, shading, anaerobic; pH = 4, 25 d: pH = 4, time = 25 d, shading, anaerobic; pH = 6, 25 d: pH = 6, time = 25 d, shading, anaerobic. | ||||||
| Initial | 0.17 | 1.36 | 11.91 | 10.90 | 75.66 | 3155.88 |
| pH = 4, 4 d | 0.00 | 0.60 | 11.03 | 7.71 | 80.66 | 2515.98 |
| pH = 6, 4 d | 0.00 | 0.59 | 11.69 | 7.75 | 79.97 | 2950.23 |
| pH = 4, 25 d | 0.17 | 1.81 | 13.09 | 11.22 | 73.71 | 2521.57 |
| pH = 6, 25 d | 0.21 | 3.43 | 13.44 | 14.09 | 68.83 | 3757.29 |
According to the relative amount of each area's representatives (Table S3†), it can be seen that, after the reaction of 4 days, the relative content of aromatic protein I, tyrosine, aromatic protein II, Biochemical Oxygen Demand (BOD5), soluble microbial by-products were all decreased, while the relative content of humic-like substances increased, and those of fulvic acid and hydrophobic acid had no consistent change; the lower the pH, the larger the increase of relative content of humic-like substances. However, the change of the relative content was quite contrary when the reaction time was 25 days. This observation mainly due to two reasons: first, some active functional groups such as –NH2 reacted with Cr(VI) in the initial period of time and then the humic-like substances began to react with Cr(VI), which caused the variation of fluorescence along with reaction time; second, HAscm had a strong absorption in long wavelength region in the case of high degree aromatization or with the existence of many unsaturated bonds.51 Therefore, the initial HAscm oxidized by Cr(VI) enhanced the degree of humification, which strengthened the absorption in long wavelength region, then in the followed reaction, Cr(VI) or Cr(III) bound to HAscm, which increased its inorganic quality, causing the enhancement of absorption in short wavelength region.
| Ari = Ai/(A3383 + A2920 + A1653 + A1510 + A1456 + A1420 + A1225 + A1126 + A1042) × 100% | (7) |
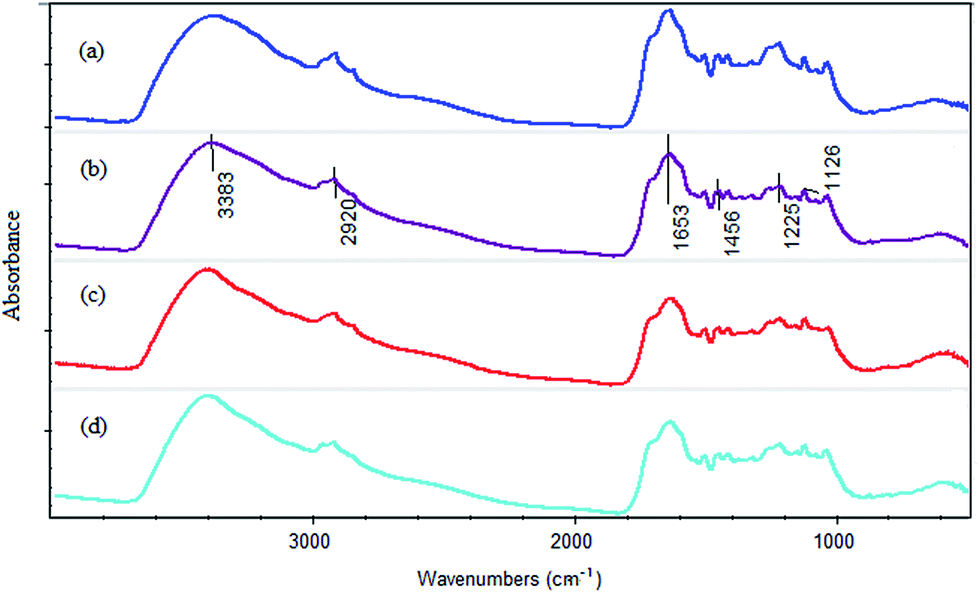 | ||
| Fig. 3 FTIR spectra under different reaction conditions: (a) HAscm: without reaction; (b) pH = 2.5, time = 4 d, shading, anaerobic; (c) pH = 4, time = 4 d, shading, anaerobic; (d) pH = 6, time = 4 d, shading, anaerobic. | ||
Ari is the relative absorbance at i wavenumber; Ai is the absorbance gotten from FTIR spectrometer at i wavenumber.
Table S4† presents the main bands and the corresponding functional groups. According to the literature, the absorption peak at 3383 cm−1 is associated with the stretching vibration of –OH group.52 The absorption peak at 1225 cm−1 is corresponded to the carboxy groups (C–O stretching vibration).53 The peaks around 2920 cm−1, 1456 cm−1 and 1126 cm−1 are attributed to aliphatic hydrocarbon.52,54 And the absorption peak at 1653 cm−1 is assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration of aromatic and CO stretching vibration.55
C stretching vibration of aromatic and CO stretching vibration.55
The calculative result is presented in Table 2. From the table, it can be seen that all the relative absorbances at 3383 cm−1 of the three samples (pH 2.5, pH 4 and pH 6) were less than that of the initial HAscm. It suggested that –OH group participated in the reduction reaction. This observation demonstrated that Cr ions were hydrated to form an outer sphere complex with HAscm. Similarly, at 1225 cm−1, the stretching vibrations of C–O in all the samples were weaker than that of initial HAscm (Table 2). The change of the relative absorbance can be recognized as the inner sphere of the complex formed from Cr and carboxyl groups. This observation supported the results of Fukushima et al. and Ohta et al.37,56 All the relative absorbances at 2920 cm−1, 1456 cm−1 and 1126 cm−1 increased after the corresponding reactions, while the relative absorbance at 1653 cm−1 became weaker (Table 2). This observation demonstrated that with the process of the reaction, some aromatic structure in HAscm decomposed, and generated the aliphatic hydrocarbon, which reduced the humification degree of HAscm. In addition, it is generally believed that quinone involves in humic respiration. The fact that the relative absorbance at 1653 cm−1 was reduced indirectly proved that quinone has reacted with Cr(VI) (Table 2). In addition, the increase of the relative absorbance at 1040 cm−1 verified that C–O–C was generated in the reaction (Table 2).
