
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA protector-free one-pot strategy to synthesize highly dispersed Pd hydrogenation catalysts in a broad loading range on polystyrene
Li-Yun Le,
Ai-Min Ren,
Xing-Heng Gao,
Dan Zhao *,
Run-Hong Xie and
Ning Zhang*
*,
Run-Hong Xie and
Ning Zhang*
Institute of Applied Chemistry, College of Chemistry, Nanchang University, Nanchang, Jiangxi 330031, China. E-mail: zhaodan@ncu.edu.cn; nzhang.ncu@163.com; Tel: +86-791-83969332
First published on 21st February 2017
Abstract
Pursuing a strategy to feasibly acquire nanoparticles in a clean state (protector-free) and desired size range (ca. 3.0–5.0 nm) is a fundamental demand for the application of state-of-the-art precious metal catalysts. Herein, vinylbenzyl trimethylammonium chloride (VTC) was copolymerized on the surface of polystyrene (PS) as a capturing group. This capturing group anchored PdCl42− via electrostatic state-of-the-art interaction to prepare PS supported Pd nanoparticles in one-pot system for the selective hydrogenation of phenylacetylene to styrene. Using a combination of inductively coupled plasma-optical emission spectrometry (ICP-OES), zeta potential analysis (ZPA), X-ray diffraction (XRD), transmission electron microscopy (TEM), and Fourier transform infrared spectra (FT-IR) techniques, it was demonstrated that introducing VTC on PS in a proper amount (VTC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :styrene molar ratio of 0.05) was a key factor to optimize the electrostatic state of the polystyrene substrate, which facilitated uniform anchoring of PdCl42− and the following in situ reduction to Pd particles under protector-free conditions, and led to the formation of particles in desired ca. 3.0–5.0 nm size range while broad Pd loading being adjusted in 20 times change (from 0.2–4.6 wt%). The catalytic measurements and corresponding kinetic analysis for the selective hydrogenation of phenylacetylene to styrene showed that the regular change of catalytic performance among the samples was directly governed by Pd particle size. In addition, the good stability of the present Pd catalyst was verified via leaching experiment and recycle use tests on typical samples. These results indicated that the present strategy was promising in acquiring clean and size-controllable metal nanoparticles, which could be inferred as a feasible system to obtain desired metal nanocatalysts for advanced heterogeneous catalysis applications.
:styrene molar ratio of 0.05) was a key factor to optimize the electrostatic state of the polystyrene substrate, which facilitated uniform anchoring of PdCl42− and the following in situ reduction to Pd particles under protector-free conditions, and led to the formation of particles in desired ca. 3.0–5.0 nm size range while broad Pd loading being adjusted in 20 times change (from 0.2–4.6 wt%). The catalytic measurements and corresponding kinetic analysis for the selective hydrogenation of phenylacetylene to styrene showed that the regular change of catalytic performance among the samples was directly governed by Pd particle size. In addition, the good stability of the present Pd catalyst was verified via leaching experiment and recycle use tests on typical samples. These results indicated that the present strategy was promising in acquiring clean and size-controllable metal nanoparticles, which could be inferred as a feasible system to obtain desired metal nanocatalysts for advanced heterogeneous catalysis applications.
1. Introduction
Precious metal (PM) catalysts have been intensively studied due to the unique properties, such as low overpotential on platinum for O2 electroreduction or small molecular electrooxidation, low-temperature on gold for CO oxidation, and high reactivity on palladium for the hydrogenation of unsaturated chemical bonds, that they have shown in many technically important heterogeneous catalysis processes.1–5 Among the factors responsible for the outstanding performance of PM catalysts, so-called size effect was highlighted as one of the most effective aspects. For instance, adjusting the particle size of PM catalysts in nanoscale was found to impact not only the state of metal surface but also the interaction between the metal and the support, which in turn directly affected the reaction behaviors and consequent performance of the catalyst.6,7 Interestingly, a common particle size range of 3.0–5.0 nm for the catalysts employing PM nanoparticles has been widely reported for the optimized catalytic performance in many different reaction systems,8–10 suggesting that designing or controllably preparing metal nanoparticles in this size range would be a basic criterion for the application of the state-of-the-art precious metal catalysts.Many efforts have been devoted to synthesizing precious metal nanoparticles in size-controllable aspect, which could be roughly differentiated into two methods. One is directly reducing the metal salt precursors with the help of protector reagents to confine the aggregation or growth of metal atomic clusters, such that to maintain the resulting sample in a definite size range. This method is advantageous in feasibly controlling the size of metal particles by employing protector reagents under widely adjustable conditions such as the nature and amount of protectors. However, the intrinsic function of the protectors is to strongly absorb on metal surface, which means they will at least occupy a part of the surface of metal particles and negatively influence the loading affinity for the support or the accessibility of reactants to the metal sites.11,12 The other method to obtain small-sized metal catalysts is introducing a support to distribute metal precursors via impregnation or deposition and then reducing the loaded precursors to metal particles. This method is beneficial for employing the features of the support such as high specific surface area or the strong interaction with metal precursor to ensure the desired dispersion of metal particles. However, note that this method could only be feasible in a low and narrow loading range of metal due to the limited surface area of the support.13,14 In addition, the randomness of impregnation or deposition processes always leads to uneven distribution of metal precursors on the supports and consequently fails to prepare metal particles in the desired narrow size range.15,16 Based on this situation, pursuing a feasible and ideal system to overcome the drawbacks of the above mentioned traditional preparation methods is still an open topic for both the design and application of precious metal catalysts.
In this study, a feasible one-pot strategy to directly synthesize highly dispersed Pd particles (ca. 3.0–5.0 nm) with adjustable and broad loading range in 20 times change on polystyrene under protector-free condition was developed via systematically adjusting the electrostatic interaction between Pd salt precursor and the surface of polystyrene. It was demonstrated that introducing vinylbenzyl trimethylammonium chloride (VTC) in a proper amount to copolymerize on the surface of polystyrene was a simple but functional way to optimize the electrostatic state of the polystyrene substrate; this consequently facilitated the uniform anchoring of Pd precursors and the following in situ reduction to narrow-size-range (ca. 3.0–5.0 nm) distributed Pd nanoparticles with broad Pd loading from 0.2 wt% to 4.6 wt% on polystyrene under protector-free condition. The mass-specific activity for the selective hydrogenation of phenylacetylene among the samples changed well along with the size or the dispersion of Pd particles, further indicated the advantages of the present system in preparing clean and well-dispersed Pd catalysts. These findings suggest that the present system is promising to acquire efficient precious metal catalysts for important heterogeneous catalysis as well as other advanced applications those depending on size-controlled nanoparticles.
2. Experimental
2.1 Materials and pretreatments
All the reagents in the study were used as received if not otherwise specified. Styrene (St, AR), potassium persulfate (KPS), and ethyl alcohol were purchased from Sinopharm Chemical Reagent Co. Before use, styrene was purified by washing with 5 wt% aqueous NaOH to remove the inhibitor and was stored at below 5 °C. Vinylbenzyl trimethylammonium chloride (VTC) was supplied by Sigma-Aldrich Inc. Sodium tetrachloropalladate(II) (Na2PdCl4) was purchased from TCI Chemicals Company. Phenylacetylene (98%) was obtained from the J&K Scientific LTD. Dodecane was purchased from Aladdin Reagent Co. Ultrapure water (18.3 MΩ cm−1) obtained via a Milli-Q water system was used throughout the experiment.2.2 Synthesis of the polystyrene-supported palladium samples
The samples were generally prepared via two steps in a one-pot system, as shown in Scheme 1. At first, desired amount of VTC was introduced to the solution containing metastable polystyrene (PS) particles to copolymerize as cationic PS. Then, aqueous Pd precursor (Na2PdCl4) was injected into the solution to form PS-supported Pd samples. The details have been described hereinafter.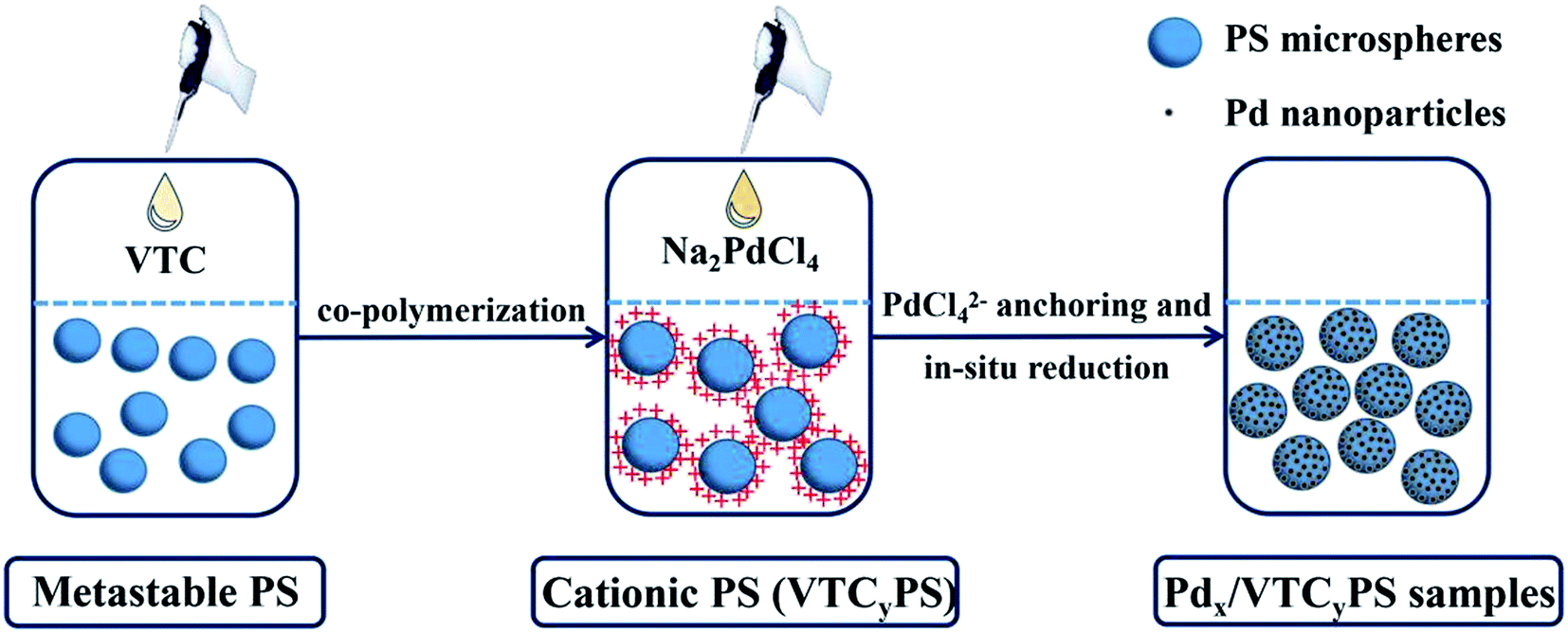 | ||
| Scheme 1 Schematic for the fabrication of Pdx/VTCyPS samples. | ||
Polymers were prepared via an emulsifier-free emulsion polymerization procedure. All the raw materials, including initiator K2S2O8 (0.08 g), ultrapure water (40 ml), and styrene monomer (4 g), were charged into a 100 ml three-necked round-bottom flask equipped with a Graham condenser and a heating mantle. The solution was purged with nitrogen for 20–30 minutes to eliminate oxygen and then polymerization was allowed to proceed for 2 hours at 70 °C under a stirring rate of 600 rpm to obtain the solution containing metastable polystyrene (PS) particles. Subsequently, a desired amount of VTC aqueous solution was injected into the solution. To ensure complete polymerization, the reaction was allowed to proceed for 12 hours at 70 °C under a stirring rate of 600 rpm to obtain a cationic PS solution.17 Then, the system was injected with desired amounts of freshly prepared aqueous solution of Na2PdCl4 and maintained at 50 °C for 6 hours. The color of the solution turned from milky white to grey, suggesting the formation of Pd particles in the system. The solid materials were separated out of the solution by centrifugation, washed several times with ethanol and water, and then dispersed in water and stored at 5 °C for use.
2.3 Characterization
The actual palladium loading on the PS microspheres was quantified using an Agilent Technologies 5100 ICP-OES inductively coupled plasma-optical emission spectrometer (ICP-OES).Zeta potential analysis was performed using Zeta Nanosizer (Nano-ZS90), which was produced by Malvern Company.
X-ray powder diffraction (XRD) experiments were carried out via a Persee XD-3 X-ray diffractometer at the scan rate of 2° min−1 in the angle range of 5–90° using Cu Kα radiation.
Transmission electron microscopy (TEM) was performed using a JEM-2100 (JEOL Co., Japan) microscope operating at 200 kV to observe the morphology and size distribution of the samples.
Fourier transform infrared spectroscopy (FT-IR) was carried out by a Nicolet 5700 FT-IR spectrometer (Thermo Fisher) using KBr pellet technique.
2.4 Catalytic measurements of the samples
The catalytic performances of the prepared samples were investigated by employing the selective hydrogenation of phenylacetylene (to styrene) as the model reaction. Ethanol (40 ml), phenylacetylene (500 μl, 4.6 mmol), and the same volume of dodecane (2.2 mmol as internal standard substance) were mixed in 100 a ml three-necked round-bottom flask, and then the mixture was injected in the given amount of sample. After degassing with H2 for several minutes at room temperature, the system was sealed by a balloon filled with pure H2 and heated to 40 °C to perform the reaction. The reaction process was monitored by filtering out a drop of the reaction solution to analyze the composition with time. The filtered solution drop was analyzed by a Gas Chromatograph (Agilent Technologies 7820A GC system) with a capillary column. For recycle tests, the catalysts were separated from the reaction solution by centrifugation, washed with ethanol, and reused for the next run.3. Results and discussion
In this study, vinylbenzyl trimethylammonium chloride (VTC) was embedded on the surface of polystyrene (PS) as a capturing group for anchoring Pd precursor via the electrostatic interaction between –NH4+ (in VTC) and PdCl42−, according to the strategy shown in Scheme 1. In view of this design, the amounts of VTC and PdCl42− could be significant factors responsible for the features of the prepared sample. To clarify this possibility, three samples with the same nominal Pd loading but varied amounts of VTC (molar ratio of VTC:styrene (y) = 0.025, 0.05, and 0.10) were prepared and investigated. ICP-OES analysis showed that the actual loadings of Pd in three samples were 0.58 wt%, 0.57 wt%, and 0.61 wt%, respectively, suggesting that the Pd loading of the three sample was all close to 0.60 wt%; thus, these three samples were denoted as Pd0.6%/VTC0.025PS, Pd0.6%/VTC0.05PS, and Pd0.6%/VTC0.10PS. Fig. 1 is the combination of the characteristic results for the three samples. The setting of VTC on polystyrene and the following Pd precursor introduction were monitored by zeta potential analysis, shown in Fig. 1A. For these examinations, all the tested solutions were redispersed aqueous solutions containing the filtered solid materials from the corresponding original solutions. Compared to the negative potential of pure polystyrene sample (Fig. 1A-o), zeta potentials for the three samples, introduced in VTC, increased in the positive potential range with the increasing the amount of VTC (Fig. 1A-a′, b′, and c′), suggesting that the cationic PS substrates were successfully formed. After the addition of PdCl42−, all the potentials subdued, indicating the obvious electrostatic interaction, i.e., anchoring of PdCl42− on cationic PS substrate present in the process. In comparison with the diffraction peaks of the PS substrate (Fig. 1B-o), the two obvious peaks at about 39.9° and 46.2° in the XRD patterns of the three Pd-containing samples, as shown in Fig. 1B, were assigned to Pd[111] and Pd[200] crystalline,18,19 respectively, further clarifying the successful anchoring of PdCl42− and in situ reduction in three samples. The morphological details of the samples are shown in Fig. 1C. With the increase in the molar ratio of VTC:styrene (y) among the three samples, although Pd loadings were similar (around 0.60 wt%), the average size of the Pd particles on the three samples was quite different: 6.2 ± 2.5 nm, 4.3 ± 1.5 nm, and 8.5 ± 4.1 nm. Only the size of the Pd particles in Pd0.6%/VTC0.05PS (with molar ratio of VTC:styrene at 0.05) was in the desired size range (ca. 3.0–5.0 nm) for state-of-the-art precious metal nanocatalysts,8–10 indicating that a proper amount of VTC on PS substrate was a key factor to control the size of Pd particles in the present preparation system, which could be attributed to the following features. The smaller amount of VTC (Pd0.6%/VTC0.025PS with the molar ratio of VTC:styrene at 0.025) might have caused the insufficient or the uneven distribution of charge on the cationic PS surface, which in turn results in a non-uniform anchoring of PdCl42−, and the following unsatisfactory size distribution of Pd particles; on the other hand, the larger amount of VTC (Pd0.6%/VTC0.10PS with the molar ratio of VTC:styrene at 0.10) would provide a higher cationic charge density on the surface of PS substrate, suggesting that the distance of anchoring group VTC would be shortened and the aggregation possibility of anchored Pd would be simultaneously enhanced, which consequently leads to the formation of larger Pd particles on the sample. In our study, it was demonstrated that by employing 0.05 molar ratio of VTC:styrene, the optimized surface charge distribution of the cationic PS substrates could be achieved for the following Pd immobilization process.
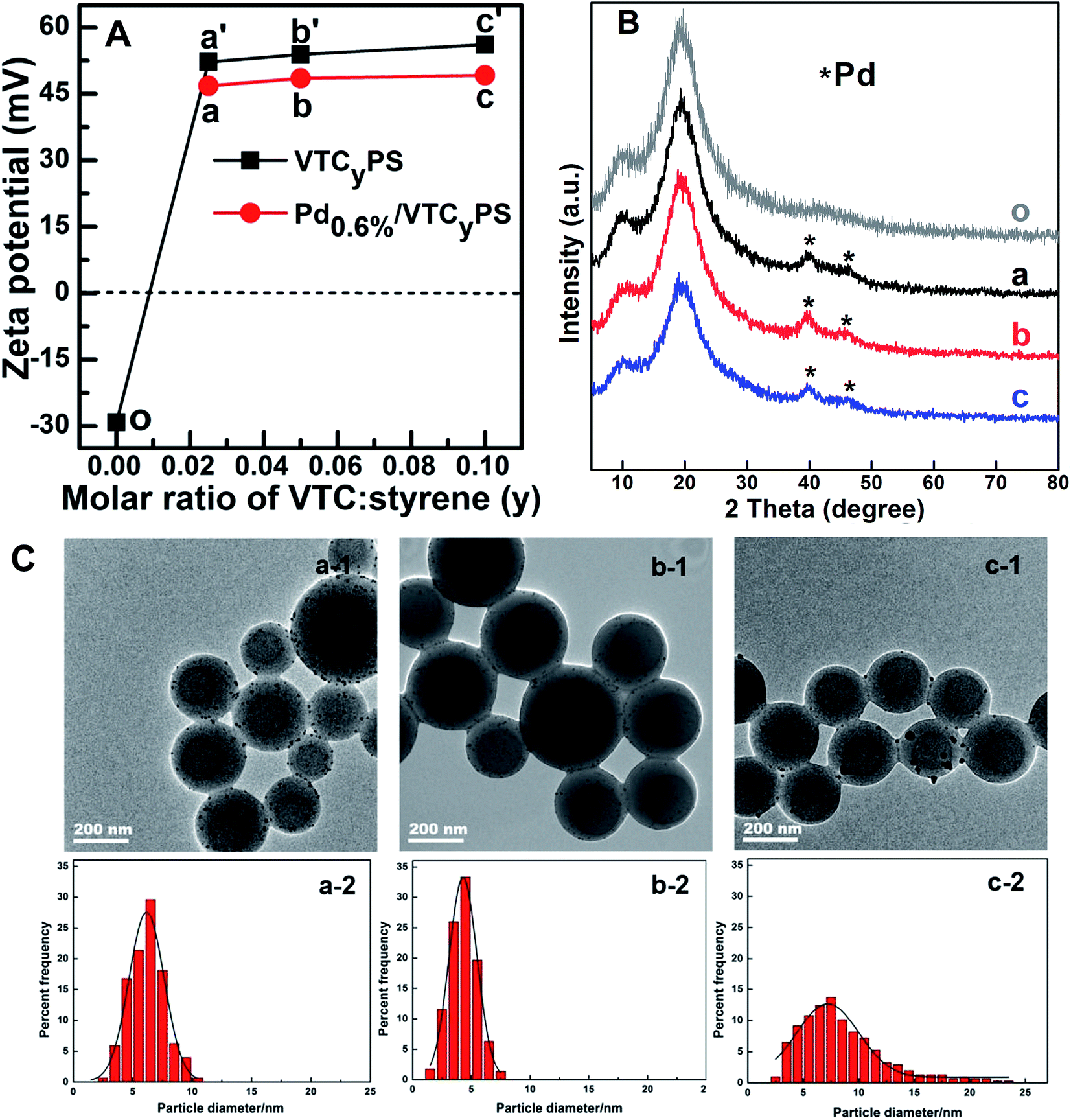 | ||
| Fig. 1 (A) Zeta potential analysis of PS, VTCyPS, and corresponding Pd0.6%/VTCyPS microspheres; (B) XRD patterns of PS and Pd0.6%/VTCyPS samples; (C) TEM images of Pd0.6%/VTCyPS samples and corresponding size distribution histograms. (o) PS, (a′) VTC0.025PS, (b′) VTC0.05PS, and (c′) VTC0.10PS; (a) Pd0.6%/VTC0.025PS, (b) Pd0.6%/VTC0.05PS, and (c) Pd0.6%/VTC0.10PS. | ||
Based on the abovementioned results, the influence of the amount of Pd precursor was further investigated by adjusting the Pd loading on the cationic PS with a constant VTC:styrene molar ratio of 0.05. Except for the prepared Pd0.6%/VTC0.05PS sample, ICP-OES analysis indicated that the actual loadings of Pd in other three Pd-loading adjusted samples were 0.2 wt%, 1.3 wt%, and 4.6 wt%, and these samples were denoted as Pd0.2%/VTC0.05PS, Pd1.3%/VTC0.05PS, and Pd4.6%/VTC0.05PS, respectively. Fig. 2 shows the results of FT-IR (Fig. 2A) and XRD (Fig. 2B) measurements for four samples. In Fig. 2A, in comparison with the slight change of C–H bending vibrations located in 1490–1400 cm−1 among the samples, the feature correlated to VTC was mainly manifested by the peak around 1600 cm−1, which was assigned to –NH stretching vibration.20,21 With the increasing Pd loadings of the samples up to 4.6 wt%, the intensity of –NH stretching vibrations among the samples gradually decreased and then disappeared (denoted by dash straight line in Fig. 2A), suggesting the continuously intensified interaction between Pd and the capturing group of –NH;21 moreover, the phenomenon was accompanied by the gradually sharpened diffraction peaks of Pd (XRD patterns of Fig. 2B). These results conjunctively indicated that Pd particles were immobilized on VTC0.05PS substrate for all the Pdx/VTC0.05PS samples. The morphological details of the samples are shown as TEM images in Fig. 3. In overview, all Pd particles were dispersed well on PS microsphere support for the samples. The observed lattice spacings of d = 0.225 nm for Pd0.2%/VTC0.05PS and d = 0.228 nm for Pd1.3%/VTC0.05PS were assigned to the Pd[111] lattice plane; and the lattice spacings of d = 0.234 nm for Pd0.6%/VTC0.05PS and d = 0.231 nm for Pd4.6%/VTC0.05PS were assigned to the Pd[100] lattice plane (JCPDS card #46-1043). These results disclosed that the palladium particles on the samples were present in completely reduced state. The analysis of size distribution of Pd particles showed that the average sizes for the three newly prepared samples were 3.6 ± 1.3 nm, 5.0 ± 1.6 nm, and 5.4 ± 1.8 nm, indicating that the strategy developed in the present study could ensure the acquiring of well dispersed Pd particles in ca. 3.0–5.0 nm size range. Considering that the Pd loadings among our samples were in 20 times variations (from 0.2–4.6 wt%), the results also suggested that the present strategy was suitable to get desired Pd nanocatalysts in a broad loading range.
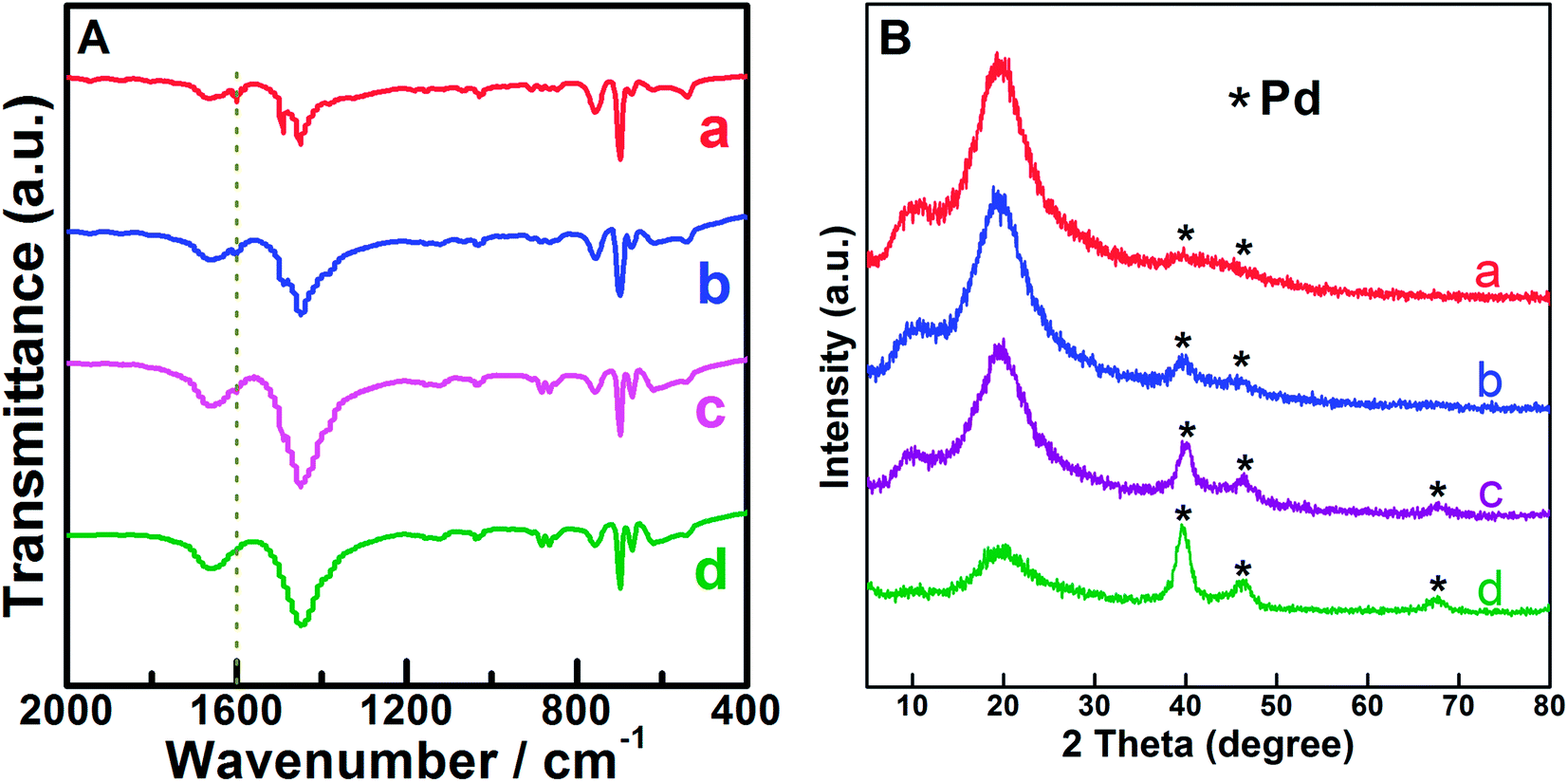 | ||
| Fig. 2 Fourier transform-infrared spectra (A) and XRD patterns (B) of Pdx/VTC0.05PS samples. (a) Pd0.2%/VTC0.05PS, (b) Pd0.6%/VTC0.05PS, (c) Pd1.3%/VTC0.05PS, and (d) Pd4.6%/VTC0.05PS. The dash straight line in (A) refers to the peak around 1600 cm−1 attributed to the –NH stretching vibration. | ||
 | ||
| Fig. 3 TEM images of the Pdx/VTC0.05PS samples and the corresponding HRTEM and size distribution histograms of supported Pd NPs. (A) Refer to Pd0.2%/VTC0.05PS; (B) refer to Pd0.6%/VTC0.05PS; (C) refer to Pd1.3%/VTC0.05PS; and (D) refer to Pd4.6%/VTC0.05PS. X-1: the overview of Pdx/VTCyPS samples; X-2: image of the single particle figures; X-3: HRTEM image of Pd NPs; X-4: histogram of the size distributions of Pd NPs (X = A, B, C, and D). | ||
According to the constructive comments of a reviewer, we have reported the features of some typical methods in literature to prepare Pd nanocatalysts and these features are shown in Table 2. In comparison with the various inorganic material-supported Pd nanocatalysts,22–28 the researches focused on the fact that Pd catalysts supported by organic materials just occupied a small fraction,29,30 which might attribute to the relatively higher thermal stability of inorganic materials compared to that of the PS-like polymer materials. However, this thermal stability of inorganic supports also implied the inconvenience in adjusting their state, especially the surface state that would significantly impact the conjunctive function with metal components. For instance, the higher temperatures were always required for preparing or processing oxide or zeolite-supported metal nanocatalysts to the desired active state.22–26 At high temperatures, there was inevitable thermodynamic tendency of isolated metals to leach or agglomerate, leading to the difficulty in controlling the particles within narrow nanoscale.23–25 In addition, although some inorganic supports such as active carbon materials possessed rich functional groups on the surface and their nature could be easily manipulated at a proper temperature for optimizing the interaction with the loaded metal, controlling the size distribution of particles was still difficult27,28 due to the uneven dispersing density of the functional groups on the surface, especially when metal loadings were altered to a relative higher amount.29,30 In contrast, in our study, the introduction of a capturing group (VTC) in a proper amount to copolymerize on the surface of polystyrene (with the molar ratio of VTC:styrene at 0.05) was verified to be a feasible way to optimize the electrostatic state of polystyrene substrate, which consequently facilitated the uniform anchoring of Pd precursors and the following in situ reduction to narrow-size-range distributed Pd nanoparticles (ca. 3.0–5.0 nm) in a broad loading range (from 0.2 wt% to 4.6 wt%) under protector-free conditions. The findings further indicated that the flexible nature of PS-like polymer materials in both design and preparation29,30 could be advantageous for conveniently synthesizing desired supported metal nanocatalysts.
The catalytic performances of the samples were investigated by employing the selective hydrogenation of phenylacetylene to styrene as a model reaction. Fig. 4A and B show the conversion curves of phenylacetylene and the corresponding yield curves of styrene with reaction time for Pd0.6%/VTC0.025PS, Pd0.6%/VTC0.05PS, and Pd0.6%/VTC0.10PS. The best catalytic performance was observed for Pd0.6%/VTC0.05PS among the three samples, which was correlated to the smaller average size of the Pd particles on this sample compared to that on the other two samples. The catalytic performance curves of four samples with constant VTC:styrene molar ratio of 0.05 and varied Pd loading were obtained and are shown in Fig. 4C (phenylacetylene conversion curves) and D (styrene yield curves). The reaction system containing pure PS sample (the Pd-absent reference sample) showed insignificant change with time; however, hydrogenation was immediately initiated when a small amount of Pdx/VTC0.05PS sample was added to the reactant solutions, and the reaction rate was accelerated in proportion to the Pd content in the catalysts. In general, each of the catalytic reaction conversion could reach 100% accompanied by a high styrene yield (average at 82%). According to the reaction data of Fig. 4 and 5A correlated ln(Ct/C0) with reaction time for each sample, and it was observed that the linear relation could be resolved for all the samples, proving that a pseudo-first-order rate kinetic dominates the reaction process for our samples. Therefore, the kinetic data such as rate constant (k), reaction rate (at the reaction time of 5 min), and the corresponding mass-specific activity of Pd (MSA-Pd) for the samples could also be resolved, as listed in Table 1. The average size of each sample is also listed in Table 1. Considering that the reciprocal of the diameter of particles in nanoscale could be used to roughly estimate the utilization or dispersion of the supported metal catalyst,31,32 Fig. 5B correlates MSA-Pd with the reciprocal of Pd NPs particle size among the samples. An approximate linear relation was observed, indicating that the catalytic performance of our samples was directly governed by the size of the Pd particles; Pd in smaller size (higher dispersion) would be more favorable for the reaction. Mimic size-dependent catalytic performance of Pd catalysts for the selective hydrogenation of phenylacetylene could be also found in literature. The styrene yields with the corresponding size of Pd particles in various catalysts22–30 are listed in Table 2; in general, employing smaller Pd particles as catalysts could be more beneficial to obtain higher styrene yields. However, note that no matter what the nature of the supports (inorganic or organic materials) was, the smallest Pd nanocatalysts such as particles around 2.0 or 1.5 nm were always present in colloid deposition systems, meaning that a significant coverage of protectors (for instance, PVP) on the metal surface was necessary for obtaining and using these predefined small Pd particles.25,29 Although the higher styrene yields could be achieved for these samples, the more intensified reaction conditions, such as longer reaction time (≥300 min)25 or higher H2 pressure (3.0 MPa),29 compared to those for other samples were simultaneously required, which should be attributed to the poor accessibility of the reactants to the metal sites on the well-protected surface of these small particles.11,12 In contrast, our samples prepared from the present protector-free system could achieve comparable catalytic performance under obviously milder reaction conditions, suggesting that the preparation of metal nanocatalysts in not only a desired size range but also in a cleaner state was desirable for their application. Therefore, the present strategy could serve as a feasible method to prepare the desired samples for heterogeneous catalysis as well as other advanced applications that depend on size-controlled and clean metal nanoparticles.
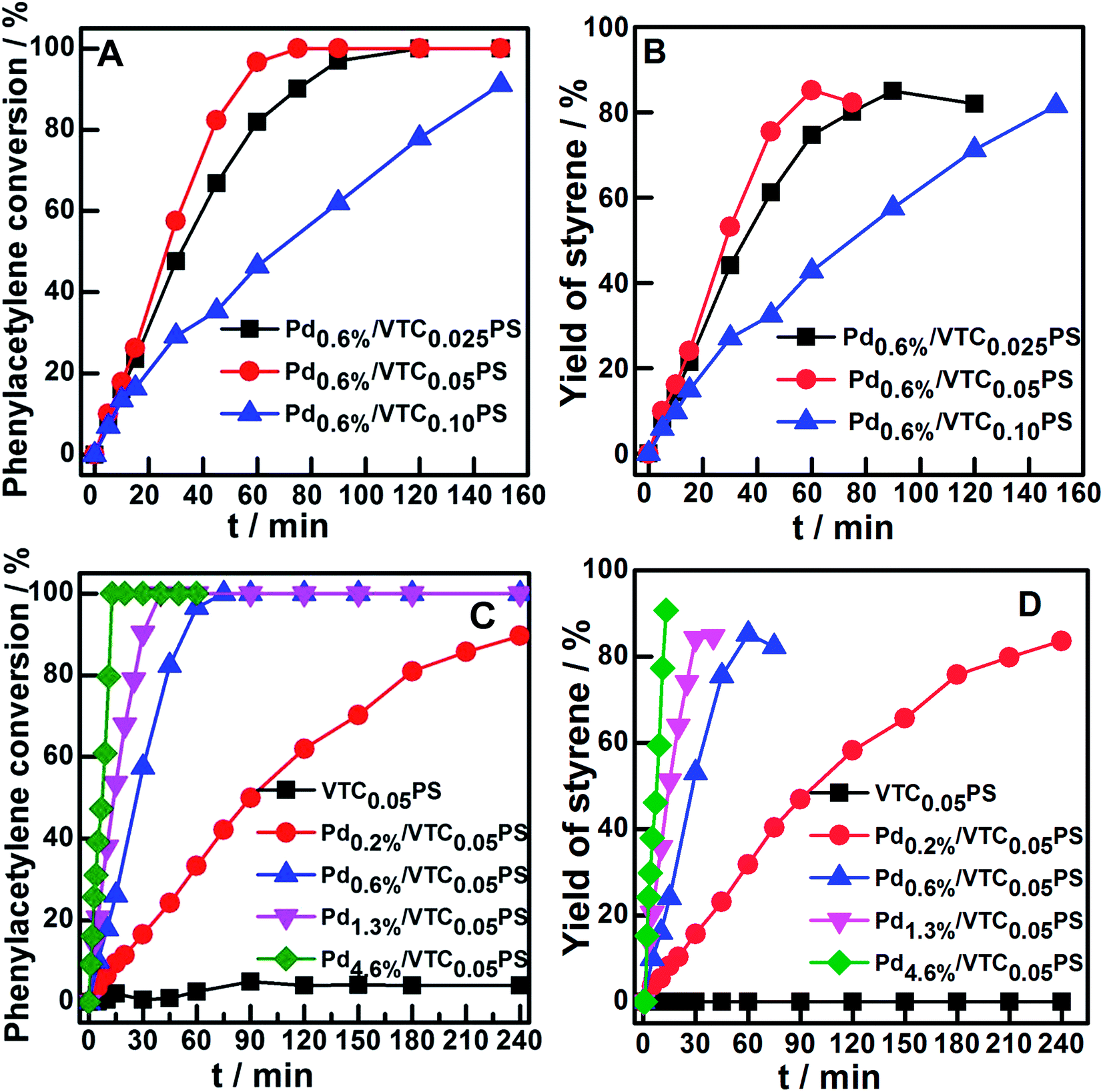 | ||
| Fig. 4 The conversion of phenylacetylene (A) and the yield of styrene (B) versus time on Pd0.6%/VTCyPS samples (y = 0.025, 0.05, and 0.10). The conversion of phenylacetylene (C) and the yield of styrene (D) versus time on VTC0.05PS and Pdx/VTC0.05PS (x = 0.2%, 0.6%, 1.3%, and 4.6%) samples. | ||
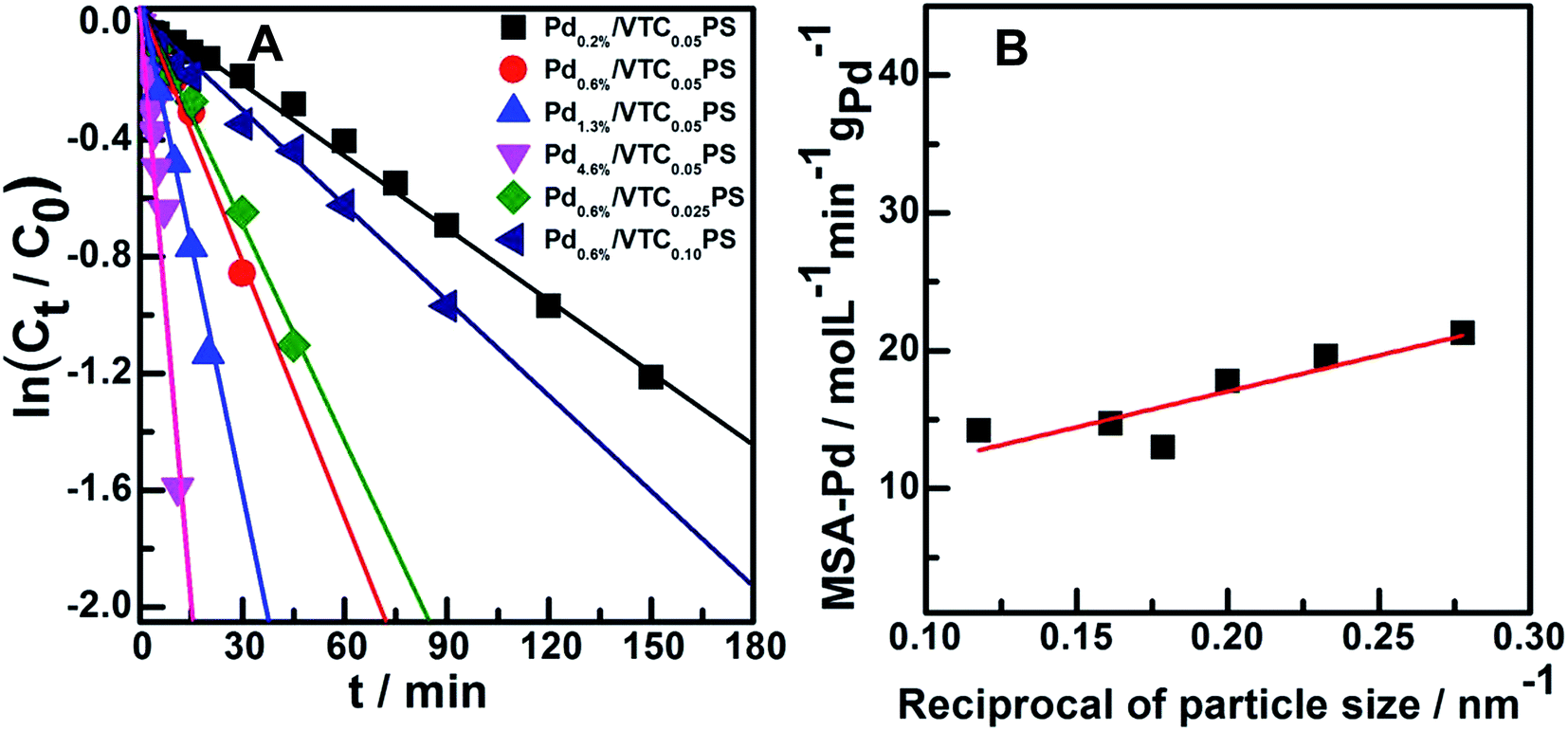 | ||
| Fig. 5 (A) Plot of ln(Ct/C0) versus reaction time for the selective hydrogenation of phenylacetylene on Pdx/VTCyPS samples. (B) The relation of mass-specific activity of Pd (MSA-Pd) for reaction with the reciprocal of Pd NPs particle size among the samples. | ||
| Catalyst | Size of Pda (nm) | Rate constant (k/min−1) | Reaction rateb (r/mol l−1 min−1) | MSA-Pdc (mol l−1 min−1 gPd−1) |
|---|---|---|---|---|
| a The size of Pd nanoparticles was measured from TEM images.b Reaction rate was adopted as the instant rate at the reaction time of 5 minutes.c The mass-specific activity of Pd nanoparticles (MSA-Pd = r/mPd) was obtained by normalizing the corresponding reaction rate to the mass of Pd used in the reaction. | ||||
| Pd0.2%/VTC0.05PS | 3.6 ± 1.3 | 0.008 | 8.2 × 10−4 | 21.3 |
| Pd0.6%/VTC0.05PS | 4.3 ± 1.5 | 0.028 | 2.3 × 10−3 | 19.6 |
| Pd1.3%/VTC0.05PS | 5.0 ± 1.6 | 0.056 | 4.7 × 10−3 | 17.8 |
| Pd4.6%/VTC0.05PS | 5.4 ± 1.8 | 0.137 | 8.9 × 10−3 | 13.0 |
| Pd0.6%/VTC0.025PS | 6.2 ± 2.5 | 0.025 | 1.8 × 10−3 | 14.7 |
| Pd0.6%/VTC0.10PS | 8.5 ± 4.1 | 0.010 | 1.8 × 10−3 | 14.2 |
| Catalysts | Preparation method | Pd particle size (ca.) | Reaction condition | Styrene yields | References |
|---|---|---|---|---|---|
| 0.21–0.42 wt% Pd/Fe3O4@SiO2 | Ion-exchange 300 °C reduced | 3.2–6.9 nm | 40 °C, 0.1 MPa, 105–150 min | 82–76% | 22 |
| 0.43–0.48 wt% Pd/α-Al2O3 | Impregnation 450 °C calcined | 4.4–15.0 nm | 50 °C, 0.1 MPa, 28 min | 78–73% | 23 |
| 1.0 wt% Pd/TiO2 | Impregnation 500 °C reduced | 3.2–5.9 nm | 30 °C, 0.5 MPa, 30–60 min | 95–60% | 24 |
| 0.70–0.90 wt% Pd/zeolite or MCM-41 | Colloid impregnation | 2.1–7.6 nm | 50 °C, 0.1 MPa, ≥300 min | 96% | 25 |
| 3.0 wt% Pd/SBA-15 | Impregnation microwave | 5.7 nm | 30 °C, 0.1 MPa, 150 min | 90% | 26 |
| 1.0 wt% Pd/C | Impregnation 150 °C calcined | Not given | 35 °C, 0.2 MPa, 10–30 min | 80–60% | 27 |
| 0.67–11.3 wt% Pd/Mpg-C3N4 | Deposition–precipitation | ≥3.3 nm | 30 °C, 0.1 Mpa, 60 min | 80–20% | 28 |
| 0.65–0.88 wt% Pd/Si–polyamine | Colloid deposition | 1.5–3.0 nm | 70 °C, 3.0 MPa, 15 min | 81–40% | 29 |
| 1.5–3.2 wt% Pd/MPF-SO3H | Impregnation | 3.3–4.8 nm | 80 °C, 1.0 MPa, 15 min | 80–35% | 30 |
| 0.20–4.6 wt% Pdx/VTC0.05PS | E-anchoring-reduction 50 °C | 3.6–5.4 nm | 40 °C, 0.1 MPa, 15–240 min | 92–80% | This work |
The stability of a typical catalyst (Pd0.6%/VTC0.05PS) was estimated by leaching experiment and recycle use test, as shown in Fig. 6. Fig. 6A compares the functions of phenylacetylene conversion with time between (a) Pd0.6%/VTC0.05PS being present during the entire reaction period and (b) Pd0.6%/VTC0.05PS being absent (removed) after 30 min of reaction. Two curves showed nearly overlapped change during 30 min of reaction; however, in contrast to the continuous increase in the phenylacetylene conversion observed in Pd0.6%/VTC0.05PS-containing mixture, nearly unchanged phenylacetylene conversion after 30 min of reaction was observed in Pd0.6%/VTC0.05PS-removed mixture. The result indicated that even if the leached metal species were present in the reaction liquid mixtures, these species would not act as a catalyst for the reaction. In addition, the reaction was repeated several times by employing fresh Pd0.6%/VTC0.05PS catalysts and all the liquid mixtures after the reaction were collected together and concentrated by evaporation. Then, the resulting liquid mixture was analyzed by ICP-OES and insignificant amount of metal components were observed; therefore, the leaching of the metal from Pd0.6%/VTC0.05PS catalysts in the reaction could be ignored in this study. Moreover, the catalytic performance of the reused Pd0.6%/VTC0.05PS catalyst was also investigated. Fig. 6B shows the conversion of phenylacetylene and the yield of styrene at 80 min employing the reused Pd0.6%/VTC0.05PS catalyst. Due to the severe loss of the solid substance during the separation process, four catalytic runs based on the collected catalysts were carried out after the fresh catalyst was used. In comparison with fresh catalyst, a slight fluctuation or decrease in the conversion of phenylacetylene and yield of styrene was observed in the subsequent catalytic runs employing reused catalysts, suggesting that our samples in the solid state acted as catalysts and can be reused for the reaction.
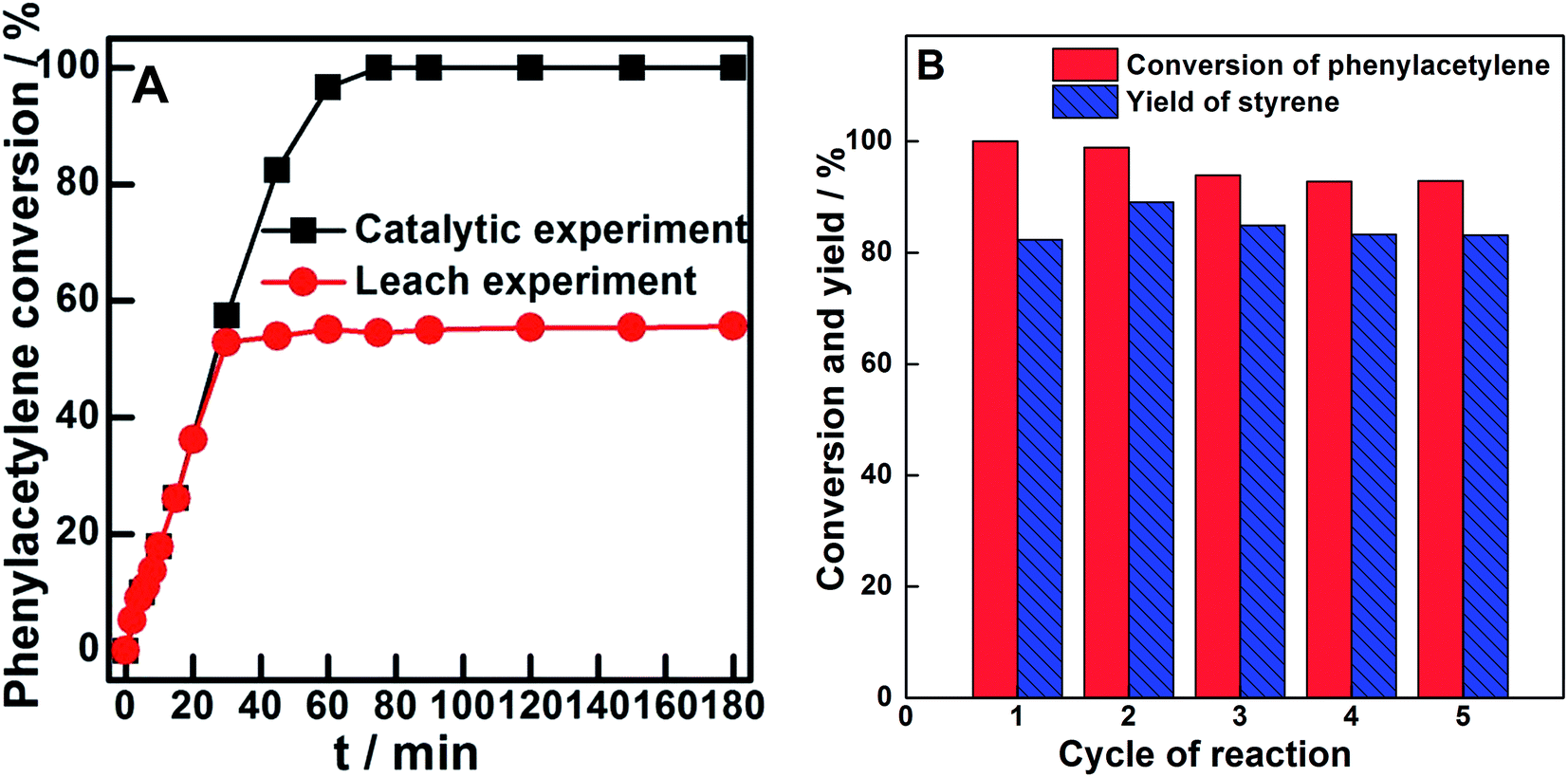 | ||
| Fig. 6 (A) Leaching experiment of Pd0.6%/VTC0.05PS sample and (B) recyclable catalytic ability of Pd0.6%/VTC0.05PS sample for the selective hydrogenation of phenylacetylene. | ||
4. Conclusions
The present method of deliberately embedding the electrostatic capturing group (for instance, VTC) in a proper amount on polystyrene for the uniform anchoring of PdCl42− and direct acquiring of protector-free Pd nanoparticles in one-pot system was demonstrated to be a feasible strategy for the preparation of clean and efficient Pd hydrogenation catalysts in the desired size range (ca. 3.0–5.0 nm) and broad Pd loading range (with 20 times change from 0.2 wt% to 4.6 wt%) in this study. The findings indicated that the outstanding flexibilities in both design and preparation of PS-like polymer materials among other popular supporters could be employed for conveniently synthesizing desired supported metal nanocatalysts. The advantages of the present strategy also suggested that it can serve as a promising system to prepare efficient precious metal catalysts for other important heterogeneous catalysis as well as other advanced applications that depend on size-controlled and clean metal nanoparticles.Acknowledgements
We gratefully acknowledge the financial support provided by the National Natural Science Foundation of China (NSFC, No. 21003071 and No. 21563018) and Doctoral Fund of Ministry of Education of China (No. 20093601120007).Notes and references
- T. Lopes, E. Antolini and E. R. Gonzalez, Int. J. Hydrogen Energy, 2008, 33, 5563–5570 CrossRef CAS.
- L. H. Yu and J. Y. Xi, Int. J. Hydrogen Energy, 2012, 37, 15938–15947 CrossRef CAS.
- L. C. Wang, Q. Liu, X. S. Huang, Y. M. Liu, Y. Cao and K. N. Fan, Appl. Catal., B, 2009, 88, 204–212 CrossRef CAS.
- S. Domínguez-Domínguez, Á. Berenguer-Murcia, B. K. Pradhan, Á. Linares-Solano and D. Cazorla-Amorós, J. Phys. Chem. C, 2008, 112, 3827–3834 Search PubMed.
- R. Chinchilla and C. Najera, Chem. Rev., 2014, 114(3), 1783–1826 CrossRef CAS PubMed.
- G. Vilé, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2015, 54, 11265–11269 CrossRef PubMed.
- J. A. Johnson, J. J. Makis, K. A. Marvin, S. E. Rodenbusch and K. J. Stevenson, J. Phys. Chem. C, 2013, 117, 22644–22651 CAS.
- A. Anastasopoulos, J. C. Davies, L. Hannah, B. E. Hayden, C. E. Lee, C. Milhano, C. Mormiche and L. Offin, ChemSusChem, 2013, 6, 1973–1982 CrossRef CAS PubMed.
- A. L. Isfahani, I. Mohammadpoor-Baltork, V. Mirkhani, A. R. Khosropour, M. Moghadam, S. Tangestaninejad and R. Kia, Adv. Synth. Catal., 2013, 355, 957–972 CrossRef CAS.
- Y. Li, E. Boone and M. A. El-Sayed, Langmuir, 2002, 18, 4921–4925 CrossRef CAS.
- Y. X. Li, Y. Wu, Y. Gao, S. S. Sha, J. F. Hao, G. Q. Cao and C. Yang, RSC Adv., 2013, 3, 26361–26366 RSC.
- Y. X. Li, Y. F. Pan, C. Yang, Y. Gao, Z. Q. Wang and G. Xue, Colloids Surf., A, 2012, 414, 504–511 CrossRef CAS.
- Y. G. Hu, T. Zhao, P. L. Zhu and R. Sun, Colloid Polym. Sci., 2012, 290, 401–409 CAS.
- J. Zhou, F. Ren, W. Wu, S. F. Zhang, X. H. Xiao, J. X. Xu and C. Z. Jiang, J. Colloid Interface Sci., 2012, 387, 47–55 CrossRef CAS PubMed.
- X. B. Jiang, W. Yu, S. J. Ding and B. Q. Li, Colloids Surf., A, 2013, 436, 579–588 CrossRef CAS.
- X. J. Cheng, L. Gao, J. P. Li, A. Q. Zhang and D. H. Zhang, Iran. Polym. J., 2012, 21, 335–341 CrossRef CAS.
- W. W. Wu, X. H. Yuan, S. S. Cao, Y. Ge, S. J. Li, Z. Y. Zhao and L. Fang, Aust. J. Chem., 2011, 64, 1541–1546 CrossRef CAS.
- T. Sun, Z. J. Wu, Q. Zhuo, X. Liu, Z. Wang and H. Y. Fan, Composites, Part A, 2014, 66, 58–64 CrossRef CAS.
- Y. X. Li, Z. F. Wu and S. J. Ye, New J. Chem., 2015, 39, 8108–8113 RSC.
- S. S. Cao, X. Jin, X. H. Yuan, W. W. Wu, J. Hu and W. C. Sheng, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1332–1338 CrossRef CAS.
- S. C. Hsiao, J. L. Ou, M. S. Huang, C. P. Chang, Y. Sung and M. D. Ger, Colloid Polym. Sci., 2010, 288, 1611–1619 CAS.
- L. Yang, X. L. Chen, Z. M. Zhou, R. Zhang, L. Li, Z. M. Cheng and X. C. Fang, ChemistrySelect, 2016, 1(18), 5599–5606 CrossRef CAS.
- J. W. Hu, Z. M. Zhou, R. Zhang, L. Li and Z. M. Cheng, J. Mol. Catal. A: Chem., 2014, 381, 61–69 CrossRef CAS.
- P. Weerachawanasak, O. Mekasuwandumrong, M. Arai, S. I. Fujita, P. Praserthdam and J. Panpranot, J. Catal., 2009, 262(2), 199–205 CrossRef CAS.
- S. Domínguez-Domínguez, Á. Berenguer-Murcia, Á. Linares-Solano and D. Cazorla-Amorós, J. Catal., 2008, 257(1), 87–95 CrossRef.
- D. Bhuyan, K. Selvaraj and L. Saikia, Microporous Mesoporous Mater., 2017, 241, 266–273 CrossRef CAS.
- S. D. Jackson and L. A. Shaw, Appl. Catal., A, 1996, 134(1), 91–99 CrossRef CAS.
- D. S. Deng, Y. Yang, Y. T. Gong, Y. Li, X. Xu and Y. Wang, Green Chem., 2013, 15(9), 2525–2531 RSC.
- E. Karakhanov, A. Maximov, Y. Kardasheva, V. Semernina, A. Zolotukhina, A. Ivanov, G. Abbott, E. Rosenberg and V. Vinokurov, ACS Appl. Mater. Interfaces, 2014, 6(11), 8807–8816 CAS.
- M. P. Boronoev, E. S. Subbotina, A. A. Kurmaeva, Y. S. Kardasheva, A. L. Maksimov and E. A. Karakhanov, Petrol. Chem., 2016, 56(2), 109–120 CrossRef CAS.
- D. Zhao, X. Xiong, C. L. Qu and N. Zhang, J. Phys. Chem. C, 2014, 118, 19007–19016 CAS.
- Z. Xu, H. M. Zhang, H. X. Zhong, Q. H. Lu, Y. F. Wang and D. S. Su, Appl. Catal., B, 2012, 111–112, 264–270 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |