DOI:
10.1039/C6RA28446A
(Paper)
RSC Adv., 2017,
7, 9373-9380
Color change of alternating copolymers with phenyl vinylethylene carbonate and N-phenylmaleimide in a solution and in the solid-state, depending on their structure†
Received
20th December 2016
, Accepted 19th January 2017
First published on 30th January 2017
Abstract
Radical copolymerization, with various monomer feeds of phenyl vinylethylene carbonate (PVEC) and N-phenylmaleimide (PMI), successfully proceeded through selective vinyl and alternating polymerization to obtain copolymers having the maleimide unit in the main-chain and the five-membered cyclic carbonate group in the side-chain. The structure of these copolymers was analyzed in detail using NMR, IR, and UV-Vis methods. The UV-Vis spectra supported the tautomerization of the maleimide unit based on the solvent effect and the alternating and/or homo sequence constructed depending on the monomer feeds. Furthermore, these copolymers exhibited sensitive and reversible color changes based on acid–base switching not only in solution but also in the solid-state. In particular, the color change in the solid-state occurred immediately after exposure to the amine vapor.
Introduction
Several types of sequence-controlled polymers have been prepared using various methods including a comonomer-selective reaction, iterative reaction and template reaction.1–8 In the comonomer-selective reaction, alternating copolymerization, the combination of the electron acceptor and electron donor monomers are commonly known to successfully achieve sequence-controlled polymerization.3–8 In particular, the maleimide derivatives are reported to be the electron acceptor monomers, which produce the AB or ABB alternating copolymer depending on the electron donor monomers such as styrene and non-conjugated olefin.3,4 Recently, we demonstrated that the radical polymerization of the vinyl ethylene carbonate (VEC) derivatives and various N-substituted maleimides (NMI) produced the sequence-controlled polymers due to the electron rich allylic moiety of VEC monomers and the electron poor maleimide moiety having a bulky and electron-withdrawing group as the N-substituents.8 Such copolymers with maleimide derivatives exhibited various properties, which include thermal stability, thermosetting, photoresponse, pH-response, nonlinear optics, molecular linkers and markers for a modification and chiral activity.4,9–14 The copolymers of VEC and NMI also exhibited a reversible color change based on an acid–base switching in solution.8,11 The color change was considered to be a result of an enolate anion tautomerization of the maleimide moiety in basic solvent and/or in the presence of a strong basic reagent.8,11,15 Although the color change and luminescence properties of poly(NMI)s with an acid–base response in solution have been shown in several reports, these properties have not been demonstrated in the solid-state. Moreover, it has been considered that the color change of these polymers in the solid-state occurs with sufficiently strong basic reagents as well as that in solution. The reversible color change in the solid-state is well known to be due to various chromisms, which are thermochromism, photochromism, electrochromism, piezochromism and vapochromism.16–22 Moreover, PVEC and PMI copolymers have the five-membered cyclic carbonate group on the side-chain that proceeds through the selective vinyl radical polymerization of the PVEC monomer. This occurs because the ring-strain energy of the five-membered cyclic carbonate is low, and the activated state of the ring-opening process has a fairly high thermodynamic energy.8,23 Such polymers are applied to thermosetting coatings and polymer electrolytes of lithium ion batteries.24 Furthermore, the polymers bearing the cyclic carbonate group on the side chain are also readily cross-linked with a diamine due to the high reactivity and chemoselectivity of the cyclic carbonate group with an amine.25 In this study, the copolymers of phenyl vinylethylene carbonate (PVEC) and N-phenylmaleimide (PMI) were prepared with various monomer feeds using a radical initiator, and the sequential structure of these copolymers was analyzed in detail using NMR, IR, and UV-Vis methods. Furthermore, the color change of PVEC and PMI copolymers in the solid-state was studied using acid and base reagents, and the relationship between the composition ratio and color depth of the copolymers was summarized. These results indicated that the PVEC and PMI copolymers exhibited reversible acid–base responsivity and vapochromism in the solid-state.
Results and discussion
Radical copolymerization of PVEC and PMI with various monomer feeds
The radical polymerization of PVEC and PMI was carried out with DTBP as an initiator in bulk at 120 °C for 24 h, respectively (Table 1, Poly-100 and Poly-0). The yield and molecular weight of PVEC homopolymer (Poly-100) was fairly low due to a sluggish monomer toward free radical polymerization that originated from essentially a substituted allylic monomer, although that of the PMI homopolymer (Poly-0) was the highest.8,23,24b On the other hand, the radical copolymerization of PVEC and PMI with various monomer feeds was also carried out under the same conditions. The yield and molecular weight increased linearly with increasing PMI feed, and the composition ratio of each monomer unit in the copolymers exhibited almost the same ratio as that of the monomer feed. In particular, the alternating copolymerization of Poly-50 (monomer feed; PVEC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PMI = 50:50) was suggested to be a result of the equivalent relationship between the monomer feed and composition ratio in the copolymers. On the other hand, the molecular weight of Poly-10 (monomer feed; PVEC:PMI = 10:90) was nonlinear out of all copolymers prepared with various monomer feeds due to the chain transfer reaction during polymerization, because the dispersity (Mw/Mn) of Poly-10 increased compared with that of the other copolymers, as well as that of the PMI homopolymer (Poly-0), as shown in Fig. S1.† Recently, we demonstrated that the copolymerization of PVEC and NMI was achieved in fairly high yield, and the molecular weight compared well with the homopolymerization of PVEC because these monomers exhibited a high reactivity that originated from the donor–accepter interaction due to the strong electron-donating character of the PVEC monomer.8,23 Therefore, the polymerization properties of PVEC monomer were improved with increases in the PMI feed.
:PMI = 50:50) was suggested to be a result of the equivalent relationship between the monomer feed and composition ratio in the copolymers. On the other hand, the molecular weight of Poly-10 (monomer feed; PVEC:PMI = 10:90) was nonlinear out of all copolymers prepared with various monomer feeds due to the chain transfer reaction during polymerization, because the dispersity (Mw/Mn) of Poly-10 increased compared with that of the other copolymers, as well as that of the PMI homopolymer (Poly-0), as shown in Fig. S1.† Recently, we demonstrated that the copolymerization of PVEC and NMI was achieved in fairly high yield, and the molecular weight compared well with the homopolymerization of PVEC because these monomers exhibited a high reactivity that originated from the donor–accepter interaction due to the strong electron-donating character of the PVEC monomer.8,23 Therefore, the polymerization properties of PVEC monomer were improved with increases in the PMI feed.
Table 1 Radical copolymerization of PVEC and PMI with various monomer feeds
| Polymer |
Monomer feed (mol%) |
Compositiona PVEC:PMI (%) |
Yieldb (%) |
Mnc (×10−4) |
Mw/Mnc |
| PVEC |
PMI |
| Determined by IR using ATR. Insoluble fraction in MeOH. Determined using GPC analysis (PSt, DMF). |
| Poly-0 |
0 |
100 |
— |
92 |
1.2 |
6.61 |
| Poly-10 |
10 |
90 |
14:86 |
82 |
1.0 |
5.12 |
| Poly-30 |
30 |
70 |
31:69 |
75 |
2.1 |
2.47 |
| Poly-50 |
50 |
50 |
50:50 |
73 |
1.2 |
2.16 |
| Poly-70 |
70 |
30 |
72:28 |
68 |
0.7 |
2.30 |
| Poly-90 |
90 |
10 |
88:12 |
48 |
0.4 |
1.91 |
| Poly-100 |
100 |
0 |
— |
39 |
0.4 |
1.86 |
The IR spectra of the produced homopolymers and copolymers are shown in Fig. 1. The IR spectra proved that the polymers having the five-membered cyclic carbonate group in the side-chain were prepared by proceeding through the selective vinyl polymerization because the characteristic peaks assigned to the five-membered ring and carbonyl moieties in the five-membered cyclic carbonate structure were observed around 1000–1200 cm−1 and 1800 cm−1, respectively. On the other hand, the five-membered ring and carbonyl moieties in the succinimide structure exhibited peaks around 1200–1400 cm−1 and 1700 cm−1, respectively. Furthermore, the peak intensity of the five-membered rings and carbonyl moieties changed linearly with the monomer feed. Therefore, these polymerization results and IR spectra indicated that the selective vinyl copolymers having the maleimide unit in the main-chain and the five-membered cyclic carbonate group in the side-chain were prepared without depending on the monomer feed, and also that these copolymers were constructed with the same composition ratio of each monomer unit as the monomer feed (Fig. 1 and S2†).
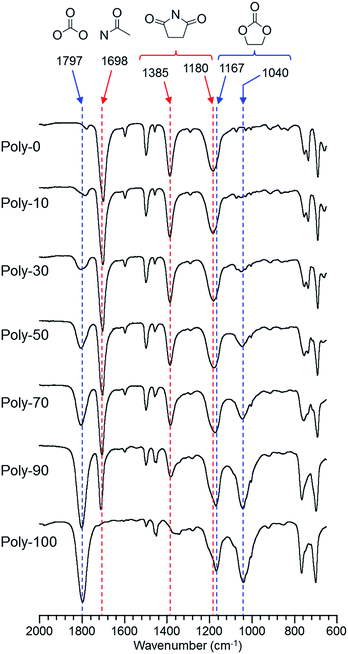 |
| | Fig. 1 IR spectra of PVEC and PMI copolymers prepared with various monomer feeds. | |
Color change of PVEC and PMI copolymers in a solution, depending on the composition ratio of each monomer unit
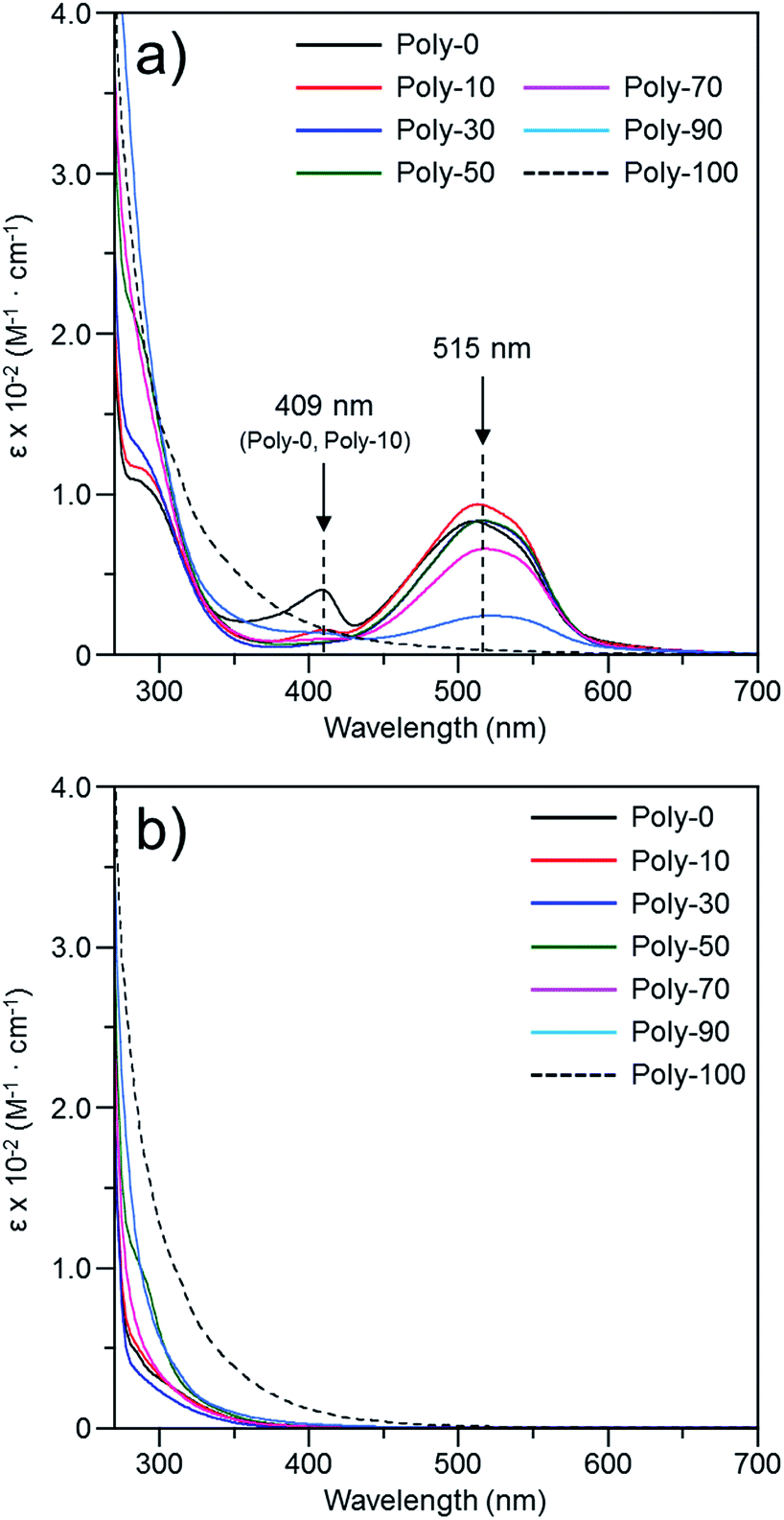
The solubility of the PVEC and/or PMI polymers in various solvents is shown in Table 2. Poly-0, Poly-100, and these copolymers were soluble in halogen, cyclic ether, ketone, and polar solvents such as carbonate, sulfoxide and amide, although Poly-0 was partly soluble in acetone and PCA. On the other hand, these polymers were insoluble in hydrocarbon, ester, nitrile, alcohol and ether. Furthermore, the color of the solution of PVEC and PMI copolymers was red in DMF. Interestingly, the color of the solution changed from red to pale red with increases in the composition ratio of the PVEC unit (Fig. 2). In addition, the red color was observed in polar and/or basic solvents such as amine, amide, sulfoxide, and cyclic carbonate, although the polymer solution was colorless in nonpolar and/or acidic solvents such as CHCl3 and CCl4. On the other hand, the solution color of Poly-100 never changed not only in DMF but also in various other solvents. Therefore, the color change depending on the composition ratio of each monomer unit indicated that at least a 10% composition ratio of the PMI unit was essential in the copolymers to exhibit the red color.
Table 2 Solubility of PVEC and/or PMI polymers prepared with various monomer feeds in common organic solventsa,b
| |
Poly-0 |
Copolymersc |
Poly-100 |
| Concentration of 2.0 mg/1.0 mL. O: soluble, X: insoluble or partly soluble. Copolymers: Poly-10, Poly-30, Poly-50, Poly-70, Poly-90. CPME: cyclopentyl methyl ether. |
| Toluene |
X |
X |
X |
| EtOAc |
X |
X |
X |
| CH3CN |
X |
X |
X |
| CHCl3 |
O |
O |
O |
| Acetone |
X |
O |
O |
| PCA |
X |
O |
O |
| i-PrOH |
X |
X |
X |
| CPMEd |
X |
X |
X |
| THF |
O |
O |
O |
| DO |
O |
O |
X |
| DMSO |
O |
O |
O |
| DMF |
O |
O |
O |
| DMAc |
O |
O |
O |
| NMP |
O |
O |
O |
 |
| | Fig. 2 Color change of PVEC and PMI copolymers in DMF depending on the composition ratio of each monomer unit. | |
Proposed main-chain structure and color change mechanism of PVEC and PMI copolymers based on tautomerization of PMI unit
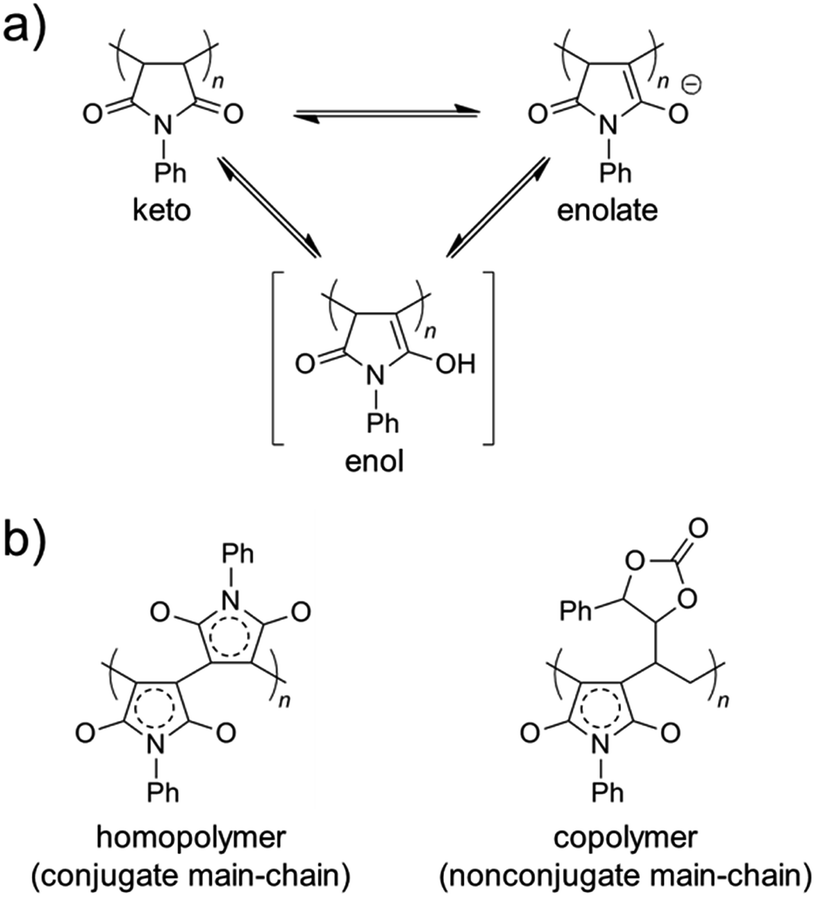
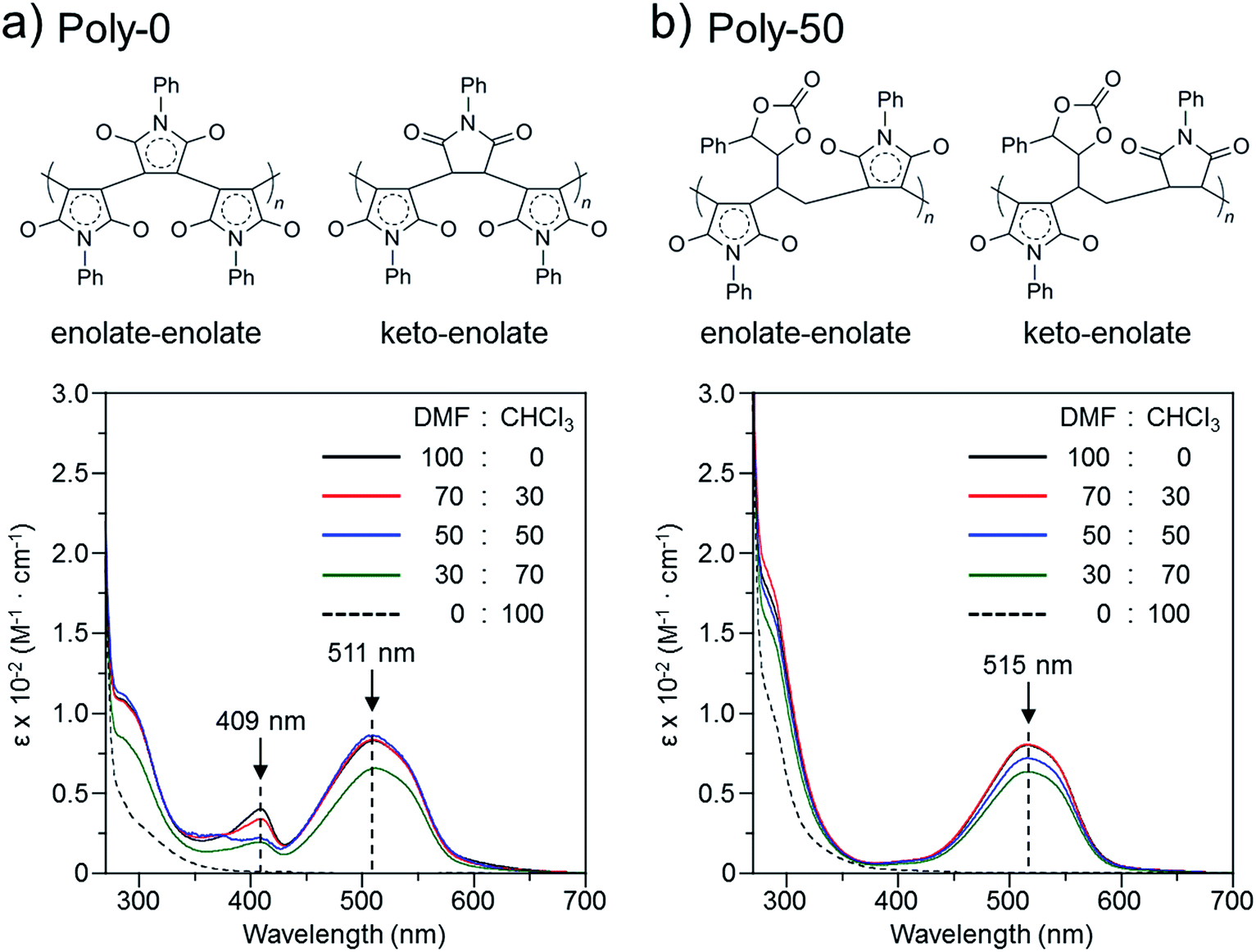
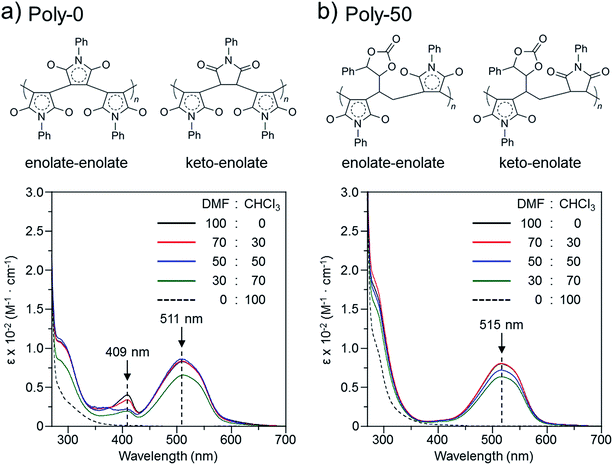
The color change was studied in detail by collecting UV-Vis spectra of the PVEC and PMI copolymers in DMF, CHCl3, and DMF/CHCl3 (Fig. 3). In the UV-Vis spectra observed in DMF, the specific maximum absorptions (λmax) were at 409 nm and 515 nm (Fig. 3a), although such absorptions were never observed in CHCl3 (Fig. 3b). The absorption observed at 515 nm was assigned to the enolate structure due to the tautomerization of the maleimide moiety, because the deprotonation occurred from the maleimide unit in the main-chain under the basic condition (Fig. 4a).8b,11,15 On the other hand, the UV-Vis spectra of Poly-10 and Poly-0 suggested that the absorption observed at 409 nm was due to the expanded conjugation of the maleimide units in the main-chain because such an absorption was never observed in the copolymer having a high composition ratio of the PVEC unit (Fig. 4b).8b Furthermore, the intensity of the λmax observed at 515 nm decreased with an increasing PVEC composition ratio in the copolymers, and this intensity change agreed with the tendency of the color change depending on the composition ratio of each monomer unit (Fig. 2 and 3a). These observations of the color change and UV-Vis spectra suggested that the main-chain structures of the PVEC and PMI copolymers were constructed from the alternating sequence and/or the block sequence of the rich monomer depending on the monomer feed (Fig. S3†). This was due to the fact that the alternating sequence of the PVEC and PMI copolymers could not be conjugated with each monomer unit. On the other hand, the copolymers having a high PMI composition ratio were constructed with part of the alternating sequence and the block sequence of the conjugated maleimide units (Fig. 4b and S3†). The UV-Vis spectra of Poly-0 and Poly-50 were measured in DMF/CHCl3 mix solvent (Fig. 5). The absorptions observed at 511 nm and 515 nm were assigned to the enolate tautomer of the maleimide unit in Poly-0 and Poly-50, respectively (Fig. 5a and b). The absorption intensity also decreased with increasing CHCl3 ratios in the solvent mixture. This intensity change suggested that the keto–enolate sequence increased with respect to the enolate–enolate sequence due to the tautomerization based on the solvent effect with increasing CHCl3 ratios in the solvent mixture (Fig. 5). On the other hand, the absorption assigned to the conjugated sequence of the maleimide unit was observed at 409 nm only in the case of Poly-0, and the absorption intensity decreased with increasing CHCl3 ratios in the solvent mixture as well as the increase of the PVEC composition ratio (Fig. 5a). This result suggested that the conjugated sequence in the main-chain disappeared together with decreases in the enolate–enolate sequence at high CHCl3 ratios (Fig. 5a). Furthermore, the UV-Vis spectrum also suggested that the alternating structure of PVEC and PMI units never formed the conjugated sequence in the main-chain because such absorptions around 409 nm were never observed in the case of Poly-50 (Fig. 5b). Therefore, these color changes and UV-Vis spectra of PVEC and PMI copolymers supported that the tautomerization of the maleimide unit, based on the solvent effect, readily occurred in the main-chain. In addition, the alternating structure was constructed in a part of the copolymer sequence depending on the composition ratio of each monomer unit.
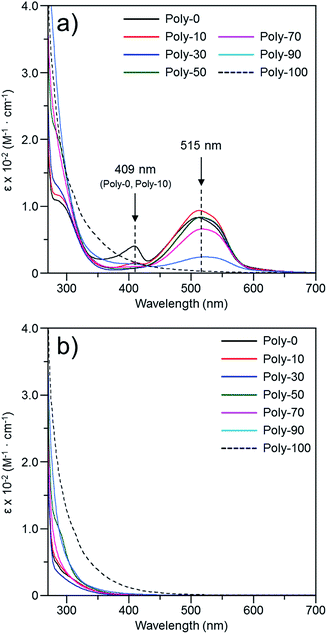 |
| | Fig. 3 UV-Vis spectra of PVEC and PMI copolymers prepared with various monomer feeds, observed in (a) DMF and (b) CHCl3 (5.0 × 10−3 mol L−1). | |
 |
| | Fig. 4 (a) Tautomer structures of PMI unit in keto, enol, and enolate forms. (b) Conjugated main-chain sequence of PMI homopolymer and nonconjugated main-chain sequences of PVEC and PMI copolymers. | |
 |
| | Fig. 5 UV-Vis spectra and main-chain structure of (a) Poly-0 and (b) Poly-50 observed in solvent mixtures of DMF and CHCl3 (5.0 × 10−3 mol L−1). | |
Color change of PVEC and PMI copolymers based on acid–base switching in solid-state
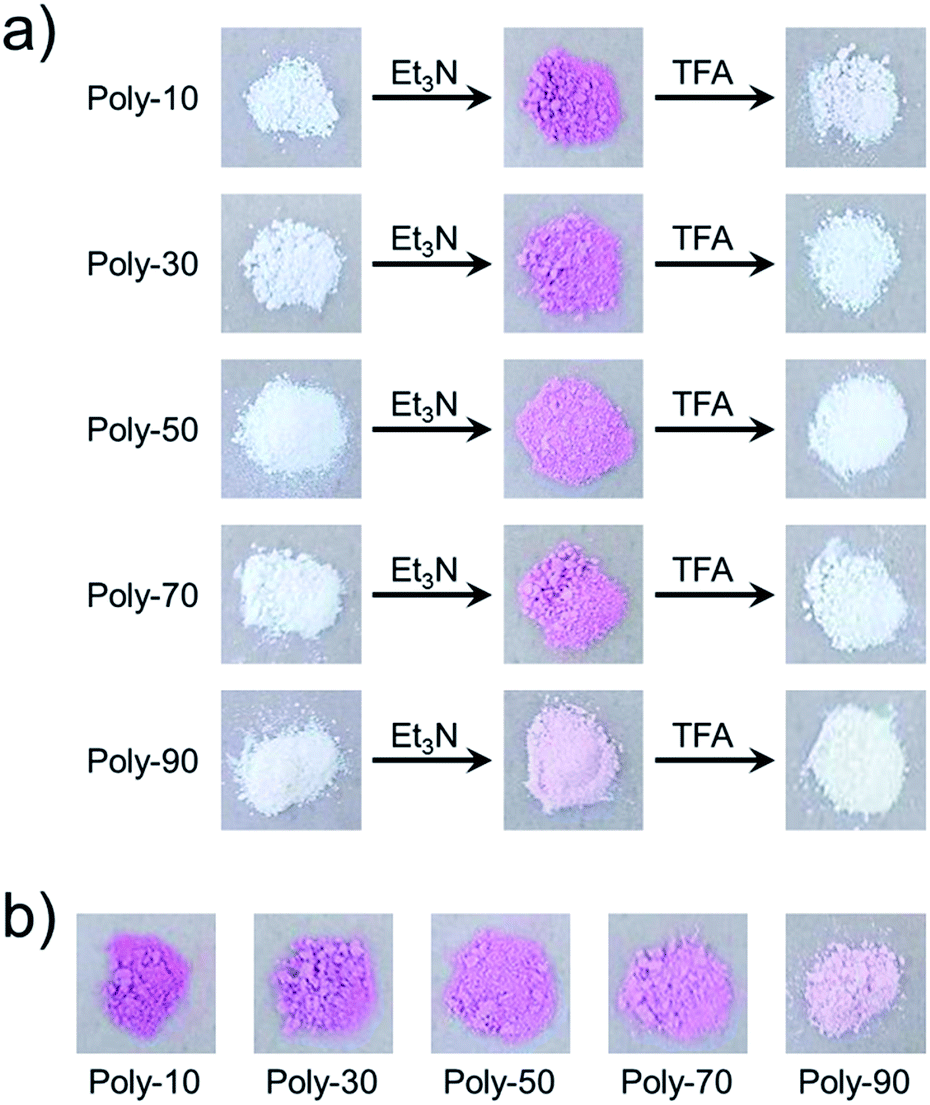
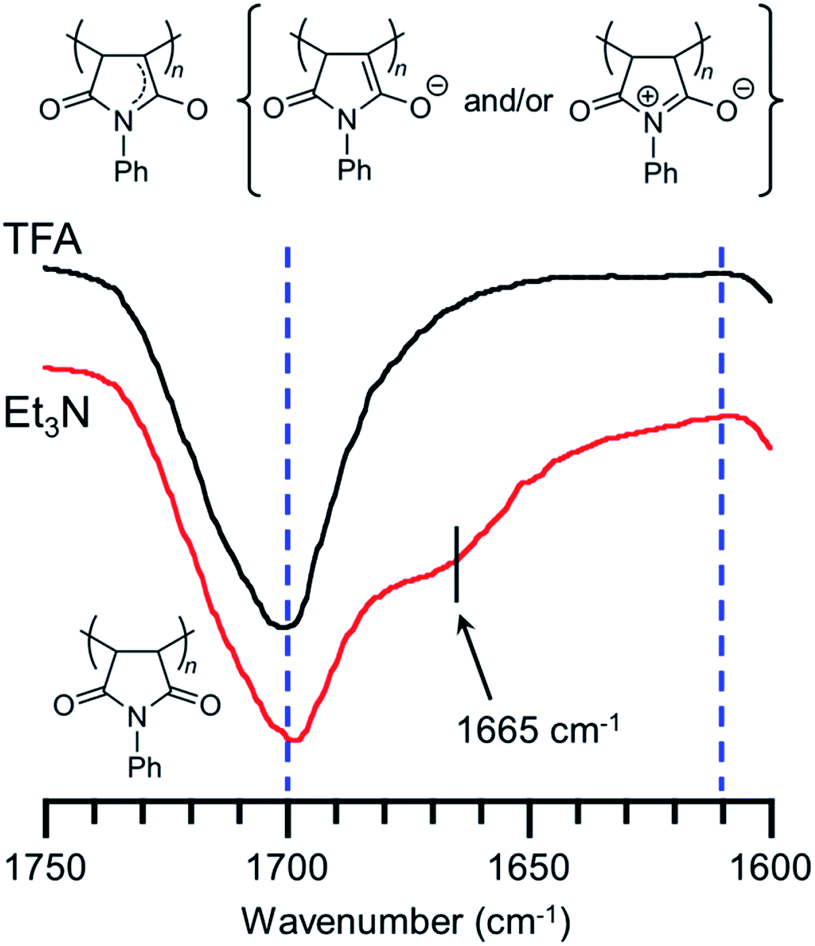
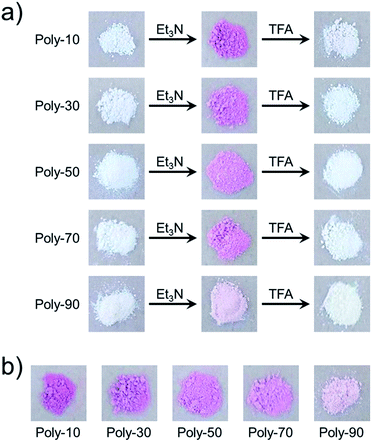
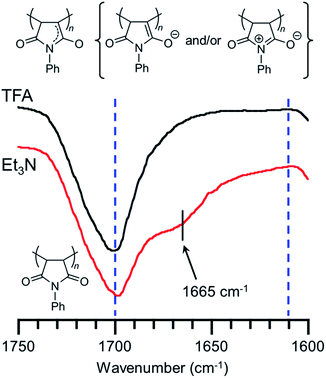
We have already reported that the solution of PVEC and NMI copolymer exhibited reversible color change between red and colorless based on acid–base switching.8 PVEC and PMI copolymers precipitated into insoluble solvent (ether) containing a slight amount of trifluoroacetic acid (TFA) or triethylamine (Et3N) to verify the color change of the copolymers based on acid–base switching in the solid-state. As shown in Fig. 6a, the white color of the synthesized copolymers immediately changed to a pink color in ether containing Et3N. Then, the pink color changed back to white color in ether containing TFA, although the precipitated copolymers never dissolved in ether containing TFA or Et3N. Furthermore, the initial white color of the copolymers changed to a pink color by exposure to Et3N vapor (Fig. 6b). Then, the pink color also changed back to the white color under the acidic atmosphere. On the other hand, the depth of the color change depended on the composition ratio of each monomer unit in the copolymers. Specifically, the PVEC composition ratio in the copolymers increased displaying a change to the pale pink color. Moreover, this color change in the solid-state and in the solution occurred due to the tautomerization of the maleimide moiety. In the IR spectra of the polymer exposed to Et3N in the solid-state, the enolate structure was observed at 1665 cm−1 as the weak shoulder assigned to the carbonyl peak of the maleimide moiety (Fig. 7). However, the conformation change was never observed in 1H and 13C NMR spectra under high concentrations of CDCl3 and DMF-d7 (Fig. S4 and S5†). In the previous reports, the shoulder peak of the NMI polymer observed around 1660–1670 cm−1 was assigned to –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– formed by the tautomerization of the maleimide moiety (Fig. 7).26,27 In addition, the tautomerization of the maleimide moiety was also supported by the broadening of the carbonyl peak in the range of 1610–1700 cm−1 (Fig. 7). Moreover, the change in the depth of the color depending on the composition ratio indicated that the copolymer of the pale pink color was formed with a high composition ratio of the PVEC unit because the color change originated from the enolate structure of the maleimide unit, as shown above. Therefore, it was found that PVEC and PMI copolymers exhibited reversible color changes based on acid–base switching in the solid-state, and the depth of the color change was controlled depending on the composition ratio of the PVEC and/or PMI units. Furthermore, it was determined that the tautomerization of the maleimide moiety gave rise to the color change of these copolymers with acid–base switching and the solvent effect as the trigger.
N– formed by the tautomerization of the maleimide moiety (Fig. 7).26,27 In addition, the tautomerization of the maleimide moiety was also supported by the broadening of the carbonyl peak in the range of 1610–1700 cm−1 (Fig. 7). Moreover, the change in the depth of the color depending on the composition ratio indicated that the copolymer of the pale pink color was formed with a high composition ratio of the PVEC unit because the color change originated from the enolate structure of the maleimide unit, as shown above. Therefore, it was found that PVEC and PMI copolymers exhibited reversible color changes based on acid–base switching in the solid-state, and the depth of the color change was controlled depending on the composition ratio of the PVEC and/or PMI units. Furthermore, it was determined that the tautomerization of the maleimide moiety gave rise to the color change of these copolymers with acid–base switching and the solvent effect as the trigger.
 |
| | Fig. 6 Color change of PVEC and PMI copolymers based on acid–base switching in the solid-state, (a) in ether with Et3N or TFA, (b) in Et3N vapor. | |
 |
| | Fig. 7 IR spectra of PVEC and PMI copolymer exposed to TFA and Et3N in the solid-state. CO peak of maleimide moiety was expanded at 1600–1750 cm−1. | |
Conclusion
The radical copolymerization with various monomer feeds of PVEC and PMI was carried out with DTBP as an initiator in bulk at 120 °C for 24 h. The yield, molecular weight, and composition ratio of the PMI unit in copolymers increased almost linearly with increasing PMI feed. IR spectra of these copolymers proved that the radical copolymerization of PVEC and PMI successfully proceeded through the selective vinyl polymerization without depending on the monomer feed. In the UV-Vis spectra, the absorptions observed at 409 nm and 515 nm were assigned to the expanded conjugation of the maleimide units in the intramolecular and the enolate structure formed from the tautomerization of the maleimide moiety, respectively. The results of UV-Vis spectra revealed that the tautomerization of the maleimide unit, based on the solvent effect, readily occurred in the main-chain. Moreover, this suggested that the alternating and/or homo sequence was constructed depending on the monomer feed. Furthermore, these copolymers exhibited a sensitive and reversible color change based on acid–base switching not only in solution but also in the solid-state. In addition, the depth of the color change was controlled depending on the composition ratio of the PVEC and/or PMI units in solution and the solid-state. It was determined that the color change of these copolymers occurred due to the tautomerization of the maleimide moiety with acid–base switching and solvent effect as the trigger.
Experimental
Measurements
Gel permeation chromatography (GPC) was carried out on a TOSOH HLC-8220 system equipped with three consecutive polystyrene gel columns [TSK-gels (bead size, exclusion limited molecular weight), super-AW4000 (6 μm, >4 × 105), super-AW3000 (4 μm, >6 × 104) and super-AW2500 (4 μm, >2 × 103)] and refractive index and ultraviolet detectors at 40 °C. The system was operated using 10 mM LiBr in DMF as eluent at a flow rate of 0.5 mL min−1. Polystyrene standards were employed for calibration. 1H and 13C NMR spectroscopy was performed with a JEOL ECS-400 NMR spectrometer operating at 400 MHz in chloroform-d (CDCl3), dimethyl sulfoxide-d6 (DMSO-d6), N,N-dimethylformamide-d7 (DMF-d7) using tetramethyl silane (TMS) as an internal reference. ATR-FTIR spectra were recorded on a Thermo Fisher Scientific Nicolet iS10 spectrometer from 650 to 4000 cm−1. UV-Vis absorption spectra were recorded on a Jasco V570 UV-Vis-NIR spectrophotometer in N,N-dimethylformamide (DMF) and chloroform (CHCl3).
Materials
CDCl3, DMSO-d6, and DMF-d7 solvents for NMR, TMS for an internal reference, and DMF and CHCl3 for spectroscopy were purchased from Wako Pure Chemical Industry. N-Phenylmaleimide (PMI) and di-tert-butyl peroxide (DTBP) were purchased from Tokyo Chemical Industry. N-Phenylmaleimide was recrystallized before polymerization.
Preparation of monomers
Phenyl butadiene monoxide (PBO) and phenyl vinylethylene carbonate (PVEC) were prepared according to the methods reported in previous literature.8,23
Polymerization
Polymerization of PVEC and/or PMI was carried out with di-tert-butyl peroxide (DTBP) as a radical initiator in bulk using a degassed and sealed tube (Scheme 1). A typical procedure of polymerization is as follows.8,23 The 1H and 13C NMR spectra of poly(PVEC-co-PMI) prepared with monomer feeds of 90:10 (Poly-90), 70:30 (Poly-70), 30:70 (Poly-30), and 10:90 (Poly-10) of PVEC and PMI, respectively, exhibited the same chemical shifts as Poly-50 (Fig. S2†).
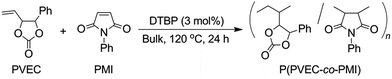 |
| | Scheme 1 Radical copolymerization of PVEC and PMI with DTBP in bulk. | |
Radical polymerization of PVEC and/or PMI
A mixture of each monomer (6.0 mmol) of PVEC and/or PMI and 26.3 mg (0.18 mmol, 3 mol%) of DTBP was frozen in liquid nitrogen and degassed under reduced pressure (2–3 mm Hg) for 30 min. After repeating this operation exactly 3 times, the tube containing the mixture of the monomer and initiator was sealed, and the mixture was stirred at 120 °C for 24 h. The resulting solid was dissolved in DMF/acetone and the solution was poured into n-hexane/ethanol. The precipitated polymer was washed with methanol and dried under dynamic vacuum at 40 °C for 24 h.
Poly(phenyl vinylethylene carbonate), Poly-100. 1H NMR (DMSO-d6, TMS): δ = 0.21–2.23 (3H, –CH2–CH–), 3.52–5.56 (2H, –CH–CH–), 6.45–7.86 (5H, C6H5–). 13C NMR (DMSO-d6, TMS): δ = 28.60, 34.18, 82.06, 83.38, 126.80, 128.89, 129.53, 135.34, 153.42.
Poly(PVEC-co-PMI) with monomer feed of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :50, Poly-50. 1H NMR (DMSO-d6, TMS): δ = 0.21–2.49 (3H, –CH2–CH–), 4.06 (2H, –CH–CH–), 4.46–6.18 (2H, –COH–COH–), 7.40 (10H, C6H5C and C6H5N). 13C NMR (DMSO-d6, TMS): δ = 28.7, 43.3, 82.3, 126.7, 128.9, 131.4, 135.1, 153.5, 175.9.
:50, Poly-50. 1H NMR (DMSO-d6, TMS): δ = 0.21–2.49 (3H, –CH2–CH–), 4.06 (2H, –CH–CH–), 4.46–6.18 (2H, –COH–COH–), 7.40 (10H, C6H5C and C6H5N). 13C NMR (DMSO-d6, TMS): δ = 28.7, 43.3, 82.3, 126.7, 128.9, 131.4, 135.1, 153.5, 175.9.
Poly(phenylmaleimide), Poly-0. 1H NMR (DMSO-d6, TMS): δ = 4.11, 4.55 (2H, –CH–CH–), 7.15, 7.41 (5H, C6H5N). 13C NMR (DMSO-d6, TMS): δ = 42.9, 126.7, 128.7, 131.5, 176.3.
Acknowledgements
This study was financially supported by JSR Co., Ltd.
Notes and references
-
(a) Y. Hibi, S. Tokuoka, T. Terashima, M. Ouchi and M. Sawamoto, Polym. Chem., 2010, 2, 341–347 RSC
 ;
(b) Y. Hibi, M. Ouchi and M. Sawamoto, Angew. Chem., Int. Ed. Engl., 2011, 50, 7434–7437 CrossRef CAS PubMed ;
(c) K. Nakatani, Y. Ogura, Y. Koda, T. Terashima and M. Sawamoto, J. Am. Chem. Soc., 2012, 134, 4373–4383 CrossRef CAS PubMed ;
(d) J.-F. Lutz, M. Ouchi, D. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef PubMed ;
(e) M. Ouchi, M. Nakano, T. Nakanishi and M. Sawamoto, Angew. Chem., Int. Ed. Engl., 2016, 128, 14804–14809 CrossRef .
;
(b) Y. Hibi, M. Ouchi and M. Sawamoto, Angew. Chem., Int. Ed. Engl., 2011, 50, 7434–7437 CrossRef CAS PubMed ;
(c) K. Nakatani, Y. Ogura, Y. Koda, T. Terashima and M. Sawamoto, J. Am. Chem. Soc., 2012, 134, 4373–4383 CrossRef CAS PubMed ;
(d) J.-F. Lutz, M. Ouchi, D. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef PubMed ;
(e) M. Ouchi, M. Nakano, T. Nakanishi and M. Sawamoto, Angew. Chem., Int. Ed. Engl., 2016, 128, 14804–14809 CrossRef . -
(a) S. Pfeifer and J. F. Lutz, J. Am. Chem. Soc., 2007, 129, 9542–9543 CrossRef CAS PubMed ;
(b) M. Zamfir and J. F. Lutz, Nat. Commun., 2012, 3, 1138 CrossRef ;
(c) J. F. Lutz, B. Schmidt and S. Pfeifer, Macromol. Rapid Commun., 2011, 32, 127–135 CrossRef CAS PubMed ;
(d) S. Srichan, D. Chan-Seng and J. F. Lutz, ACS Macro Lett., 2012, 1, 589–592 CrossRef CAS ;
(e) S. Srichan, H. Mutlu and J. F. Lutz, Eur. Polym. J., 2015, 62, 338–346 CrossRef CAS .
-
(a) M. Matsuda, K. Satoh and M. Kamigaito, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1774–1785 CrossRef CAS ;
(b) T. Soejima, K. Satoh and M. Kamigaito, J. Am. Chem. Soc., 2016, 138, 944–954 CrossRef CAS PubMed ;
(c) T. Soejima, K. Satoh and M. Kamigaito, Polym. Chem., 2016, 7, 4833–4841 RSC ;
(d) K. Satoh, Polym. J., 2015, 47, 527–536 CrossRef CAS ;
(e) K. Satoh and M. Kamigaito, Kobunshi Ronbunshu, 2015, 72, 421–432 CrossRef CAS .
-
(a) T. Otsu, A. Matsumoto, T. Kubota and S. Mori, Polym. Bull., 1990, 23, 43–50 CrossRef CAS ;
(b) K. Takeda and A. Matsumoto, Macromol. Chem. Phys., 2010, 211, 782–790 CrossRef CAS ;
(c) M. Hisano, K. Takeda, T. Takashima, Z. Jin, A. Shiibashi and A. Matsumoto, Macromolecules, 2013, 46, 7733–7744 CrossRef CAS ;
(d) M. Hisano, T. Takashima, Z. Jin, A. Shiibashi and A. Matsumoto, Macromol. Chem. Phys., 2013, 214, 1612–1620 CrossRef CAS ;
(e) D. Yamamoto and A. Matsumoto, Macromolecules, 2013, 46, 9526–9536 CrossRef CAS ;
(f) A. Tsujii, M. Namba, H. Okamura and A. Matsumoto, Macromolecules, 2014, 47, 6619–6626 CrossRef CAS ;
(g) A. Tsujii, L. Lou, M. Nagashima, H. Okamura and A. Matsumoto, J. Adhes. Soc. Jpn., 2015, 51, 336–341 CrossRef CAS .
- Y. Abiko, A. Matsumura, K. Nakabayashi and H. Mori, React. Funct. Polym., 2015, 93, 170–177 CrossRef CAS .
- G. Q. Chen, Z. Q. Wu, J. R. Wu, Z. C. Li and F. M. Li, Macromolecules, 2000, 33, 232–234 CrossRef CAS .
- Y. Ren, Z. Zhu and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3828–3835 CrossRef CAS .
-
(a) Y. Yoshida and T. Endo, Polymer, 2016, 102, 167–175 CrossRef CAS ;
(b) Y. Yoshida and T. Endo, Polym. Chem., 2016, 7, 6770–6778 RSC .
- I. Takase, K. Kawazu, H. Aida and H. Kohkame, Kobunshi Ronbunshu, 1990, 47(7), 569–574 CrossRef CAS .
- M. Mohamed, K. C. Hsu, J. L. Hong and S. W. Kuo, Polym. Chem., 2015, 7, 135–145 RSC .
- H. Ahn, J. Hong, S. Kim, I. Choi and M. Park, ACS Appl. Mater. Interfaces, 2015, 7, 704–712 CAS .
-
(a) P. Sung, C. Chen, S. Wu and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 2189–2194 CrossRef CAS ;
(b) İ. Serhatli, Y. Yagci, E. Hattemer, R. Zentel, E. Schmälzlin, S. Hohenadl, C. Braüchle and K. Meerholz, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1589–1595 CrossRef ;
(c) F. A. Nicolescu, V. V. Jerca, I. Dancus, A. Petris, T. V. Nicolescu, I. B. Rau, V. I. Vlad, D. S. Vasilescu and D. M. Vuluga, J. Polym. Res., 2011, 18, 1009–1016 CrossRef CAS .
-
(a) K. Koehler, K. Anseth and C. Bowman, Biomacromolecules, 2013, 14, 538–547 CrossRef CAS PubMed ;
(b) R. Klein, F. Übel and H. Frey, Macromol. Rapid Commun., 2015, 36, 1822–1828 CrossRef CAS PubMed .
-
(a) T. Oishi, H. Yamasaki and M. Fujimoto, Polym. J., 1991, 23, 795–804 CrossRef CAS ;
(b) K. Onimura and T. Oishi, Polymeric Chiral Catalyst Design and Chiral Polymer Synthesis, wiley, 2011, p. 356 Search PubMed ;
(c) Y. Okamoto, T. Nakano, H. Kobayashi and K. Hatada, Polym. Bull., 1991, 25, 5–8 CAS .
-
(a) H. C. Haas and R. L. MacDonald, J. Polym. Sci., Polym. Chem. Ed., 1973, 11, 327–343 CrossRef CAS ;
(b) H. C. Haas and R. D. Moreau, J. Polym. Sci., Polym. Chem. Ed., 1975, 13, 2327–2334 CrossRef CAS .
-
(a) E. Hadjoudis and I. Mavridis, Chem. Soc. Rev., 2004, 33, 579–588 CAS ;
(b) R. Klajn, Chem. Soc. Rev., 2013, 43, 148–184 RSC ;
(c) A. Seeboth, D. Lötzsch, R. Ruhmann and O. Muehling, Chem. Rev., 2014, 114, 3037–3068 CrossRef CAS PubMed .
-
(a) T. Yamase, Chem. Rev., 1998, 98, 307–326 CrossRef CAS PubMed ;
(b) M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed ;
(c) A. Fihey, A. Perrier, W. Browne and D. Jacquemin, Chem. Soc. Rev., 2015, 44, 3719–3759 RSC .
-
(a) R. Mortimer, Chem. Soc. Rev., 1997, 26, 147–156 RSC ;
(b) P. Beaujuge and J. Reynolds, Chem. Rev., 2010, 110, 268–320 CrossRef CAS PubMed ;
(c) Z. Liu, W. Qi and G. Xu, Chem. Soc. Rev., 2015, 44, 3117–3142 RSC .
-
(a) O. Wenger, Chem. Rev., 2013, 113, 3686–3733 CrossRef CAS PubMed ;
(b) J. Askim, M. Mahmoudi and K. Suslick, Chem. Soc. Rev., 2013, 42, 8649–8682 RSC .
-
(a) C. Reichardt, Chem. Soc. Rev., 1992, 21, 147–153 RSC ;
(b) V. Machado, R. Stock and C. Reichardt, Chem. Rev., 2014, 114, 10429–10475 CrossRef CAS PubMed .
-
(a) V. Minkin, Chem. Rev., 2004, 104, 2751–2776 CrossRef CAS PubMed ;
(b) E. Cariati, E. Lucenti, C. Botta, U. Giovanella, D. Marinotto and S. Righetto, Coord. Chem. Rev., 2016, 306, 566–614 CrossRef CAS .
-
(a) R. Motoshige, Y. Mawatari, A. Motoshige, Y. Yoshida, T. Sasaki, H. Yoshimizu, T. Suzuki, Y. Tsujita and M. Tabata, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 752–759 CrossRef CAS ;
(b) Y. Mawatari, A. Motoshige, Y. Yoshida, R. Motoshige, T. Sasaki and M. Tabata, Polymer, 2014, 55, 2356–2361 CrossRef CAS .
-
(a) B. Ochiai, M. Matsuki, D. Nagai, T. Miyagawa and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 584–592 CrossRef CAS ;
(b) B. Ochiai and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2827–2834 CrossRef CAS .
-
(a) D. Webster and A. Crain, Prog. Org. Coat., 2000, 40, 275–282 CrossRef CAS ;
(b) D. Webster, Prog. Org. Coat., 2003, 47, 77–86 CrossRef CAS ;
(c) Y. Wang, F. Fan, A. L. Agapov, T. Saito, J. Yang, X. Yu, K. Hong, J. Mays and A. P. Sokolov, Polymer, 2014, 55, 4067–4076 CrossRef CAS .
-
(a) N. Kihara and T. Endo, Makromol. Chem., 1992, 193, 1481–1492 CrossRef CAS ;
(b) A. Suzuki, D. Nagai, B. Ochiai and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5983–5989 CrossRef CAS ;
(c) A. Sudo, H. Morishita and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3896–3902 CrossRef CAS .
- G. V. Paesschen and D. Timmerman, Makromol. Chem., 1964, 78, 112–120 CrossRef .
-
(a) M. Yamada and I. Takase, Kobunshi Kagaku, 1966, 23, 348–353 CrossRef CAS ;
(b) M. Yamada, I. Takase and T. Mishima, Kobunshi Kagaku, 1967, 24, 326–333 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28446a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*