| Wavenumbers (cm−1) | Relative absorbance (%) | |||
|---|---|---|---|---|
| HAscm | pH = 2.5 | pH = 4 | pH = 6 | |
| a HAscm: without reaction; pH = 2.5: pH = 2.5, time = 4 d, shading, anaerobic; pH = 4: pH = 4, time = 4 d, shading, anaerobic; pH = 6: pH = 6, time = 4 d, shading, anaerobic. | ||||
| 3383 | 13.42 | 12.77 | 12.28 | 12.96 |
| 2920 | 10.53 | 11.17 | 11.14 | 11.03 |
| 1653 | 13.89 | 12.29 | 11.55 | 11.86 |
| 1510 | 10.36 | 10.61 | 10.71 | 10.45 |
| 1456 | 10.33 | 10.61 | 10.78 | 10.58 |
| 1420 | 10.31 | 10.61 | 10.78 | 10.58 |
| 1225 | 11.21 | 10.85 | 10.99 | 10.93 |
| 1126 | 10.18 | 10.67 | 10.99 | 10.91 |
| 1042 | 9.77 | 10.41 | 10.76 | 10.68 |
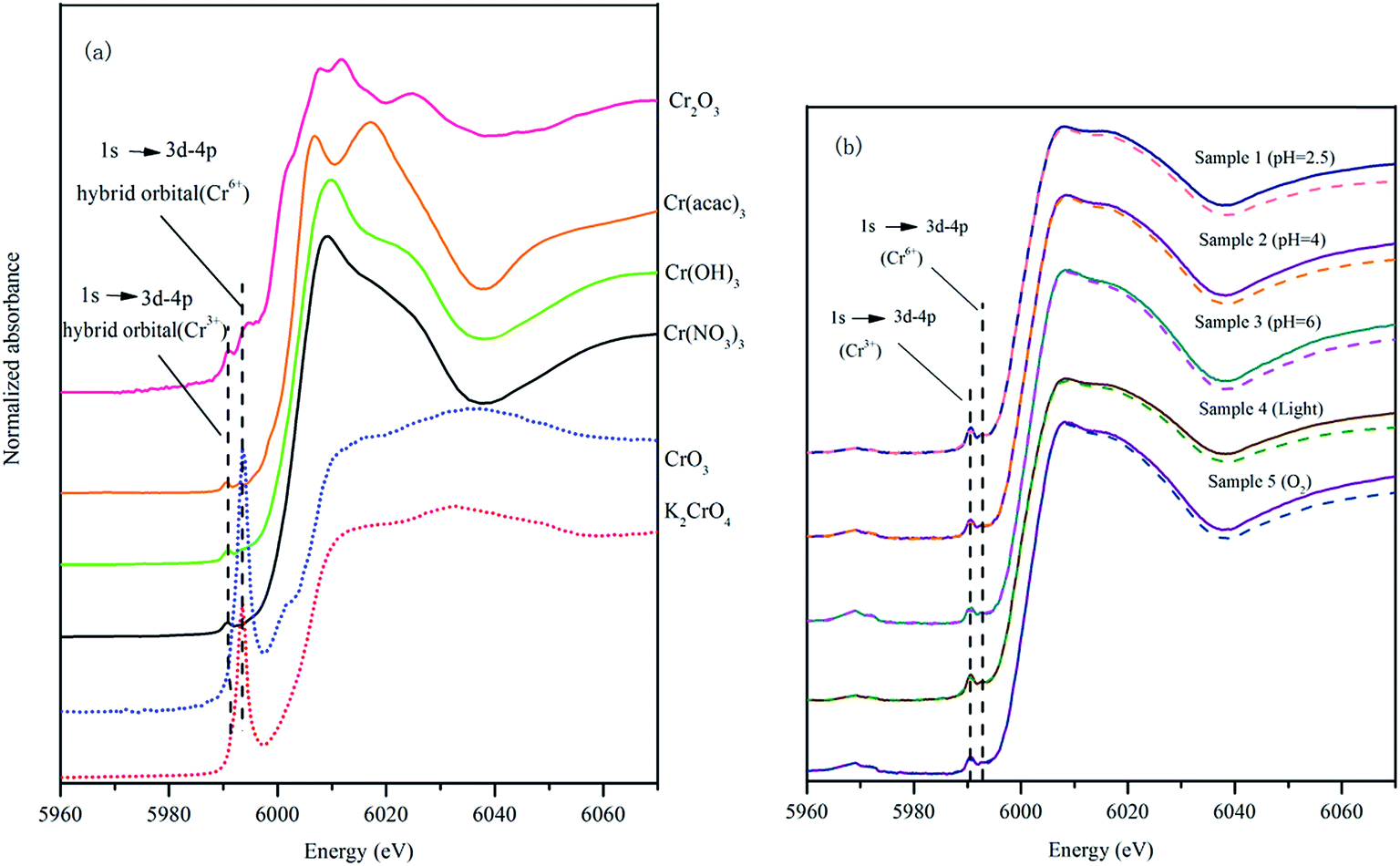 | ||
| Fig. 4 XANES spectra of: (a) standard substance; (b) the reaction samples: sample 1: pH = 2.5, time = 4 d, shading, anaerobic; sample 2: pH = 4, time = 4 d, shading, anaerobic; sample 3: pH = 6, time = 4 d, shading, anaerobic; sample 4: pH = 2.5, time = 4 d, lighting, anaerobic; sample 5: pH = 2.5, time = 4 d, shading, aerobic. The dotted line shows the linear combination fitting results of XANES spectra of samples. | ||
The XANES spectra of K2CrO4 and CrO3 are similar, especially the broad peak at 6032 eV (Fig. 4(a)). For Cr(III), although Cr(acac)3, Cr(OH)3 and Cr(NO3)3 are all the Cr(III) compounds, their XANES spectra were different. Cr(acac)3 had bimodal peaks at 6006 eV and 6017 eV (Fig. 4(a)). The inorganic Cr compounds Cr(OH)3 and Cr(NO3)3 had the similar spectra. Cr(OH)3 had a peak at 6009 eV and Cr(NO3)3 had a narrower peak at 6008 eV (Fig. 4(a)). The XANES spectrum of Cr2O3 differ with Cr(OH)3 and Cr(NO3)3, it had two small and narrow peaks at 6008 eV and 6011 eV, respectively (Fig. 4(a)).
