
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyethyleneimine-mediated seed growth approach for synthesis of silver-shell silica-core nanocomposites and their application as a versatile SERS platform†
Chongwen Wang‡
ab,
Min Li‡bc,
Qingjun Li‡bc,
Kehan Zhangb,
Chaoguang Wangb,
Rui Xiao*b and
Shengqi Wang*abc
aCollege of Life Sciences & Bio-Engineering, Beijing University of Technology, Beijing 100124, PR China. E-mail: sqwang@bmi.ac.cn
bBeijing Institute of Radiation Medicine, Beijing 100850, PR China. E-mail: ruixiao203@sina.com
cHenan University of Chinese Medicine, Zhengzhou, Henan 450008, PR China
First published on 24th February 2017
Abstract
We synthesized silver-shell silica-core nanoparticles (SiO2@PEI@Ag NPs) with complete silver shells through the proposed polyethyleneimine (PEI)-mediated seed growth method. Cationic PEI rapidly self-assembled on the silica spheres through sonication to form a thin multifunctional interlayer, which can adsorb Au seeds densely and uniformly and stabilize the entire structure. Ag shell formation was completed within 2 min, and generated SiO2@PEI@Ag NPs were highly uniform in size and shape with nanoscale roughness. The improved seed growth method can be generally used to coat Ag shells of different thicknesses on the SiO2 cores with any particle size to form a well-dispersed core/shell nanostructure with high SERS activity. SiO2@PEI@Ag NPs of different sizes could be versatile SERS platforms for different applications. The potential of these particles as SERS substrates was verified by detection of the pesticide thiram and potential as SERS tags was verified by detection of human IgG, with detection limits as low as 10−9 M and 10 pg mL−1, respectively. Hence, SiO2@PEI@Ag NPs are powerful tools for practical SERS detection.
Introduction
Ag nanoshells have attracted extensive attention because of their outstanding chemical and optical properties and potential in the fields of catalysis,1 antibacterial,2 sensing,3 bioimaging,4 and surface-enhanced Raman scattering (SERS).5,6 Monodisperse nanoparticles with high-performance and controllable Ag shells should be used to attain good effectiveness. The use of dielectric silica nanoparticles (SiO2 NPs) as core templates for fabricating core–shell composite spheres with Ag nanoshells (SiO2@Ag) is an efficient strategy because of their uniform spherical shape, high stability, easy preparation, low cost, and large particle size range.7 However, direct coating of Ag shell on the bare SiO2 core is difficult because of mutual incompatibility between these materials; therefore, the SiO2 core should be functionalized with special functional groups to increase the degree of surface coverage or change its surface properties.8Over the past years, scholars have developed various methods for the synthesis of SiO2@Ag NPs with controllable morphology; these methods include electroless plating,9 layer by layer (LBL),10 direct deposition,11 and seed growth-mediated methods.12 The SnCl2-mediated electroless plating method, which was proposed by Liz-Marzán et al., can be used to homogeneously deposit Ag NPs on the SiO2 core but requires repeated adsorption of Sn2+ and Ag+ ions onto the surface of the silica particles.13 LBL method readily produces Ag nanoshells by stacking Ag NPs on silica core surfaces through electrostatic interaction; this method is time consuming because of multistep deposition and difficulty in producing complete Ag shells.14 Several attempts have been made to directly deposit Ag NPs on SiO2 particles by soaking them in ethanolic solutions of AgNO3 and butylamine;15 the results are unsatisfactory, that is, complete and controllable thickness Ag shells were not produced on the core surface. Of these methods, seed-mediated growth method is the most promising approach to grow uniform and complete Ag nanoshells by adding new atoms onto the existing nuclei.16 Therefore, additional presynthetic steps are required for absorption or deposition of metal seeds on the silica core surface before subsequent growth of Ag shells. To date, amine-functionalized silica particles have been widely investigated because amine groups act as attachment points for small Au or Ag seeds; silane coupling agents are commonly used for synthesis of amino-modified silica spheres.17,18 However, full surface amino modification is sometimes difficult to achieve; thus uniform and dense seed absorption on the core particles cannot be achieved, eventually affecting the complete Ag shell formation.
Basing on our previous studies,19–21 we proposed a polyethyleneimine (PEI)-mediated seed growth method to achieve highly reproducible and controllable coating silica spheres with complete Ag shells in this study. PEI was used to form a bifunctional thin interlayer to achieve the absorption of dense Au NPs as seeds and keep the nanostructure stable during shell growth. The entire reaction process for Ag shell formation was completed within 2 min; the resulting SiO2@PEI@Ag NPs were highly uniform in size and shape with high SERS activity. The SERS activities of the as-prepared SiO2@PEI@Ag NPs were tested using p-aminothiophenol (PATP) as probe molecule. Moreover, Ag shells of different thicknesses easily formed outside the silica cores of different sizes depending on different detection demands. On one hand, high-sensitive detection of the pesticide thiram was achieved using large-sized SiO2@PEI@Ag NPs (300 nm core) as SERS substrates at a detection limit of 10−9 M, which is lower than the standard safety value (7 ppm) prescribed by the U.S. Environmental Protection Agency (EPA).22 On the other hand, small-sized SiO2@PEI@Ag NPs (80 nm core) as high-quality SERS tags were successfully applied to SERS-based immunoassay for human immunoglobulin (IgG) detection with a low detection limit of 10 pg mL−1. Hence, SiO2@PEI@Ag NPs can be potentially used as effective and versatile SERS tools in practical applications, including pollutant detection, food safety, and immunodetection.
Methods
Materials and chemicals
Silver nitrate (AgNO3), chlorauric acid tetrahydrate (HAuCl4·4H2O), ammonia solution (28%), and formaldehyde (37%) were purchased from Sinopharm Chemical Reagent Co. Branched PEI (MW 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), tetraethoxysylane (TEOS), polyvinylpyrolidone (PVP, MW 40000), aminothiophenol PATP, 2-nitrobenzoic acid (DTNB), 11-mercaptoundecanoic acid (MUA), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), thiram, human IgG, and goat anti-human IgG were purchased from Sigma-Aldrich. All other chemicals were purchased from Shanghai Chemical Reagent Co., Ltd. Ultrapure water with resistivity of 18.2 MΩ cm−1 was used to prepare aqueous solutions.
000), tetraethoxysylane (TEOS), polyvinylpyrolidone (PVP, MW 40000), aminothiophenol PATP, 2-nitrobenzoic acid (DTNB), 11-mercaptoundecanoic acid (MUA), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), thiram, human IgG, and goat anti-human IgG were purchased from Sigma-Aldrich. All other chemicals were purchased from Shanghai Chemical Reagent Co., Ltd. Ultrapure water with resistivity of 18.2 MΩ cm−1 was used to prepare aqueous solutions.
