
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and biological evaluation of novel technetium-99m-labeled phenylquinoxaline derivatives as single photon emission computed tomography imaging probes targeting β-amyloid plaques in Alzheimer's disease†
Shimpei Iikuni,
Masahiro Ono *,
Keiichi Tanimura,
Hiroyuki Watanabe,
Masashi Yoshimura and
Hideo Saji
*,
Keiichi Tanimura,
Hiroyuki Watanabe,
Masashi Yoshimura and
Hideo Saji
Department of Patho-Functional Bioanalysis, Graduate School of Pharmaceutical Sciences, Kyoto University, 46-29 Yoshida Shimoadachi-cho, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: ono@pharm.kyoto-u.ac.jp; Fax: +81-75-753-4568; Tel: +81-75-753-4608
First published on 10th April 2017
Abstract
The development of an imaging probe targeting β-amyloid (Aβ) plaques in Alzheimer's disease labeled with technetium-99m, the most commonly used radioisotope for clinical diagnoses, has been strongly anticipated. In this study, we synthesized three novel 99mTc complexes with the phenylquinoxaline scaffold and evaluated their properties for imaging Aβ plaques. The 99mTc and corresponding Re complexes were synthesized with bis(aminoethanethiol) (BAT) as a chelating ligand. In a binding affinity assay using recombinant Aβ(1–42) aggregates in vitro, the 99mTc-labeled N,N-dimethylated phenylquinoxaline derivative (99mTc-BAT-C3-PQ-1) and the corresponding Re complex showed sufficient affinity for Aβ(1–42) aggregates. An in vivo biodistribution study in normal mice revealed that 99mTc-BAT-C3-PQ-1 showed a moderate initial brain uptake and a reasonable clearance from the brain. An ex vivo autoradiographic examination with 99mTc-BAT-C3-PQ-1 showed the marked labeling of Aβ plaques in brain sections from Tg2576 transgenic mice but not age-matched controls. 99mTc-BAT-C3-PQ-1 may be a potential single photon emission computed tomography probe for imaging Aβ plaques in Alzheimer's disease.
Introduction
Alzheimer's disease (AD) is the most common neurodegenerative disorder of the brain, with symptoms of memory loss, continuous cognitive decline, and behavioral disturbance. AD is pathologically characterized by the extracellular deposition of senile plaques consisting of β-amyloid (Aβ) and the intracellular deposition of neurofibrillary tangles consisting of hyperphosphorylated tau. AD is responsible for 60–70% of dementia in the elderly.1,2 Currently, there is no medical treatment that cures or prevents AD.1 Since Aβ deposition is believed to play a key role in neurodegeneration and begin prior to the onset of the disease, the development of an Aβ-targeted drug for AD has attracted much interest.3 The early diagnosis of AD will be needed for early intervention and appropriate treatment of the disease.Nuclear medical diagnoses with positron emission tomography (PET) have been utilized as major in vivo imaging modalities to carry out noninvasive diagnosis. Over the past few decades, research on the imaging of Aβ aggregates in AD brains with PET has made marked progress. Many clinical studies using [11C]PIB, which is the main Aβ imaging probe, have proved this utility for AD diagnosis.4–7 More recently, [18F]florbetapir (Amyvid),7–9 [18F]flutemetamol (Vizamyl),4,10,11 and [18F]florbetaben (Neuraceq)7,12,13 have been approved by the U.S. Food and Drug Administration for clinical AD diagnosis. However, the routine clinical use of PET probes labeled with 11C and 18F might be limited by their short physical half-lives (t1/2 = 20 and 110 min, respectively).
To address the problem of an increase in AD patients in the world, more generally versatile Aβ imaging with single photon emission computed tomography (SPECT) is needed. In exploratory research on the development of Aβ imaging probes for SPECT, a number of radioiodinated compounds with affinity for Aβ aggregates have been reported.14–17 Among them, a recent report suggested that [123I]ABC577 may be a clinically useful tracer for SPECT imaging of Aβ plaques in the brains of AD patients.18
Among clinically utilized radioisotopes for SPECT, 99mTc is the most commonly used in nuclear medical diagnoses for several reasons, including easy production by a 99Mo/99mTc generator, its physical half-life (6.01 h) being compatible with the biological localization, and its γ-ray energy (141 keV) suitable for in vivo imaging. Thus, the development of 99mTc-labeled Aβ imaging probes will provide simple, convenient, and widespread SPECT-based imaging methods for the detection of Aβ plaques in the living brain.
To develop useful 99mTc-labeled imaging agents targeting Aβ depositions in AD brains, compact and neutral 99mTc-labeled ligands have been reported, such as derivatives of biphenyl,19 phenylbenzothiazole,20 chalcone,21 flavone, aurone,22 curcumin,23 arylbenzofuran,24,25 dibenzylideneacetone,26 and arylbenzoxazole.27,28 However, there have been no reports on the development of clinically useful 99mTc-labeled probes due to either low affinity for Aβ aggregates or low brain uptake.
We previously reported phenylquinoxaline derivatives labeled with 125I or 18F targeting Aβ in AD brains (Fig. 1).29–31 These derivatives displayed high binding affinity for Aβ aggregates with Ki values in the nM range and showed specific labeling of Aβ plaques in sections of brain tissue from AD patients. Notably, [18F]PQ-6 showed an excellent Ki value (0.895 nM) and intensive specific labeling of Aβ in the AD brain section.30 These properties were superior to those of clinically used Aβ imaging probes, such as [11C]PIB and [18F]florbetapir (Ki = 9.00 and 12.8 nM, respectively). Based on these encouraging results, we designed novel 99mTc-labeled Aβ imaging probes with the scaffold of phenylquinoxaline (Fig. 1). Unlike radiohalogens, a chelating ligand is necessary for the introduction of metal 99mTc to the organic molecule. Considering the permeability at the blood–brain barrier (BBB), we selected bis(aminoethanethiol) (BAT) as a compact chelating ligand to form a neutral 99mTc complex. We herein synthesized novel 99mTc complexes based on phenylquinoxaline with BAT and evaluated their biological properties as potential SPECT probes targeting Aβ plaques in AD.
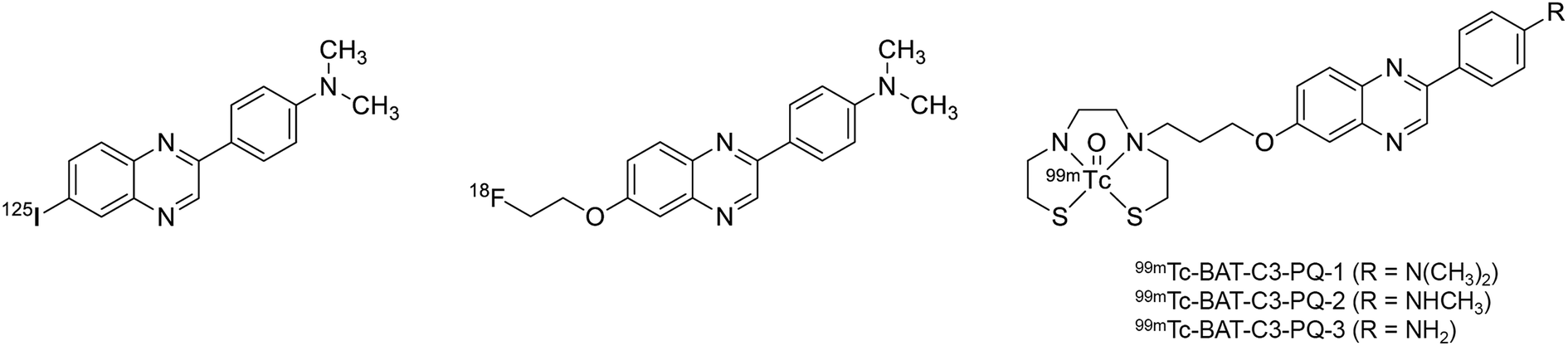 | ||
| Fig. 1 Chemical structures of the radioiodinated or radiofluorinated phenylquinoxaline derivatives reported in our previous study, and 99mTc-labeled phenylquinoxaline derivatives in the present study. | ||
Materials and methods
General
All reagents were obtained commercially and used without further purification unless otherwise indicated. Na99mTcO4 was purchased from Nihon Medi-Physics Co., Ltd. (Tokyo, Japan) or obtained from a commercial 99Mo/99mTc generator (Ultra-Techne Kow; FUJIFILM RI Pharma Co., Ltd., Tokyo, Japan). W-Prep 2XY (Yamazen, Osaka, Japan) was used for silica gel column chromatography on a Hi Flash silica gel column (40 μm, 60 Å, Yamazen). 1H NMR spectra were recorded on a JNM-ECS400 (JEOL, Tokyo, Japan) with tetramethylsilane (TMS) as an internal standard. Coupling constants are reported in Hertz. Multiplicity was defined as singlet (s), doublet (d), triplet (t), or multiplet (m). Mass spectra were obtained on a SHIMADZU LCMS-2020 EV (SHIMADZU, Kyoto, Japan). High-resolution mass spectrometry (HRMS) was conducted with a LCMS-IT-TOF (SHIMADZU). Reversed-phase high-performance liquid chromatography (RP-HPLC) was performed with a Shimadzu system (SHIMADZU, an LC-20AT pump with an SPD-20A UV detector, λ = 254 nm) with a Cosmosil C18 column (Nacalai Tesque, Kyoto, Japan, 5C18-AR-II, 4.6 × 150 mm) delivered at a flow rate of 1.0 mL min−1 using a solvent of H2O/MeCN [11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 (0 min) to 1:3 (30 min)] as the mobile phase.
:9 (0 min) to 1:3 (30 min)] as the mobile phase.
Animals
Animal experiments were conducted in accordance with our institutional guidelines and were approved by the Kyoto University Animal Care Committee. Male ddY mice were purchased from Japan SLC, Inc. (Shizuoka, Japan). Female Tg2576 and wild-type mice were purchased from Taconic Farms, Inc. (New York, U.S.A.). Animals were fed standard chow and had free access to water. All efforts were made to minimize suffering.Chemistry
:5) to give 302 mg of 1 (26%). 1H NMR (400 MHz, CDCl3) δ 7.55 (d, J = 6.8 Hz, 2H), 6.45 (d, J = 6.8 Hz, 2H), 3.86 (s, broad, 1H), 2.69 (s, 3H), 1.21 (s, 12H). MS (ESI): m/z calculated for C13H21BNO2+ [M + H]+, 234; found, 234.:5) to give 454 mg of 2 (26%). 1H NMR (400 MHz, CDCl3) δ 7.41–7.39 (m, 12H), 7.29–7.23 (m, 12H), 7.22–7.17 (m, 6H), 3.32 (s, 2H), 3.00–2.82 (m, 4H), 2.38–2.16 (m, 10H), 1.76–1.70 (m, 2H), 1.37 (s, 9H). MS (ESI): m/z calculated for C52H58BrN2O2S2+ [M + H]+, 887; found, 887.:1, 16 mL) was added Pd(PPh3)4 (115 mg, 0.10 mmol). The mixture was heated to reflux for 1 h. After cooling to room temperature, H2O (50 mL) was added. After the extraction of the mixture with AcOEt (50 mL × 2), the organic phases were combined and dried over MgSO4. Evaporation of the solvent afforded a residue, which was purified by silica gel chromatography (CHCl3/MeOH = 10:1) to give 236 mg of 4 (89%). 1H NMR (400 MHz, DMSO-d6) δ 9.31 (s, 1H), 8.13 (d, J = 7.6 Hz, 2H), 7.87 (d, J = 8.4 Hz, 1H), 7.35 (d, J = 8.4 Hz, 1H), 7.23 (s, 1H), 6.86 (d, J = 7.6 Hz, 2H), 3.01 (s, 6H). MS (ESI): m/z calculated for C16H16N3O+ [M + H]+, 266; found, 266.:2, 30 mL) was added Pd(PPh3)4 (115 mg, 0.10 mmol). The mixture was heated to reflux for 30 min. After cooling to room temperature, H2O (200 mL) was added. The mixture was extracted with AcOEt (100 mL × 2). The organic phases were combined and dried over MgSO4. Evaporation of the solvent afforded a residue, which was purified by silica gel chromatography (AcOEt/hexane = 1:1) to give 153 mg of 5 (61%). 1H NMR (400 MHz, CDCl3) δ 9.06 (s, 1H), 7.98 (d, J = 8.8 Hz, 2H), 7.90 (d, J = 9.2 Hz, 1H), 7.37 (s, 1H), 7.33–7.29 (m, 1H), 6.70 (d, J = 8.8 Hz, 2H), 2.90 (s, 3H). MS (ESI): m/z calculated for C15H14N3O+ [M + H]+, 252; found, 252.:1, 24 mL) was added Pd(PPh3)4 (231 mg, 0.20 mmol). The mixture was heated to reflux for 2 h. After cooling to room temperature, H2O (60 mL) was added. The mixture was extracted with AcOEt (60 mL × 2). The organic phases were combined and dried over MgSO4. Evaporation of the solvent afforded a residue, which was purified by silica gel chromatography (AcOEt/hexane = 3:1) to give 49 mg of 6 (10%). 1H NMR (400 MHz, CD3OD) δ 9.14 (s, 1H), 7.94 (d, J = 8.8 Hz, 2H), 7.91 (d, J = 9.2 Hz, 1H), 7.37 (dd, J = 9.2, 2.8 Hz, 1H), 7.26 (d, J = 2.8 Hz, 1H), 6.83 (d, J = 8.8 Hz, 2H). MS (ESI): m/z calculated for C14H12N3O+ [M + H]+, 238; found, 238.:1) to give 87 mg of 7 (62%). 1H NMR (400 MHz, CDCl3) δ 9.18 (s, 1H), 8.09 (d, J = 8.8 Hz, 2H), 7.92 (d, J = 9.2 Hz, 1H), 7.40–7.37 (m, 14H), 7.30–7.27 (m, 6H), 7.23–7.16 (m, 12H), 6.85 (d, J = 8.8 Hz, 2H), 4.03 (t, J = 6.0 Hz, 2H), 3.07 (s, 6H), 2.95–2.84 (m, 4H), 2.41–2.25 (m, 10H), 1.76 (t, J = 6.4 Hz, 2H), 1.37 (s, 9H). MS (ESI): m/z calculated for C68H72N5O3S2+ [M + H]+, 1070; found, 1070.:1) to give 38 mg of 8 (27%). 1H NMR (400 MHz, CDCl3) δ 9.17 (s, 1H), 8.05 (d, J = 8.4 Hz, 2H), 7.92 (d, J = 8.8 Hz, 1H), 7.40–7.37 (m, 12H), 7.30–7.28 (m, 6H), 7.23–7.16 (m, 14H), 6.74 (d, J = 8.4 Hz, 2H), 4.03 (t, J = 6.4 Hz, 2H), 3.02–2.96 (m, 2H), 2.92 (s, 3H), 2.42–2.25 (m, 12H), 1.77 (t, J = 6.4 Hz, 2H), 1.37 (s, 9H). MS (ESI): m/z calculated for C67H70N5O3S2+ [M + H]+, 1056; found, 1056.:1, 22 mL), to which were added ReOCl3(PPh3)2 (70 mg, 0.084 mmol) and 1 M AcONa in MeOH (4 mL). The reaction mixture was heated to reflux for 8 h. After cooling to room temperature, CHCl3 (60 mL) was added and filtered. Evaporation of the filtrate gave a residue, which was purified by silica gel chromatography (CHCl3/MeOH = 10:1) and then RP-HPLC (MeCN/H2O = 1:1) to give 24 mg of 9 (84%). 1H NMR (400 MHz, DMSO-d6) δ 9.42 (s, 1H), 8.17 (d, J = 8.8 Hz, 2H), 7.95 (d, J = 8.4 Hz, 1H), 7.49 (dd, J = 8.4, 2.4 Hz, 1H), 7.46 (d, J = 2.4 Hz, 1H), 6.86 (d, J = 8.8 Hz, 2H), 4.30 (t, J = 6.4 Hz, 2H), 4.18–3.83 (m, 4H), 3.64–3.16 (m, 6H), 3.02 (s, 6H), 2.72–1.68 (m, 6H). HRMS (ESI): m/z calculated for C25H32N5NaO2187ReS2+ [M + Na]+, 708.1447; found, 708.1446.:1, 22 mL), to which were added ReOCl3(PPh3)2 (32 mg, 0.038 mmol) and 1 M AcONa in MeOH (4 mL). The reaction mixture was heated to reflux for 2 h. After cooling to room temperature, H2O (50 mL) was added. The mixture was extracted with CHCl3 (50 mL × 2) and dried over MgSO4. Evaporation of the solution gave a residue, which was purified by silica gel chromatography (CHCl3/MeOH = 10:1) and then RP-HPLC (MeCN/H2O = 1:1) to give 5 mg of 10 (39%). 1H NMR (400 MHz, DMSO-d6) δ 9.38 (s, 1H), 8.10 (d, J = 8.8 Hz, 2H), 7.93 (d, J = 8.4 Hz, 1H), 7.48 (dd, J = 8.4, 2.4 Hz, 1H), 7.45 (d, J = 2.4 Hz, 1H), 6.69 (d, J = 8.8 Hz, 2H), 4.30 (t, J = 6.4 Hz, 2H), 4.14–3.82 (m, 4H), 3.64–3.17 (m, 6H), 2.76 (s, 3H), 2.72–1.68 (m, 6H). HRMS (ESI): m/z calculated for C24H31N5O2187ReS2+ [M + H]+, 672.1471; found, 672.1475.:1) to give 46 mg of 11 (62%). 1H NMR (400 MHz, CDCl3) δ 9.18 (s, 1H), 8.01 (d, J = 8.4 Hz, 2H), 7.98 (d, J = 10.0 Hz, 1H), 7.42–7.35 (m, 2H), 6.83 (d, J = 8.4 Hz, 2H), 4.28 (t, J = 6.0 Hz, 2H), 3.66 (t, J = 6.4 Hz, 2H), 2.42 (t, J = 6.4 Hz, 2H). MS (ESI): m/z calculated for C18H16BrN3O+ [M + H]+, 358; found, 358.:1, 60 mL × 2). The organic phases were combined and dried over MgSO4. Evaporation of the solvent afforded a residue, which was purified by silica gel chromatography (AcOEt/hexane = 3:1) to give 18 mg of 12 (13%). 1H NMR (400 MHz, CDCl3) δ 9.17 (s, 1H), 8.01 (d, J = 8.4 Hz, 2H), 7.93 (d, J = 9.2 Hz, 1H), 7.41–7.36, (m, 14H), 7.33–7.27 (m, 4H), 7.24–7.15 (m, 12H), 4.15–3.80 (m, 4H), 2.42–2.20 (m, 10H). MS (ESI): m/z calculated for C66H68N5O3S2+ [M + H]+, 1043; found, 1043.:1, 5 mL), to which were added ReOCl3(PPh3)2 (18 mg, 0.022 mmol) and 1 M AcONa in MeOH (4 mL). The reaction mixture was heated to reflux overnight. After cooling to room temperature, CHCl3 (60 mL) was added and the mixture was filtered. Evaporation of the filtrate gave a residue, which was purified by silica gel chromatography (CHCl3/MeOH = 9:1) and then RP-HPLC (MeCN/H2O = 1:1) to give 4.1 mg of 13 (59%). 1H NMR (400 MHz, DMSO-d6) δ 9.35 (s, 1H), 8.02 (d, J = 8.8 Hz, 2H), 7.92 (d, J = 8.4 Hz, 1H), 7.47 (d, J = 8.4 Hz, 1H), 7.44 (s, 1H), 6.70 (d, J = 8.8 Hz, 2H), 5.69 (s, 2H), 4.29 (t, J = 5.6 Hz, 2H), 4.18–3.82 (m, 4H), 3.64–3.06 (m, 6H), 2.72–1.68 (m, 6H). HRMS (ESI): m/z calculated for C23H28N5NaO2187ReS2+ [M + Na]+, 680.1134; found, 680.1134.99mTc-labeling reaction
To a solution of sodium glucoheptonate dehydrate (2.0 g, 7.0 mmol) in H2O (25 mL) was added 0.75 mL of an SnCl2·2H2O solution [12 mg of tin(II) chloride dehydrate (53 mmol) dissolved in 15 mL of 0.1 M HCl]. This solution was adjusted to pH 8.5–9.0 using a small amount of 0.1 M NaOH. After being lyophilized to give the pulverulent Sn glucoheptonate (SnGH) kit, 1.0 mg of this was weighed and added to an Na99mTcO4 solution (200 μL). The solution was reacted at room temperature for 10 min to give a 99mTcGH solution. A solution of the precursor (7, 8, or 12) (0.3 mg) in TFA (400 μL) was mixed with triethylsilane (20 μL), and the solvents were removed under a stream of argon gas. The residue was resolved in MeCN (100 μL), followed by the addition of 0.1 M HCl (10 μL) and the 99mTcGH solution (100 μL). The reaction mixture was heated to 110 °C for 20 min. After cooling to room temperature, saturated NaHCO3 (aq.) (30 μL) was added. The mixture was purified by RP-HPLC. The 99mTc-labeled phenylquinoxaline complex was analyzed by analytical RP-HPLC on a Cosmosil C18 column (5C18-AR-II, 4.6 × 150 mm) with a solvent of H2O/MeCN [11:9 (0 min) to 1:3 (30 min)] as the mobile phase at a flow rate of 1.0 mL min−1. The radioactivity of the 99mTc-labeled form was recorded for 30 min.
Measurement of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P
P
The experimental determination of partition coefficients was performed in 1-octanol and phosphate-buffered saline (PBS) (pH 7.4). The two phases were presaturated with each other. 1-Octanol (3.0 mL) and PBS (3.0 mL) were pipetted into a 15 mL test tube containing the 99mTc complex. The test tube was vortexed for 2 min and centrifuged (5 min, 4000g). Aliquots (500 μL) from the 1-octanol and PBS phases were transferred into two test tubes for counting. One milliliter of the remaining 1-octanol phase, new 1-octanol (2.0 mL), and PBS (3.0 mL) were pipetted into a new test tube. The vortexing, centrifuging, and counting were repeated until consistent partition coefficient values were obtained (usually the sixth partition). The amount of radioactivity in each tube was measured with a γ counter (Wallac 1470 Wizard; PerkinElmer, Massachusetts, U.S.A.). The partition coefficient was calculated using the equation: logP = log[count1-octanol/countPBS].
Binding assay using Aβ(1–42) aggregates in solution
A solid form of Aβ(1–42) was purchased from the Peptide Institute (Osaka, Japan). Aggregation was carried out by gently dissolving the peptide (0.25 mg mL−1) in PBS (pH 7.4). The solution was incubated at 37 °C for 42 h with gentle and constant shaking. Aggregated Aβ(1–42) was used after being diluted to a concentration appropriate for the assays with PBS. The binding assay was performed by mixing 50 μL of Aβ(1–42) aggregates (final conc., 0.125, 1.25, 2.5, and 5.0 μg mL−1), 50 μL of [99mTc]14, [99mTc]15, or [99mTc]16 (8.3 kBq in 30% EtOH), and 900 μL of 30% EtOH. After incubation at room temperature for 3 h, the mixture was filtered through Whatman GF/B filters (Whatman, Kent, U.K.) using a Brandel M-24 cell harvester (Brandel, Maryland, U.S.A.), and the radioactivity of the filters containing the bound 99mTc complex was measured using a γ counter (PerkinElmer).Competitive inhibition assay using Aβ(1–42) aggregates in solution
A mixture containing 50 μL of Re complex (9 or 10) or 6-iodo-2-(4′-dimethylamino)phenyl-imidazo[1,2-a]-pyridine (IMPY) (final conc., 10 pM to 20 μM in DMSO), 50 μL of [125I]IMPY (final conc., 2.5 kBq mL−1 in EtOH), 50 μL of Aβ(1–42) aggregates (final conc., 0.125 μg mL−1), and 850 μL of 10% DMSO was incubated at room temperature for 3 h. The mixture was then filtered through Whatman GF/B filters (Whatman) using a Brandel M-24 cell harvester (Brandel), and the radioactivity of the filters containing the bound 125I ligand was measured using a γ counter (PerkinElmer). Values for the half-maximal inhibitory concentration (IC50) were determined from displacement curves of three independent experiments using GraphPad Prism 5.0 (GraphPad Software, Inc., California, U.S.A.), and those for the inhibition constant (Ki) were calculated using the Cheng–Prusoff equation: Ki = IC50/(1 + [L]/Kd),32 where [L] is the concentration of [125I]IMPY used in the assay and Kd is the dissociation constant of IMPY (4.2 nM).30In vivo biodistribution assessment in normal mice
A saline solution (100 μL) of [99mTc]14 (20 kBq) containing EtOH (20 μL) and Tween80 (0.1 mg) was injected directly into the tail vein of ddY mice (male, 5 weeks old). The mice were sacrificed at 2, 10, 30, and 60 min postinjection. The organs of interest were removed and weighed, and radioactivity was measured using a γ counter (PerkinElmer). The % injected dose per g (% ID per g) of samples was calculated by comparing the sample counts with the count of the initial dose.Ex vivo autoradiography using Tg2576 and wild-type mice
Tg2576 transgenic mice (female, 32 months old) and wild-type mice (female, 32 months old) were used as the AD model and age-matched control, respectively. A saline solution (150 μL) of [99mTc]14 (25.9 MBq) containing EtOH (30 μL) and Tween80 (0.15 mg) was injected through the tail vein. After the mice were sacrificed at 30 min postinjection by decapitation, the brain was immediately removed, embedded in Super Cryoembedding Medium (SCEM) compound (SECTION-LAB Co., Ltd., Hiroshima, Japan), and then frozen in a dry ice/hexane bath. Frozen sections were prepared at a 30 μm thickness and exposed to a BAS imaging plate (Fuji Film, Tokyo, Japan) overnight. Autoradiographic images were obtained using a BAS5000 scanner system (Fuji Film). After autoradiographic examination, the same sections were stained by thioflavin-S to confirm the presence of Aβ depositions. For thioflavin-S fluorescent staining, the sections were immersed in a 100 μM thioflavin-S solution containing 50% EtOH for 3 min, washed in 50% EtOH for 1 min two times, and examined using a microscope (BIOREVO BZ-9000; Keyence Corp., Osaka, Japan) equipped with a GFP-BP filter set.Results and discussion
Chemistry
The synthesis of the precursors for the 99mTc-labeling and the corresponding Re complex based on phenylquinoxaline is outlined in Scheme 1. A methoxy group of 6-methoxy-2-chloroquinoxaline was converted to a hydroxyl group using AlCl3/toluene according to our previous report,30 which afforded 3. The phenylquinoxaline backbone was formed by the Suzuki coupling reaction between 3 and the corresponding boronate ester, which afforded 4, 5, and 6 in 89, 61, and 10% yields, respectively. The thiol-protected chelation ligand (Trt-Boc-BAT) was synthesized as reported previously.21 A trimethylene group was introduced into Trt-Boc-BAT and 6 as a linker by reacting them with 1,3-dibromopropane, which afforded 2 and 11 in 26 and 62% yields, respectively.24 The precursors for 99mTc-labeling, compounds 7, 8, and 12, were synthesized by conjugating the chelating ligand and phenylquinoxaline scaffold.24 Technetium has no stable isotopes; therefore, direct structural characterization is very challenging. Since rhenium, the group VIIB congener of technetium, generally produces complexes with similar physical and biological properties to those of technetium, it is often used as a nonradioactive surrogate of technetium for large-scale synthesis and structural characterization.19,21,22,24–27 We prepared the corresponding Re complexes with phenylquinoxaline. After deprotection of the thiol groups of 7, 8, and 12 in TFA and triethylsilane, the Re complexes 9 (Re-BAT-C3-PQ-1), 10 (Re-BAT-C3-PQ-2), and 13 (Re-BAT-C3-PQ-3) were synthesized, respectively, through a reaction with ReOCl3(PPh3)2. 1H NMR spectra and the HPLC profiles for three Re complexes (9, 10, and 13) proved their high-level purity (ESI Fig. S1–S4†).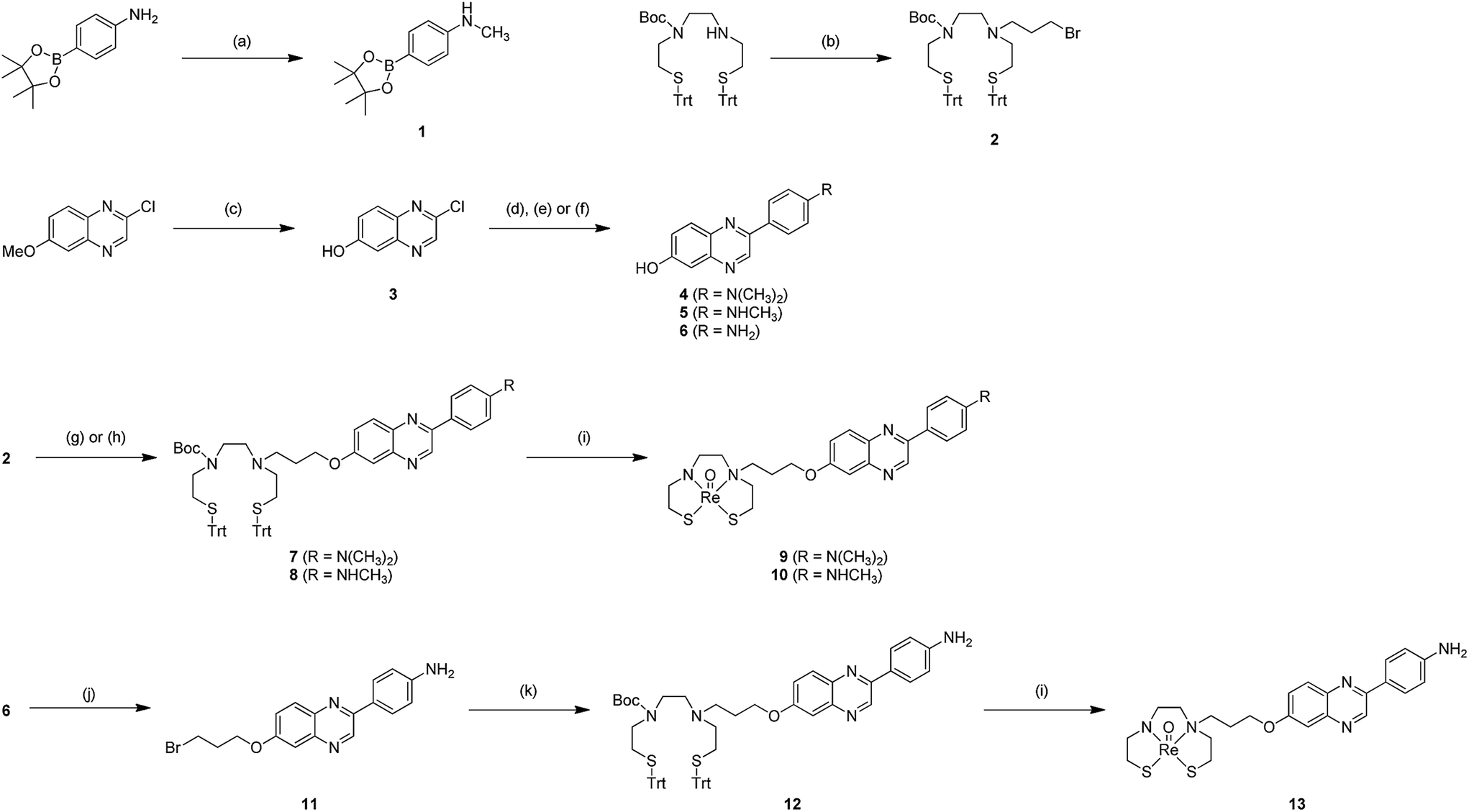 | ||
| Scheme 1 Synthetic route for phenylquinoxaline derivatives. Reagents and conditions: (a) (1) paraformaldehyde, sodium methoxide, MeOH, reflux, (2) NaBH4, reflux; (b) 1,3-dibromopropane, N,N-diisopropylethylamine, DMF, 90 °C; (c) AlCl3, toluene, 0 °C to 80 °C; (d) N,N-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline, Pd(PPh3)4, dioxane, 2 M Na2CO3 (aq.), reflux; (e) 1, Pd(PPh3)4, dioxane, 2 M Na2CO3 (aq.), reflux; (f) 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline, Pd(PPh3)4, dioxane, 2 M Na2CO3 (aq.), reflux; (g) 4, Cs2CO3, DMF, 70 °C; (h) 5, Cs2CO3, DMF, 70 °C; (i) (1) triethylsilane, TFA, room temperature, (2) ReOCl3(PPh3)2, AcONa, CH2Cl2, MeOH, reflux; (j) 1,3-dibromopropane, K2CO3, MeCN, reflux; (k) Trt-Boc-BAT, N,N-diisopropylethylamine, DMF, 110 °C. | ||
99mTc-labeling reaction
The 99mTc-labeling reaction was performed by a ligand exchange reaction employing the precursors 7, 8, and 12 with 99mTcGH, which afforded [99mTc]14 (99mTc-BAT-C3-PQ-1), [99mTc]15 (99mTc-BAT-C3-PQ-2), and [99mTc]16 (99mTc-BAT-C3-PQ-3), respectively (Scheme 2). The radiochemical yields for [99mTc]14, [99mTc]15, and [99mTc]16 were 56, 62, and 44%, respectively. The resulting mixture was analyzed by RP-HPLC, showing that a single radioactive complex was formed with radiochemical purity higher than 99% after purification by HPLC. The radiochemical identity of the 99mTc complex was verified by comparative HPLC using the corresponding Re complex as a reference (Table 1). The retention times on RP-HPLC (radioactivity) for [99mTc]14 (99mTc-BAT-C3-PQ-1), [99mTc]15 (99mTc-BAT-C3-PQ-2), and [99mTc]16 (99mTc-BAT-C3-PQ-3) were 23.3, 16.6, and 10.8 min, respectively. Moreover, the retention times on RP-HPLC (absorption at 254 nm) for 9 (Re-BAT-C3-PQ-1), 10 (Re-BAT-C3-PQ-2), and 13 (Re-BAT-C3-PQ-3) were 22.1, 15.6 and 10.1 min, respectively. The retention times between 99mTc-labeled tracers and the corresponding Re complexes suggest that the desired 99mTc-labeled phenylquinoxaline derivatives were successfully synthesized.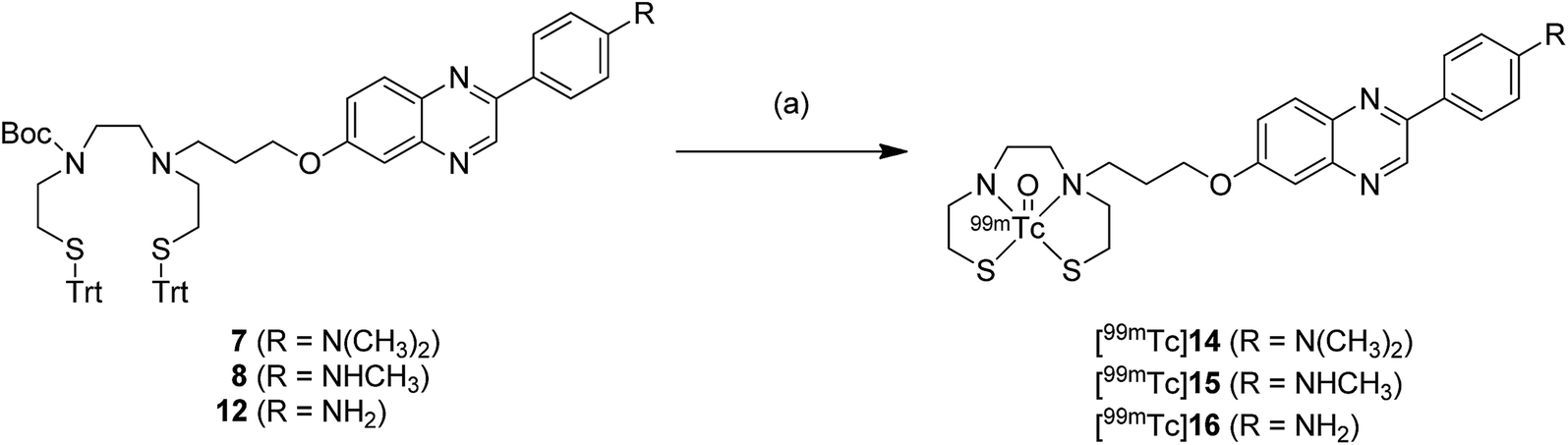 | ||
| Scheme 2 99mTc-labeling of phenylquinoxaline derivatives. Reagents and conditions: (a) (1) triethylsilane, TFA, room temperature, (2) 99mTc-glucoheptonate, 0.1 N HCl, MeCN, 110 °C. | ||
| Re complex | Retention time | 99mTc complex | Retention time |
|---|---|---|---|
| a RP-HPLC using a solution of H2O/MeCN [11:9 (0 min) to 1:3 (30 min)] as a mobile phase. |
|||
| 9 | 22.1 min | [99mTc]14 | 23.3 min |
| 10 | 15.6 min | [99mTc]15 | 16.6 min |
| 13 | 10.1 min | [99mTc]16 | 10.8 min |
Binding assay using Aβ(1–42) aggregates in solution
To evaluate the binding affinity for Aβ(1–42) aggregates of [99mTc]14 (99mTc-BAT-C3-PQ-1), [99mTc]15 (99mTc-BAT-C3-PQ-2), and [99mTc]16 (99mTc-BAT-C3-PQ-3), an in vitro binding experiment in solution was carried out. Fig. 2 shows the binding affinity of [99mTc]14, [99mTc]15, and [99mTc]16 as specific Aβ(1–42) aggregate-bound radioactivity (%) at different concentrations of Aβ(1–42) aggregates. The percent radioactivity of [99mTc]14 (99mTc-BAT-C3-PQ-1) and [99mTc]15 (99mTc-BAT-C3-PQ-2) bound to aggregates increased dependent on the dose of Aβ(1–42), while [99mTc]16 (99mTc-BAT-C3-PQ-3) showed no marked affinity for the aggregates. In terms of Aβ(1–42) aggregate-bound radioactivity, the derivatives ranked in the following order: [99mTc]14 > [99mTc]15 > [99mTc]16, at all concentrations of Aβ(1–42) aggregates. The order in terms of the strength of binding corresponded with those of radioiodinated and radiofluorinated quinoxaline derivatives; in other words, the affinity increased in the order of the N,N-dimethylated derivative > N-monomethylated derivative > primary amino derivative.29,30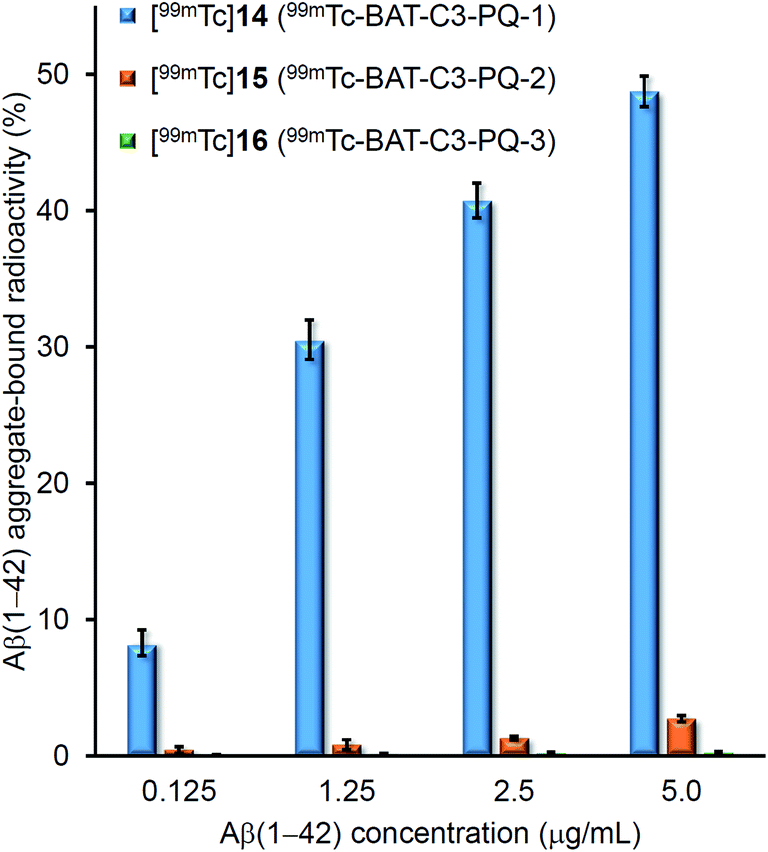 | ||
| Fig. 2 Binding assay of 99mTc-labeled phenylquinoxaline derivatives using Aβ(1–42) aggregates. Values are the mean ± standard deviation of the mean of three independent experiments. | ||
Competitive inhibition assay using Aβ(1–42) aggregates in solution
Since [99mTc]14 and [99mTc]15 showed affinity for Aβ aggregates in the Aβ(1–42) binding assay, quantitative binding affinity for Aβ aggregates was evaluated with rhenium complexes 9 (Re-BAT-C3-PQ-1) and 10 (Re-BAT-C3-PQ-2) according to conventional methods using [125I]IMPY.33 Both Re complexes 9 and 10 inhibited the binding of [125I]IMPY in a dose-dependent manner, with Ki values of 53.7 and 296 nM, respectively (Table 2). The affinity for Aβ(1–42) aggregates of Re complexes well reflected that of the corresponding 99mTc complexes, confirmed above. These Re complexes showed the Ki values, which were comparable to those of the corresponding Re complexes of the 99mTc-labeled probes reported previously (13.6–393 nM).24,26,27 The binding affinity of the N,N-dimethylated derivative (9) was slightly lower than that of PIB (9.00 nM)34 and IMPY (12.3 nM), indicating that it may exhibit sufficient affinity for the imaging of Aβ(1–42) aggregates in vivo. However, the Ki values of these Re complexes were higher than those of quinoxaline derivatives with introduced iodine (4.1 nM) and fluorine (0.895 nM) reported previously.29,30 This result suggests that the introduction of the bulky 99mTc-BAT complex into the phenylquinoxaline scaffold interferes with the binding to Aβ(1–42) aggregates, as shown in some reports on other Aβ imaging probes conjugated with the 99mTc-BAT complex.24 According to the result of binding affinity for Aβ(1–42) aggregates in vitro, further studies were conducted using [99mTc]14 (99mTc-BAT-C3-PQ-1) with the highest binding affinity.In vivo biodistribution assessment in normal mice
To evaluate the brain uptake of [99mTc]14 (99mTc-BAT-C3-PQ-1), a biodistribution experiment was performed with normal mice (Table 3). An ideal Aβ imaging probe should show high-level efficiency along with BBB penetration to deliver an adequate dose into the brain, and then achieve rapid clearance of the non-bound tracer from the brain to reduce the background signal in the AD brain. Previous studies suggested that the optimal measured logP-value of compounds to pass freely across the BBB ranged from 0.9 to 2.5.35 The logP-value for [99mTc]14 was 2.45, indicating that this complex might penetrate the BBB. [99mTc]14 showed the highest initial uptake at 2 min postinjection (0.88% ID per g), which was comparable or slightly lower than those of 99mTc-labeled probes reported previously (0.2–1.80% ID per g).19–22,24–27 The radioactivity of [99mTc]14 in the brain was rapidly eliminated (0.25% ID per g at 60 min postinjection). The brain2 min/brain60 min ratio is generally used as an index for evaluating pharmacokinetics in vivo. The brain2 min/brain60 min ratio of 99mTc-BAT-C3-PQ-1 was 3.52, indicating that it displayed one of the best profiles of radioactivity in the brain among 99mTc complexes reported previously.19–22,24–27 Although the brain2 min/brain60 min ratio of 99mTc-BAT-C3-PQ-1 was lower than those of [11C]PIB (11.7)6 and [123I]IMPY (14.4),33 it was comparable to that of [18F]florbetapir (3.80).30 These indicate its promising property for the imaging of Aβ in the brain. Unfortunately, radioactivity in the stomach suggests the partial decomposition to pertechnetate in vivo.
| Tissue | Time after injection (min) | |||
|---|---|---|---|---|
| 2 | 10 | 30 | 60 | |
| a Expressed as % injected dose per gram of tissue. Each value is the mean (standard deviation) of five animals at each interval.b Expressed as % injected dose. | ||||
| Blood | 10.97 (0.90) | 5.39 (0.32) | 3.04 (0.20) | 2.38 (0.17) |
| Liver | 27.07 (0.60) | 29.66 (1.96) | 31.41 (1.63) | 26.25 (1.47) |
| Kidney | 12.51 (1.18) | 9.12 (0.33) | 6.47 (0.36) | 4.84 (0.58) |
| Intestine | 2.35 (0.33) | 6.06 (0.48) | 14.22 (1.96) | 16.88 (2.92) |
| Spleen | 4.53 (1.23) | 3.32 (0.19) | 2.38 (0.26) | 1.84 (0.25) |
| Pancreas | 4.92 (0.22) | 3.95 (0.51) | 2.82 (0.19) | 2.09 (0.93) |
| Heart | 10.40 (1.00) | 4.17 (0.25) | 2.64 (0.18) | 1.88 (0.21) |
| Lung | 12.72 (1.12) | 6.82 (0.50) | 4.97 (0.56) | 3.38 (0.57) |
| Stomachb | 1.90 (0.17) | 4.89 (1.31) | 8.44 (1.90) | 6.79 (0.82) |
| Brain | 0.88 (0.08) | 0.77 (0.08) | 0.52 (0.03) | 0.25 (0.02) |
Ex vivo autoradiography using Tg2576 and wild-type mice
According to the results of the binding affinity assay in vitro and biodistribution in normal mice, we further evaluated the potential of [99mTc]14 (99mTc-BAT-C3-PQ-1) for imaging Aβ plaques in the AD model mouse. An ex vivo autoradiographic examination was carried out using Tg2576 transgenic mouse (Fig. 3). Since Tg2576 mice are known to overproduce Aβ aggregates in the brain, they have been commonly used to evaluate the specific binding to Aβ aggregates in experiments in vitro and in vivo.24,30,36 The brain was removed at 30 min postinjection for autoradiography. [99mTc]14 showed intensive radioactive spots in sections from the Tg2576 mouse, but not the age-matched control (Fig. 3A and C). Furthermore, these spots corresponded with Aβ depositions confirmed by fluorescent staining in the same sections with thioflavin-S, a histochemical dye commonly used to stain Aβ plaques (Fig. 3B, red arrowheads). This suggests that 99mTc-BAT-C3-PQ-1 had sufficient properties to detect Aβ plaques in the transgenic mouse, although it displayed a lower affinity for Aβ aggregates and lower brain uptake in normal mice than the clinically utilized probes, such as [11C]PIB and [18F]florbetapir. In addition, the nonspecific accumulation of radioactivity within the white matter, which was demonstrated in our previous reports,30 was not observed for 99mTc-BAT-C3-PQ-1, as shown in Fig. 3A and C, probably due to its low brain uptake. However, some Aβ depositions were not labeled with 99mTc-BAT-C3-PQ-1. This may derive from its low brain uptake, low affinity, and low in vivo stability, which are insufficient to bind to Aβ aggregates deposited in the whole brain. Therefore, further modifications are required to improve the properties of 99mTc-labeled phenylquinoxaline derivatives for imaging Aβ plaques in AD brains. | ||
| Fig. 3 Ex vivo autoradiograms from a Tg2576 mouse (A) and a wild-type mouse (C) with [99mTc]14 (99mTc-BAT-C3-PQ-1). The same sections are also stained with thioflavin-S (B). | ||
Conclusion
We successfully synthesized three 99mTc-labeled phenylquinoxaline derivatives and their corresponding Re complexes. Among them, 99mTc-BAT-C3-PQ-1 and its corresponding Re complex showed sufficient binding affinity for Aβ aggregates in an in vitro binding assay. An in vivo biodistribution study with 99mTc-BAT-C3-PQ-1 in normal mice indicated its moderate initial brain uptake and reasonable clearance from the brain. Moreover, it showed extensive labeling of Aβ plaques in the transgenic mouse brain. Taken together, the findings in the present study provide important information on developing clinically useful 99mTc-labeled Aβ imaging probes for the diagnosis of AD based on quinoxaline derivatives. Additional modifications to further improve the pharmacokinetics in the brain and the binding affinity for Aβ aggregates of 99mTc complexes are underway.Acknowledgements
The study was supported by a Grant-in-Aid for Scientific Research (B) (Grant Number 26293274), the Nakatani Foundation for Advancement of Measuring Technologies in Biomedical Engineering, Japan Research Foundation for Clinical Pharmacology, and a JSPS Research Fellowship for Young Scientists (16J05493).References
- A. Nordberg, Lancet Neurol., 2004, 3, 519–527 CrossRef CAS PubMed.
- R. L. Nussbaum and C. E. Ellis, N. Engl. J. Med., 2003, 348, 1356–1364 CrossRef CAS PubMed.
- E. Giacobini and R. E. Becker, J. Alzheimer's Dis., 2007, 12, 37–52 CrossRef CAS PubMed.
- S. Hatashita, H. Yamasaki, Y. Suzuki, K. Tanaka, D. Wakebe and H. Hayakawa, Eur. J. Nucl. Med. Mol. Imaging, 2014, 41, 290–300 CrossRef CAS PubMed.
- W. E. Klunk, H. Engler, A. Nordberg, Y. Wang, G. Blomqvist, D. P. Holt, M. Bergstrom, I. Savitcheva, G. F. Huang, S. Estrada, B. Ausen, M. L. Debnath, J. Barletta, J. C. Price, J. Sandell, B. J. Lopresti, A. Wall, P. Koivisto, G. Antoni, C. A. Mathis and B. Langstrom, Ann. Neurol., 2004, 55, 306–319 CrossRef CAS PubMed.
- C. A. Mathis, Y. Wang, D. P. Holt, G. F. Huang, M. L. Debnath and W. E. Klunk, J. Med. Chem., 2003, 46, 2740–2754 CrossRef CAS PubMed.
- R. Ni, P. G. Gillberg, A. Bergfors, A. Marutle and A. Nordberg, Brain, 2013, 136, 2217–2227 CrossRef PubMed.
- K. J. Lin, W. C. Hsu, I. T. Hsiao, S. P. Wey, L. W. Jin, D. Skovronsky, Y. Y. Wai, H. P. Chang, C. W. Lo, C. H. Yao, T. C. Yen and M. P. Kung, Nucl. Med. Biol., 2010, 37, 497–508 CrossRef CAS PubMed.
- D. F. Wong, P. B. Rosenberg, Y. Zhou, A. Kumar, V. Raymont, H. T. Ravert, R. F. Dannals, A. Nandi, J. R. Brasic, W. Ye, J. Hilton, C. Lyketsos, H. F. Kung, A. D. Joshi, D. M. Skovronsky and M. J. Pontecorvo, J. Nucl. Med., 2010, 51, 913–920 CrossRef CAS PubMed.
- R. Lundqvist, J. Lilja, B. A. Thomas, J. Lotjonen, V. L. Villemagne, C. C. Rowe and L. Thurfjell, J. Nucl. Med., 2013, 54, 1472–1478 CrossRef CAS PubMed.
- N. Nelissen, K. Van Laere, L. Thurfjell, R. Owenius, M. Vandenbulcke, M. Koole, G. Bormans, D. J. Brooks and R. Vandenberghe, J. Nucl. Med., 2009, 50, 1251–1259 CrossRef CAS PubMed.
- G. A. Becker, M. Ichise, H. Barthel, J. Luthardt, M. Patt, A. Seese, M. Schultze-Mosgau, B. Rohde, H. J. Gertz, C. Reininger and O. Sabri, J. Nucl. Med., 2013, 54, 723–731 CrossRef CAS PubMed.
- W. Zhang, S. Oya, M. P. Kung, C. Hou, D. L. Maier and H. F. Kung, Nucl. Med. Biol., 2005, 32, 799–809 CrossRef CAS PubMed.
- H. F. Kung, M. P. Kung, Z. P. Zhuang, C. Hou, C. W. Lee, K. Plössl, B. Zhuang, D. M. Skovronsky, V. M. Lee and J. Q. Trojanowski, Mol. Imaging Biol., 2003, 5, 418–426 CrossRef PubMed.
- W. Qu, M. P. Kung, C. Hou, T. E. Benedum and H. F. Kung, J. Med. Chem., 2007, 50, 2157–2165 CrossRef CAS PubMed.
- M. Ono, Y. Cheng, H. Kimura, H. Watanabe, K. Matsumura, M. Yoshimura, S. Iikuni, Y. Okamoto, M. Ihara, R. Takahashi and H. Saji, PLoS One, 2013, 8, e74104 CAS.
- C. J. Chen, K. Bando, H. Ashino, K. Taguchi, H. Shiraishi, K. Shima, O. Fujimoto, C. Kitamura, S. Matsushima, K. Uchida, Y. Nakahara, H. Kasahara, T. Minamizawa, C. Jiang, M. R. Zhang, M. Ono, M. Tokunaga, T. Suhara, M. Higuchi, K. Yamada and B. Ji, J. Nucl. Med., 2015, 56, 120–126 CrossRef PubMed.
- Y. Maya, Y. Okumura, R. Kobayashi, T. Onishi, Y. Shoyama, O. Barret, D. Alagille, D. Jennings, K. Marek, J. Seibyl, G. Tamagnan, A. Tanaka and Y. Shirakami, Brain, 2016, 139, 193–203 CrossRef PubMed.
- Z. P. Zhuang, M. P. Kung, C. Hou, K. Ploessl and H. F. Kung, Nucl. Med. Biol., 2005, 32, 171–184 CrossRef CAS PubMed.
- K. Serdons, T. Verduyckt, J. Cleynhens, C. Terwinghe, L. Mortelmans, G. Bormans and A. Verbruggen, Bioorg. Med. Chem. Lett., 2007, 17, 6086–6090 CrossRef CAS PubMed.
- M. Ono, R. Ikeoka, H. Watanabe, H. Kimura, T. Fuchigami, M. Haratake, H. Saji and M. Nakayama, ACS Chem. Neurosci., 2010, 1, 598–607 CrossRef CAS PubMed.
- M. Ono, R. Ikeoka, H. Watanabe, H. Kimura, T. Fuchigami, M. Haratake, H. Saji and M. Nakayama, Bioorg. Med. Chem. Lett., 2010, 20, 5743–5748 CrossRef CAS PubMed.
- M. Sagnou, D. Benaki, C. Triantis, T. Tsotakos, V. Psycharis, C. P. Raptopoulou, I. Pirmettis, M. Papadopoulos and M. Pelecanou, Inorg. Chem., 2011, 50, 1295–1303 CrossRef CAS PubMed.
- Y. Cheng, M. Ono, H. Kimura, M. Ueda and H. Saji, J. Med. Chem., 2012, 55, 2279–2286 CrossRef CAS PubMed.
- M. Ono, Y. Fuchi, T. Fuchigami, N. Kobashi, H. Kimura, M. Haratake, H. Saji and M. Nakayama, ACS Med. Chem. Lett., 2010, 1, 443–447 CrossRef CAS PubMed.
- Y. Yang, M. Cui, B. Jin, X. Wang, Z. Li, P. Yu, J. Jia, H. Fu, H. Jia and B. Liu, Eur. J. Med. Chem., 2013, 64, 90–98 CrossRef CAS PubMed.
- X. Wang, M. Cui, J. Jia and B. Liu, Eur. J. Med. Chem., 2015, 89, 331–339 CrossRef CAS PubMed.
- X. Wang, M. Cui, P. Yu, Z. Li, Y. Yang, H. Jia and B. Liu, Bioorg. Med. Chem. Lett., 2012, 22, 4327–4331 CrossRef CAS PubMed.
- M. Cui, M. Ono, H. Kimura, B. Liu and H. Saji, Bioorg. Med. Chem. Lett., 2011, 21, 4193–4196 CrossRef CAS PubMed.
- M. Yoshimura, M. Ono, K. Matsumura, H. Watanabe, H. Kimura, M. Cui, Y. Nakamoto, K. Togashi, Y. Okamoto, M. Ihara, R. Takahashi and H. Saji, ACS Med. Chem. Lett., 2013, 4, 596–600 CrossRef CAS PubMed.
- P. Yu, M. Cui, X. Wang, X. Zhang, Z. Li, Y. Yang, J. Jia, J. Zhang, M. Ono, H. Saji, H. Jia and B. Liu, Eur. J. Med. Chem., 2012, 57, 51–58 CrossRef CAS PubMed.
- Y. Cheng and W. H. Prusoff, Biochem. Pharmacol., 1973, 22, 3099–3108 CrossRef CAS PubMed.
- M. P. Kung, C. Hou, Z. P. Zhuang, B. Zhang, D. Skovronsky, J. Q. Trojanowski, V. M. Lee and H. F. Kung, Brain Res., 2002, 956, 202–210 CrossRef CAS PubMed.
- M. Ono, Y. Cheng, H. Kimura, M. Cui, S. Kagawa, R. Nishii and H. Saji, J. Med. Chem., 2011, 54, 2971–2979 CrossRef CAS PubMed.
- D. D. Dishino, M. J. Welch, M. R. Kilbourn and M. E. Raichle, J. Nucl. Med., 1983, 24, 1030–1038 CAS.
- A. Alpar, U. Ueberham, M. K. Bruckner, G. Seeger, T. Arendt and U. Gartner, Brain Res., 2006, 1099, 189–198 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28395k |
| This journal is © The Royal Society of Chemistry 2017 |