
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Arylidene indanone scaffold: medicinal chemistry and structure–activity relationship view
José C. J. M. D. S. Menezes†

Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal
First published on 31st January 2017
Abstract
Arylidene indanone (AI) scaffolds are considered as the rigid cousins of chalcones, incorporating the α,β-unsaturated ketone system of chalcones forming a cyclic 5 membered ring. They are generally synthesized from 1-indanone and benzaldehydes via an aldol reaction. The furnished molecules have been explored as inhibitors of cholinesterases towards the treatment of Alzheimer’s disease, as tubulin depolymerizing agents, as inhibitors of breast cancer and leukemia, inhibitors of dual specificity phosphatase (DUSP), as antimalarials, and for many other uses. This review is an effort to highlight the biochemical effects of arylidene indanones designed from natural or known drug compounds, discuss their structure–activity relationships (SAR), and correlate them with related chalcones providing insights for further development of this scaffold.
 José C. J. M. D. S. Menezes | José C. J. M. D. S. Menezes completed his PhD in synthetic Organic Chemistry from Goa University, India in 2009 with Prof. S. P. Kamat and a post-doc with Prof. José A. S. Cavaleiro working on natural and synthetic porphyrins from October 2010–2013, at the Department of Chemistry, University of Aveiro, Portugal. He has published 20 papers in peer reviewed international journals. His research interests are the design of new molecules using natural molecule templates, synthesis and applications in biology. Presently he is working as a Tokyo Biochemical Research Foundation post-doctoral fellow at the Department of Pharmacy, Nagasaki International University, Japan. |
1. Introduction
Natural indanones like pterosin P 1 from fern (Pteridium) species,1–3 4-hydroxy-7-methylindanone 2 and 7-formyl-3-methoxy-5-methylindanone 3 from marine cyanobacterium4,5 have shown antibacterial,4 antispasmodic,6 inhibition of hypoxia-induced activation of vascular endothelial growth factor (VEGF)5 and cytotoxic activities.3 The 4,5,6-trimethoxyindanone-estradiol hybrid 4 was more potent against MCF-7 breast cancer cells in comparison to the chalcone–estradiol precursor 5 (see Fig. 1 for structures).7 | ||
| Fig. 1 Natural and synthetic indanones and the basic similarities between arylidene indanone and its cousin, the chalcone. | ||
Arylidene indanones (AI) are mostly synthetic molecules, consisting of the natural/synthetic indanone core (see the part of the structure in green lines) and an aryl group linked through an exocyclic double bond at the 2-position of this indanone core (for the numbering system see Fig. 1). This AI scaffold often connoted as cyclic chalcone can be considered as rigid cousins or homologs of chalcones, incorporating the α,β-unsaturated ketone (red dashed circles) into a cyclic 5 member ring which is attached to the 2-position of chalcone ring (red arrow) and to the α-position of the unsaturated bond (blue arrow), making it a planar/rigid exocyclic α,β-unsaturated ketone system. Chalcones, belong to the class of flavonoids and have been explored as anti-infective, anti-inflammatory and anti-cancer agents.8,9 The interaction of natural or synthetic chalcones with molecular targets like tubulin, kinases, oxidoreductases, hydrolases, esterases,10 and cell cycle blockers have been reviewed.11 Recent developments of the synthetic chalcone scaffold with different heterocyclic rings or hybrids with other natural compounds for various therapeutic roles have been compiled.12
The position of the arylidene ring with respect to the indanone core results in a planar structure and allows transmission of the electron donor effects of the substituents via the double bond onto the carbonyl group of indanone, thus lowering the wavenumber-frequency more effectively compared to chalcones.13 Higher values were observed in the 13C NMR of the α,β-unsaturated ketone carbons of AIs compared with chalcones which had similar electron donating substituents in the aryl ring.13,14 The position of absorption maxima (UV-vis) for AI is higher in comparison with chalcones having similar para-substituents in the aryl ring indicating the strongest conjugation due to its planar structure.15 The lipophilicity of AI can be increased by geometric inversion of the double bond from the E to Z-isomer which occurs due to UV-light.16 In the case of chalcones, the double bond can have either the s-cis or s-trans conformation while in the case of AI the molecule is forced to adopt the s-cis conformation due to the cyclic ring. The introduction of an α-substituent on the double bond makes the chalcone adopt the s-trans conformation and this was explored to design potent tubulin inhibitors.17,18
Retrosynthesis of AI leads to 1-indanones (natural or synthetic) which are assembled from phenyl/aryl propionic acid via cyclization reactions using polyphosphoric acid,19 methane sulphonic acid,20 Friedel–Crafts acylation under microwave heating and ultrasonication,21 and Nazarov cyclization of chalcones.7 Further reaction of suitably substituted 1-indanone with benzaldehyde derivatives under aldol conditions or potassium carbonate and microwave irradiation leads to the synthesis of AI.22–24 Substitution in the indanone ring as well as in the benzaldehyde moiety has furnished AI scaffolds having therapeutic effects against breast cancer,25 leukemia,26 and Alzheimer’s disease,27 AI scaffolds causing inhibition of DUSP28,29 and AI scaffolds acting as potential antimalarial agents.30,31 This review discusses the various therapeutic roles of AIs, their structure–activity relationships and correlates them wherever possible with structurally similar or related chalcones. The web-based Molinspiration software and the related molecular properties (using the Lipinski rule) were utilized to predict theoretical milog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P (octanol–water partition coefficient) of selected groups of molecules.32,33
P (octanol–water partition coefficient) of selected groups of molecules.32,33
2. Effect of AI in Alzheimer’s disease
Alzheimer’s disease (AD) is a neurodegenerative disorder which is the cause of dementia (loss of memory) in the later stages of adult life and its prevalence is estimated to rise up to 106 million by 2050.34 The neuropathological features of AD are extracellular deposits of amyloid beta peptide (Aβ) called amyloidal plaques and hyperphosphorylated neurofibrillary tangles (NFTs).35 Cleavage of the amyloid protein precursor (APP) by secretases (α-,β-, and γ-secretase) results in the production of various species of Aβ which due to non-clearance from the brain form Aβ42 oligomers which hydrophobically self-aggregate and give rise to diffuse senile plaques (SPs).35The human brain has two major cholinesterases; acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) both differing in their genetics, structure, and kinetics. In patients with AD, BuChE is found in neurons and glial cells as well as in neuritic plaques and tangles.36 Acetylcholine, a neurotransmitter molecule linked with intellectual abilities decreases in the brain due to enhanced acetylcholinesterase activity resulting from AD or old age. The acetylcholinesterase (AChE) enzyme structure determined for Torpedo californica (Tc) consists of a catalytic triad (having residues Ser200, His440, and Glu327) and a peripheral anionic site (PAS).37 The active site also contains an anionic subside consisting of the key residue Trp84 located near the bottom of the cavity. This site interacts with the quaternary ammonium group of acetylcholine substrates and other ligands via cation–π interaction.37 While the PAS of the acetylcholinesterase enzyme is located at the entrance of the narrow catalytic gorge which serves as a transient binding site for substrates entering the active site. The Aβ peptide interacts with the PAS resulting in the folding of these proteins and the formation of plaques.37 Donepezil hydrochloride 6 (see Fig. 2) interacts with these dual binding sites of the AChE enzyme38 and is the presently used drug (Aricept) for all stages of Alzheimer’s disease.39 Several other drugs like rivastigmine 7, galantamine 8, memantine 9 and the combination of donepezil and memantine have been approved.39 Hybrid compounds for the treatment of AD have been derived by combining fragments or drug molecules like 6 and tacrine 10;40,41 8 and 9;42 and natural molecules like curcumin, berberine, ferulic and caffeic acid,42,43 chalcones and coumarin derivatives,43 and resveratrol and clioquinol.44
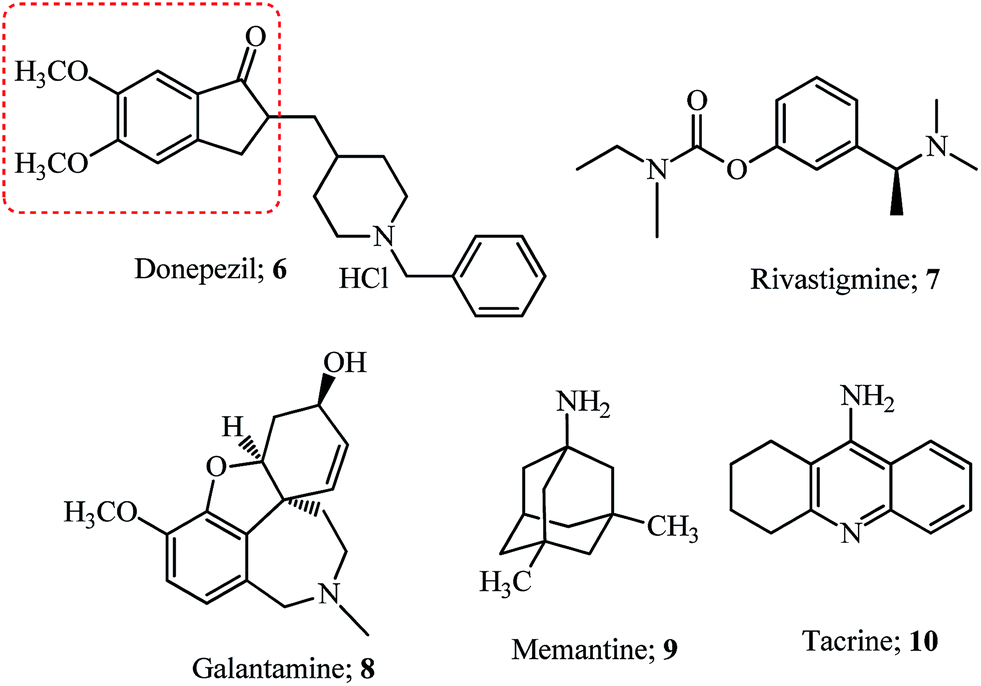 | ||
| Fig. 2 Drugs approved for the treatment of Alzheimer’s disease. | ||
Most of the compounds discussed in this section have been derived from the indanone core of 6 with modification in the 5,6-position or substitution of arylidene with fragments from other drugs. Hybrid molecules designed by combining the phenyl-N-methylbenzylamine fragment of AP2238 (see the structure and fragment in the blue ellipsoid; Fig. 3), with the indanone core of donepezil 6 (see the red dashed box in Fig. 2) resulted in several new indanone derivatives with potencies against AChE (5 examples).45
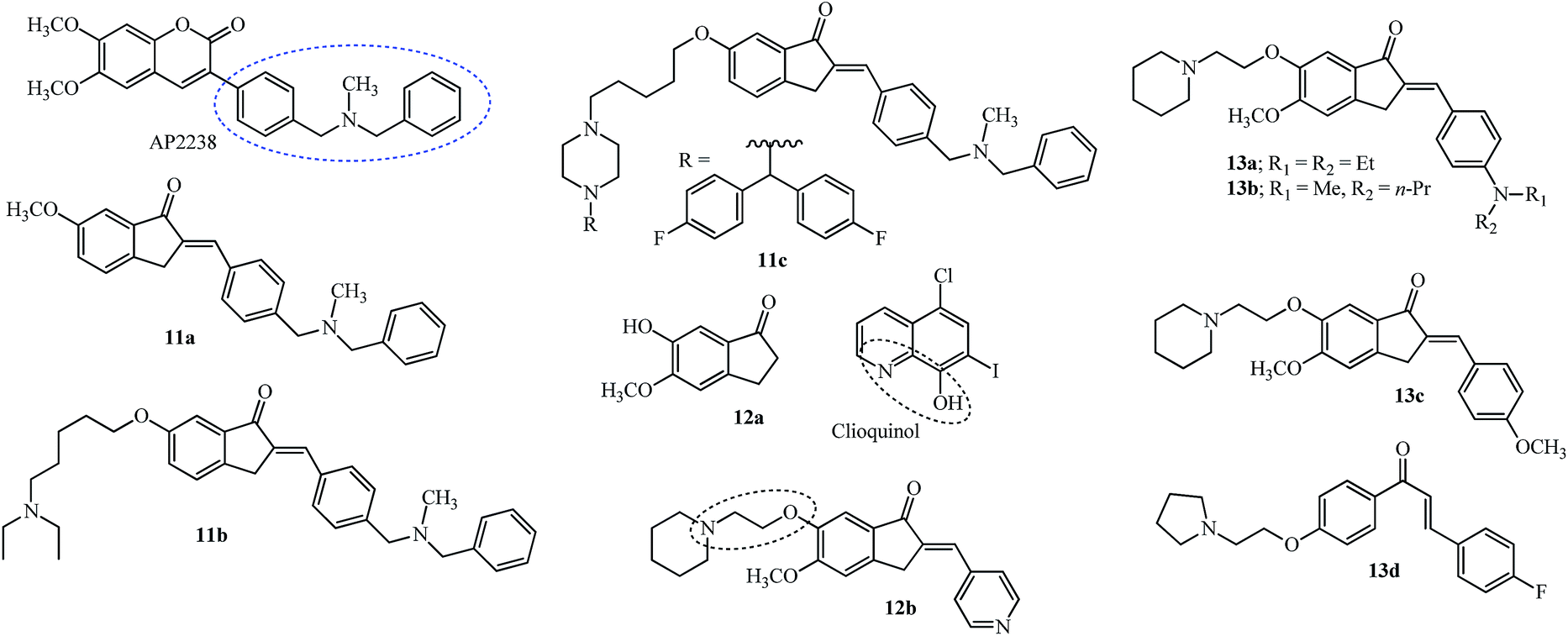 | ||
| Fig. 3 Cholinesterase inhibitor hybrids derived from donepezil and AP2238. | ||
Although the 5,6-dimethoxy groups of donepezil have efficient interaction with the PAS of the cholinesterase enzyme, the methoxy at 6-position of indanone 11a gave a hydrophobic interaction with the side chain of the Leu289 residue and was more potent for AChE inhibition.45 Further modification of the 6-position by introducing alkyl chains of varying lengths having different terminal amine groups was studied for efficient interaction with the PAS of AChE (12 examples). The optimum length of the alkyl spacer was 5 carbons, with the terminal N,N-diethyl substituent (11b). The ability of 11b to prevent AChE-induced and self Aβ aggregation was noted.45
APP is cleaved by a β-secretase known as the β-site APP cleaving enzyme, BACE1, and this is the rate-limiting step in the generation of Aβ peptide fragments.35 Inhibition of this enzyme was attempted by retaining the basic structure of 11b and introducing piperazine on the pentyl side chain with a di-substituent on the second nitrogen (8 examples). Substitution with bis(4-fluorophenyl)methyl resulted in the molecule 11c which showed potent activity against BACE1 (IC50 = 2.49 μM).46 Docking studies of 11c with the BACE1 enzyme showed interaction with amino acid residues outside the binding pocket but it was unable to enter the narrow gorge of hAChE due to the bulky nature of the bis(4-fluorophenyl)methyl group and it had low neurotoxicity in human neuronal SH-SY5Y cells.46
Meng and co-workers evaluated the modification of the 6-position of indanone by derivatives of the type 12 (13 examples).49 The introduction of the piperidinyl ethoxy substituent at the 6-position of indanone 12a and pyridinyl methylene at the 2-position made indanone 12b more potent than donepezil 6 and the starting indanone 12a (see Fig. 3 and Table 1 for IC50 values). The introduction of the piperidinyl ethoxy group in the 6-position of the starting indanone 12a had no improvement on the inhibition efficiency indicating the crucial nature of the double bond for the activity. Chelation of biometals like Cu2+, Fe3+ and Zn2+ is an important strategy for treatment of AD since these metals accumulate on the formation of amyloidal plaques and SPs.53,54 Metal chelation studies using 12b and a solution of Cu2+, Fe3+ and Zn2+ indicated a blue shift using UV-vis spectrophotometry (a decrease in wavelength or a move towards the blue end of the visible spectrum) from 360 nm to 352 nm, 346 nm, and 357 nm respectively.49 The piperidinyl ethoxy arm of 12b (2 carbon spacer) functions/mimics as the biometal chelator group is also present in clioquinol (see the dashed oval shape for the clioquinol structure in Fig. 3) and is used as a reference for these studies. This fact was fortified by using a compound with a carbon spacer of 5 (C = 5) which did not show any metal chelation.49 In the case of clioquinol, the donor groups N and O may form a bidentate chelate which can form a five-membered ring/complex with bivalent metals and the five-membered ring complex formed during chelation is more effective, compared to any other ring size.55 The benefits/challenges of using chelating therapy for AD have been given an overview, which includes clioquinol among other molecules.53,54
| Compound | IC50 hAChE (μM) | IC50 hBuChE (μM) | Method/substrate and Ref | Compound | IC50 hAChE (μM) | IC50 hBuChE (μM) | Method/substrate and Ref |
|---|---|---|---|---|---|---|---|
| a Human recombinant BACE1 (hBACE1) using FRET assay; ND-not determined. | |||||||
| 11a | 0.63 | 7.93 | Ellman45 | 7 | 10.54 | 0.26 | Ellman47 |
| 11b | 0.18 | 10.0 | Ellman | 14a | 0.045 | 3.015 | eeAChE/eqBuChe48 |
| 6 | 0.02 | 7.42 | Ellman45 | 14b | 0.025 | 2.434 | eeAChE/eqBuChe |
| AP2238 | 0.044 | 48.9 | Ellman45 | 14c | 0.034 | 1.969 | eeAChE/eqBuChe |
| 11ca | 2.49a | ND | hBACE1 | 6 | 0.039 | 3.36 | eeAChE/eqBuChe48 |
| 12a | 91.96 | 258.03 | Ellman49 | 14d | 4.895 | — | eeAChE/Ellman50 |
| 12b | 0.0018 | 9.5 | Ellman | 6 | 0.2197 | — | eeAChE/Ellman50 |
| 6 | 0.026 | 4.66 | Ellman49 | 10 | 0.3715 | — | eeAChE/Ellman50 |
| 13a | 14.8 nM | 1384.9 | eeAChE/eqBuChe51 | 15a | 0.42 | 315 | Ellman52 |
| 13b | 18.6 nM | 1417.1 | eeAChE/eqBuChe | 15b | 0.035 | 82 | Ellman |
| 13c | 14.8 nM | 3213.6 | eeAChE/eqBuChe | 15c | 0.045 | 26.6 | Ellman |
| 6 | 38.2 nM | 3365.4 | eeAChE/eqBuChe51 | 15d | 0.10 | 84.6 | Ellman |
| 10 | 109 nM | 15.2 | eeAChE/eqBuChe51 | 7 | 2.07 | 1.41 | Ellman52 |
| 13d | 0.21 | 13.65 | Ellman47 | — | — | — | — |
The 6-piperidinylethoxy-5-methoxy indanone moiety from 12b was retained while the 4-position on the arylidene ring was varied to design “multi-target one ligand” molecules (11 examples). Derivatives 13a–13c (Fig. 3) indicated a higher potency for inhibition of AChE with nanomolar IC50 values compared with 10 and similar ones to 6 (see Table 1 for IC50 values).51 The substituent at the 4-position of the phenyl ring showed no relevant role as the potencies were similar for compounds 13a–13c.51 Inhibition of Aβ aggregation by 13a and 13b was noted. Antioxidant activity was highly diminished for 13c highlighting the importance of the amino group at the 4-position on the phenyl ring.51 The chalcone derivative 13d(Fig. 3) having 2-pyrrolidinyl ethoxy at the 4′-position and a fluoro group at the 4-position showed potency for AChE compared to BuChE and this was correlated by docking analysis which showed a dual interaction with the catalytic site and the PAS of the AChE enzyme.47
The bis(4-fluorophenyl)methyl piperazine group (see R of 11c in Fig. 3) was introduced on the 4-position of the arylidene group (14a; Fig. 4) providing the most potent acetylcholinesterase inhibition amongst the 15 tested compounds.48 The pyrimidine 14b and 3-furoyl groups 14c also gave potent acetylcholinesterase and butyrylcholinesterase inhibition compared with 6 (see Table 1).48 The substitution on the piperazine ring by N,N-dimethyl aminopropyl group gave the most potent molecule 14d having AChE inhibition activity (among 24 examples) and was compared with standard reference donepezil 6 and tacrine 10.50
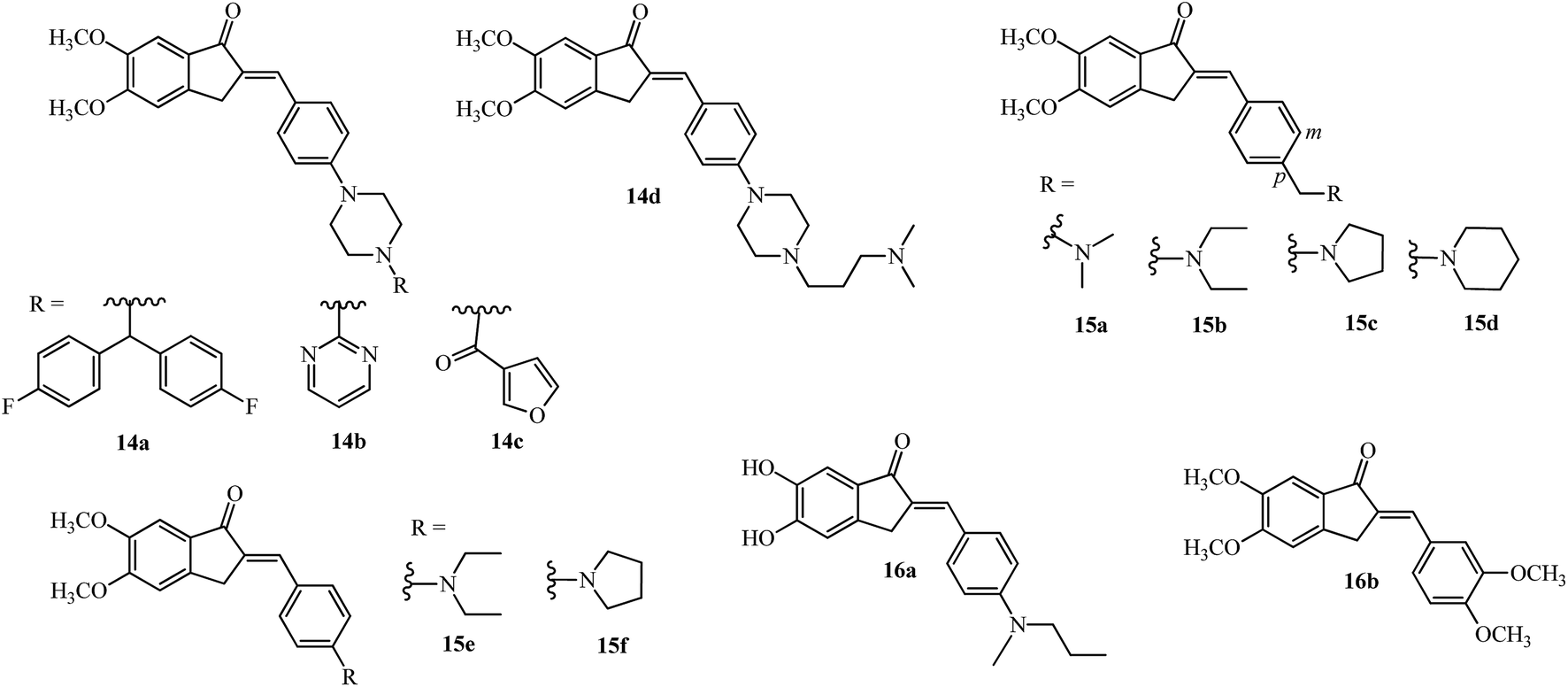 | ||
| Fig. 4 Inhibitors of acetylcholinesterase and Aβ aggregation and metal chelation agents. | ||
Incorporating the N,N-dimethyl amino side chain similar to that found in rivastigmine 7 (Fig. 2), at the para position of the benzene ring and varying the substituents to diethyl, pyrrolidine and piperidine resulted in 20 compounds of the type 15a–d (see Fig. 4). The para isomers were more effective than their meta isomers. The diethyl 15b and pyrrolidine 15c AIs were more potent compared with dimethyl amino 15a, piperidine 15d, and rivastigmine 7 (see IC50 values in Table 1). In general, the AIs were more potent than their reduced counterparts and selective for AChE over BuChE.52 The active indanone derivative 15b improved the acquisition and memory loss induced by scopolamine52,56 sodium nitrite, and ethanol56 and showed neuroprotective functions in PC12 cells.56
Interaction of the diethylamine nitrogen of 15b with the Trp84 residue situated at the bottom of the AChE gorge and of the indanone ring with the indole ring of Trp279 at the top of the gorge was shown by docking studies based on the X-ray crystal structure of the TcAChE–E2020 (donepezil) complex (see Fig. 5 reproduced with permission from Elsevier Masson SAS).52 These docking interactions were verified by independent QSAR and docking studies of 15b which revealed similar findings.57 Another recent docking study also verified these finding and found the pyrrolidinyl derivative 15c which is a non-classical bioisostere of 15b to have a similar potency.58 A recent study with the AI derivatives 15e and 15f having the N,N-diethylamino and pyrrolidine groups directly attached to the 4-position of aryl ring didn’t show promising AChE or BuChE inhibition activities,50 pointing out the importance of methylene substitution at the 4-position of aryl ring as seen in 15b and 15c.52
 | ||
| Fig. 5 Docking of 15b (carbon atoms in black and bonds in purple), into the gorge of the TcAChE enzyme. This figure has been reproduced from ref. 52 with permission from Elsevier Masson SAS. | ||
Self-mediated Aβ1–42 aggregation which is implicated in the progression of AD, was targeted by 16a (Fig. 4; highest potency of 80.1% at a concentration of 20 μM) which was among 19 derivatives having the 5,6-position of the indanone core substituted by methoxy and hydroxy groups while the arylidene ring had methoxy and alkylamino or dialkylamino substitution at the 3′,4′-position.27 Stoichiometric interaction of 16a (ratio of 1:1) with Cu2+ solutions showed a red shift (437 nm to 451 nm) by UV-vis spectrophotometry, while no significant change for Fe3+ and Zn2+ metals were observed.27 The ortho-dihydroxy group is also present in natural flavonoids like quercetin and myricetin and has shown chelation of copper ions at physiological pH.59
Monoamine oxidases (MAO) A and B are enzymes which belong to the family of flavin adenine dinucleotides (FADs) responsible for catalyzing the oxidative deamination of neurotransmitters like serotonin, dopamine and adrenaline in the CNS due to the progression of AD.60 Inhibitors of MAO-A/B may increase the levels of these neurotransmitters improving the learning and memory deficit of patients having AD. Selective inhibition of MAO-B by 16a was observed (7.50 μM); while 5-methoxy-6-hydroxy substitution diminished this activity (40.5 μM), indicating the importance of the dihydroxyl substitution.27 Various scaffolds like coumarins and indole have been synthesized and tested as dual MAO-B and AChE inhibitors,60 indicating the unexplored potential of the arylidene indanone scaffold.
Although the dihydroxy-substituted 16a showed MAO-B inhibition, several new compounds like 16c–16h (Fig. 6) similar to this scaffold have shown inhibition with mono hydroxy/methoxyl substitution at the 5-position.61,62 The 5-monohydroxyl compound 16c showed better inhibition of MAO-B compared to the 6-hydroxy isomer 16d (see IC50 values in Table 2). In a previous study, the appropriate benzyloxy substituent on the 5 or 6-position of the indanone ring was shown to have influence in the inhibition of MAO-B over MAO-A.63 Moreover, a comparison of the MAO-B inhibition data of the 5 and 6-hydroxyindanones 16c′ and 16d′ (ref. 63) with the arylidene indanones 16c and 16d indicates that the inhibition potency improved with the introduction of the benzylidene moiety.61 The calculated milogP values of 16c and 16d (3.24) are higher than 16c′ and 16d′ (1.22) indicating an increase of lipophilicity due to the benzylidene moiety.32
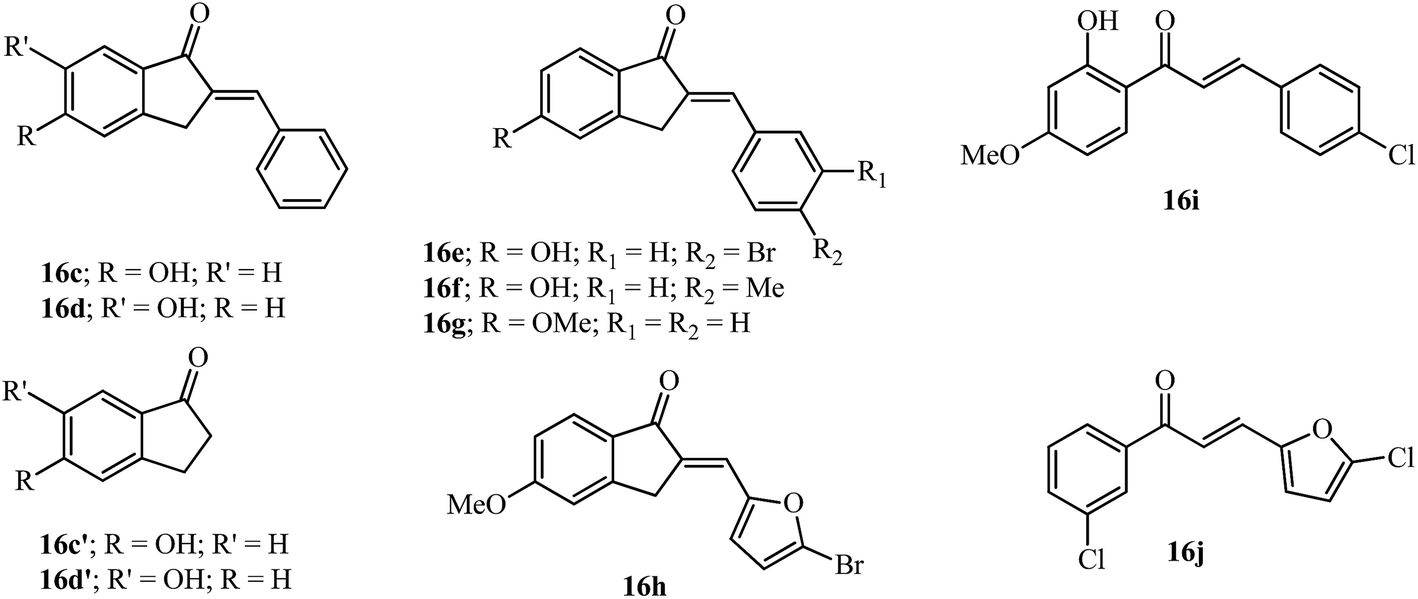 | ||
| Fig. 6 AI and chalcones tested as inhibitors of MAO-A/B. | ||
| Compound | milogP |
IC50 MAO-A (μM) | IC50 MAO-B (μM) |
|---|---|---|---|
| a NI-no inhibition at maximum tested concentration of 100 μM. | |||
| 16c | 3.24 | 1.94 | 0.376 |
| 16d | 3.24 | 0.440 | 2.42 |
| 16c′ | 1.22 | 61.7 | >100 |
| 16d′ | 1.22 | 64.7 | 85.4 |
| 16e | 4.05 | 1.05 | 0.0053 |
| 16f | 3.69 | 3.22 | 0.0052 |
| 16g | 3.77 | NIa | 0.0092 |
| 16h | 3.79 | 0.183 | 0.0044 |
| 16i | 4.46 | — | 0.0044 |
| 16j | 4.34 | 28.6 | 0.174 |
Substitution on the arylidene ring at the 4′-position with bromo (16e) and methyl (16f) gave potent results among 22 tested compounds. These compounds were more potent for MAO-B than as MAO-A inhibitors. The lipophilic 5-methoxyindanone with the benzylidene moiety 16g improved the MAO-B inhibition value compared to that observed for 16c. The modification of the arylidene group using hetero arylidene moieties like pyridine, furan and thiophene gave the equipotent compound 16h which has a 5-bromo-2-furan substituent.62 Chalcones 16i and 16j having some structural similarities with the AIs discussed above were more potent for MAO-B compared to their use as MAO-A inhibitors (see Table 2).64,65
Detection of senile plaques (SPs) which are composed of Aβ protein aggregates and NFTs may help in the diagnosis of AD.66 The importance of single photon emission computed tomography (SPECT) radiotracers for imaging amyloid plaques in the brain of AD patients has been an active area of research using different types of molecules.66 Amyloid imaging probes e.g., the radioactive labeled Pittsburgh compound B (PIB), and its 18F analog are routinely used for detection (Fig. 7). AIs 17a–17d (Fig. 7) were evaluated as amyloid imaging agents in vitro and in vivo since they qualify as a neutral molecule possessing at least two phenyls (or heterocyclic) rings and a linker and are in conjugation with each other, and the molecule as a whole has a hydrophobic, planarized π system.67
 | ||
| Fig. 7 Amyloid imaging probes and potential AI and chalcones as imaging probes for β-amyloid plaques. | ||
A binding affinity assay with human AD brain homogenates showed that 17a has the highest binding constant (Ki = 16 ± 1 nM) while the 5-methoxy 17b showed an increase in the binding constant (Ki = 21 ± 1 nM). The iodinated derivatives 17c–17d were bound to the SPs in less than 20 minutes but were unusable for in vivo experiments due to their high binding constants (>20 nM) while 17d had excellent fluorescence staining in SPs. Radioiodination derivatives 17c [125I] and 17d [125I] showed good brain uptake/penetration (2.74% and 4.58% ID g−1 (injected dose per gram) respectively; 2 minutes after intravenous injection), clearance rates (0.40% and 0.45% ID g−1 respectively; 60 minutes) and labeling of SPs with as intense signals as those of 4-(6-iodoimidazo[1,2-a]pyridin-2-yl)-N,N-dimethylaniline (IMPY). They were also more stable in human brain homogenates (17c [125I]; 98% for 60 min) compared to those in mice (17c [125I]; 13% for 30 min).67
Modification of the same scaffold of 17a–17d, with the incorporation of a methoxy group at the 6-position and iodine at the 5-position, improved the binding affinity (Ki = 28.63 nM) of 17e, in comparison to 17c and 17d (see discussion in the previous paragraph and Fig. 7). Furthermore, 17e showed good brain uptake/penetration (5.29% ID g−1; 2 minutes after intravenous injection) which can be explained by the increase of lipophilicity due to the methoxy group and correlated with the partition coefficient value of logP 3.45. The stability of radioiodinated 17e in human AD brain homogenates was higher (17e [125I]; 98% for 60 min) compared to those in mice (17e [125I]; 9.9% for 30 min).68
The radio labelled chalcone 17f having similar substitution of N,N-dimethyl and an iodine group at the 4 and 4′-positions also showed excellent binding affinities (Kd = 4.2 ± 1.1 nM), rapid uptake (2.04% ID g−1; 2 minutes after intravenous injection) and clearance (0.40% ID g−1; 30 min) from the brain.69 Based on the same substitution pattern fluoro-polyethyleneglycol (PEG) chalcones 17g, 17h and 17i were evaluated for detection of plaques and showed binding affinities (inhibition constant; Ki = 20–50 nM), [11C] radio labelled 17i had rapid uptake (4.31% ID g−1; 2 minutes after intravenous injection) and clearance (0.35% ID g−1; 60 min) from mice brains and [18F] radio labelled chalcone 17i intensely labeled existing amyloid plaques in the brain tissues from AD patient thus having potential as positron emission computed tomography (PET) probe.70
3. Arylidene indanones as inhibitors of tubulin assembly
Microtubules are dynamic polymers, stiff hollow cylindrical structures with an outer diameter of about 25 nm and a wall approximately 5 nm thick, formed from globular protein subunits of α-tubulin and β-tubulin of about 55 kDa each that are tightly bound together by noncovalent bonds.71 They assemble/disassemble continuously in the cell filling the cytoplasm and are equipped to transmit signals throughout the cell and interact with signaling proteins and organelles. The nucleation–elongation pathway for the polymerization of αβ-tubulin dimers occurs by forming a short polymer nucleus to which subunits are added non-covalently. The association of tubulin subunits to the growing polymer is faster compared to the depolymerization and at the steady state; there is a balance between these two processes. This dynamic steady state, wherein the microtubules alternate between a period of formation and disassembly is termed dynamic instability. Chemical agents interact with microtubules promoting either disassembly or assembly, thereby blocking microtubule dynamics, modifying biological processes and signaling pathways and leading ultimately to apoptosis.71Indanocine 18 was identified as a potent microtubule-depolymerizing agent with anti-proliferative activity by the National Cancer Institute’s (NCI) Therapeutic Program (see Fig. 8).25 Indanocine 18 blocked tubulin polymerization, induced apoptosis in stationary phase multidrug resistant cancer cells and didn’t affect the viability of non-proliferating cells.25 Assessment of the in vivo antitumor activity of 18 was hampered due to poor water solubility (soluble in DMSO up to 20 mM and up to 100 μM in aqueous solutions). This solubility issue was overcome by designing the 4-phosphate analog 18a, having similar in vitro activity but reduced comparatively to 18 which is maybe due to the size of a phosphate group, low penetration of the drug in cell membranes and dephosphorylation kinetics.72
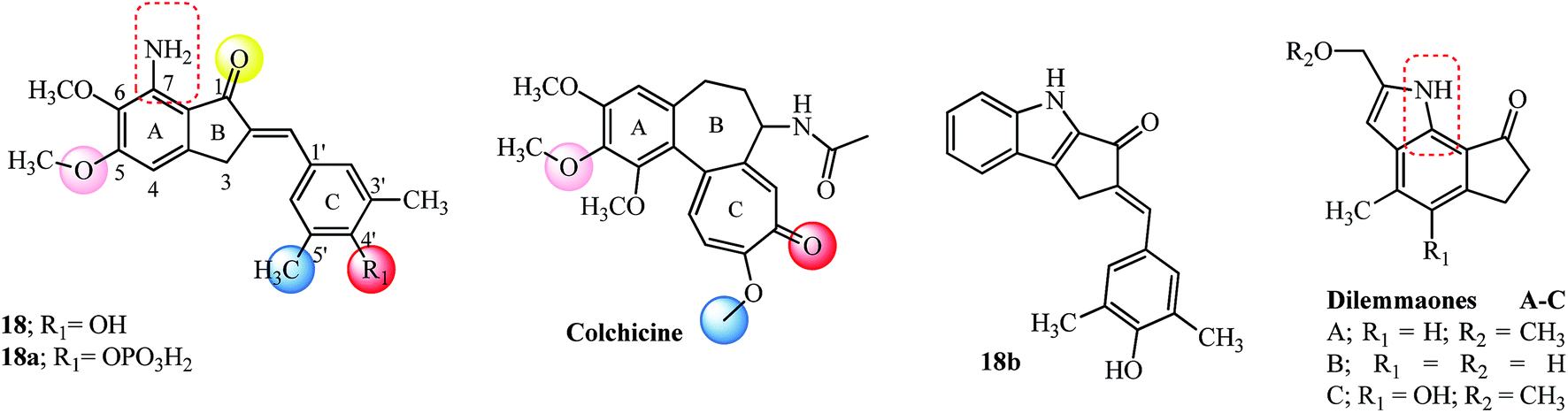 | ||
| Fig. 8 Indanocine and its derivatives, colchicine as tubulin binding agents. | ||
The importance and structure–activity relationships of the 4′-hydroxy and 3′,5′-dimethyl substitution in the arylidene moiety and amino group in the 7-position were confirmed by designing appropriate derivatives (24 examples) and using them against Jurkat cells.73 A docking based study of various agents that bind to the colchicine site on tubulin was evaluated to get a seven point structure-based pharmacophore which is required for effective binding. The red, pink and yellow spheres (hydrogen bond acceptors; see Fig. 8) and blue sphere (hydrophobic center) mark the similar type of binding sites on the colchicine molecule as well as on indanocine 18.74 The overall geometric and steric requirements of binding with the colchicine molecule were satisfied by the rigid portion of the indanocine scaffold which is caused by the hydrophobic ring A and planar ring C (18 has six out of the seven-point pharmacophores).74 These results fortified the SAR derived earlier for indanocine derivatives.73 The theoretically calculated milogP and TPSA values for 18 and its 4-phosphate derivative 18a (see Table 3) corroborate well with the observed IC50 values reinforcing the basic structural requirements. Based on these theoretical milogP calculations development of the monohydrochloride salt of 18 may have better cell penetration and bioavailability as observed for a millepachine derivative (see 22d in Fig. 11).75 At a concentration of 5 nM, 18 inhibited and strongly suppressed microtubule dynamics and resulted in concentration-dependent slow wound healing of MDA-MB-231 cells in the scratch wounds healing assay indicating a reduction in the migration of cells.76
P calculation of AI as tubulin inhibitors
| Compound | M. wt | milogP |
n-ROTB | n-ON acceptors | n-OHNH donors | n-Violations | Volume | TPSA | IC50 μM | Cell lines |
|---|---|---|---|---|---|---|---|---|---|---|
| a Non-Small Cell Lung NCI-H522.b Properties calculated with/without chloride counter ion mass gave same results.c PDT-podophyllotoxin. | ||||||||||
| 18 | 339.39 | 3.51 | 3 | 5 | 3 | 0 | 311.33 | 81.79 | 0.001 | Jurkat |
| 18a | 419.37 | 2.50 | 5 | 8 | 4 | 0 | 356.68 | 128.32 | 30 nM | HUVEC |
| 18ba | 303.36 | 4.71 | 1 | 3 | 2 | 0 | 277.93 | 53.09 | 0.093 | NCI-H522 |
| 18.HClb | 340.40 | 0.77 | 3 | 5 | 4 | 0 | 312.13 | 83.41 | — | — |
| 19 | 388.42 | 2.54 | 7 | 7 | 0 | 0 | 350.50 | 72.47 | 2.20 (1.88) | MCF-7 (tubulin) |
| 19a | 566.60 | 4.19 | 11 | 10 | 0 | 1 | 509.16 | 100.18 | 1.0 (3.04) | MCF-7 (tubulin) |
| 19b | 520.23 | 4.45 | 8 | 9 | 0 | 1 | 456.45 | 90.94 | 10 nM (0.63) | MCF-7 (tubulin) |
| 19c | 494.51 | 4.73 | 8 | 7 | 0 | 0 | 437.45 | 72.47 | 0.68 (0.73) | MCF-7 (tubulin) |
| 19d | 521.52 | 4.52 | 9 | 10 | 0 | 1 | 455.86 | 118.30 | 4.0 (1.54) | MCF-7 (tubulin) |
| PDTc | 412.44 | 2.31 | 4 | 7 | 1 | 0 | 362.25 | 83.47 | 3.5 (0.74) | MCF-7 (tubulin) |
The incorporation of indole motif as bioisostere for the dimethoxyaniline group of 18 provided with indole indanone derivative called indolocine 18b (Fig. 8), among 8 other derivatives studied as tubulin interacting agents.77 Indolocine 18b tested positive against the Non-Small Cell Lung NCI-H522 cell line in a nanomolar concentration. Retention of the 4′-hydroxy and 3′,5′-dimethyl substitution in the arylidene moiety of the new indolocine derivatives indicated that most derivatives were equipotent, and complemented the previous SAR73 as well as a pharmacophoric model.74 A striking resemblance to the 7-amine substitution from 18 is seen by incorporation of natural motifs like indole into the indanone structure like dilemmaones A–C (see red dashed box on structures) from South African sponge Ectyonanchora flabellate.78
3.1 Potential of tri-methoxy substitution on the indanone core for inhibiting cancer cell growth via tubulin interaction
The tri-methoxy benzene substituted moiety is common to several important tubulin binding natural molecules like colchicine, combretastatins-A4 and A-1 and podophyllotoxin (see Fig. 8 and 9), forming the hydrophobic center and interacting with the major binding site on tubulin.74 The red and pink spheres on combretastatin A-4 and pink and yellow spheres on podophyllotoxin are hydrogen bond acceptors (Fig. 9) while the blue sphere (hydrophobic center) marks the similar type of binding sites seen on the colchicine molecule in tubulin by pharmacophore modeling.74 The incorporation of the 3,4,5-trimethoxyphenyl group found in ring A of colchicine into the ring A of indanone 19 and modification at the 3-position with another aryl ring having a tri-methoxy substitution pattern (see the E-ring in podophyllotoxin; Fig. 9) imparts anticancer properties to the indanone 19 scaffold against breast cancer cells (IC50 = 2.20 μM).79,80 The introduction of the arylidene moiety resulted in AIs of the type 19a–19c among the 15 derivatives tested against human cancer cell lines.81 Compounds 19a, 19b, and 19c were superior to the parent indanone, indicating an improvement in activity on the introduction of the arylidene moiety. Indanone 19b inhibited tubulin polymerization in the same range when compared to podophyllotoxin (see Table 3 for comparison of IC50 values). Further in vivo acute oral toxicity tests in Swiss albino mice indicated 1000 mg kg−1 body weight as a single acute oral dose.81 Indanone 19b showed antiproliferative activity against the ER + breast cancer cell line (MCF-7, IC50 = 0.010 μM) and didn’t allow the cells to form colonies in a concentration-dependent manner. The in vivo oral toxicity test using 19b in Swiss albino mice indicated a 100 mg kg−1 body weight dose once in 28 days as a single acute dose and a 300 mg kg−1 body weight dose as a single dose.82,83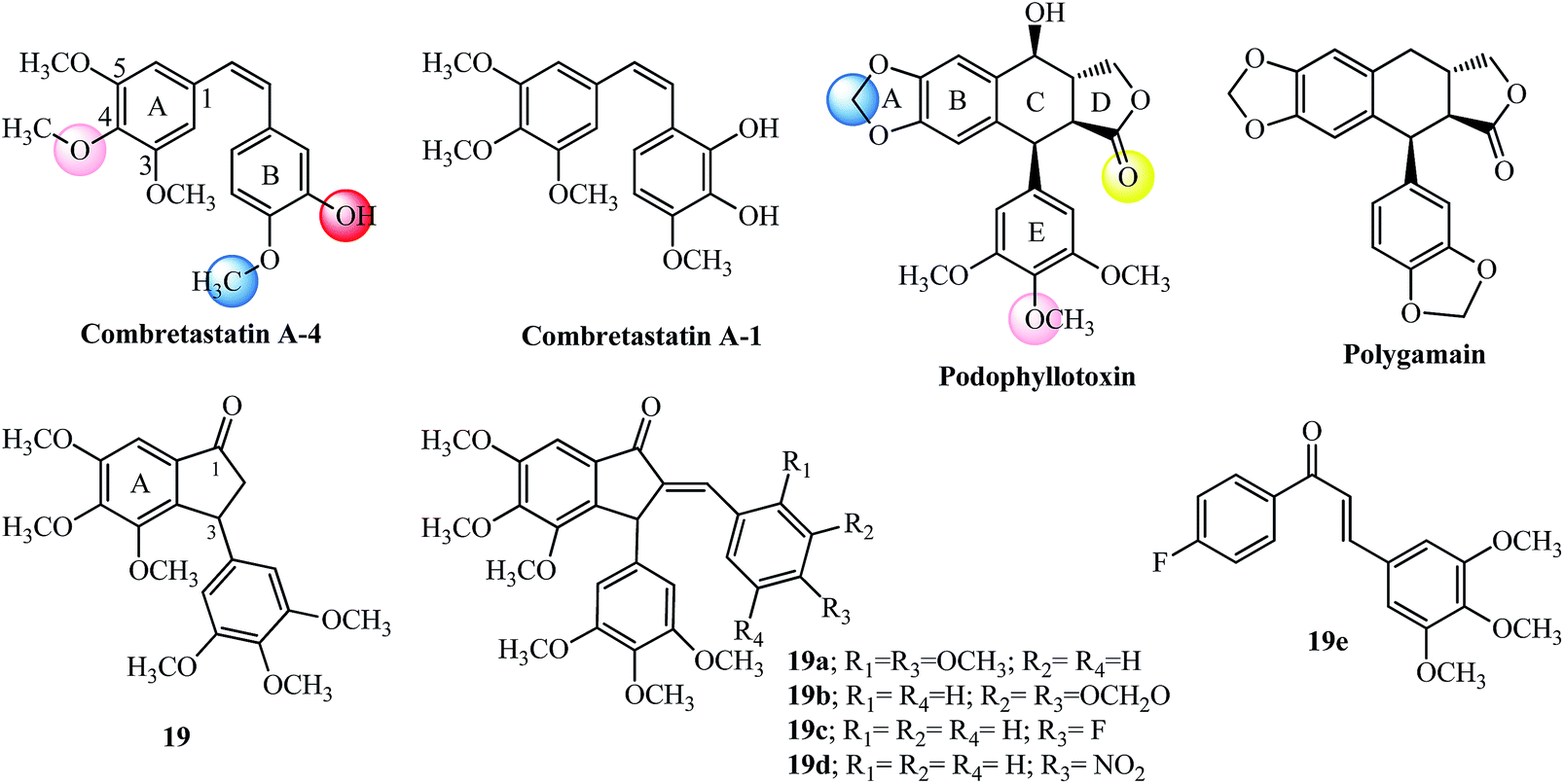 | ||
| Fig. 9 Natural anti-tubulin agents and AI designed based on tri-methoxy substitution. | ||
Structurally the methylenedioxy group is an important moiety for cancer drug design which may interact favorably with the molecular targets in cancer cells and function as a prodrug by the action of cytochrome p450 enzymes.84–86 The 3,4-methylenedioxyphenyl moiety in polygamain (see Fig. 9), a natural product isolated from Amyris madrensis shares the same space occupied by the tri-methoxy benzene moiety found in podophyllotoxin and shows a hydrophobic interaction within the colchicine-binding site on tubulin.87 The presence of a fluorine atom in compound 19c shows better experimental potency in both tests, and plays a critical role in the design of several anti-cancer agents.88 Chalcone 19e having a similar substitution pattern with respect to the positioning of the fluorine and tri-methoxy groups had the highest potency (30 nM) towards inhibition of NO production in RAW macrophages.89
Further development of the indanone scaffold (7 examples) with the introduction of the nitro group in the para-position gave 19d which has potent activity in several cancer cell lines and was tolerated up to 300 mg kg−1 doses in acute oral toxicity experiments.90 In spite of 19d having an almost similar molar mass and calculated theoretical volume as 19b, it violates Lipinski rule but was found to effectively cross the blood–brain barrier due to its lipophilic nature (milogP-4.52; Table 3).90
Recently AI incorporating methoxy groups in the indanone core similar to the colchicine A-ring were evaluated in several human cancer cell lines as antitumor agents (18 examples). The studies showed that AI with 4,5,6-trimethoxyl substitution similar to the colchicine A-ring (20a–20b; Fig. 10) was more potent compared to the isomeric 5,6,7-trimethoxyl, 5-methoxy-6-hydroxy and 5,6-dimethoxy indanone moieties tested.91 The synthetic aurone derivative 20c (an oxygen analog of active indanone 20a; Fig. 10) gave similar results against the K562 human chronic myelogenous leukemia cell line.92 It is worthwhile pointing out that the most active indanone derivative 20a shares the same tri-methoxy and isovanillin type substitution pattern as synthetic aurone and chalcone SD400 (ref. 92 and 93) (see Fig. 10 for structures and a comparison of the colored spheres with the binding sites seen on the colchicine molecule in tubulin by pharmacophore modelling74 in Fig. 8). Several tri-methoxy substituted chalcones among others with diverse substitution patterns and the ability to inhibit tubulin have been reviewed.10
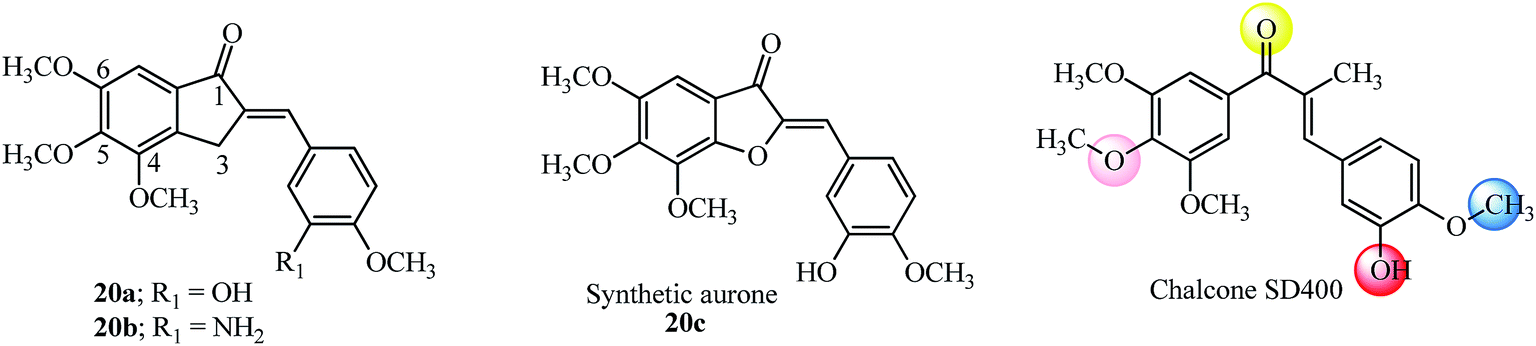 | ||
| Fig. 10 Anti-proliferative AI, aurone and chalcone SD400 based on colchicine tri methoxyl substitution. | ||
Incorporating medicinally important scaffolds like indoles94 with 4,5,6-trimethoxy indanones gave hybrid compounds with promising anti-cancer activity (14 examples). The linkage with the indole moiety was varied between the 3, 4 and 5 positions. The potent compounds (21a–21b and 22a–22b; Fig. 11) gave promising results in several cancer cell lines and inhibited tubulin with no influence of the position of contact with the indole moiety.95 Similarly, millepachine-indolyl chalcone 22c with substitution at the 5-position of N-methyl indole was the most potent among others and showed cytotoxicity against all tested cancer cells.96 The bioavailability of millepachine derivative 22d was improved by 47% after forming its hydrochloride salt.75 This rationale may also increase the bioavailability of derivative 20b which is a potent anti-tubulin agent, equipotent with 20a (see Fig. 10) and has structural similarity with the aryl ring in 22d.
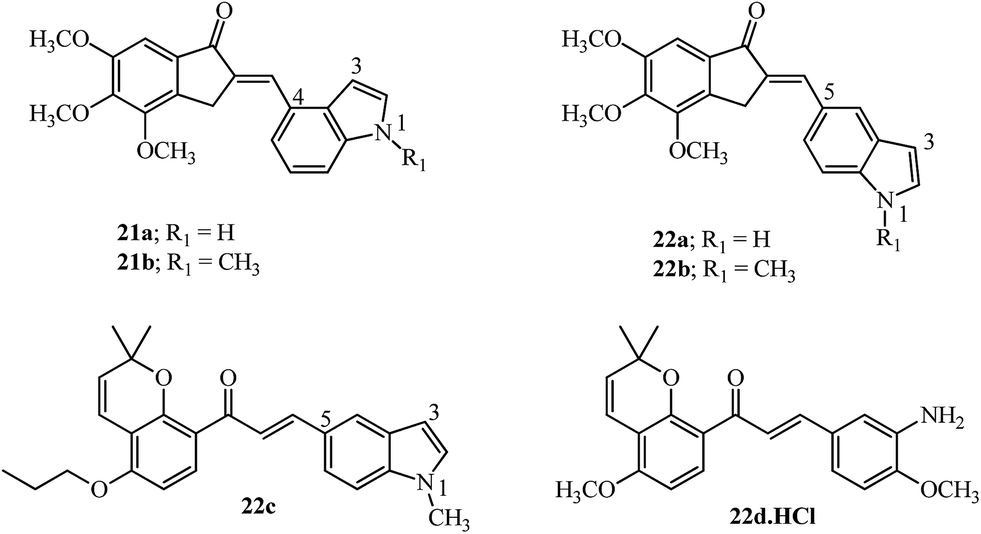 | ||
| Fig. 11 Indolyl-indanones, indolyl-millepachine hybrid, and millepachine hydrochloride. | ||
Several chalcone derivatives attached to the 3-position of the indole ring have shown induction of methuosis, a non-apoptotic cell death,97 use as microtubule depolymerizing agents98,99 and have anti-inflammatory activities100 while when connected to the 5-position of indole ring function as imaging agents by competitively binding to the Aβ fibers for detection of β-amyloid plaques in the brains of AD patients.101
4. Therapeutic potential of AI against breast cancer
Aromatase is a cytochrome P450 enzyme, which functions as a rate-limiting enzyme in the conversion of androgens to estrogens. Nonsteroidal aromatase inhibitors of the type; 2-(4-pyridylmethylene)-1-indanone 23 (Fig. 12) showed selectivity and increased potency toward aromatase as compared to aminoglutethimide.102,103 On similar lines, the relative potency of imidazolyl substituted derivative 24 against aromatase inhibition was 54 times more potent than aminoglutethimide (4 examples) and fared better in comparison with indanone 23. This potency may be attributed to the orientation of the side chain which influences the binding at the active site of aromatase.104 2-Benzylidene-4,7-dimethyl indan-1-one 25 showed inhibition against the MCF-7 cell line and was non-toxic to the normal breast cell line HBL-100 up to 100 μM.105 The 2-arylidene-1-indanones 25 and 26 (Fig. 12) were evaluated in MCF-7 cells showing superior activity (7.3 μM) in a dose-dependent manner.106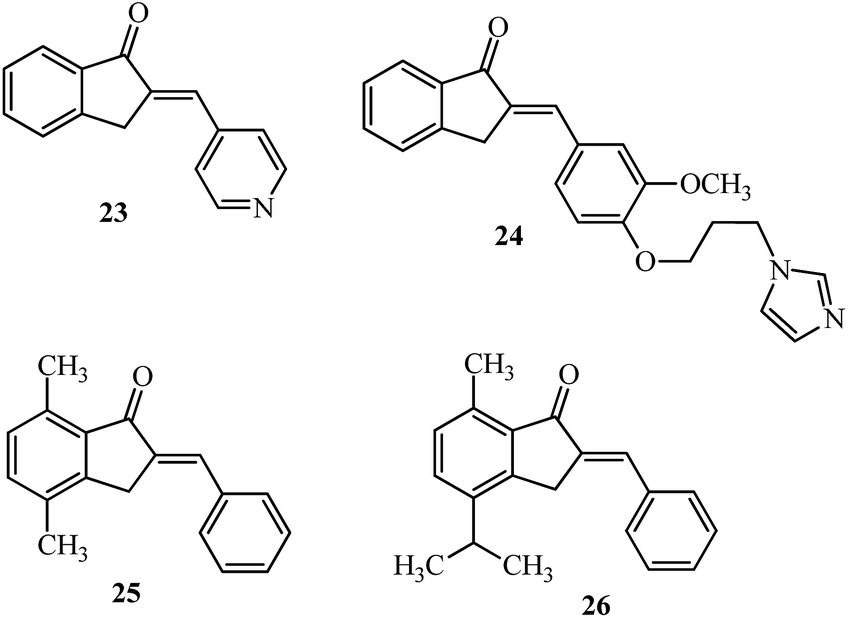 | ||
| Fig. 12 AI and their derivatives inhibiting aromatase, MCF-7 cell growth and lung cancer. | ||
5. Arylidene indanones in treatment of lung cancer
Akt activation and phosphorylation is the key to the survival of cancer cells and has been linked to lung cancer resistant to chemotherapy. The over-expression of Akt provides resistance to chemotherapy and makes it an interesting therapeutic tool for developing anti-cancer agents. Indan-1-one 25 inhibited Akt enzyme activity by 0.20 μM and did not alter the IC50 value on increasing the ATP concentration, indicating allosteric binding of the molecule to the enzyme. Indan-1-one 25 reduced cell proliferation of non-small cell lung cancer cell lines, NCI-H 460 and A549 cells with a GI50 of 0.28 μM and 0.77 μM via the caspase-activated apoptosis pathway.1076. Arylidene indanones in treatment of leukemia
The therapeutic effects of natural and synthetic flavonoids including chalcones with respect to structure activity relationships on various forms of leukemia has been recently reviewed.108 AIs being the rigid cousins of chalcones also play important roles in the therapy of leukemia. Indanocine 18 (Fig. 8) a potent tubulin binding agent, induces apoptosis in malignant B cells from patients with chronic lymphocytic leukemia (CLL) but remains ineffective in resistant CEM human lymphoblastoid cells due to a single point mutation of the nucleotide close to the colchicine binding site of tubulin.26 The combination of an equimolar concentration of 18, an anti-tubulin agent and mizoribine, a prodrug, synergistically led to a decrease in the viability of chronic lymphocytic leukemia (CLL) cells.1097. Arylidene indanones as dual specificity phosphatase (DUSP) inhibitors
Dual specificity phosphatases (DUSP) are enzymes that dephosphorylate and inactivate mitogen-activated protein kinases (MAPKs). DUSP6, also known as mitogen-activated protein kinase phosphatase 3 (MKP3), functions as a feedback regulator of fibroblast growth factor (FGF) signaling to limit the activity of extracellular signal-regulated kinase (ERK) 1 and 2. The catalytic activity of members of the DUSP family like DUSP6 and DUSP1 is substrate specific which on interaction with ERK is enhanced, while DUSP5 is active and independent of substrate binding.28 FGFs play crucial roles in development, proliferation, and cellular homeostasis.28 These FGFs also bind to tyrosine kinase receptor proteins to activate the signaling of the rat sarcoma homolog RAS/MAPK cascade among others.110BCI 27 was identified as a specific inhibitor of DUSP6 and DUSP1 but not of DUSP5 using a zebrafish chemical screen which is an animal model well suited for small molecule screening (Fig. 13).28,29 BCI 27 at a concentration of 10 μM, functions as an inhibitor of DUSP6 with increased expression of FGF in treated embryos. Furthermore, using analogs 27a and 27b it was shown that cyclohexylamine, as well as the benzylidene moiety, plays an important role in the inhibition (compare EC50 values in Table 4).28
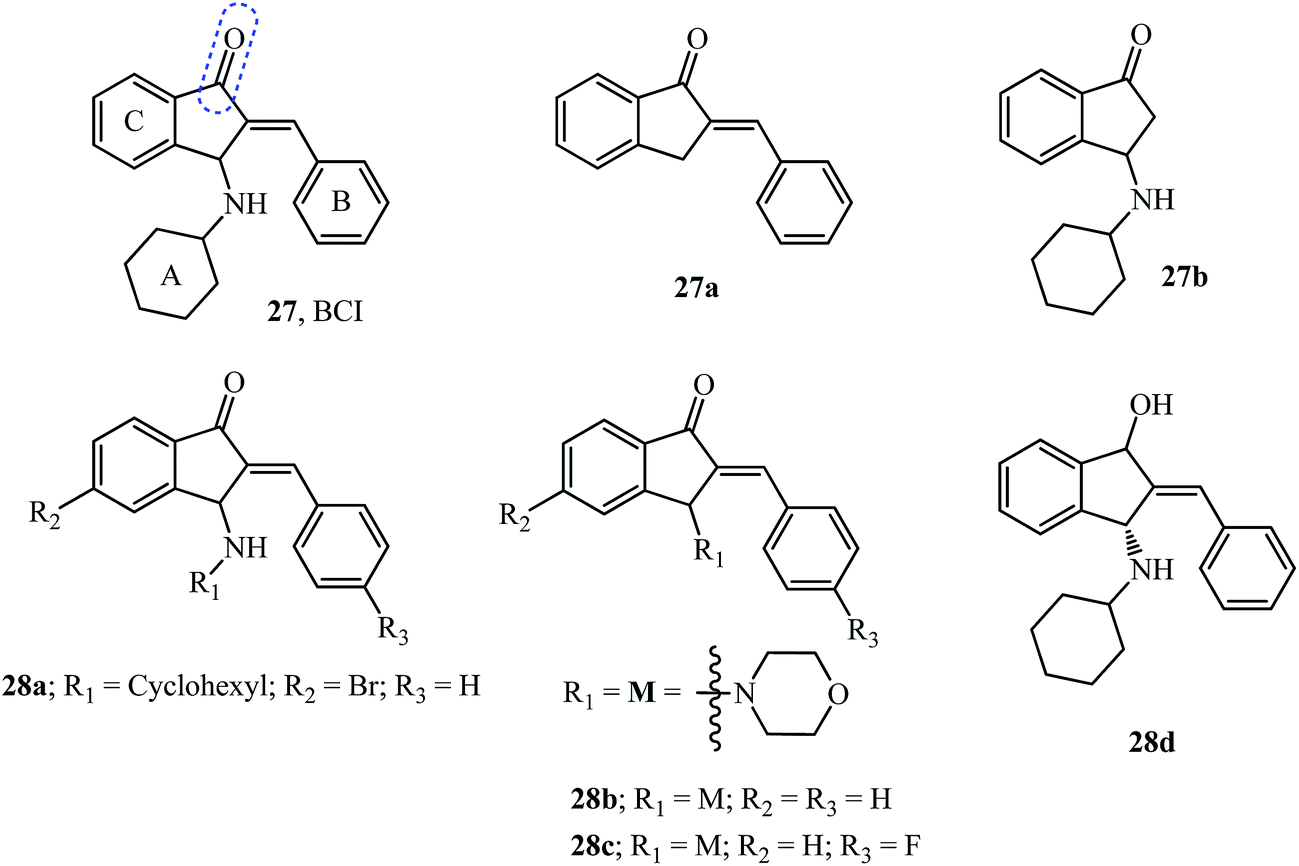 | ||
| Fig. 13 BCI and its analogs tested as DUSP inhibitors. | ||
P calculation of AI tested as DUSP inhibitors
| Compound | Mol. weight | milogP |
n-ROTB | n-ON acceptors | n-OHNH donors | n-Violations | Volume | TPSA | EC50 μMa | IC50 of DUSP6 inhibition | IC50 of DUSP1 inhibition |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a EC50-concentration required for FGF activation in zebrafish assay; ND-not determined. | |||||||||||
| 27 | 317.43 | 5.21 | 3 | 2 | 1 | 1 | 310.24 | 29.10 | 10.6 ± 0.8 | 13.3 ± 1.8 | 8.0 ± 0.6 |
| 27a | 220.27 | 3.74 | 1 | 1 | 0 | 0 | 207.81 | 17.07 | Inactive | ND | ND |
| 27b | 229.32 | 3.19 | 2 | 2 | 1 | 0 | 228.81 | 29.10 | Inactive | ND | ND |
| 28a | 396.33 | 6.00 | 3 | 2 | 1 | 1 | 328.12 | 29.10 | 12.0 ± 3.0 | 55.9 ± 8.6 | 28.6 ± 3.8 |
| 28b | 305.38 | 3.40 | 2 | 3 | 0 | 0 | 285.97 | 29.54 | 4.5 ± 0.6 | 50.8 ± 4.1 | 28.4 ± 1.8 |
| 28c | 323.37 | 3.56 | 2 | 3 | 0 | 0 | 290.90 | 29.54 | 10.3 | ND | ND |
| 28d | 319.45 | 4.88 | 3 | 2 | 2 | 0 | 316.10 | 32.26 | ≫25 | ND | ND |
A total of 11 compounds (from 29 examples) effectively activated FGF signaling in a concentration-dependent manner in Tg(dusp6:EGFP)pt6 embryos with most of them being as potent as the starting pharmacophore 27.29 Toxicity studies of the compounds indicated BCI analog 28a showed no toxicity even at twice the EC50 (20 μM) required for activation of FGF signaling indicating an attractive and clean molecule to investigate DUSPs.29 The theoretical milogP calculations (Table 4) provide results which would disqualify the active molecules 27 and 28a but substantiate the result for 28b, which follows the Lipinski “rule of five” with no violations. However, it is not a necessity to qualify the “rule of five” to have drug-likeness.111
The study revealed the more potent derivative 28b due to the introduction of a heteroatom in the ring at the 3-position of indanone and the fluoro substituent in 28c compared to 27.29 The importance of the carbonyl functionality was assayed by testing its reduced form as hydroxyl derivative 28d (Fig. 12), which showed no activity.29
The selective DUSP6 inhibitor, indanone 27, induced cell death in patient-derived pre-B acute lymphoblastic leukemia (ALL) cells and also overcame mechanisms of drug-resistance. Treatment of patient-derived ALL cells led to hyperactivation of Erk and compromised the negative control of Erk signaling causing cell death.112
8. Quinolinylmethylidene indanones as antimalarial agents
Malaria is an infectious disease which causes deaths worldwide and is caused by transmission of Plasmodium strains by mosquito bites. Natural chalcones, like licochalcone A isolated from Chinese licorice roots protected mice infected with P. yoelii for 3 to 6 days.113 The quinoline moiety is important in drugs like chloroquine used for the treatment of malaria infections.114 Hybrids resulting from substituted indanones and quinolines led to indanones 29a and 29b (Fig. 14) as potent antimalarial agents (13 examples).30 SAR studies indicated that monosubstitution in the quinoline ring at position 8 were effective inhibitors of haem polymerization (29a and 29b). The in vivo tests using mice infected with Plasmodium berghei ANKA, showed survival up to 12 days after infection by treatment with indanone 29b (at a concentration of 20 mg kg−1 intraperitoneal once daily for 4 days post infection).30 | ||
| Fig. 14 Quinolinylmethylidene indanones and licochalcone A as antimalarial agents. | ||
The SAR was carried out by substitution in the indanone ring at positions 5 and 7 by a methoxy group as well as positions 6, 7 and 8 of the quinoline moiety (10 examples). In vivo tests in mice infected with Plasmodium berghei, showed a survival rate of 11 days for indanones 30a and 30b (at concentration of 20 mg kg−1 intraperitoneal once daily for 4 days post infection).31
9. Arylidene indanone as an aryl hydrocarbon receptor antagonist
Recently, the simple E/Z-2-benzylindene-5,6-dimethoxy-3,3-dimethylindan-1-one, 31 (Fig. 15) was shown to transiently act as an aryl hydrocarbon receptor (AhR) antagonist and selectively inhibit the UVB-induced expression of the CYP1A1 gene in normal human epidermal keratinocytes (NHEK). Results of the topical application of ointment containing 0.5% 31 protected human skin from UV-B radiation by reduced expression of CYP1A1, COX-2 and MMP-1 genes.115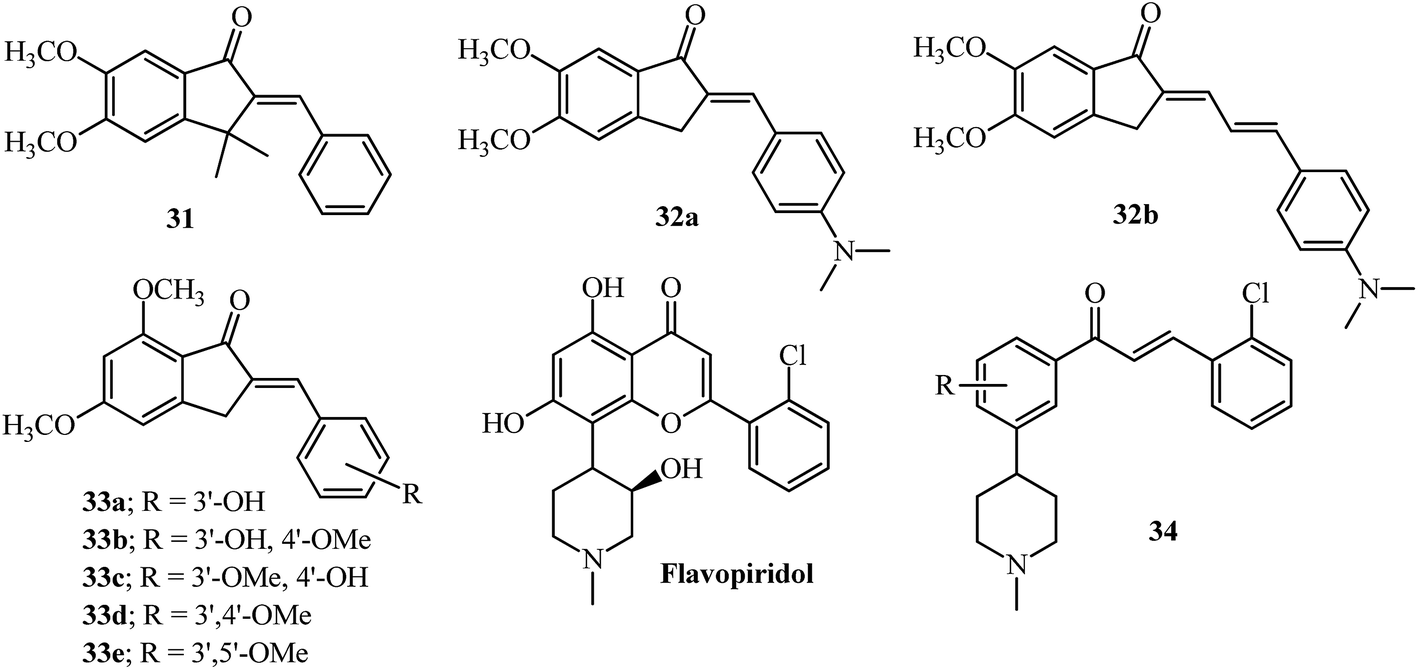 | ||
| Fig. 15 Aryl hydrocarbon Receptor (AhR) antagonist, p-glycoprotein and ABCG2 inhibitors based on AI; flavopiridol and its chalcone derivative as inhibitors of cell cycle progression. | ||
10. Bioavailability and SAR considerations
Although no bioavailability studies are reported for the AIs mentioned in this review, a concise description of the analysis, degradation and metabolism of acetylcholinesterase inhibitors i.e., donepezil, the most structurally related molecule to the AI scaffold has been reviewed.116 The current status of clinical trials using disease modifying drugs in AD has also been reviewed.117 The increased lipophilicity of 6-substituted derivatives 12b, 13a–13c which were equipotent to 6 (Table 1) may improve their bioavailability by delaying metabolic demethylation.116Bioavailability of chalcone and arylidene indanone was shown to be dependent on the formation of ionized species of molecules (pKa) which have hydroxyl or N,N-dimethyl amino groups, which may inhibit their passive diffusion across cell membranes.118 The dimethylamino containing derivatives remain uncharged at pH 7.4; therefore, they may easily pass through the cellular membrane while the hydroxyl group may get ionized with the negative charge which may inhibit their diffusion. Inclusion complexes with cyclodextrins have shown great potential for drug delivery systems by increasing their bioavailability.119,120
Although, the 4′-phosphate derivative of indanocine showed reduced cytotoxicity compared to 18,72 the phosphate derivatives of combretastatin-like chalcones have functioned as prodrugs and were equipotent to the parent molecules with respect to cell proliferation. While the same compounds were inactive for tubulin inhibition probably due to the in vitro assay not being cell-based and no hydrolysis by phosphatases can occur.93 Several combretastatin analogs like A-1 and A-4 phosphates are in various stages of clinical trials.121 The ethoxy derivative had a better half-life compared to its methoxy derivative and also improved the potency by delaying possible metabolic inactivation.122 The lipophilic acetate derivative of the natural flavonoid quercetin increased uptake in cells and delayed metabolism.123
Inhibition of P-glycoprotein (P-gp), the drug efflux transporter in cancer cells can help increase the bioavailability of anti-cancer drugs. Natural flavonoids have been shown to be inhibitors of P-gp and have been co-administered with anti-cancer drugs.108 Indanone derivatives like 32a and 32b have shown inhibition activity in the nanomolar range towards efflux of daunomycin using the CCRF vcr1000 cell line (Fig. 15).124 The SAR provides insights into the higher activity of 32b based on the presence of one more rotatable bond than 32a while the remaining structure is identical.124 Indanones 33a–33e have shown modulation of the ABCG2 transporter which is responsible for the efflux of a wide variety of drugs resulting in drug resistance.125,126 The resulting SAR indicated the presence of the dimethoxylated A-ring, adjacent to the carbonyl group and the exocyclic double bond to be key features for such activity. The activity was also dependent on the mono or disubstituted aryl ring having hydroxyl or methoxyl groups (33b–33e).125
The majority of publications discussed in this review have a substitution at the para position of the arylidene ring, with ortho positions effectively not explored. The substitution of an ortho position, in comparison to the para isomer, can be differentiated by ESI-MS in AI as well as chalcones.23,127 Moreover the o-substituted chalcone derivative has higher potency compared to p-substituted when the other structural substitutions are maintained.84,128 The ring B of flavopiridol and combretastatin A-1 are classical examples of ortho substituents influencing the potential of these scaffolds as therapeutic drugs.121 The potency of combretastatin derivatives having a 2-amino substitution in the B-ring was improved as inhibitors of tubulin assembly, cytotoxicity and in vivo reduction of blood flow to tumors.129
Incorporation of the N-methyl-piperidinyl alkaloid moiety found in flavopiridol without the hydroxyl group in chalcones 34 has shown anti-cancer properties by inhibiting cell cycle progression11,130 and can be explored on the AI scaffold. Incorporation of indole or quinoline moieties with indanone may be beneficial in the design of new antimicrobial or antimalarial agents,114 which may become more effective for resistant strains of bacteria on quaternization of nitrogen and improved solubility in the physiological medium.131
11. Conclusions
Arylidene indanones tested as acetylcholinesterase inhibitors have shown that the 6-position of the indanone core is important for interactions with the PAS of the enzyme45 and subsequent modifications improve the efficiency of interaction.45,49,51 Chelation of metals like copper is critical for Alzheimer’s therapy and chelating groups like piperidinyl ethoxy mimicking clioquinol in 12b or the catechol moiety in 16a serve as an important aspect for new drug design.27,49 Incorporation of bulky groups like bis(4-fluorophenyl)methyl on piperazine either on the indanone side 11c or arylidene ring 14a can modulate either as a BACE1 inhibitor or acetylcholinesterase inhibitor.46,48 Tubulin depolymerization agents require the tri-methoxy substitution on the indanone ring to mimic the colchicine type activity.74,80,81,91,95Tunability of the α,β-unsaturated ketone system as a Michael acceptor is paramount to the type of biological activity,132 since these systems are present in many natural products like chalcones as well as AI, where the molecule is more planar comparatively and the transmission of electronic effects of the aryl substituents are directed towards the carbonyl group.13 The importance of the double bond at the 2-position in arylidene indanone has been verified by SAR studies, indicating no DUSP inhibition activity if removed28 or improved tubulin activity compared to unsubstituted compounds79,81 or diminished AChE activity if removed or hydrogenated.49,52 Similarly increased MAO-B inhibition was seen for benzylidene substituted 5-hydroxyindanones.61,63
The chalcones have been merited with a lot of attention by structural modification of the position of the hydroxyl, methoxyl and other substitutions such as incorporation of heteroaryl moieties like coumarins, naphthyl, furan etc. displaying diverse activities.10–12 Similarly, in-depth studies of novel arylidene indanone scaffolds by pharmacophore modeling may give better insights into the unexplored potential of this scaffold.
Acknowledgements
The University of Aveiro, Fundação para a Ciência e a Tecnologia (FCT, Portugal), European Union, QREN, FEDER and COMPETE for funding the QOPNA research unit (project PEst-C/QUI/UI0062/2013). The author thanks QOPNA for a research grant and Dr Amparo Faustino, Department of Chemistry, University of Aveiro, J M G Pereira, Aveiro Portugal and Dr Marc Diederich, Seoul National University, South Korea for scientific access support.References
- A. I. Syrchina and A. A. Semenov, Chem. Nat. Compd., 1983, 18, 1–11 CrossRef.
- N. V. Kovganko, Z. N. Kashkan and S. N. Krivenok, Chem. Nat. Compd., 2004, 40, 227–229 CrossRef CAS.
- X. Ge, G. Ye, P. Li, W.-J. Tang, J.-L. Gao and W.-M. Zhao, J. Nat. Prod., 2008, 71, 227–231 CrossRef CAS PubMed.
- B. Jaki, J. Heilmann and O. Sticher, J. Nat. Prod., 2000, 63, 1283–1285 CrossRef CAS.
- D. G. Nagle, Y.-D. Zhou, P. U. Park, V. J. Paul, I. Rajbhandari, C. J. G. Duncan and D. S. Pasco, J. Nat. Prod., 2000, 63, 1431–1433 CrossRef CAS.
- H. Sheridan, N. Frankish and R. Farrell, Eur. J. Med. Chem., 1999, 34, 953–966 CrossRef CAS PubMed.
- H. O. Saxena, U. Faridi, J. K. Kumar, S. Luqman, M. P. Darokar, K. Shanker, C. S. Chanotiya, M. M. Gupta and A. S. Negi, Steroids, 2007, 72, 892–900 CrossRef CAS PubMed.
- Z. Nowakowska, Eur. J. Med. Chem., 2007, 42, 125–137 CrossRef CAS PubMed.
- M. Das and K. Manna, J. Toxicol., 2016, 2016, 7651047 Search PubMed.
- B. Zhou and C. Xing, Med. Chem., 2015, 5, 388–404 Search PubMed.
- A. Boumendjel, X. Ronot and J. Boutonnat, Curr. Drug Targets, 2009, 10, 363–371 CrossRef PubMed.
- P. Singh, A. Anand and V. Kumar, Eur. J. Med. Chem., 2014, 85, 758–777 CrossRef CAS PubMed.
- P. Perjési, A. Perjéssy, E. Kolehmainen, E. Ősz, M. Samaliková, J. Linnanto and E. Virtanen, J. Mol. Struct., 2004, 697, 41–47 CrossRef.
- P. Perjési, J. Linnanto, E. Kolehmainen, E. Ősz and E. Virtanen, J. Mol. Struct., 2005, 740, 81–89 CrossRef.
- K. Fodor, V. Tomescova, T. Kőszegi, I. Kron and P. Perjési, Monatsh. Chem., 2011, 142, 463–468 CrossRef CAS.
- P. Perjési, Monatsh. Chem., 2015, 146, 1275–1281 CrossRef.
- S. Ducki, Anti-Cancer Agents Med. Chem., 2009, 9, 336–347 CrossRef CAS PubMed.
- N. J. Lawrence, R. P. Patterson, L.-L. Ooi, D. Cook and S. Ducki, Bioorg. Med. Chem. Lett., 2006, 16, 5844–5848 CrossRef CAS PubMed.
- S. P. Kamat, J. C. Menezes, B. M. Siddhaye and S. K. Paknikar, Indian J. Chem., 2008, 47B, 1597–1599 CAS.
- V. Premasagar, V. A. Palaniswamy and E. J. Eisenbraun, J. Org. Chem., 1981, 46, 2974–2976 CrossRef CAS.
- M. Oliverio, M. Nardi, P. Costanzo, L. Cariati, G. Cravotto, S. V. Giofrè and A. Procopio, Molecules, 2014, 19, 5599–5610 CrossRef PubMed.
- A. Lévai, ARKIVOC, 2004, 15–33 Search PubMed.
- J. C. J. M. D. S. Menezes, J. A. S. Cavaleiro and M. R. M. Domingues, Rapid Commun. Mass Spectrom., 2013, 27, 2461–2471 CrossRef CAS PubMed.
- R. Mondal, T. K. Mandal and A. K. Mallik, Org. Chem. Int., 2012, 456097 Search PubMed.
- L. M. Leoni, E. Hamel, D. Genini, H. Shih, C. J. Carrera, H. B. Cottam and D. A. Carson, J. Natl. Cancer Inst., 2000, 92, 217–224 CrossRef CAS PubMed.
- X. H. Hua, D. Genini, R. Gussio, R. Tawatao, H. Shih, T. J. Kipps, D. A. Carson and L. M. Leoni, Cancer Res., 2001, 61, 7248–7254 CAS.
- L. Huang, C. Lu, Y. Sun, F. Mao, Z. Luo, T. Su, H. Jiang, W. Shan and X. Li, J. Med. Chem., 2012, 55, 8483–8492 CrossRef CAS PubMed.
- G. Molina, A. Vogt, A. Bakan, W. Dai, P. Q. D. Oliveira, W. Znosko, T. E. Smithgall, I. Bahar, J. S. Lazo, B. W. Day and M. Tsang, Nat. Chem. Biol., 2009, 5, 680–687 CrossRef CAS PubMed and references cited therein.
- V. N. Korotchenko, M. Saydmohammed, L. L. Vollmer, A. Bakan, K. Sheetz, K. T. Debiec, K. A. Greene, C. S. Agliori, I. Bahar, B. W. Day, A. Vogt and M. Tsang, ChemBioChem, 2014, 15, 1436–1445 CrossRef CAS PubMed.
- J. E. Charris, J. N. Domínguez, N. Gamboa, J. R. Rodrigues and J. E. Angel, Eur. J. Med. Chem., 2005, 40, 875–881 CrossRef CAS PubMed.
- J. E. Charris, G. M. Lobo, J. Camacho, R. Ferrer, A. Barazarte, J. N. Domínguez, N. Gamboa, J. R. Rodrigues and J. E. Angel, Lett. Drug Des. Discovery, 2007, 4, 49–54 CrossRef CAS.
- http://www.molinspiration.com.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- H. W. Querfurth and F. M. LaFerla, N. Engl. J. Med., 2010, 362, 329–344 CrossRef CAS PubMed.
- A. Kumar, A. Singh and Ekavali, Pharmacol. Rep., 2015, 67, 195–203 CrossRef CAS PubMed.
- E. Giacobini, Pharmacol. Res., 2004, 50, 433–440 CrossRef CAS PubMed.
- M. Singh, M. Kaur, H. Kukreja, R. Chugh, O. Silakari and D. Singh, Eur. J. Med. Chem., 2013, 70, 165–188 CrossRef CAS PubMed and references cited therein.
- Y. Kawakami, A. Inoue, T. Kawai, M. Wakita, H. Sugimoto and A. J. Hopfinger, Bioorg. Med. Chem., 1996, 4, 1429–1446 CrossRef CAS PubMed.
- Current Alzheimer’s Treatments, http://www.alz.org/research/science/alzheimers_disease_treatments.asp, accessed 13 October, 2015.
- P. Camps, X. Formosa, C. Galdeano, T. Gómez, D. Munoz-Torrero, M. Scarpellini, E. Viayna, A. Badia, M. V. Clos, A. Camins, M. Pallas, M. Bartolini, F. Mancini, V. Andrisano, J. Estelrich, M. Lizondo, A. Bidon-Chanal and F. J. Luque, J. Med. Chem., 2008, 51, 3588–3598 CrossRef CAS PubMed.
- P. Camps, X. Formosa, C. Galdeano, T. Gómez, D. Munoz-Torrero, L. Ramírez, E. Viayna, E. Gómez, N. Isambert, R. Lavilla, A. Badia, M. V. Clos, M. Bartolini, F. Mancini, V. Andrisano, A. Bidon-Chanal, Ó. Huertas, T. Dafni and F. J. Luque, Chem.-Biol. Interact., 2010, 187, 411–415 CrossRef CAS PubMed.
- K. S. T. Dias and C. Viegas-Junior, Curr. Neuropharmacol., 2014, 12, 239–255 CrossRef PubMed.
- N. Guzior, A. Więckowska, D. Panek and B. Malawska, Curr. Med. Chem., 2015, 22, 373–404 CrossRef CAS PubMed.
- F. Mao, J. Yan, J. Li, X. Jia, H. Miao, Y. Sun, L. Huang and X. Li, Org. Biomol. Chem., 2014, 12, 5936–5944 CAS.
- S. Rizzo, M. Bartolini, L. Ceccarini, L. Piazzi, S. Gobbi, A. Cavalli, M. Recanatini, V. Andrisano and A. Rampa, Bioorg. Med. Chem., 2010, 18, 1749–1760 CrossRef CAS PubMed.
- A. Rampa, F. Mancini, A. D. Simone, F. Falchi, F. Belluti, R. M. C. D. Martino, S. Gobbi, V. Andrisano, A. Tarozzi, M. Bartolini, A. Cavalli and A. Bisi, Bioorg. Med. Chem. Lett., 2015, 25, 2804–2808 CrossRef CAS PubMed.
- H.-R. Liu, C. Zhou, H.-Q. Fan, J.-J. Tang, L.-B. Liu, X.-H. Gao, Q.-A. Wang and W.-K. Liu, Chem. Biol. Drug Des., 2015, 86, 517–522 CAS.
- C. B. Mishra, S. Kumari, A. Manral, A. Prakash, V. Saini, A. M. Lynn and M. Tiwari, Eur. J. Med. Chem., 2017, 125, 736–750 CrossRef CAS PubMed.
- F.-C. Meng, F. Mao, W.-J. Shan, F. Qin, L. Huang and X.-S. Li, Bioorg. Med. Chem. Lett., 2012, 22, 4462–4466 CrossRef CAS PubMed.
- B. N. Sağlık, S. Ilgın and Y. Özkay, Eur. J. Med. Chem., 2016, 124, 1026–1040 CrossRef PubMed.
- L. Huang, H. Miao, Y. Sun, F. Meng and X. Li, Eur. J. Med. Chem., 2014, 87, 429–439 CrossRef CAS PubMed.
- R. Sheng, Y. Xu, C. Hu, J. Zhang, X. Lin, J. Li, B. Yang, Q. He and Y. Hu, Eur. J. Med. Chem., 2009, 44, 07–17 CrossRef CAS PubMed.
- M. L. Hegde, P. Bharathi, A. Suram, C. Venugopal, R. Jagannathan, P. Poddar, P. Srinivas, K. Sambamurti, K. J. Rao, J. Scancar, L. Messor, L. Zecca and P. Zatta, J. Alzheimer’s Dis., 2009, 17, 457–468 CAS.
- M. A. Santos, K. Chand and S. Chaves, Coord. Chem. Rev., 2016, 327–328, 287–303 CrossRef CAS.
- S. J. S. Flora and V. Pachauri, Int. J. Environ. Res. Public Health, 2010, 7, 2745–2788 CrossRef CAS PubMed.
- J. Zhang, D. Zhu, R. Sheng, H. Wu, Y. Hu, F. Wang, T. Cai, B. Yang and Q. He, Eur. J. Pharmacol., 2009, 613, 1–9 CrossRef CAS PubMed and references cited therein.
- L.-l. Shen, G.-x. Liu and Y. Tang, Acta Pharmacol. Sin., 2007, 28, 2053–2063 CrossRef CAS PubMed.
- N. Chitranshi, S. Gupta, P. K. Tripathi and P. K. Seth, Med. Chem. Res., 2013, 22, 2328–2345 CrossRef CAS.
- L. Mira, M. T. Fernandez, M. Santos, R. Rocha, M. H. Florencio and K. R. Jennings, Free Radical Res., 2002, 36, 1199–1208 CrossRef CAS PubMed.
- J. Joubert, J. P. Petzer, L. H. A. Prins, B. P. Repsold and S. F. Malan, Drug Design and Discovery in Alzheimer’s Disease, 2014, ch. 5, pp. 291–365 Search PubMed.
- M. S. Nel, A. Petzer, J. P. Petzer and L. J. Legoabe, Bioorg. Med. Chem. Lett., 2016, 26, 4599–4605 CrossRef CAS PubMed.
- M. S. Nel, A. Petzer, J. P. Petzer and L. J. Legoabe, Bioorg. Chem., 2016, 69, 20–28 CrossRef CAS PubMed.
- S. Mostert, A. Petzer and J. P. Petzer, ChemMedChem, 2015, 10, 862–873 CrossRef CAS PubMed.
- F. Chimenti, R. Fioravanti, A. Bolasco, P. Chimenti, D. Secci, F. Rossi, M. Yánez, F. Orallo, F. Ortuso and S. Alcaro, J. Med. Chem., 2009, 52, 2818–2824 CrossRef CAS PubMed.
- S. J. Robinson, J. P. Petzer, A. Petzer, J. J. Bergh and A. C. U. Lourens, Bioorg. Med. Chem. Lett., 2013, 23, 4985–4989 CrossRef CAS PubMed.
- M. Ono and H. Saji, Int. J. Mol. Imaging, 2011, 2011, 543267 Search PubMed.
- J.-P. Qiao, C.-S. Gan, C.-W. Wang, J.-F. Ge, D.-D. Nan, J. Pan and J.-N. Zhou, ChemBioChem, 2012, 13, 1652–1662 CrossRef CAS PubMed.
- D.-D. Nan, C.-S. Gan, C.-W. Wang, J.-P. Qiao, X.-M. Wang and J.-N. Zhou, Eur. J. Med. Chem., 2016, 124, 117–128 CrossRef CAS PubMed.
- M. Ono, M. Haratake, H. Mori and M. Nakayama, Bioorg. Med. Chem., 2007, 15, 6802–6809 CrossRef CAS PubMed.
- M. Ono, R. Watanabe, H. Kawashima, Y. Cheng, H. Kimura, H. Watanabe, M. Haratake, H. Saji and M. Nakayama, J. Med. Chem., 2009, 52, 6394–6401 CrossRef CAS PubMed.
- F. Mollinedo and C. Gajate, Apoptosis, 2003, 8, 413–450 CrossRef CAS PubMed and references cited therein.
- L. M. Leoni, 2001, http://www.dtic.mil/dtic/tr/fulltext/u2/a424665.pdf.
- H. Shih, L. Deng, C. J. Carrera, S. Adachi, H. B. Cottam and D. A. Carson, Bioorg. Med. Chem. Lett., 2000, 10, 487–490 CrossRef CAS PubMed.
- T. L. Nguyen, C. McGrath, A. R. Hermone, J. C. Burnett, D. W. Zaharevitz, B. W. Day, P. Wipf, E. Hamel and R. Gussio, J. Med. Chem., 2005, 48, 6107–6116 CrossRef CAS PubMed.
- Z. Yang, W. Wu, J. Wang, L. Liu, L. Li, J. Yang, G. Wang, D. Cao, R. Zhang, M. Tang, J. Wen, J. Zhu, W. Xiang, F. Wang, L. Ma, M. Xiang, J. You and L. Chen, J. Med. Chem., 2014, 57, 7977–7989 CrossRef CAS PubMed.
- S. Kapoor and D. Panda, Biochem. Pharmacol., 2012, 83, 1495–1506 CrossRef CAS PubMed and references cited therein.
- G. A. Tunbridge, J. Oram and L. Caggiano, MedChemComm, 2013, 4, 1452–1456 RSC.
- D. R. Beukes, M. T. Davies-Coleman, M. Kelly-Borges, M. K. Harper and D. J. Faulkner, J. Nat. Prod., 1998, 61, 699–701 CrossRef CAS PubMed.
- H. O. Saxena, U. Faridi, S. Srivastava, J. K. Kumar, M. P. Darokar, S. Luqman, C. S. Chanotiya, V. Krishna, A. S. Negi and S. P. S. Khanuja, Bioorg. Med. Chem. Lett., 2008, 18, 3914–3918 CrossRef CAS PubMed.
- D. Chanda, S. Bhushan, S. K. Guru, K. Shanker, Z. A. Wani, B. A. Rah, S. Luqman, D. M. Mondhe, A. Pal and A. S. Negi, Eur. J. Pharm. Sci., 2012, 47, 988–995 CrossRef CAS PubMed.
- A. P. Prakasham, A. K. Saxena, S. Luqman, D. Chanda, T. Kaur, A. Gupta, D. K. Yadav, C. S. Chanotiya, K. Shanker, F. Khan and A. S. Negi, Bioorg. Med. Chem., 2012, 20, 3049–3057 CrossRef CAS PubMed.
- A. Singh, K. Fatima, A. Singh, A. Behl, M. J. Mintoo, M. Hasanain, R. Ashraf, S. Luqman, K. Shanker, D. M. Mondhe, J. Sarkar, D. Chanda and A. S. Negi, Eur. J. Pharm. Sci., 2015, 76, 57–67 CrossRef CAS PubMed.
- A. S. Negi, A. P. Prakasham, A. K. Saxena, S. Luqman, D. Chanda, T. Kaur and A. Gupta, US Pat., US 8,633,242 B2, 2014.
- B. Orlikova, J. C. J. M. D. S. Menezes, S. Ji, S. P. Kamat, J. A. S. Cavaleiro and M. Diederich, Eur. J. Med. Chem., 2014, 84, 173–180 CrossRef CAS PubMed and references cited therein.
- S. Sale, R. G. Tunstall, K. C. Ruparelia, P. C. Butler, G. A. Potter, W. P. Steward and A. J. Gescher, Invest. New Drugs, 2006, 24, 459–464 CrossRef CAS PubMed.
- R. D. Bruno and V. C. O. Njar, Bioorg. Med. Chem., 2007, 15, 5047–5060 CrossRef CAS PubMed.
- R. M. Hartley, J. Peng, G. A. Fest, S. Dakshanamurthy, D. E. Frantz, M. L. Brown and S. L. Mooberry, Mol. Pharmacol., 2012, 81, 431–439 CrossRef CAS PubMed.
- C. Isanbor and D. O’Hagan, J. Fluorine Chem., 2006, 127, 303–319 CrossRef CAS.
- J. Rojas, M. Payá, J. N. Dominguez and M. L. Ferrándiz, Bioorg. Med. Chem. Lett., 2002, 12, 1951–1954 CrossRef CAS PubMed.
- A. Singh, K. Fatima, A. Srivastava, S. Khwaja, D. Priya, A. Singh, G. Mahajan, S. Alam, A. K. Saxena, D. M. Mondhe, S. Luqman, D. Chanda, F. Khan and A. S. Negi, Chem. Biol. Drug Des., 2016, 88, 625–634 CAS.
- J. Hu, J. Yan, J. Chen, Y. Pang, L. Huang and X. Li, MedChemComm, 2015, 6, 1318–1327 RSC.
- N. J. Lawrence, D. Rennison, A. T. McGown and J. A. Hadfield, Bioorg. Med. Chem. Lett., 2003, 13, 3759–3763 CrossRef CAS PubMed.
- S. Ducki, D. Rennison, M. Woo, A. Kendall, J. F. D. Chabert, A. T. McGown and N. J. Lawrence, Bioorg. Med. Chem., 2009, 17, 7698–7710 CrossRef CAS PubMed.
- N. K. Kaushik, N. Kaushik, P. Attri, N. Kumar, C. H. Kim, A. K. Verma and E. H. Choi, Molecules, 2013, 18, 6620–6662 CrossRef CAS PubMed.
- J. Chen, J. Yan, J. Hu, Y. Pang, L. Huang and X. Li, RSC Adv., 2015, 5, 68128–68135 RSC.
- G. Wang, C. Li, L. He, K. Lei, F. Wang, Y. Pu, Z. Yang, D. Cao, L. Ma, J. Chen, Y. Sang, X. Liang, M. Xiang, A. Peng, Y. Wei and L. Chen, Bioorg. Med. Chem., 2014, 22, 2060–2079 CrossRef CAS PubMed.
- M. W. Robinson, J. H. Overmeyer, A. M. Young, P. W. Erhardt and W. A. Maltese, J. Med. Chem., 2012, 55, 1940–1956 CrossRef CAS PubMed.
- A. Boumendjel, A. McLeer-Florin, P. Champelovier, D. Allegro, D. Muhammad, F. Souard, M. Derouazi, V. Peyrot, B. Toussaint and J. Boutonnat, BMC Cancer, 2009, 9, 242 CrossRef PubMed.
- V. Martel-Frachet, M. Keramidas, A. Nurisso, S. DeBonis, C. Rome, J.-L. Coll, A. Boumendjel, D. A. Skoufias and X. Ronot, Oncotarget, 2015, 6, 14669–14686 CrossRef PubMed.
- A. Özdemir, M. D. Altıntop, G. Turan-Zitouni, G. A. Çiftçi, I. Ertorun, Ö. Alataş and Z. A. Kaplancıklı, Eur. J. Med. Chem., 2015, 89, 304–309 CrossRef PubMed.
- M. Cui, M. Ono, H. Kimura, B. L. Liu and H. Saji, Bioorg. Med. Chem. Lett., 2011, 21, 980–982 CrossRef CAS PubMed.
- R. W. Hartmann, H. Bayer and G. Grün, J. Med. Chem., 1994, 37, 1275–1281 CrossRef CAS PubMed.
- R. Gupta, D. P. Jindal, B. Jit, G. Narang, A. Palusczak and R. W. Hartmann, Arch. Pharm., 2004, 337, 398–401 CrossRef CAS PubMed.
- R. Bansal, G. Narang, C. Zimmer and R. W. Hartmann, Med. Chem. Res., 2011, 20, 661–669 CrossRef CAS.
- R. Prasanna and C. C. Harish, Oncol. Res., 2010, 18, 1–8 Search PubMed.
- R. Suresha, C. C. Kanakam, K. Dineshkumar and W. Hopper, Am. J. Biomed. Sci. Eng., 2015, 1, 32–38 Search PubMed.
- P. Rajagopalan, K. A. Alahmari, A. A. Elbessoumy, M. Balasubramaniam, R. Suresh, M. E. A. Shariff and H. C. Chandramoorthy, Cancer Chemother. Pharmacol., 2016, 77, 393–404 CrossRef CAS PubMed.
- J. C. J. M. D. S. Menezes, B. Orlikova, F. Morceau and M. Diederich, Crit. Rev. Food Sci. Nutr., 2015, 56, S4–S28 CrossRef PubMed and references cited therein.
- D. A. Carson, M. D. Rosenbach, C. J. Carrera and L. M. Leoni, US Pat., US 2004/0096436 A1, 2004.
- B. W. Day, M. W. Tsang, V. N. Korotchenko, WO 2010/108058 A2, 2010.
- G. Vistoli, A. Pedretti and B. Testa, Drug Discovery Today, 2008, 13, 285–294 CrossRef CAS PubMed.
- S. Shojaee, R. Caeser, M. Buchner, E. Park, S. Swaminathan, C. Hurtz, H. Geng, L. N. Chan, L. Klemm, W.-K. Hofmann, Y. H. Qiu, N. Zhang, K. R. Coombes, E. Paietta, J. Molkentin, H. P. Koeffler, C. L. Willman, S. P. Hunger, A. Melnick, S. M. Kornblau and M. Müschen, Cancer Cell, 2015, 28, 114–128 CrossRef CAS PubMed.
- M. Chen, T. G. Theander, S. B. Christensen, L. Hviid, L. Zhai and A. Kharazmi, Antimicrob. Agents Chemother., 1994, 38, 1470–1475 CrossRef CAS PubMed.
- K. Kaur, M. Jain, R. P. Reddy and R. Jain, Eur. J. Med. Chem., 2010, 45, 3245–3264 CrossRef CAS PubMed.
- J. Tigges, T. Haarmann-Stemmann, C. F. A. Vogel, A. Grindel, U. Hübenthal, H. Brenden, S. Grether-Beck, G. Vielhaber, W. Johncock, J. Krutmann and E. Fritsche, J. Invest. Dermatol., 2014, 134, 556–559 CrossRef CAS PubMed.
- L. A. Marques, M. Giera, H. Lingeman and W. M. Niessen, Biomed. Chromatogr., 2011, 25, 278–299 CrossRef CAS PubMed.
- S. Salomone, F. Caraci, G. M. Leggio, J. Fedotova and F. Drago, Br. J. Clin. Pharmacol., 2011, 73, 504–517 CrossRef PubMed.
- I. Kron, Z. Pudychová-Chovanová, B. Veliká, J. Guzy and P. Perjési, Monatsh. Chem., 2012, 143, 13–17 CrossRef CAS.
- R. L. Carrier, L. A. Miller and I. Ahmed, J. Controlled Release, 2007, 123, 78–99 CrossRef CAS PubMed.
- G. Tiwari, R. Tiwari and A. K. Rai, J. Pharm. BioAllied Sci., 2010, 2, 72–79 CrossRef CAS PubMed.
- G. C. Tron, T. Pirali, G. Sorba, F. Pagliai, S. Busacca and A. A. Genazzani, J. Med. Chem., 2006, 49, 3033–3044 CrossRef CAS PubMed.
- Y.-S. Wu, M. S. Coumar, J.-Y. Chang, H.-Y. Sun, F.-M. Kuo, C.-C. Kuo, Y.-J. Chen, C.-Y. Chang, C.-L. Hsiao, J.-P. Liou, C.-P. Chen, H.-T. Yao, Y.-K. Chiang, U.-K. Tan, C.-T. Chen, C.-Y. Chu, S.-Y. Wu, T.-K. Yeh, C.-Y. Lin and H.-P. Hsieh, J. Med. Chem., 2009, 52, 4941–4945 CrossRef CAS PubMed.
- K. Sakao, M. Fujii and D.-X. Hou, BioFactors, 2009, 35, 399–405 CrossRef CAS PubMed.
- Z. Parveen, G. Brunhofer, I. Jabeen, T. Erker, P. Chiba and G. F. Ecker, Bioorg. Med. Chem., 2014, 22, 2311–2319 CrossRef CAS PubMed.
- H. M. Sim, K. Y. Loh, W. K. Yeo, C. Y. Lee and M. L. Go, ChemMedChem, 2011, 6, 713–724 CrossRef CAS PubMed.
- H.-M. Sim, C.-P. Wu, S. V. Ambudkar and M.-L. Go, Biochem. Pharmacol., 2011, 82, 1562–1571 CrossRef CAS PubMed.
- J. C. J. M. D. S. Menezes, J. A. S. Cavaleiro, S. P. Kamat, C. M. R. F. Barros and M. R. M. Domingues, Rapid Commun. Mass Spectrom., 2013, 27, 1303–1310 CrossRef CAS PubMed.
- P. Boeck, P. C. Leal, R. A. Yunes, V. C. Filho, S. López, M. Sortino, A. Escalante, R. L. E. Furlán and S. Zacchino, Arch. Pharm., 2005, 338, 87–95 CrossRef CAS PubMed.
- K. A. Monk, R. Siles, M. B. Hadimani, B. E. Mugabe, J. F. Ackley, S. W. Studerus, K. Edvardsen, M. L. Trawick, C. M. Garner, M. R. Rhodes, G. R. Pettit and K. G. Pinney, Bioorg. Med. Chem., 2006, 14, 3231–3244 CrossRef CAS PubMed.
- X. Liu and M.-L. Go, Bioorg. Med. Chem., 2006, 14, 153–163 CrossRef CAS PubMed.
- M. C. Jennings, K. P. C. Minbiole and W. M. Wuest, ACS Infect. Dis., 2015, 1, 288–303 CrossRef CAS PubMed.
- S. Amslinger, ChemMedChem, 2010, 5, 351–356 CrossRef CAS PubMed.
Footnote |
| † Present address: Department of Pharmacy, Nagasaki International University, Huis Ten Bosch Cho 2825-7 Sasebo, Nagasaki Japan 859-3298, Correspondence to: Dr José C. J. M. D. S. Menezes, E-mail: E-mail: jose_menz@yahoo.co.in; , josemenezes@ua.pt. |
| This journal is © The Royal Society of Chemistry 2017 |