Preparation of high-performance SiO2@PEI@Ag NPs
SiO2 NPs were prepared according to a modified Stöber method, which involves the ammonia-catalyzed hydrolysis and condensation of TEOS in a mixture of ethanol and water.17 The size of SiO2 NPs can be controlled by modulating the molar ratio of TEOS, water, and ammonia. The SiO2@PEI NPs were synthesized through a PEI self-assembly process under sonication condition. First, 0.5 g PEI was dissolved in 100 mL of deionized water by ultrasonication for 10 min. Next, 0.1 mL of prepared SiO2 NPs (10 mg mL−1) was dispersed in the PEI solution under sonication at room temperature for 1 h, during which PEI gradually self-assembled on the silica cores. Then, the obtained SiO2@PEI NPs were centrifuged and rinsed for five times with deionized water to remove the excess PEI. Colloidal 3–5 nm Au NPs were prepared according to the method of YouXing Fang.23 After preparation, PEI-modified SiO2 NPs were mixed with 3–5 nm Au NPs and sonicated for 30 min to form SiO2@PEI–Au seed NPs. Briefly, 5 mg of SiO2@PEI–Au seed was dispersed in 200 mL of 0.2 mM silver nitrate aqueous solution containing 0.2 wt% PVP. The mixture was added with excess amounts of 37% formaldehyde (150 μL) and 28% ammonia solution (300 μL) in sequence. The SiO2@PEI@Ag core–shell NPs were obtained within 2 min under sonication at 30 °C. The products were centrifuged and re-dispersed with deionized water to 1 mg mL−1.SERS detection of thiram
In a typical experiment, a standard thiram solution from 10−5 M to 10−10 M was prepared using a thiram stock solution. For the SERS measurement, 500 μL of thiram solution of different concentrations was mixed with 50 μL of SiO2@PEI@Ag NPs (300 nm core) in 1.5 mL centrifuge tube, and the mixture was incubated under sonication for 1 h. Then, SiO2@PEI@Ag NPs was separated by centrifugation and rinsed twice with ethanol to remove excess thiram. After washing, the precipitate was resuspended in 5 μL of ethanol. The suspension was dropped on a silicon substrate and air dried. The SERS spectra of the sample were then recorded.Preparation of SiO2@PEI@Ag NPs SERS tags for the SERS immunoassay
Briefly, 100 μL of 10 mM DTNB was added to 10 mL of the as-prepared SiO2@PEI@Ag NPs (80 nm) solution. The mixture was vigorously stirred at 30 °C for 2 h. After purification (5000 rpm for 5 min), the SiO2@PEI@Ag/DTNB was linked with goat anti-human IgG (10 μL, 10 mg mL−1) by covalent bonding through the EDC/NHS chemistry. For blocking the unreacted carboxyl sites on the SiO2@PEI@Ag surface, 100 μL of BSA (10%, v/v) was added to the mixture and allowed to react for an additional 1 h. To remove any excess unbound antibody, the solution was centrifuged at 4000 rpm for three times. Finally, SiO2@PEI@Ag SERS tags were obtained by re-dispersing the precipitates into 5 mL of phosphate-buffered saline (PBS, 10 mM, pH 7.4) and stored at 4 °C before use.Preparation of immunomagnetic beads
Magnetic microspheres (a-Fe2O3, 1.5 μm) were synthesized following the method proposed by Gan et al.24 Briefly, 100 mg of the as-prepared Fe2O3 particles were dispersed in a deionized water–ammonia–ethanol mixture (10 mL/8 mL/180 mL) by sonication for 10 min to prepare a-Fe2O3@SiO2 microspheres. Subsequently, 100 μL of TEOS in ethanol was gradually dropped into the solution by vigorous sonication at room temperature for 60 min. The obtained a-Fe2O3@SiO2 microspheres were collected using a magnet, followed by washing with deionized water and ethanol for three times each.For preparation of immunomagnetic beads, a-Fe2O3@SiO2 microspheres were carboxyl group-functionalized and conjugated with antibody according to a previous method reported by Li et al.25,26 Briefly, 100 μL of triethoxysilylpropyl succinic anhydride (TEPSA) was added to 10 mL of the a-Fe2O3@SiO2 microsphere aqueous solution and incubated overnight. Carboxyl-terminated a-Fe2O3@SiO2 microspheres were obtained after magnetic separation and redispersed in 1 mL of PBS solution (10 mM, pH 7.4). Subsequently, EDC (100 μL, 10 mM) and NHS (20 μL, 100 mM) were added to 100 μL of the carboxylated a-Fe2O3@SiO2 microspheres (5 mg mL−1) solution. The mixture was added with PBS buffer (10 mM, pH 7.4) until the total volume reached 1 mL. The mixture was shaken for 20 min (800 rpm, 37 °C), added with 5 μL of goat anti-human IgG (10 mg mL−1 in PBS), and shaken for another 1 h. The products were magnetically separated and rinsed three times with PBS (10 mM, pH 7.4) to remove excess antibodies. The antibody-conjugated a-Fe2O3@SiO2 microspheres were blocked with 100 μL of BSA (10%, v/v) for 1 h under the same shaking condition to avoid nonspecific absorption. The resulting immunomagnetic beads were stored at 4 °C before use.
Immunoassay protocol
In a typical experiment, 500 μL of PBS buffer containing the target human IgG at various concentrations was prepared and incubated with 10 μL of the capture antibody-conjugated immunomagnetic beads for 30 min at 37 °C with gentle shaking. After immunocapture of the target human IgG, the immune complexes were magnetically collected in the mixture by applying an external magnet and washed twice with PBS containing Tween-20 buffer (PBST, 0.05%, v/v). Subsequently, 200 μL of the SiO2@PEI@Ag SERS tags were added and incubated for another 45 min at 37 °C with gentle shaking. The resulting sandwich immune complexes were washed three times with PBST (0.05%, v/v), resuspended in 2 μL of deionized water, and transferred to a silicon substrate for SERS measurements.Instruments
Transmission electron microscopy (TEM) images were recorded on a Hitachi H-7650 microscope at an accelerating voltage of 200 kV. The samples for HRTEM imaging recorded on a Philips Tecnai G2 F20 microscope equipped with energy-dispersive X-ray analysis system. Scanning electron microscopy (SEM) analysis was performed with a JEOL JSM-7001F microscopy at an accelerating voltage of 5 kV. Elemental mapping images were acquired by energy-dispersive X-ray spectroscopy (EDS) using another JEOL JEM-2100 electron microscope equipped with a STEM unit. Powder X-ray diffraction (XRD) patterns of the products were investigated on a Japan Rigaku D/max 2550 VB/PC rotation anode X-ray diffractometer. UV-visible spectra were recorded with a Shimadzu 2600 spectrometer. Zeta potential was measured by dynamic light scattering with Zetasizer Nano ZS (Malvern, UK). Raman measurement was performed at room temperature on a Renishaw inVia plus Raman system with a 785 nm excitation wavelength. Incident radiation was coupled into an Olympus BX51 optical microscope and focused to a 2 μm-diameter spot through a 50× objective. Data acquisition time was usually 1 s, and the peak intensities of the samples were normalized with respect to the silicon wafer at 520 cm−1.Results and discussion
Characterization of SiO2@PEI@Ag nanoparticles
Fig. 1a shows the fabrication of high-performance SiO2@PEI@Ag NPs. The first step is the formation of the SiO2 NPs used as cores through Stöber method. Second, SiO2@PEI NPs were prepared with the cationic PEI coated on the silica core under sonication. PEI possesses plenty of primary amine groups and positive charges; thus, PEI can assemble quickly on the surface of the negatively charged particles to achieve surface amino group functionalization. Third, dense negatively charged 3–5 nm Au NPs were absorbed on the surface of SiO2@PEI NPs as seeds by positive electricity of PEI. These small Au NPs were attached firmly to SiO2@PEI NPs through sonication because of the covalent binding among the –NH2 groups of the PEI and Au NPs. Finally, high-performance SiO2@PEI@Ag NPs were quickly obtained by adopting our previously reported seed-mediated growth method.19 The complete Ag shells were formed within several seconds through the isotropic growth of all Au seeds and the stabilization brought about by PVP. | ||
| Fig. 1 Schematic of synthesis of SiO2@PEI@Ag NPs (a). TEM images of (b) SiO2@PEI NPs, (d) SiO2@PEI–Au seed, (f) SiO2@PEI@Ag NPs, and their corresponding magnified TEM images in (c), (e), and (g), respectively. (h) HRTEM picture of a part of Ag shell in (g), and (i) SEM image of SiO2@PEI@Ag NPs. The inset in (c) is the corresponding magnified HRTEM image of its edge (arrows indicate a thin PEI shell). | ||
TEM and SEM analyses were employed to examine the morphology of the as-obtained samples in different stages. Monodispersed SiO2 NPs (80 nm) were prepared using the modified Stöber method, and the SiO2 NPs were uniform with smooth surface (Fig. S1†). The hydrophilic polymer PEI self-assembled easily on the surface of SiO2 NPs by sonication and could not affect the dispersity of SiO2 NPs (Fig. 1b). The HRTEM image confirms the uniform coating of PEI with 1 nm thickness (Fig. 1c). After the SiO2@PEI with 3–5 nm Au seed was coated, the TEM image in Fig. 1d shows that the dense small Au NPs spread uniformly on the surface of the SiO2@PEI NPs. The Au seeds on the SiO2 NPs served as nucleation sites for subsequent fabrication of the complete Ag shell. Fig. 1f shows the TEM image of the prepared SiO2@PEI@Ag NPs, and the size of the particles increased from 80 nm to approximately 100 nm. The thickness of the Ag shell was approximately 10 nm. A strong contrast difference between the magnified TEM images in Fig. 1e (discontinuous dots) and Fig. 1g (continuous shell) was observed, indicating that the complete Ag shell was formed successfully on SiO2 NPs. The HRTEM image of a part of the Ag shell is shown in Fig. 1h; the space between two crystal planes is 0.236 nm, corresponding to the (111) plane of Ag with face-centered cubic (fcc) phase.27 To investigate the structure of the shell, we characterized the obtained samples by SEM. The SEM image (Fig. 1i) shows that numerous large Ag NPs covered the entire surface of the nanocomposite, forming a continuous shell with nanoscale roughness.
SiO2 NPs function as supporting core that effectively stabilizes nanocomposites in the solution to prevent aggregation; the as-obtained products at different stages exhibited excellent water dispersibility (Fig. 2a). As shown in Fig. 2b, the SiO2@PEI–Au seed displayed an absorption peak at about 528 nm (curve c), whereas SiO2 and SiO2@PEI NPs showed no characteristic absorption peaks (curves a and b); this finding suggests that large amounts of Au NPs were successfully absorbed on the SiO2 NPs. After the Ag shell was formed, an obvious broad plasmonic resonance peak appeared at approximately 384 nm because of the Mie plasmonic resonance from the Ag NPs (curve d).28 The zeta potentials of SiO2@PEI@Ag NPs were measured at different stages of synthesis. Fig. 2c shows that the zeta potentials of the SiO2 NPs increased from −35.7 mV to +45.2 mV when PEI was modified. This change implies that positively charged PEI completely assembled onto the surface of SiO2 particles. After coating of the Au seeds and the formation of the Ag shell, the zeta potential became negative again. The detailed data of the zeta potentials are shown in Fig. S2.†
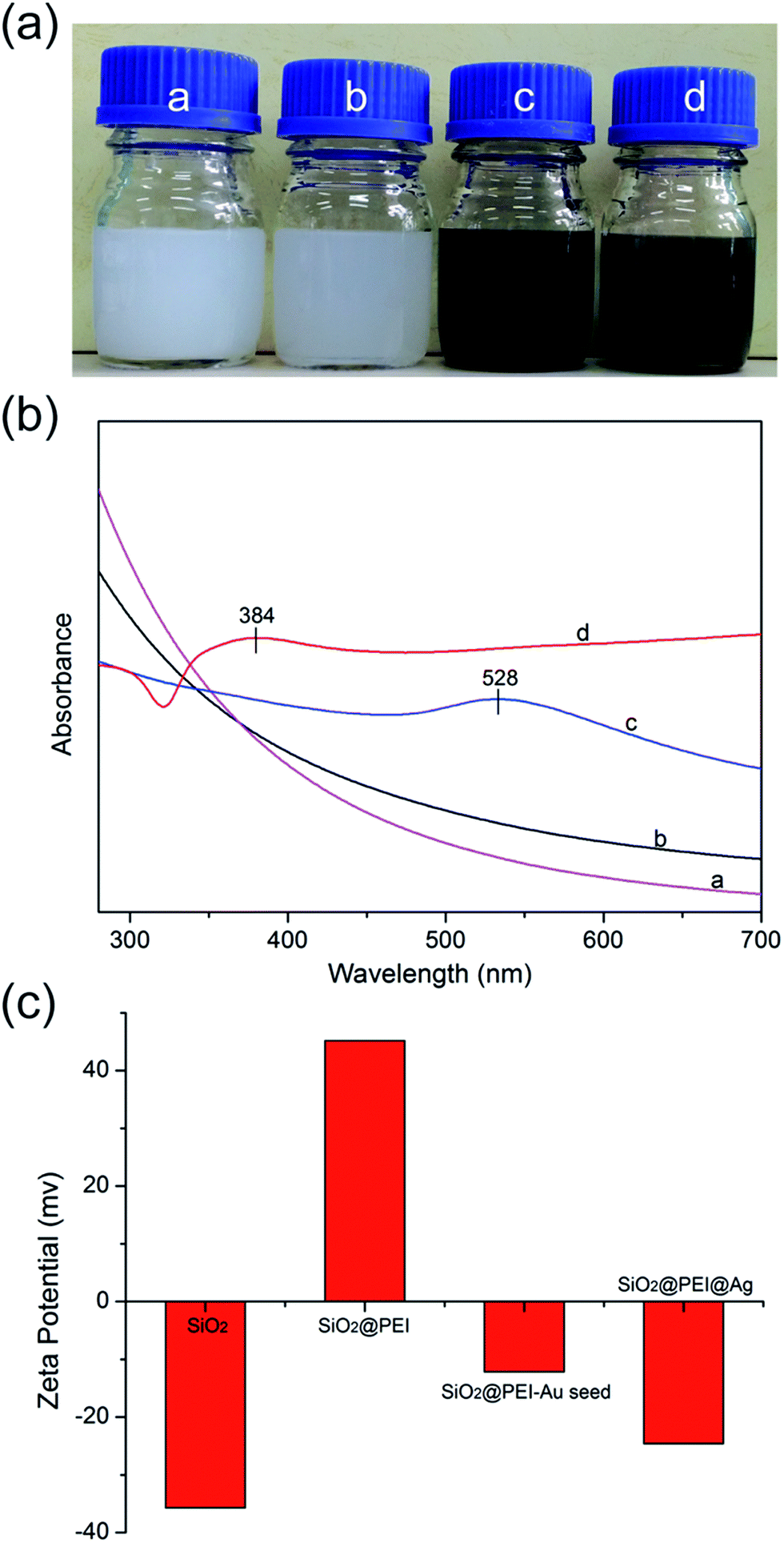 | ||
| Fig. 2 Photographs (a), UV-vis spectra (b), and statistical results of the zeta potentials (c) of the as-obtained samples during different stages: a-SiO2, b-SiO2@PEI, c-SiO2@PEI–Au seed, and d-SiO2@PEI@Ag NPs. | ||
Powder XRD was applied to evaluate the crystalline properties of the as-prepared samples. As displayed in Fig. 3a, the synthesized SiO2@PEI–Au seed exhibited X-ray diffraction peaks, which could be indexed to the cubic structure of the Au crystals (JCPDS No. 04-0784).29 No diffraction peaks corresponding to SiO2 and PEI were observed because of their amorphous form. Further examination of the SiO2@PEI@Ag NPs showed no evident variation in peak positions, but a pronounced increase in intensity was observed when the Ag shells were formed. Four diffraction peaks were observed at 38.1°, 44.2°, 64.4°, and 77.4°, which correspond to the (111), (200), (220), and (311) planes of Ag with the fcc phase, respectively.30 This finding is acceptable because the Au and Ag nanocrystals showed minimal differences in the XRD pattern.31 X-ray elemental mapping analysis was performed to provide additional insights into the definite morphology and formation mechanism of SiO2@PEI@Ag NP synthesis. Fig. 3c–f shows the elemental mapping of a single SiO2@PEI@Ag NP. The Si and C elements are distributed throughout the particles, whereas the Au and Ag signals are mainly enriched on the surface of the nanocomposites.
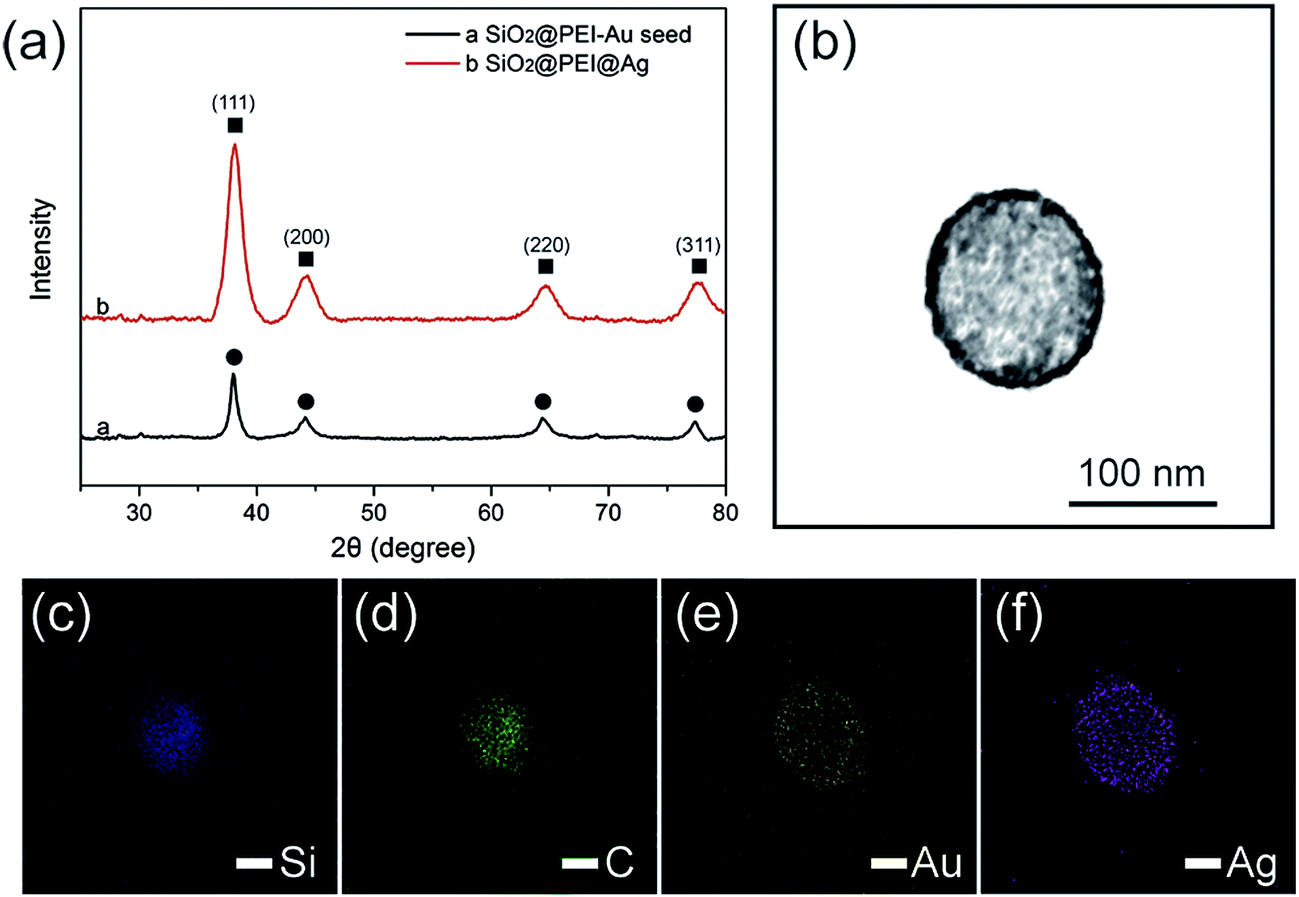 | ||
| Fig. 3 (a) Typical XRD patterns of the as-synthesized SiO2@PEI–Au seed and SiO2@PEI@Ag NPs. The circles label the peaks of cubic Au, whereas the squares label the peaks of cubic Ag. (b) Bright-field TEM image of a single SiO2@PEI@Ag NP and (c–f) the associated elemental mappings showing the element distributions of Si (c, blue), C (d, green), Au (e, yellow), and Ag (f, violet) in the nanocomposite. | ||
The strategy of Ag shell formation involves seed-mediated growth. Uniform Ag shells were produced by a one-step redox reaction in the presence of small and dense Au NPs absorbed on the SiO2 NPs. When ammonia was added, Ag+ was reduced by a general reductant (CH2O) within several seconds and was deposited on the Au seeds of SiO2@PEI, resulting in a discontinuous Ag NPs shell. With increasing AgNO3 concentration, the Ag NPs gradually became large, finally intersecting with each other and forming a continuous Ag shell. Notably, PVP plays important roles in this process. PVP covers the facets of the Au seeds; PVP not only prevents the aggregation of nanoparticles but also facilitates the isotropic growth of Ag particles to form uniform Ag shells.
Based on our proposed PEI-mediated seed growth method, controlling the size and coating condition of Ag shells can be easily realized by changing the concentration of AgNO3. Fig. 4a–e shows the morphologies of the SiO2@PEI@Ag NPs synthesized with different concentrations of AgNO3 while the other parameters remained constant. When low concentrations of AgNO3 (such as 0.05 or 0.1 mM) was added, Ag+ was reduced on the surface of the absorbed Au seeds but not in amounts sufficiently large to form a continuous shell Fig. 4b and c. When the concentration of AgNO3 was increased to 0.2 mM, the resulting Ag shell became slightly rough but basically continuous (Fig. 4d). Furthermore, the Ag shell became absolutely continuous when the AgNO3 concentration reached 0.3 mM (Fig. 4e). These experimental results indicate that the coverage and thickness of the Ag shells can be well controlled by increasing the concentration of AgNO3 from 0 mM to 0.3 mM while keeping all other parameters fixed.
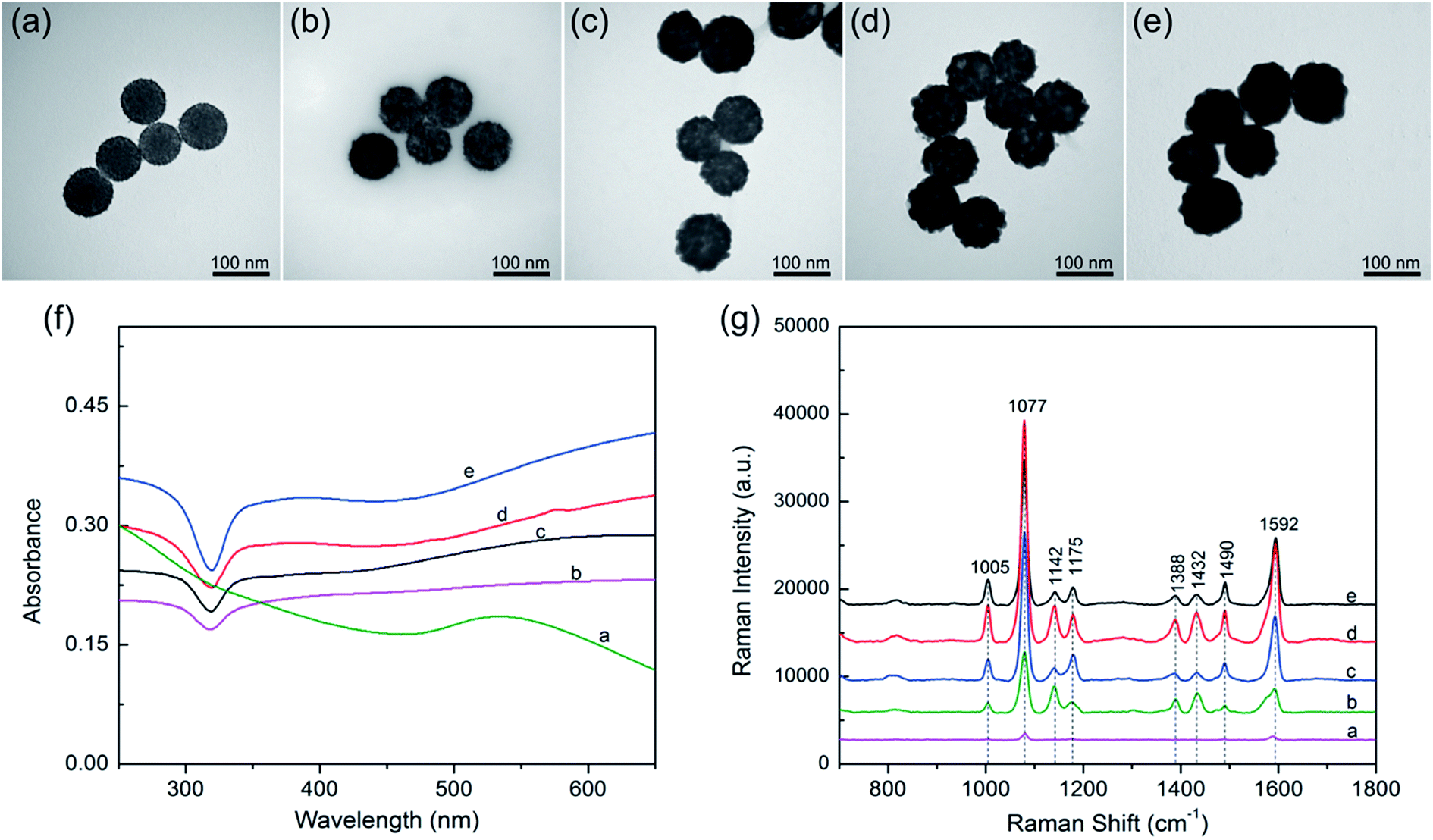 | ||
| Fig. 4 (a–e) TEM images of SiO2@PEI@Ag synthesized with different concentrations of AgNO3. (a) 0, (b) 0.05, (c) 0.1, (d) 0.2, and (e) 0.3 mM. (f) UV-visible spectra of SiO2@PEI@Ag synthesized with different concentrations of AgNO3: (a) −0, (b) −0.05, (c) −0.1, (d) −0.2, and (e) −0.3 mM, and their corresponding Raman spectra of PATP (g). | ||
The UV-vis spectra of the SiO2@PEI@Ag NPs synthesized with different concentrations of AgNO3 are shown in Fig. 4f. All products were diluted with deionized water for adsorption spectrum measurement. The broad absorption peak of Ag shell was observed from the SiO2@PEI@Ag, likely due to the coupling between neighboring Ag nanoparticles inside the shell region, and large scattering from the SiO2 and Ag shell.32 Similar observation was also reported by Liu and co-workers.12 Moreover, as the Ag shells on the SiO2 cores became continuous and thick, the plasmon resonance peaks slightly red-shifted, and the peak width became narrower. The change in plasmon resonance peak can be explained by plasmon resonance excitation from the close Ag NPs; this phenomenon indicates that the Ag shell changed from incomplete to complete.
SERS was first observed in the 1970s on an electrochemically roughened Ag electrode, which showed an anomalous millionfold enhancement in Raman scattering intensity.33 Ag NPs and Ag nanoshells enjoy a reputation as an ultrasensitive substrate for SERS.34 Considering the Ag shell with nanoscale roughness and controllable morphology, these SiO2@PEI@Ag NPs must be efficient SERS substrates under the irradiation of a visible laser. We evaluated the SERS activity of SiO2@PEI@Ag NPs by using PATP as the model SERS probe. According to the theory proposed by Wu et al., the PATP molecules adsorbed on the nanoscale Ag surfaces undergo catalytic coupling reaction to selectively produce a new surface species p,p′-dimercaptoazobenzene (DMAB), an aromatic azo compound.35,36 The Raman shifts at 1432 and 1388 cm−1 are related to the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibrations of DMAB, whereas typical SERS peaks of PATP are primarily located at 1592 and 1077 cm−1. As revealed in Fig. 4g, the SiO2@PEI–Au seed possesses the lowest Raman intensities because of the presence of small Au NPs on the silica surface (curve a). The SERS activity of SiO2@PEI@Ag NPs was gradually enhanced as the AgNO3 concentration increased (curves b–d) because the Ag shell thickness and coverage increased through subsequent growth. However, the overgrowth of Ag shell decreased the SERS activity of SiO2@PEI@Ag NPs (curve e) because the nanogaps on the Ag shells disappeared. By contrast, we can conclude that the SiO2@PEI@Ag NPs with a nearly complete Ag shell prepared with 0.2 mM AgNO3 were the most efficient SERS active substrates in series. Furthermore, we examined the detection sensitivity of the SiO2@PEI@Ag NPs (0.2 mM AgNO3) for detection of PATP with different concentrations. As shown in Fig. S3a,† the limit of detection (LOD) for PATP with SiO2@PEI@Ag NPs was as low as 1 × 10−10 M. The accumulation time was set at 1 s and the incident power at 1% of the laser power when recording the Raman spectra. We conducted five tests for each sample, and the results were statistically analyzed to obtain an intensity–concentration calibration curve (polynomial fitting); the curve shows the plot of the intensity of the SERS peak at 1077 cm−1 versus the logarithmic concentration of the sample (Fig. S3b†). Furthermore, the SERS enhancement factors (EF) for PATP were roughly estimated by an adjusted method, as described in detail in ESI S4.† The EF value of the peak at 1077 cm−1 was estimated to be approximately 6.53 × 107.
N stretching vibrations of DMAB, whereas typical SERS peaks of PATP are primarily located at 1592 and 1077 cm−1. As revealed in Fig. 4g, the SiO2@PEI–Au seed possesses the lowest Raman intensities because of the presence of small Au NPs on the silica surface (curve a). The SERS activity of SiO2@PEI@Ag NPs was gradually enhanced as the AgNO3 concentration increased (curves b–d) because the Ag shell thickness and coverage increased through subsequent growth. However, the overgrowth of Ag shell decreased the SERS activity of SiO2@PEI@Ag NPs (curve e) because the nanogaps on the Ag shells disappeared. By contrast, we can conclude that the SiO2@PEI@Ag NPs with a nearly complete Ag shell prepared with 0.2 mM AgNO3 were the most efficient SERS active substrates in series. Furthermore, we examined the detection sensitivity of the SiO2@PEI@Ag NPs (0.2 mM AgNO3) for detection of PATP with different concentrations. As shown in Fig. S3a,† the limit of detection (LOD) for PATP with SiO2@PEI@Ag NPs was as low as 1 × 10−10 M. The accumulation time was set at 1 s and the incident power at 1% of the laser power when recording the Raman spectra. We conducted five tests for each sample, and the results were statistically analyzed to obtain an intensity–concentration calibration curve (polynomial fitting); the curve shows the plot of the intensity of the SERS peak at 1077 cm−1 versus the logarithmic concentration of the sample (Fig. S3b†). Furthermore, the SERS enhancement factors (EF) for PATP were roughly estimated by an adjusted method, as described in detail in ESI S4.† The EF value of the peak at 1077 cm−1 was estimated to be approximately 6.53 × 107.
The proposed PEI-mediated seed growth method can be generally used to synthesize various Ag-coated silica core–shell NPs, ranging from nanoscale to microscale levels. Fig. 5a–c show the TEM images of SiO2 NPs with different sizes (120–300 nm), whereas Fig. 5d–f and Fig. 5g–i show their corresponding fabricated SiO2@PEI–Au seed and SiO2@PEI@Ag NPs, respectively. The obtained SiO2@PEI@Ag NPs with different sizes possess good dispersity, uniform Ag shells, and highly reproducible structure; these particles can be used for different purposes. For example, large-size SiO2@PEI@Ag NPs (300–500 nm) can act as high-performance SERS substrates, and small NPs (60–120 nm) can be used as effective SERS tags.
 | ||
| Fig. 5 (a–c) TEM images of SiO2@PEI NPs with different sizes: (a) 120, (b) 200, and (c) 300 nm, and their corresponding fabricated SiO2@PEI–Au seed (d), (e), and (f), and SiO2@PEI@Ag NPs in (g), (h), and (i), respectively. | ||
We selected large SiO2@PEI@Ag NPs (SiO2 300 nm, Ag shell 50 nm) as SERS substrate because these particles are large enough to be completely separated by centrifugation and provide sufficient enhancement for direct detection. As illustrated in Scheme 1a, the target analyte with specific affinity could be enriched with SiO2@PEI@Ag NPs; the formed complexes were separated by centrifugation and dropped on a silicon chip for SERS measurement. We detected the Raman signals of trace thiram, a widely used sulfur-containing fungicide,37,38 to prove the practicability of the SiO2@PEI@Ag NPs. The Raman mapping of the thiram at the 1383 cm−1 band confirms the good reproducibility and reliability of Raman detection with SiO2@PEI@Ag NPs as SERS substrate (Fig. 6a). Fig. 6b shows the SEM image of the SiO2@PEI@Ag NPs covered Si chips from the square area of the 2D-Raman map. Fig. 6c shows the SERS spectra of thiram in ethanol, with concentrations ranging from 1.0 × 10−5 M to 1.0 × 10−10 M with SiO2@PEI@Ag NPs (300 nm core) as SERS substrate; the intensity of the Raman peaks of thiram located at 932, 1148, 1383, and 1508 cm−1 were observed at concentrations as low as 10−8 M. The most prominent peak around 1385 cm−1 was observed at concentrations as low as 10−9 M. All of the major vibrational modes of thiram observed are consistent with previous reports,39 as categorized in Table S1.† The detection limit of thiram in the experiment (10−9 M) is lower than the maximum residue limit of 7 ppm (0.03 mM) in fruit, as prescribed by the U.S. EPA. The detection is based on the main Raman peak (1383 cm−1) and then quantified by its intensity. We conducted five tests for each of the samples and conducted statistical analysis of the results to obtain an intensity–concentration calibration curve (polynomial fitting), which plots the intensity of the SERS peak at 1383 cm−1 versus the logarithmic concentration of the sample (Fig. 6d).
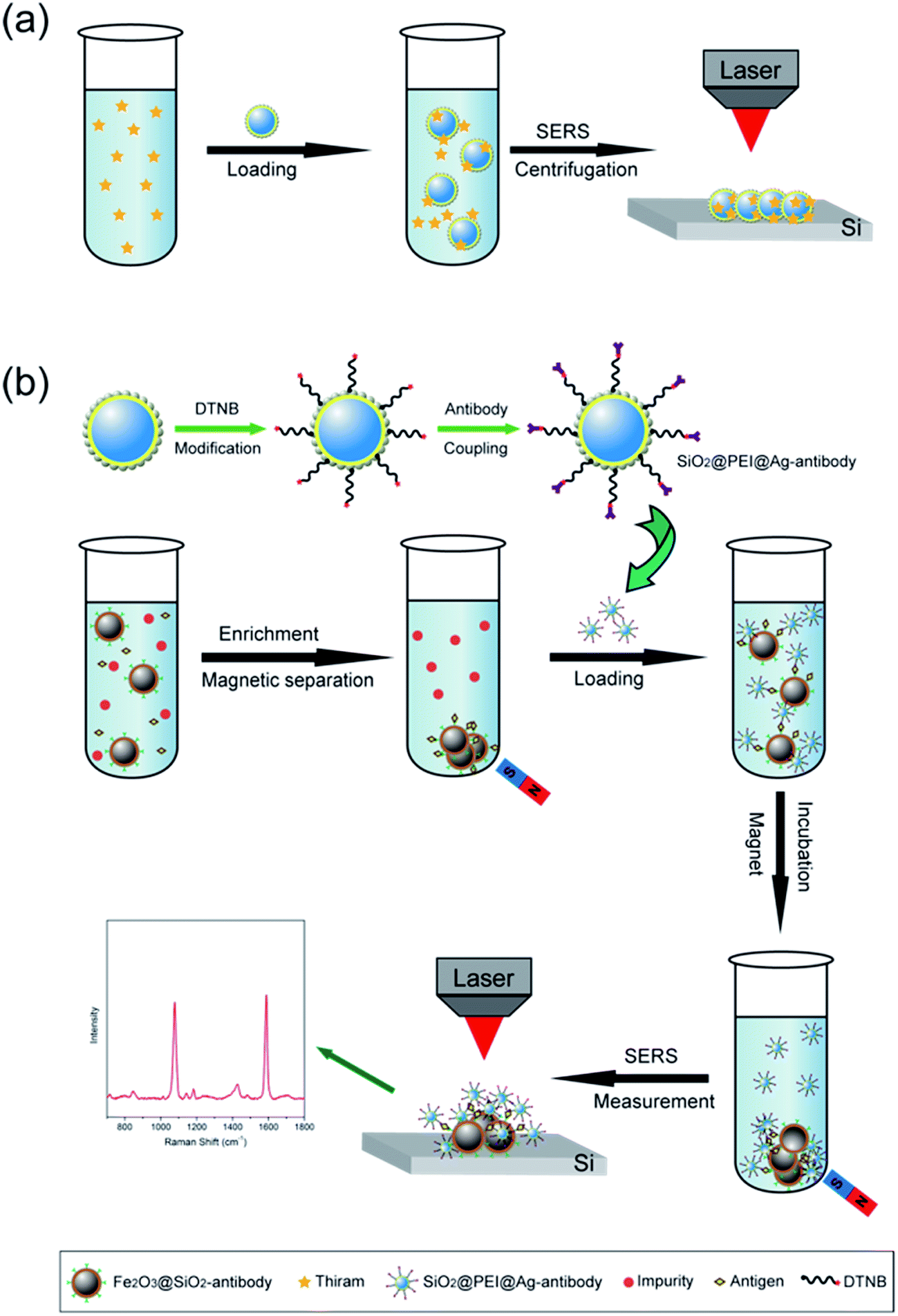 | ||
| Scheme 1 (a) SERS detection protocol for specific binding molecule (thiram) by using the SiO2@PEI@Ag NPs as SERS substrates. (b) Sketch maps of the preparation of SiO2@PEI@Ag SERS tags and the SERS immune protocol for human IgG detection. | ||
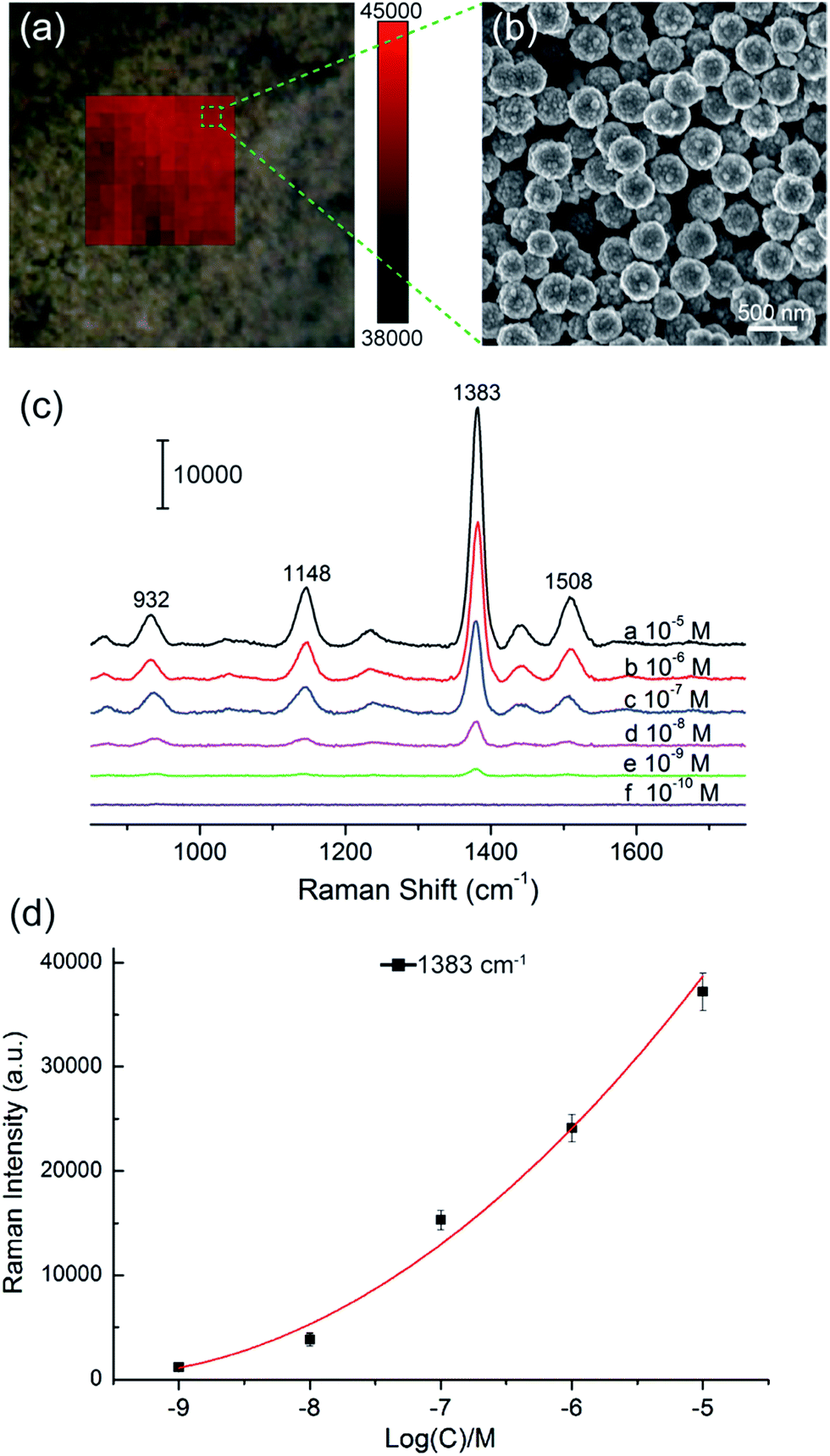 | ||
| Fig. 6 SiO2@PEI@Ag NPs (300 nm core) as high-performance SERS substrates for the in situ detection of pesticide thiram. (a) Area mapping of the SERS signal on SiO2@PEI@Ag NPs with thiram (10−5 M). The Raman map was recorded with the Raman peak at 1383 cm−1 and a 1 μm step. (b) SEM image of the SiO2@PEI@Ag NPs on a Si chip. (c) SERS spectra of thiram with different concentrations on the SiO2@PEI@Ag NPs. (d) The intensity–concentration calibration curve for thiram at a concentration range of 1.0 × 10−5 to 1.0 × 10−9 M by using SERS intensity at 1383 cm−1. Each spectrum is the average of five independently collected spectra. | ||
Small SiO2@PEI@Ag NPs (60–100 nm) combine the advantages of high SERS activity of Ag and the homogeneous superiority of SiO2 NPs and show greater surface roughness compared with Au/Ag spherical particles of similar size. These NPs can be used as efficient SERS tags for highly reproducible SERS immunoassays. Scheme 1b represents the operating principle of the SERS immunoassays using SiO2@PEI@Ag SERS tags and immunomagnetic beads, and human IgG is selected as the model target biomolecule to explore the sensitivity of the presented immunoassay protocol. A sandwich coupled plasmonic nanostructure of immunomagnetic beads/IgG/SiO2@PEI@Ag SERS tags was used for human IgG detection. The capture antibody-coated immunomagnetic beads were mainly utilized to magnetically separate and enrich the human IgG in the solution. Impurities were removed, and the detection antibody-coated SiO2@PEI@Ag SERS tags were incubated. These procedures led to the formation of sandwich immune complexes according to immunity affinity. Finally, the formed sandwich immune complexes could be collected quickly and compressed into one dense dot by magnetic attraction, and analyzed with the Raman spectrometer.
SiO2@PEI@Ag (80 nm core, 10 nm Ag shell) was utilized in the design of efficient SERS tags for SERS immunoassay by labeling DTNB molecules and further coupling detection antibodies via the amidation reaction between the amino groups and preactivated carboxyl groups by EDC and NHS. The micron-sized immunomagnetic beads were obtained by a three step synthetic route. The a-Fe2O3 particles (diameter of ∼1.5 μm) possess superparamagnetic property was synthesized first (Fig. S5a†). Well-dispersed a-Fe2O3@SiO2 microspheres with a silica shell thickness of 50 nm were obtained by using Stöber method.40 The transparent edge of the core–shell microstructure in Fig. S5b† implies the successful coating of silica around the magnetic core. Finally, the a-Fe2O3@SiO2 microspheres were carboxyl-modified with TEPSA; the capture antibodies were conjugated on the magnetic microspheres by covalent bonding via the EDC/NHS chemistry.
Fig. 7a Illustrates the SERS spectra of the sandwich immunocomplex for various concentrations of human IgG. The concentration of human IgG was varied from 10 pg mL−1 to 1 μg mL−1. Each spectrum is the average of five SERS spectra and has undergone baseline subtraction. The power at the samples was 1% of the full laser power and the integration time was 1 s. The Raman peak of DTNB centered at 1329 cm−1 (symmetric NO2 stretching bands) was used for quantitative evaluation of human IgG. The calibration curve is shown in Fig. 7b, where the error bars indicate standard deviations from four measurements. The limit of detection (LOD) is defined as the analyte concentration that produces a signal three times larger than the standard deviation of the blank control;41 hence, the LOD of the immunoassay protocol was 10 pg mL−1. The SEM images demonstrated that the SERS signal of the immunocomplex originated from the SiO2@PEI@Ag SERS tags linked to the surface of the immunomagnetic beads. As shown in Fig. 7c, a large number of SiO2@PEI@Ag SERS tags (80 nm core) were coupled with immunomagnetic beads because of immunity affinity in the presence of the target human IgG, whereas the blank control experiment indicated no binding of the SERS tags to immunomagnetic beads (Fig. 7d). Basing on the results, we demonstrated the great potential of these SiO2@PEI@Ag NPs (80 nm core) as effective SERS tag for highly selective and sensitive protein detection.
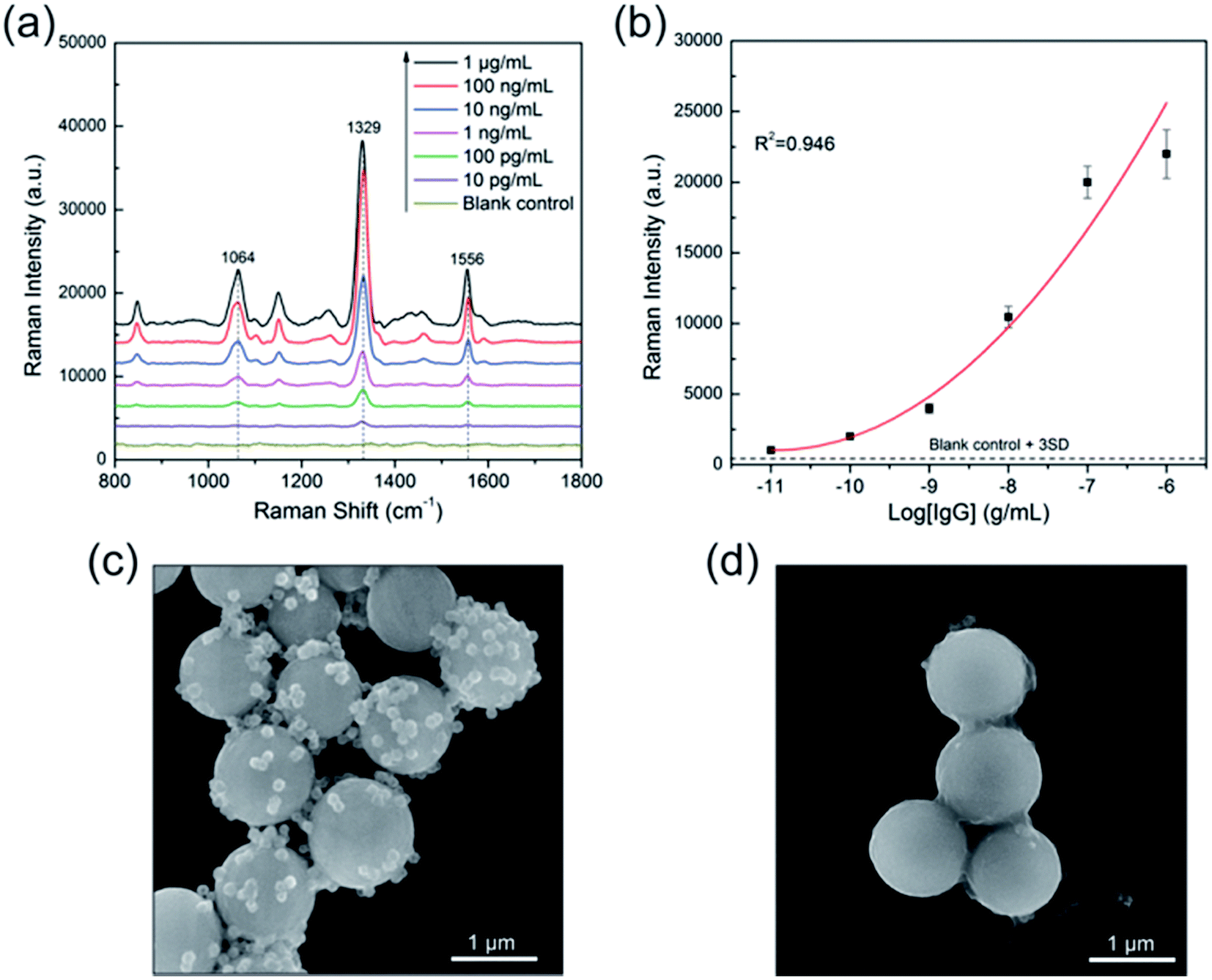 | ||
| Fig. 7 (a) Concentration-dependent SERS spectra of human IgG ranging from 1 μg mL−1 to 10 pg mL−1 and the blank control. (b) Dose–response curve of the above SERS-based immunoassay (R2 = 0.946). Error bars indicate the standard deviation obtained from four measurements. SEM images of sandwich immunocomplexes with (c) 1 μg mL−1 of human IgG and (d) without human IgG. | ||
Conclusions
We demonstrate a facile and effective polyethyleneimine-mediated seed growth method for fabrication of novel SiO2@PEI@Ag NPs with monodispersity, high homogeneity, and a complete Ag shell. In these NPs, PEI was skillfully used to absorb dense Au seeds and prevent particle aggregation. The coverage lever and thickness of the Ag nanoshell can be easily tailored by varying the concentration of AgNO3. The detailed nanostructures of the SiO2@PEI@Ag NPs were characterized by TEM, SEM, XRD, X-ray elemental mapping technique, and UV-visible spectroscopy. To the best of our knowledge, this study is the first to report the use of polymer PEI to grow complete Ag shells with controllable thickness on silica spheres. Moreover, the newly developed PEI-mediated seed growth method can be generally used to synthesize various SiO2@PEI@Ag NPs, ranging from nanoscale to microscale. These obtained SiO2@PEI@Ag NPs with different sizes could produce highly enhanced Raman signals with good uniformity and reproducibility because of its uniform Ag shells and highly reproducible structure, which can be used as versatile SERS platforms for different purposes. We further demonstrated that large SiO2@PEI@Ag NPs (300 nm core) can be used as effective SERS substrates, as verified by the detection of pesticide thiram with a detection limit of 10−9 M. Small SiO2@PEI@Ag NPs (80 nm core) as high-quality SERS tags were successfully applied to SERS-based immunoassay for human IgG detection, with detection limit as low as 10 pg mL−1. Therefore, SiO2@PEI@Ag NPs can be used as an excellent SERS platform for chemical and biological analyses.Acknowledgements
This work was supported by Grants from the National Natural Science Foundation of China (No. 81230089 and 51605486), and Beijing Municipal Science & Technology Commission (No. Z161100000116040).Notes and references
- T. Ung, L. M. Liz-Marzán and P. Mulvaney, J. Phys. Chem. B, 1999, 103, 6770–6773 CrossRef CAS
.
- M. Lv, S. Su, Y. He, Q. Huang, W. Hu, D. Li, C. Fan and S. T. Lee, Adv. Mater., 2010, 22, 5463–5467 CrossRef CAS PubMed
- J.-B. Zeng, Y.-Y. Cao, J.-J. Chen, X.-D. Wang, J.-F. Yu, B.-B. Yu, Z.-F. Yan and X. Chen, Nanoscale, 2014, 6, 9939–9943 RSC
- B. Rodríguez-González, F. Attouchi, M. F. Cardinal, V. Myroshnychenko, O. Stéphan, F. J. García de Abajo, L. M. Liz-Marzán and M. Kociak, Langmuir, 2012, 28, 9063–9070 CrossRef PubMed
- A. Sánchez-Iglesias, P. Aldeanueva-Potel, W. Ni, J. Pérez-Juste, I. Pastoriza-Santos, R. A. Alvarez-Puebla, B. N. Mbenkum and L. M. Liz-Marzán, Nano Today, 2010, 5, 21–27 CrossRef
- C. Y. Li, M. Meng, S. C. Huang, L. Li, S. R. Huang, S. Chen, L. Y. Meng, R. Panneerselvam, S. J. Zhang, B. Ren, Z. L. Yang, J. F. Li and Z. Q. Tian, J. Am. Chem. Soc., 2015, 137, 13784–13787 CrossRef CAS PubMed
- K. Wang, X. Zhang, C. Niu and Y. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 1272–1278 CAS
- J. K. Yang, H. Kang, H. Lee, A. Jo, S. Jeong, S. J. Jeon, H. I. Kim, H. Y. Lee, D. H. Jeong, J. H. Kim and Y. S. Lee, ACS Appl. Mater. Interfaces, 2014, 6, 12541–12549 CAS
- J. B. Liu, W. Dong, P. Zhan, S. Z. Wang, J. H. Zhang and Z. L. Wang, Langmuir, 2005, 21, 1683–1686 CrossRef CAS PubMed
- T.-H. Chen, R.-D. Jean, K.-C. Chiu, C.-H. Chen and D.-M. Liu, Appl. Phys. Lett., 2012, 100, 223101 CrossRef
- K. Kim, H. B. Lee, Y. M. Lee and K. S. Shin, Biosens. Bioelectron., 2009, 24, 1864–1869 CrossRef CAS PubMed
- T. Liu, D. Li, D. Yang and M. Jiang, Colloids Surf., A, 2011, 387, 17–22 CrossRef CAS
- Y. Kobayashi, V. Salgueiriño-Maceira and L. M. Liz-Marzán, Chem. Mater., 2001, 13, 1630–1633 CrossRef CAS
- G. Ren, W. Wang, M. Shang, H. Zou and S. Cheng, Langmuir, 2015, 31, 10517–10523 CrossRef CAS PubMed
- K. Kim, H. S. Kim and H. K. Park, Langmuir, 2006, 22, 8083–8088 CrossRef CAS PubMed
- Z.-J. Jiang and C.-Y. Liu, J. Phys. Chem. B, 2003, 107, 12411–12415 CrossRef CAS
- R. Wang, X. Ji, Z. Huang, Y. Xue, D. Wang and W. Yang, J. Phys. Chem. C, 2016, 120, 377–385 CAS
- L. Gomez, M. Arruebo, V. Sebastian, L. Gutierrez and J. Santamaria, J. Mater. Chem., 2012, 22, 21420–21425 RSC
- C. Wang, J. Xu, J. Wang, Z. Rong, P. Li, R. Xiao and S. Wang, J. Mater. Chem. C, 2015, 3, 8684–8693 RSC
- C. Wang, P. Li, J. Wang, Z. Rong, Y. Pang, J. Xu, P. Dong, R. Xiao and S. Wang, Nanoscale, 2015, 7, 18694–18707 RSC
- J. Wang, X. Wu, C. Wang, Z. Rong, H. Ding, H. Li, S. Li, N. Shao, P. Dong, R. Xiao and S. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 19958–19967 CAS
- P. Guo, D. Sikdar, X. Huang, K. J. Si, W. Xiong, S. Gong, L. W. Yap, M. Premaratne and W. Cheng, Nanoscale, 2015, 7, 2862–2868 RSC
- Y. Fang, S. Guo, C. Zhu, Y. Zhai and E. Wang, Langmuir, 2010, 26, 11277–11282 CrossRef CAS PubMed
- Z. Gan, A. Zhao, Q. Gao, M. Zhang, D. Wang, H. Guo, W. Tao, D. Li, E. Liu and R. Mao, RSC Adv., 2012, 2, 8681 RSC
- M. Li, J. Zhang, S. Suri, L. J. Sooter, D. Ma and N. Wu, Anal. Chem., 2012, 84, 2837–2842 CrossRef CAS PubMed
- M. Li, J. W. Kang, S. Sukumar, R. R. Dasari and I. Barman, Chem. Sci., 2015, 6, 3906–3914 RSC
- T. Liu, D. Li, D. Yang and M. Jiang, Chem. Commun., 2011, 47, 5169–5171 RSC
- Q. An, P. Zhang, J. M. Li, W. F. Ma, J. Guo, J. Hu and C. C. Wang, Nanoscale, 2012, 4, 5210–5216 RSC
- F. Ke, L. Wang and J. Zhu, Nanoscale, 2015, 7, 1201–1208 RSC
- J. Ma, K. Wang and M. Zhan, ACS Appl. Mater. Interfaces, 2015, 7, 16027–16039 CAS
- Q. An, M. Yu, Y. Zhang, W. Ma, J. Guo and C. Wang, J. Phys. Chem. C, 2012, 116, 22432–22440 CAS
- N. Gao, T. Yang, T. Liu, Y. Zou and J. Jiang, RSC Adv., 2015, 5, 55801–55807 RSC
- J. F. Li, X. D. Tian, S. B. Li, J. R. Anema, Z. L. Yang, Y. Ding, Y. F. Wu, Y. M. Zeng, Q. Z. Chen, B. Ren, Z. L. Wang and Z. Q. Tian, Nat. Protoc., 2013, 8, 52–65 CrossRef CAS PubMed
- Y. Zhao, Y.-J. Zhang, J.-H. Meng, S. Chen, R. Panneerselvam, C.-Y. Li, S. B. Jamali, X. Li, Z.-L. Yang, J.-F. Li and Z.-Q. Tian, J. Raman Spectrosc., 2016, 47, 662–667 CrossRef CAS
- D.-Y. Wu, X.-M. Liu, Y.-F. Huang, B. Ren, X. Xu and Z.-Q. Tian, J. Phys. Chem. C, 2009, 113, 18212–18222 CAS
- Y. F. Huang, D. Y. Wu, H. P. Zhu, L. B. Zhao, G. K. Liu, B. Ren and Z. Q. Tian, PCCP Phys. Chem. Chem. Phys., 2012, 14, 8485–8497 RSC
- S. Liu, C. Jiang, B. Yang, Z. Zhang and M. Han, RSC Adv., 2014, 4, 42358–42363 RSC
- X. Zhang, C. Niu, Y. Wang, S. Zhou and J. Liu, Nanoscale, 2014, 6, 12618–12625 RSC
- C. Niu, B. Zou, Y. Wang, L. Cheng, H. Zheng and S. Zhou, Langmuir, 2016, 32, 858–863 CrossRef CAS PubMed
- Y. Wang, K. Wang, B. Zou, T. Gao, X. Zhang, Z. Du and S. Zhou, J. Mater. Chem. C, 2013, 1, 2441 RSC
- B. Zhao, J. Shen, S. Chen, D. Wang, F. Li, S. Mathur, S. Song and C. Fan, Chem. Sci., 2014, 5, 4460–4466 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28629a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |