
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient strategy toward saturated N,P-heterocycles. Synthesis of l,2-azaphospholidines and extension to the preparation of azaphosphacane and azaphosphanane higher homologues†
H. Boufrouraa,
A. Abdelliab,
F. Bourdreuxa,
A. Gauchera,
G. Clavierc,
M. L. Efritb,
H. M'rabet*b and
D. Prim *a
*a
aUniversité de Versailles Saint Quentin-en-Yvelines, Institut Lavoisier de Versailles-UMR CNRS 8180, 45 avenue des Etats-Unis, 78035 Versailles, France. E-mail: damien.prim@uvsq.fr
bUniversité de Tunis El Manar, Faculté des Sciences de Tunis, Laboratoire de Synthèse Organique et Hétérocyclique, 2092 Tunis, Tunisia. E-mail: mrabet.fst@gmail.com
cENS-Cachan, PPSM-UMR CNRS 853, Bâtiment d'Alembert, 61, avenue du Président Wilson 94235 CACHAN, Cedex, France
First published on 24th March 2017
Abstract
Access to a new family of saturated N,P-heterocycles is described. Allylphosphonochloridates react with primary amines through a single synthetic operation involving the formation of N–P and N–C bonds to afford l,2-azaphospholidines. The N–P bond formation/aza-Michael cyclization sequence is extended to eight- and nine-membered N,N,P-heterocycles. The first lines of a conformational study of diazaphosphacanes are described. DFT calculations allowed the identification of two diastereomers that display preferred boat-chair and twisted-boat-chair conformations.
Building and functionalizing heterocycles has stimulated the research of methodological synthetic tools for many years. This thriving field is constantly evolving as the properties of these molecules are highly dependent on the presence of heteroatoms and the way they are connected with each other, the ring size as well as its aromatic or saturated nature. The state of saturation of heterocyclic compounds has been even considered recently as an additional descriptor of the molecular complexity in drug design.1 The development of reliable synthetic methods, easy to implement, which allow selective access to saturated heterocycles of various sizes and comprising two or more heteroatoms becomes thus an issue of major importance.2
In this context, azaphosphacyclanes represent attractive targets that have focused much attention over the last decades mostly due to their biological and therapeutic properties.3 Indeed, members of this class of P-heterocycles display a wide panel of pharmaceutical and biological activities such as anticancer,4 cytotoxicity5 or antitumor effects.6 N,P-Heterocycles were also used as ligands for complexation of metals.7 The modification of their properties can be envisioned through the association of additional substituents at the heterocyclic core or the modulation of the ring size. Both strategies represent synthetic challenges that directed the interest of the scientific community until recently.3 In this context, l,2-azaphospholidines, are accessible from main synthetic routes based on the formation of N–P or C–P bonds during the final cyclisation step requiring the preparation of advanced precursors such as γ-amino phosphonochloridates, ammonium-phosphate salts, or Arbuzov-based starting material.8 Although more scarcely reported, combination phosphorochloridite derivatives and γ-chloropropylamines, ring expansion of polymethylated 1-azidophosphetanes account for the formation of l,2-azaphospholidines.9 In addition, the ring closure metathesis (RCM) recently emerged as a convenient tool to prepare dihydro-phosphinine oxides that can be considered, using a further functionalization or reduction step, as advanced precursors in sequences leading to l,2-azaphospholidine targets.10
In this communication, we disclose a versatile single step methodology towards the preparation of new l,2-azaphospholidines. We envisioned the preparation of such saturated targets by taking advantage of the propensity of activated allylphosphono derivatives to undergo selective Michael-type additions γ to the P-atom11 and the dual reactivity of easily accessible phosphonochloridate 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 (Fig. 1).
12 (Fig. 1).
 | ||
| Fig. 1 One pot N–P bond formation – aza-Michael addition sequence. | ||
This new strategy would thus involve the formation of two N–P and N–C bonds in a single operation using a primary amine and allow the installation of unprecedented substituents at both the nitrogen and carbon centers in minimizing the production of waste (–HCl). Our strategy will also be extended to the synthesis of new eight- and nine-membered saturated N,P-heterocycles. First lines of the conformational study of diazaphosphacanes are described.
We started studying the reactivity of phosphonochloridate 112 with a first set of primary amines. Indeed, the reactions of benzyl amine, aniline, 4-aminoindan, 1-adamantylamine and 1-adamantylmethylamine with the phosphonochloridate 1 were successively examined (Table 1).
|
||||
|---|---|---|---|---|
| Entry | Amine 2 | Time | 3 (%) | 4 (%) |
| 1 | 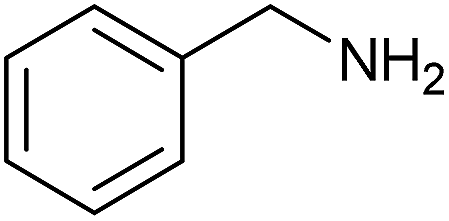 |
16 h |  |
|
| 2 |  |
28 h |  |
|
| 3 |  |
48 h |  |
|
| 4 |  |
72 h |  |
|
| 5 |  |
22 h |  |
|

In refluxing toluene, the use of 2 eq. of benzylamine was shown optimal and led to the selective formation of 4-ethoxycarbonyl-1,2-azaphospholidines 4a in 70% yield (entry 1). Attempts to increase yields using an additional base such as Et3N, DBU to trap HCl or a larger excess of the reacting amine failed. Interestingly, neither the possible aza-Michael adduct nor the phosphoramidate 3 could be detected in the crude material. In contrast, moving from benzylamine to the less nucleophilic aniline (entry 2), gave the corresponding phosphoramidate 3a in 60% yield. Although anilines may afford the corresponding aza-Michael adduct,13 the latter could not be identified under such reaction conditions. Similarly, the reaction of 4-aminoindan under identical reaction conditions led to the formation of phosphoramidate 3b as a unique product in 58% yield (entry 3). Comparison of adamantyl- and adamantylmethyl-amine gave a similar trend (compare entries 4 and 5). Indeed, phosphonochloridate 1 undergoes selective nucleophilic substitution at the P atom leading to the phosphoramidate 3c in the presence of the bulky adamantylamine. In sharp contrast, the less sterically constrained adamantylmethyl-amine selectively afforded the expected 4-ethoxycarbonyl-1,2-azaphospholidine 4b in 39% yield. Although several scenarios could rule the first step of the sequence, the coexistence of two competitive reactions involving the formation of the P–N bond vs. the formation of the C–N bond remains plausible. The most nucleophilic amines might preferentially afford the aza-Michael adduct prior the formation of the N–P bond. In the presence of lesser nucleophilic or bulky and sterically demanding amines such as aniline derivatives, reaction at the more electrophilic phosphorus atom might occur leading to the formation of the P–N phosphoramide bond. Once the P–N bond formed, the decrease of nucleophilicity of the nitrogen atom lone pair plausibly precludes further aza-Michael cyclisation.
The methodology was next extended to the preparation of several new 4-ethoxycarbonyl-1,2-azaphospholidines 4c–f as shown in Fig. 2. Under the aforementioned reaction conditions, we were able to cleanly isolate phospholidine targets from methylbenzylamine, 2-pyridinemethylamine, 2-thiophenemethylamine and 2-furanemethylamine in high yields ranging from 58 to 91% regardless of the electronic contribution of the heterocycle.14 Indeed electron rich thiophene and furane derivatives were isolated in similar yields by comparison with the electron deficient pyridine analogue. Phosphoramide 3 and 1,2-azaphospholidines 4 display characteristic 31P NMR chemical shifts. Indeed, P atoms resonated at 25.1 to 27.5 ppm and 43.3 to 44.2 ppm for compounds 3 and 4 respectively.15
 | ||
| Fig. 2 Access to variously 1,2-azaphospholidines 4. | ||
Our next goal was to react phosphonochloridate 1 with diamines in order to prepare N,P-heterocycles of larger size (Scheme 1). In this context, ethylene- and propylene-diamines would generate unprecedented eight- and nine-membered heterocycles “respectively”.
 | ||
| Scheme 1 First synthesis of diazaphosphocane 5 and diazaphosphonane 6. | ||
We focused on N,N′-dimethyl secondary amines in order to force each nitrogen atom to either generate the P–N or the C–N bond of the nucleophilic substitution – aza-Michael sequence and avoid the formation of phospholidines. Disappointingly, the use of the aforementioned conditions (in toluene at reflux) led to intractable mixtures of starting material and several P-based products. However, moving from toluene to t-BuOH was beneficial to the formation of cyclic products. Indeed, refluxing 1 and 2 eq. N,N′-dimethylethylenediamine for 20 h afforded the unprecedented diazaphosphocane 5 in 72% yield. Although several side reactions might take place under these conditions and the formation of ethylene bisphosphoramides could not be ruled out, only compound 5 was cleanly isolated and identified after purification. Our strategy was further extended to the use of N,N′-dimethyl-1,3-propylenediamine in order to prepare the corresponding diazaphosphonane 6. If the reaction tediously led to a mixture of products, the nine-membered N–P architecture was gratifyingly isolated in a fair 34% yield and fully characterized. In contrast to 6, which showed limited stability over a few days even when stored under careful conditions, diazaphosphocane 5 revealed stable. Both N,N,P-heterocycles exhibit characteristic 31P NMR chemical shifts. Indeed, P atoms in 5 and 6 resonate at 35 and 34 ppm in good accordance with a decrease of the ring strain observed when moving from nearly 44 ppm for the five-membered N,P analogues. Both 13C and 31P NMR data suggest the presence of diastereomers compatible with the presence of two chiral centers that are the P and C2 atoms. Compounds 5 and 6 were obtained as mixtures of two diastereomers that explain enlarged signals displayed in 1H NMR spectra. If fact, the complexity of 1H NMR spectra most probably results from the contribution of several fingerprints intrinsic to the molecular structure of 5 and 6. Gratifyingly, in the case of the stable compound 5, both diastereomers 5dia1 and 5dia2 could be painfully separated on silica gel chromatography. 1H NMR spectra of both diastereomer and comparison between them (ESI†) showed complementary signals but still display enlarged signals corresponding to the protons supported by the heterocyclic core. Classical variable-temperature NMR experiments were not conclusive and did not allow gaining further structural informations, but questioned about the impact of the joint presence of three heteroatoms within a medium-sized heterocycle.
Indeed, the presence of (i) one phosphorus atoms inducing P–H couplings, (ii) two chiral centers generating diastereomers, (iii) possible inversion of nitrogen atoms lone pairs and (iv) several potential conformations of the saturated flexible medium-sized ring, might all lead to an overlapped contribution of all these structural features. We thus turned our attention to theoretical calculations with the aim to understand the complexity of the NMR data and give the first lines of the conformation space study of novel medium-sized heterocycles comprising one P and two N atoms. To this end, we focused in this paper on the diazaphosphocane 5 as the first representative example of this series of heterocycles. Examination of literature data showed that conformational analysis of medium rings has been the subject of sustained attention over the last decades. Within this area the conformation of cyclooctane,16 cyclononane,17 fused azocines and azonines18 but also eight-membered benzoannulated lactams19 or silicon- and boron-based heterocycles20 have been examined using NMR and DFT techniques. In the latter studies, the shape of the saturated rings are driven by the presence of one or several fragments such as fused aromatic systems, amides or ketones, able to impose a partial rigidity to the backbone. In deep contrast with such relevant eight-membered ring systems described in the literature, unreported diazaphosphocane 5 do not display shape constraints. Apart, from the phosphoramide P(O)N fragment, for which a possible contribution of a planar PN double bond cannot be excluded, the entire structure remains flexible. Our objectives were to try identifying both diastereomers and further gaining a first set of information towards a more complete study of the entire conformation space of such heterocycles.
We thus started by generating a distribution of conformations using molecular mechanics (MM) for both diastereomers 5a1 and 5a2 (see ESI†). We selected the most occurring conformations of lowest energy, for each diastereomers 5a1 and 5a2. Those conformations were then used for quantum mechanics (QM) structure refinement at B3LYP/6-311++(d,p) level of theory. This basis set has been shown to be an excellent compromise between accuracy and computational time for this particular kind of calculations.21 As shown in Fig. 3, both families of computed enantiomers (5a1/5a1′ and 5a2/5a2′) display a close but not identical 3D geometry. Indeed, a preferred boat-chair (BC) conformation is calculated for 5a1 instead of a preferred twisted-boat-chair (TBC) conformation for 5a2. Both preferred conformations are in good agreement with recent conformational studies dealing with medium-sized heterocycles.16–20
 | ||
| Fig. 3 Computed preferred conformations for diastereomers 5a1, 5a1′, 5a2 and 5a2′ (protons are omitted for clarity). | ||
We next tried to assign each of them to their respective experimental data. To this end predictive chemical shifts were computed within the GIAO approximation using DFT at the PBE0/6-311+G(2d,p) level of theory on the preferred conformations 5a1 and 5a2.22,23 Computed details for each diastereomer can be found in the ESI.† A careful comparison of scaled experimental and calculated chemical shifts has been carried out for H and C nuclei (see Table ESI†).24 Data collected in Table 1 reflect the complexity of both 1H and 13C NMR experimental spectra. Indeed, both separated diastereomer 5dia1 and 5dia2 display a close set of chemical shifts (compare columns 1/2 and 5/6). Computed data (columns 3/4 and 7/8) revealed a similar trend for nuclei 1–3, 7, 8, 11 and 12. In contrast, calculated chemical shifts for nuclei 4–6 and 9–10, exhibit marked deviations between 5a1 and 5a2. Fig. 4 allowed visualizing Δδ between experimental data for 5dia1 and 5dia2 and between computed data for 5a1 and 5a2. In both nuclei series, Δδ between experimental and computed data are strongly depending on their location at the diazaphosphacane core. Indeed, protons and carbons 4, 5 and 6 are representatives of a clear overestimation of the computed chemical shifts.25
 | ||
| Fig. 4 Comparison of Δδ between experimental data for 5dia1 and 5dia2 in blue and between computed data for 5a1 and 5a2 in red. | ||
In these cases, variations of Δδ ranging from 0.00 to 0.27 ppm for protons and from 0.00 to 6.17 ppm for carbons were observed. In fact these nuclei belong to the more flexible fragment of the diazaphosphacane. Their chemical shifts are thus deeply impacted by tiny conformations modifications and may result from a contribution of several conformations. Other nuclei, such as 1, 2, 3 located at more rigid fragments of the heterocycle or at pendant ethyl groups exhibit lower variation of Δδ being thus less affected by conformational changes. Nuclei 9 and 10 are also worthy to observed. If comparison of experimental and computed protons chemical shifts seemed more accurate, the corresponding carbons display largest variation of Δδ. The differences observed for the carbon of the methyl groups positioned on both nitrogen atoms of the heterocycle perfectly illustrate the joint impact of conformational changes of the cycle, the inversion of the nitrogen atom lone pairs and the presence of the phosphorus atom. The presence of the phosphorus atom and the possible formation of a planar phosphoramide moiety limit the degree of freedom of the nitrogen atoms substituted by C10, which consequently conducted to a smaller Δδ deviation than for C9.
With the aim to identify which experimental data set could match the calculated data, we undertook to compare computed and experimental chemical shifts by pairing NMR data and to calculate corrected mean absolute error (CMAD) as shown in Table 2 (ESI†).26 The CMAD are very close in each case. Indeed, the latter range between 0.13 and 0.16 for 1H (see Tables 2 and 3 ESI†) and between 1.52 and 2.57 for 13C. A first trend shows that the combination dia2–5a2 seems the most consistent for both nuclei. This first approximation should also lead to lower CMAD for the dia1–5a1 pair. Unfortunately, CMAD of 1.73 and 0.14 calculated for 13C and 1H respectively do not allow a complete discrimination of both experimental and calculated pairs.
Conclusions
In summary, we have described an efficient approach for the preparation of a novel family of l,2-azaphospholidines. The synthetic method combines the formation of an N–P and a N–C bond from a phosphonochloridate precursor and an intra-molecular aza-Michael cyclization. This methodology was next successfully extended to prepare eight- and nine-membered saturated N,N,P-heterocycles by reacting the corresponding phosphonochloridate with 1,2- and 1,3-diamines respectively. The overlap of several conformational contributions led to complex analytical data. These observations encouraged us to complement the synthesis of these heterocycles with a first approach to the study of their conformational space. This first set of evidence arising from DFT calculations is in complete agreement with the presence of multiple conformations for both diastereomers of 5. A preferred boat-chair (BC) as well as a preferred twisted-boat-chair (TBC) conformation was evidenced for 5a1 and 5a2 respectively. Our study represents a major challenge because these fully saturated heterocycles do not display any planar pattern but are set by an important degree of flexibility, possible interconversions at both nitrogen atoms, and further include one phosphorus atom which induces additional couplings. In this communication, we describe the basis for further study of the conformational space of medium-sized saturated heterocycles comprising several heteroatoms. The latter will require taking into account the contribution of all populations of conformers through a further statistical analysis in order to refine the correlations with experimental data. A more complete study of conformations in the diazaphosphacane and diazaphosphonane series is currently underway and will be reported in due course.Notes and references
- T. J. Ritchie, S. J. F. MacDonald, R. J. Young and S. D. Pickett, Drug Discovery Today, 2011, 16, 164 CrossRef CAS PubMed; T. J. Ritchie and S. J. F. MacDonald, Drug Discovery Today, 2009, 14, 1011 CrossRef PubMed; F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef PubMed.
- C.-V. T. Vo, M. U. Luescher and J. W. Bode, Nat. Chem., 2014, 6, 310 CrossRef CAS PubMed.
- For recent overview see: R. Brewster, M.-C. Vandergeten and F. Montel, Eur. J. Org. Chem., 2014, 905 CrossRef CAS; H. Akbas, A. Okumus, Z. Kılıç, T. Hökelek, Y. Süzen, L. Y. Koç, L. Açık and Z. B. Çelik, Eur. J. Med. Chem., 2013, 70, 294 CrossRef PubMed; V. Point, R. K. Malla, F. Carriere, S. Canaan, C. D. Spilling and J.-F. Cavalier, J. Med. Chem., 2013, 56, 4393 CrossRef PubMed.
- A. Paci, T. Martens and J. Royer, Bioorg. Med. Chem. Lett., 2001, 11, 1347 CrossRef CAS PubMed; G. Zon, Prog. Med. Chem., 1982, 19, 205 CrossRef PubMed.
- P. Maynard-Faure, C. Gonser, V. Vaime and D. Bouchu, Tetrahedron Lett., 1998, 39, 2315 CrossRef CAS.
- Q. Sun, R.-T. Li, W. Guo, J.-R. Cui, T.-M. Cheng and Z.-M. Ge, Bioorg. Med. Chem. Lett., 2006, 16, 3727 CrossRef CAS PubMed.
- S. S. Krishnamurthy, V. Sreenivasa Reddy, A. Chandrasekaran and M. Nethaji, Phosphorus, Sulfur Silicon Relat. Elem., 1992, 64, 99 CrossRef CAS.
- I. Natchev, Bull. Chem. Soc. Jpn., 1988, 61, 3699 CrossRef CAS; D. J. Collins, P. F. Drygala and J. M. Swan, Tetrahedron Lett., 1982, 23, 1117 CrossRef; D. J. Collins, J. W. Hetherington and J. M. Swan, Aust. J. Chem., 1974, 27, 1759 CrossRef; O. V. Bykhovskaya, I. M. Aladzheva, D. I. Lobanov, P. V. Petrovskii, K. A. Lyssenko, I. V. Fedyanin and T. A. Mastryukova, Russ. Chem. Bull., 2005, 54, 2642 CrossRef; I. M. Aladzheva, D. I. Lobanov, O. G. V. Bykhovskaya, P. V. Petrovskii, K. A. Lyssenko and T. A. Mastryukova, Heteroat. Chem., 2003, 14, 596 CrossRef.
- I. M. Aladzheva, O. V. Bykhovskaya, P. V. Petrovskii, K. A. Lyssenko, M. Y. Antipin and T. A. Mastryukova, Russ. Chem. Bull., 2005, 54, 2635 CrossRef CAS; M. J. P. Harger, J. Chem. Soc. D, 1971, 442 RSC.
- A. Abdelli, F. Bourdreux, A. Gaucher, M. L. Efrit, H. M'rabet and D. Prim, Tetrahedron Lett., 2016, 57, 379 CrossRef CAS and reference cited therein; P. R. Hanson and D. S. Stoianova, Tetrahedron Lett., 1999, 40, 3297 CrossRef; D. S. Stoianova and P. R. Hanson, Org. Lett., 2000, 2, 1769 CrossRef PubMed.
- A. Abdelli, M. L. Efrit, A. Gaucher, H. M'rabet and D. Prim, Tetrahedron Lett., 2015, 56, 5397 CrossRef CAS; A. Abdelli, A. Gaucher, M. L. Efrit, H. M'rabet and D. Prim, Tetrahedron Lett., 2015, 56, 1679 CrossRef; A. Abdelli, H. M'rabet, M. L. Efrit, A. Gaucher and D. Prim, J. Sulfur Chem., 2014, 35, 674 CrossRef.
- Phosphonochloridate 1, was readily prepared in 91% yield from allylphosphonate precursor with oxalyl chloride in dichloromethane according to known procedures (see ESI†): M. Lewandowska, P. Ruszkowski, D. Baraniak, A. Czarnecka, N. Kleczewska and L. Celewicz, Eur. J. Med. Chem., 2013, 67, 188 CrossRef CAS PubMed; N. Coşkun and M. Çetin, Tetrahedron, 2000, 56, 2053 CrossRef; K. T. Sprott and P. R. Hanson, J. Org. Chem., 2000, 65, 4721 CrossRef PubMed.
- For a recent example see: Z. Amara, E. Drege, C. Trouffard, P. Retailleau, M.-E. Tran Huu-Dau and D. Joseph, Chem.–Eur. J., 2014, 20, 15840 CrossRef CAS PubMed.
- Phospholidine derivatives are sensitive to air moisture generating the corresponding phosphoric acids by hydrolysis of the P–N bond and have thus to be carefully stored under Ar atmosphere and at low temperature as already observed and described in the literature. See: P. Fourgeaud, C. Midrier, J.-P. Vors, J.-N. Volle, J.-L. Pirat and D. Virieux, Tetrahedron, 2010, 66, 758 CrossRef CAS; Y. Machida and I. Saito, J. Org. Chem., 1979, 44, 865 CrossRef.
- The presence of a set of two 31P chemical shifts between 41 and 46 ppm was in complete accordance with the formation of mixtures of diastereomers. In some cases, diastereomers could be separated. See ESI.†.
- J. P. Fitzgerald, J. Chem. Educ., 1993, 70, 988 CrossRef CAS; W. C. Still and I. Galynker, Tetrahedron, 1981, 37, 3981 CrossRef.
- F. A. L. Anet and J. J. Wagner, J. Am. Chem. Soc., 1971, 93, 5266 CrossRef CAS.
- M. Qadir, J. Cobb, P. W. Sheldrake, N. Whittall, A. P. J. White, K. K. Hii, P. N. Horton and M. B. Hursthouse, J. Org. Chem., 2005, 70, 1552 CrossRef CAS PubMed.
- A. Witosinska, B. Musielak, P. Serda, M. Owinska and B. Rys, J. Org. Chem., 2012, 77, 9784 CrossRef CAS PubMed.
- L. P. Burke, A. D. DeBellis, H. Fuhrer, H. Meier, S. D. Pastor, G. Rihs, G. Rist, R. K. Rodebaugh and S. P. Shum, J. Am. Chem. Soc., 1997, 119, 8313 CrossRef CAS; H. Braunschweig, I. Krummenacher, L. Mailänder and F. Rauch, Chem. Commun., 2015, 51, 14513 RSC.
- For recent uses of this basis set see: P. R. Rablen, S. A. Pearlman and J. Finkbiner, J. Phys. Chem. A, 1999, 103, 7357 CrossRef CAS; M. A. Muñoz and P. Joseph-Nathan, Magn. Reson. Chem., 2009, 47, 578 CrossRef PubMed; B. Musielak, T. A. Holak and B. Rys, J. Org. Chem., 2015, 80, 9231 CrossRef PubMed.
- PBE0 method revealed recently useful in the elucidation of complex natural heterocyclic molecules. See: K. M. Snyder, J. Sikorska, T. Ye, L. Fang, W. Su, R. G. Carter, K. L. McPhail and P. H.-Y. Cheong, Org. Biomol. Chem., 2016, 14, 5826 CAS.
- Comparison of calculated and experimental chemical shifts have been realized see Table ESI.†.
- For 1H and 13C empirical scaling see: G. K. Pierens, J. Comput. Chem., 2014, 35, 1388 CrossRef CAS PubMed . For 31P empirical scaling see: S. K. Latypov, F. M. Polyancev, D. G. Yakhvarov and O. G. Sinyashin, Phys. Chem. Chem. Phys., 2015, 17, 6976 RSC.
- Additional study at B3LYP/6-311++G(d,p) level of theory as well as full details of comparison of Δδ between experimental data for 5dia1 and 5dia2 in bleu and between computed data for 5a1 and 5a2 in red are presented in ESI.†.
- For recent application of CMAD see: M. W. Lodewyk, C. Soldi, P. J. Jones, M. M. Olmstead, J. Rita, J. T. Shaw and D. J. Tantillo, J. Am. Chem. Soc., 2012, 134, 18550 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra and computational details. See DOI: 10.1039/c6ra28420e |
| This journal is © The Royal Society of Chemistry 2017 |