Fig. 4(b) presents the XANES spectra of the samples after the reaction of 4 days. The XANES spectra of the 5 experimental samples had a similar pattern. All the spectra had a small peak characteristic for Cr(III) at 5990 eV, however there was no intense peak representing Cr(VI) at 5993 eV (Fig. 4(b)). This result clearly indicated that the solid precipitated in the low pH condition from the experimental solutions did not contain Cr(VI). Only Cr(III) that was reduced from Cr(VI) bounds to HAscm.
A computer program Artemis was used to carry out XANES analysis in this research. Linear combination fitting for HA–Cr used XANES spectra of equal-weighted reference samples containing K2CrO4, CrO3, Cr(acac)3, Cr(OH)3, Cr(NO3)3 and Cr2O3. When the fitting result of any reference compound was 0, deleted it and fitted again. At the end, three standard substances (Cr(acac)3, Cr(OH)3 and Cr2O3) were left. The linear combination fitting results are summarized in Table 3.
| Sample | Sample 1 (pH = 2.5) | Sample 2 (pH = 4) | Sample 3 (pH = 6) | Sample 4 (lighting) | Sample 5 (aerobic) |
|---|---|---|---|---|---|
| a Sample 1 (pH = 2.5): pH = 2.5, time = 4 d, shading, anaerobic; sample 2 (pH = 4): pH = 4, time = 4 d, shading, anaerobic; sample 3 (pH = 6): pH = 6, time = 4 d, shading, anaerobic; sample 4 (lighting): pH = 2.5, time = 4 d, lighting, anaerobic; sample 5 (aerobic): pH = 2.5, time = 4 d, shading, aerobic. | |||||
| R-factor | 0.003 | 0.002 | 0.003 | 0.003 | 0.002 |
| Chi-square | 0.315 | 0.301 | 0.339 | 0.351 | 0.279 |
| Cr(acac)3 | 30.3% (2.0%) | 38.5% (3.8%) | 41.6% (4.0%) | 25.9% (2.1%) | 42.7% (3.6%) |
| Cr(OH)3 | 0% | 14.4% (4.8%) | 22.5% (5.1%) | 0% | 19.2% (4.6%) |
| Cr2O3 | 69.7% (2.0%) | 47.1% (6.1%) | 35.9% (6.5%) | 74.1% (2.1%) | 38.1% (5.9%) |
From this table, it can be seen that the content of Cr(VI) in all the 5 samples are 0%. This result was in good agreement with the researches of Ohta et al. and Park et al., they reported that the Cr(III) reduced from Cr(VI) bounds to HAs, and the unreacted Cr(VI) remained in the experimental solutions.37,58
With the increase of pH, the relative content of Cr(acac)3 and Cr(OH)3 were increasing, however, the effect of pH on the variation of the relative content of Cr2O3 was opposite (Table 3). In the solution Cr(III) was easy to be hydrolyzed to generate chromium hydroxide.59 In an experiment of Cr(VI) reduction in soil, Kappen et al. found that Cr(OH)3·nH2O presented under pH condition of 4–6.60 However, Ohta et al. believed that there was not Cr(OH)3 generated in Cr(VI) reduction solution.37 In Table 3, the contents of Cr(OH)3 were higher than 5% when pH was 4 and 6, the higher the content of OH− in the solution, the more the Cr(OH)3 adsorbed by HAscm. So our results were in agreement with the results of Kappen et al.60 The Cr(acac)3 was regarded as the main components of the inner sphere complex.37 With the increase of pH, more and more of inner sphere complex was formed. It was possibly because: the structure of HAs in acid solution was randomly cowered.61,62 This was also why the high pH was helpful to stretch the HAs. On the other hand, the linear combination fitting result by Artemis was only the relative one. Although the reduction capacity of Cr(VI) at pH 2.5 was much higher than that at pH 4 and 6, the content of the inner sphere complex at pH 2.5 was lower than that at the other two pH conditions. This was mainly due to the functional groups which can produce inner sphere complex with Cr(III) had been reacted completely, only the outer Cr(III) precipitation formed by electrostatic adsorption with OH was in effect.
The relative content of inner sphere complexes in the sample from illumination condition was less than that under shading condition (Table 3). Besides illumination made the reduction capacity of HAscm to Cr(VI) be increased by 93% from the single factor experiment results. This was largely because the complexing functional group was limited.
From Table 3 it can be seen that the relative content of inner sphere complexes in the samples from the aerobic condition was the highest comparing with other 4 samples. This elucidated that O2 promoted not only the chelating of the Cr(III) but also the reduction of Cr(VI). This observation was different from what was observed by Fulda et al., they reported that O2 promoted the Cu(II) complexation and inhibits reduction of Cu(II).49 Since the FTIR spectra showed that both hydroxyl and carboxyl groups were involved in the reaction, and the removal rate of Cr(VI) under aerobic condition was more than 90%. It was possibly that the reaction of O2 with HAscm creates carboxyl group, increasing the content of the coordination group. Furthermore, Cr(VI) can not bind with HAscm. So O2 can promote the complexation of the Cr(III) with HAscm and does not affect the reduction of Cr(VI).
4. Conclusion
HAscm had the same properties with HAs from soil, peat and lignite, but a lower degree of humification and more functional groups such as phenolic hydroxyl and carboxyl groups. This was why HAscm had a good reduction ability to heavy metals.The results obtained in this study can be concluded that the functional groups in HAscm played a decisive role in the reduction of heavy metals. After Cr(VI) was reduced, the Cr(III) generated forms an outer sphere complex with –OH and inner sphere complex with carboxyl groups in HAscm. Without the reduction reaction the free Cr(VI) stayed in its unreacted from in the solutions. The HAscm–Cr(III) complexes were consist of Cr(acac)3, Cr(OH)3 and Cr2O3, and these components in the HAscm–Cr(III) complexes had different fractionations under different conditions.
Acknowledgements
This work was supported by the Natural Science Foundation of Shanghai [grant numbers 14ZR1443400]; International Science and Technology Cooperation Foundation of Shanghai [grant numbers 12230700800]. We gratefully acknowledge Shanghai Synchrotron Radiation Facility (SSRF) support. We would like to thank the professor Xiaohong Guan (Tongji University) for the help in XANES analysis.References
- M. Chrysochoou and C. P. Johnston, GeoCongress 2012: State of the Art and Practice, in Geotechnical Engineering, 2012, pp. 3959–3967 Search PubMed.
- M. Costa, Toxicol. Appl. Pharmacol., 2003, 188, 1–5 CrossRef CAS PubMed.
- N. Unceta, M. Astorkia, Z. Abrego, A. Gomez-Caballero, M. A. Goicolea and R. J. Barrio, Talanta, 2016, 154, 255–262 CrossRef CAS PubMed.
- Y. Li, W. Wang, L. Zhou, Y. Liu, Z. A. Mirza and X. Lin, Chemosphere, 2017, 169, 131–138 CrossRef CAS PubMed.
- Y. Q. Lan, B. L. Deng, C. Kim, E. C. Thornton and H. F. Xu, Environ. Sci. Technol., 2005, 39, 2087–2094 CrossRef CAS PubMed.
- M. Chrysochoou, D. R. Ferreira and C. P. Johnston, J. Hazard. Mater., 2010, 179, 650–657 CrossRef CAS PubMed.
- L. Di Palma, M. T. Gueye and E. Petrucci, J. Hazard. Mater., 2015, 281, 70–76 CrossRef CAS PubMed.
- R. Singh, V. Misra and R. P. Singh, J. Nanopart. Res., 2011, 13, 4063–4073 CrossRef CAS.
- Y. Li, J. Liang, X. He, L. Zhang and Y. Liu, J. Hazard. Mater., 2016, 320, 216–225 CrossRef CAS PubMed.
- L. N. Dossing, K. Dideriksen, S. L. S. Stipp and R. Frei, Chem. Geol., 2011, 285, 157–166 CrossRef CAS.
- M. Mullet, F. Demoisson, B. Humbert, L. J. Michot and D. Vantelon, Geochim. Cosmochim. Acta, 2007, 71, 3257–3271 CrossRef CAS.
- Y. Jung, J. Choi and W. Lee, Chemosphere, 2007, 68, 1968–1975 CrossRef CAS PubMed.
- P. Zhou, Y. Li, Y. Shen, Y. Lan and L. Zhou, J. Hazard. Mater., 2012, 237, 194–198 CrossRef PubMed.
- B. Pakzadeh and J. R. Batista, Water Res., 2011, 45, 3055–3064 CrossRef CAS PubMed.
- A. Reyhanitabar, L. Alidokht, A. R. Khataee and S. Oustan, Eur. J. Soil Sci., 2012, 63, 724–732 CrossRef CAS.
- J. Nemecek, O. Lhotski and T. Cajthaml, Sci. Total Environ., 2014, 485, 739–747 CrossRef PubMed.
- M. Stefaniuk, P. Oleszczuk and Y. S. Ok, Chem. Eng. J., 2016, 287, 618–632 CrossRef CAS.
- P. R. Wittbrodt and C. D. Palmer, Environ. Sci. Technol., 1996, 30, 2470–2477 CrossRef CAS.
- P. R. Wittbrodt and C. D. Palmer, Eur. J. Soil Sci., 1997, 48, 151–162 CrossRef CAS.
- L. E. Eary and D. Rai, Soil Sci. Soc. Am. J., 1991, 55 Search PubMed.
- Z. Struyk and G. Sposito, Geoderma, 2001, 102, 329–346 CrossRef CAS.
- P. Quagliotto, E. Montoneri, F. Tambone, F. Adani, A. Roberto Gobetto and G. Viscardi, Environ. Sci. Technol., 2006, 40, 1686–1692 CrossRef CAS PubMed.
- L. Yaoling, J. Xiaolin, N. mingxing, X. Qianqian, J. Yulei and W. Min, China Water Supply and Wastewater, 2016, 32, 119–122 Search PubMed.
- K. M. Abd El-Rheem, A. A. Afifi and R. A. Youssef, Life Sci. J., 2012, 9, 356–362 Search PubMed.
- E. W. D. Huffman Jr and H. A. Stuber, Analytical methodology for elemental analysis of humic substances, in Humic substances in soil, sediment, and water-Geochemistry, isolation, and characterization, ed. G. R. Aiken, D. M. McKnight, R. L. Wershaw and P. MacCarthy, John Wiley and Sons, New York, 1985, pp. 433–455 Search PubMed.
- D. Fabbri, M. Mongardi, L. Montanari, G. C. Galletti, G. Chiavari and R. Scotti, Fresenius. J. Anal. Chem., 1998, 362, 299–306 CrossRef CAS.
- J. O. Skjemstad, Aust. J. Soil Res., 1994, 32, 1215–1229 CrossRef CAS.
- J. Peuravuori and K. Pihlaja, Anal. Chim. Acta, 1998, 363, 235–247 CrossRef CAS.
- Z. M. Gu, X. R. Wang and X. Y. Gu, Chin. J. Anal. Chem., 2000, 28, 314–317 CAS.
- L. S. Clescerl, Freshwater Science, 1998, 56, 387 Search PubMed.
- B. Lv, M. Xing, J. Yang, W. Qi and Y. Lu, Bioresour. Technol., 2013, 132, 320–326 CrossRef CAS PubMed.
- G. Haberhauer, B. Rafferty, F. Strebl and M. H. Gerzabek, Geoderma, 1998, 83, 331–342 CrossRef CAS.
- J. G. Parsons, M. Hejazi, K. J. Tiemann, J. Henning and J. L. Gardea-Torresdey, Microchem. J., 2002, 71, 211–219 CrossRef CAS.
- A. Ohta, Geostand. Geoanal. Res., 2015, 39, 87–103 CrossRef CAS.
- Y. K. Sun, G. M. Zhou, X. M. Xiong, X. H. Guan, L. N. Li and H. L. Bao, Water Res., 2013, 47, 4340–4348 CrossRef CAS PubMed.
- B. Scaglia, F. Tambone and F. Adani, J. Environ. Sci., 2013, 25, 487–494 CrossRef CAS.
- A. Ohta, H. Kagi, H. Tsuno, M. Nomura and T. Okai, Geochem. J., 2012, 46, 409–420 CrossRef CAS.
- M. S. Elovitz and W. Fish, Environ. Sci. Technol., 1995, 29, 1933–1943 CrossRef CAS PubMed.
- D. M. Zhilin, P. Schmitt-Kopplin and I. V. Perminova, Environ. Chem. Lett., 2004, 2, 141–145 CrossRef CAS.
- N. H. Hsu, S. L. Wang, Y. C. Lin, G. D. Sheng and J. F. Lee, Environ. Sci. Technol., 2009, 43, 8801–8806 CrossRef CAS PubMed.
- H. Zhang and S. E. Lindberg, Environ. Sci. Technol., 2001, 35, 928–935 CrossRef CAS PubMed.
- L. Deng, N. S. Deng, L. W. Mou and F. T. Zhu, J. Environ. Sci., 2010, 22, 76–83 CrossRef CAS.
- P. P. Vaughan and N. V. Blough, Environ. Sci. Technol., 1998, 32, 2947–2953 CrossRef.
- M. Lipski, J. Slawinski and D. Zych, J. Fluoresc., 1999, 9, 133–138 CrossRef CAS.
- L. C. Hsu, S. L. Wang, Y. C. Lin, M. K. Wang, P. N. Chiang, J. C. Liu, W. H. Kuan, C. C. Chen and Y. M. Tzou, Environ. Sci. Technol., 2010, 44, 6202–6208 CrossRef CAS PubMed.
- M. Gaberell, Y. P. Chin, S. J. Hug and B. Sulzberger, Environ. Sci. Technol., 2003, 37, 4403–4409 CrossRef CAS PubMed.
- E. Kaiser and B. Sulzberger, Limnol. Oceanogr., 2004, 49, 540–554 CrossRef CAS.
- W. Zheng and H. Hintelmann, Geochim. Cosmochim. Acta, 2009, 73, 6704–6715 CrossRef CAS.
- B. Fulda, A. Voegelin, F. Maurer, I. Christl and R. Kretzschmar, Environ. Sci. Technol., 2013, 47, 10903–10911 CrossRef CAS PubMed.
- S. E. Page, M. Sander, W. A. Arnold and K. McNeill, Environ. Sci. Technol., 2012, 46, 1590–1597 CrossRef CAS PubMed.
- N. Senesi, T. M. Miano, M. R. Provenzano and G. Brunetti, Sci. Total Environ., 1989, 81–82, 143–156 CrossRef.
- M. Perez-Rodriguez, I. Horak-Terra, L. Rodriguez-Lado and A. M. Cortizas, Spectrochim. Acta, Part A, 2016, 168, 65–72 CrossRef CAS PubMed.
- P. Zaccheo, G. Cabassi, G. Ricca and L. Crippa, Org. Geochem., 2002, 33, 327–345 CrossRef CAS.
- A. Fakhry, O. Osman, H. Ezzat and M. Ibrahim, Spectrochim. Acta, Part A, 2016, 168, 244–252 CrossRef CAS PubMed.
- M. Ibrahim, A. J. Hameed and A. Jalbout, Appl. Spectrosc., 2008, 62, 306–311 CrossRef CAS PubMed.
- M. Fukushima, K. Nakayasu, S. Tanaka and H. Nakamura, Anal. Chim. Acta, 1995, 317, 195–206 CrossRef CAS.
- F. de Groot, G. Vanko and P. Glatzel, J. Phys.: Condens. Matter, 2009, 21 Search PubMed.
- D. Park, Y.-S. Yun and J. M. Park, J. Colloid Interface Sci., 2008, 317, 54–61 CrossRef CAS PubMed.
- F. C. Richard and A. C. Bourg, Water Res., 1991, 25, 807–816 CrossRef CAS.
- P. Kappen, E. Welter, P. H. Beck, J. M. McNamara, K. A. Moroney, G. M. Roe, A. Read and P. J. Pigram, Talanta, 2008, 75, 1284–1292 CrossRef CAS.
- R. R. Engebretson and R. Vonwandruszka, Environ. Sci. Technol., 1994, 28, 1934–1941 CrossRef CAS PubMed.
- K. Ghosh and M. Schnitzer, Can. J. Soil Sci., 1980, 60, 373–379 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28253a |
| This journal is © The Royal Society of Chemistry 2017 |