
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fabrication of SiO2@silicalite-1 and its use as a catalyst support
Xiaobing Yang,
Liuqing Huang,
Jintang Li,
Xueyuan Tang and
Xuetao Luo *
*
Fujian Key Laboratory of Advanced Materials, College of Materials, Xiamen University, Xiamen 361005, China. E-mail: xuetao@xmu.edu.cn
First published on 21st February 2017
Abstract
SiO2@silicalite-1, using silica sol (pH = 9.47, SiO2 ≈ 30 wt%) as the silica source, was directly synthesized in a eutectic mixture where silicalite-1 grains were formed in the three-dimensional net structure of silica gel, grown in situ by transforming amorphous SiO2 into an MFI-type structure and coated with amorphous SiO2. The alkalinity, template agent, and crystallization time strongly affect the physicochemical properties of SiO2@silicalite-1. The physicochemical properties of these samples were characterized by scanning electron microscopy (SEM), X-ray diffraction (XRD), transmission electron microscopy (TEM), UV-vis diffuse reflectance spectra and nitrogen adsorption. The results show that the SiO2@silicalite-1 is synthesized in a eutectic mixture and its physicochemical properties can be tuned by controlling the content of sodium hydroxide and tetrapropyl ammonium bromide (TPABr). A time-dependent study reveals that the formation process obeys an in situ epitaxial growth and phase transformation mechanism. Finally, SiO2@silicalite-1 was used as the support to prepare TiO2-loaded SiO2@silicalite-1 (TiO2@SiO2@silicalite-1). After five loading procedures, it could load 0.44% TiO2 nanoparticles, which is higher than the TiO2 nanoparticle loading in traditional silicalite-1 (0.13%). When the catalysts were used as a catalyst for the degradation of rhodamine B (RhB) aqueous solution under UV light, the photocatalytic efficiency of TiO2@SiO2@silicalite-1 (89.2%) is higher than TiO2@silicalite-1 (only 34.6%). The rate of degradation using TiO2@SiO2@silicalite-1 is 4.3 times faster than that using TiO2@silicalite-1. Furthermore, SiO2@TiO2@silicalite-1 exhibits high stability of photocatalytic performance. After five repeated cycles, the photocatalytic efficiency of TiO2@SiO2@silicalite-1 is 88.09%, which reduces only by 1.1%.
1. Introduction
Zeolites are microporous crystalline materials, which have high surface areas, excellent hydrothermal stability, controllable porosity and acid/basic/redox properties. Many zeolites have been discovered and used in industrial applications,1–5 such as NaX,6 MCM-41,7 5A,8 zeolite Y,9 ZSM-5.10 They are composed of SiO4 and AlO4 tetrahedral structures and have been widely used as adsorbents,11–13 ion exchangers and catalysts in the chemical industry.14,15 Among these applications, zeolites were widely used as supporters in many fields. For example, Yang et al. employed SBA-15 as a support to load nanocrystalline titanium dioxide (TiO2) in its pores, which showed much higher photodegradation ability for methylene blue than commercial pure P25 TiO2 nanoparticles.16 Panpranot et al. used MCM-41 as the support to develop MCM-41-supported CoRu catalysts for CO hydrogenation, in which Co and Ru were well loaded on the pores of MCM-41 and showed high activity.17ZSM-5, a kind of crystalline microporous aluminosilicate with the MFI-type framework, has a strong and tunable acidity, excellent shape selectivity and good hydrothermal stability, leading to their wide use as sorbents,18–20 catalysts, such as petrochemical processing for improving gasoline quality and producing important chemical materials.21–23 The properties of ZSM-5 zeolite are great influenced by the Al2O3/SiO2 ratio and the morphology.24 Recently, the high-silica ZSM-5 zeolite (silicalite-1) has received much attention because of its high shape selectivity for molecular sorption and diffusion, catalytic applications.25–27
Generally, silicalite-1 is synthesized as powders. It consists of pure SiO2 and has high surface areas. Accordingly, it is a potential to load some materials on silicalite-1, such as TiO2 nanoparticles. In our previous research, the surface of silicalite-1 is smooth and the cavity of silicalite-1 is about 0.5–0.55 nm.28 So it is difficult to load some materials on its surface firmly or in its pores due to its smooth surface and small cavity. What is more, when silicalite-1 is loaded with some materials in its pores, its surface area will decrease obviously. It can influence its catalytic activity significantly. In order to meet growing environmental challenges, it is important to explore new technologies to treat or synthesize silicalite-1 which can load a large amount of other materials and keep its large surface area.
In this work, we demonstrate a sol–gel method for one-step synthesis of SiO2@silicate-1 which has high crystallinity and rough surface. We also explored the formation of silicalite-1. Silicalite-1 grains are formed by transforming amorphous SiO2 into MFI-type structure. The surface of SiO2@silicalite-1 is coated with amorphous SiO2 nanoparticles and full with grooves. The amorphous SiO2 and grooves can provide more spaces for some materials to load on the surface of silicalite-1. Finally, SiO2@silicalite-1 was used as support to load TiO2 nanoparticles. The catalytic activity of the catalyst was investigated by the photocatalytic degradation of RhB.
2. Experimental
2.1. Synthesis of SiO2@silicalite-1
All chemicals were purchased from Sinopharm Chemical Reagent Co.,Ltd (analytical grade) and used as received without further purification. The samples were synthesized by first adding X g (where X is 0.5, 1, 1.5, respectively) tetrapropyl ammonium bromide (TPABr) and then Y g (where Y is 0.1, 0.2, 0.3, respectively) sodium hydroxide to 30 ml silica sol (30 wt%, pH = 9.46) under vigorous stirring for 2 h at room temperature. After that, the gel mixture was transferred to the Teflon-lined stainless-steel autoclave and heated at 180 °C in the baking oven for different time (3 h, 12 h, 24 h). The schematic diagram of the synthetic procedure of core–shell SiO2@silicalite-1 is shown in Fig. 1. The obtained gelatinous monoliths were slightly poured out and dried at 60 °C for 48 h. Finally, all samples were calcined at 550 °C for 6 h with the heating rate of 5 °C min−1 in the air atmosphere to remove the templates. And then, the core–shell SiO2@silicalite-1 was grinded into powders.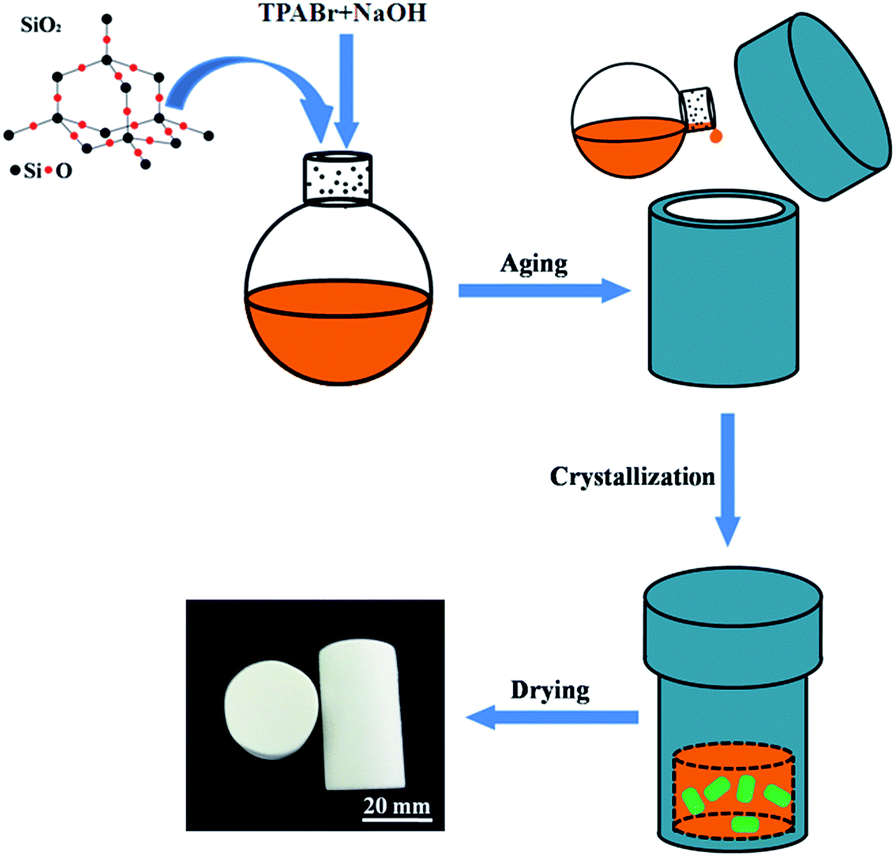 | ||
| Fig. 1 Schematic diagram of the synthetic procedure and the synthesized SiO2@silicalite-1. | ||
2.2. Synthesis of TiO2@SiO2@silicalite-1
SiO2@silicalite-1 was used as support to load TiO2 nanocrystalline by the sol–gel method. The precursor solution of TiO2 was prepared as follows: 11.2 ml tetrabutyl titanate (TBOT) and 2 ml acetic acid were dissolved in 33.8 ml absolute ethanol. The solution was stirred vigorously at room temperature for 20 min, followed by the addition of a mixture containing 4.5 ml distilled water, 10 ml ethanol and 0.5 ml nitric acid. Then, the solution was stirred continuously at room temperature to hydrolyze for 5 minutes to obtain a TiO2 sol. After that, 5 g SiO2@silicalite-1 powders was immersed into the above solution. And the solution kept stirring for 20 minutes at room temperature. Then, the TiO2-loaded SiO2@silicalite-1 (TiO2@SiO2@silicalite-1) was filtrated and dried at drying oven and, subsequently, the process was repeated from immersion to drying for 5 times. Finally, TiO2@SiO2@silicalite-1 was calcined at 550 °C in air for 2 h, which resulted in the preparation of TiO2@SiO2@silicalite-1 catalyst. In order to draw a comparison, the traditional silicalite-1 was used as support to load TiO2 at the same process to prepare the TiO2@silicalite-1 catalyst.2.3. Catalyst characterization
The surface morphology of all ground samples was observed using scanning electron microscopy (SEM) on Hitachi SU-70 microscope at acceleration voltage of 5 kV. The X-ray powder diffraction patterns were recorded on a Bruker-AxsD8 diffractometer using Cu-Kα radiation (40 kV and 40 mA) in the angular range (2θ) from 5 to 60°. Transmission electron microscopy (TEM) images were taken using a TEM (JEOL Model JEM-2100) operated at an accelerating voltage of 100 kV using a 20 μm aperture. UV-vis diffuse reflectance spectra was recorded on a UV-101 PC scanning spectrophotometer. The nitrogen adsorption–desorption isotherm were recorded using TriStar 3020 (Micromeritics, USA) analyzer. The specific surface areas were calculated using the multiple-point Brunauer–Emmett–Teller (BET) method by the Barrett–Joyner–Halenda (BJH) model. The micropore size distribution was measured using the Horvath–Kawazoe (HK) analysis method.2.4. Catalytic evaluation
In order to evaluate the catalytic activity of the catalyst, all samples were used to degrade the rhodamine B (RhB) solution in an inclosed box with side the volume of 25 × 25 × 25 cm3. A Philips lamp irradiating UV light with the power of 40 W was fixed on the top of the box. The distance between the UV light and the reactor is 26 cm. And a magnetic stirrer was located at the bottom of the box. Before UV light irradiation, the solution was stirred continuously in the dark for 30 min to ensure the adsorption–desorption equilibrium between RhB and the catalyst. Afterwards, the UV light was turned on and the solution was irradiated for 90 min. In order to determine the residual dye concentration in solution during this time, a few milliliters of the solution was withdrawn from the reactor and centrifuged to remove the photocatalyst before measuring. The fractional degradation efficiency (DR) of RhB was calculated by the following equation:where C0 is the initial concentration of RhB and Ct is the concentration at definite interval of time, respectively.

3. Results and discussion
3.1. Preparation of SiO2@silicalite-1 and its characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9, it is composed of silica and silicalite-1 crystals. The particle size of silicalite-1 is about 40 μm (Fig. 2a). With the increase of the NaOH/SiO2 mass ratio (from 0.1:9 to 0.3:9), the particle size of silicalite-1 particles decreases (from 40 μm to 25 μm). It indicates that more nuclei are formed at the higher sodium hydroxide content and relative smaller crystals are produced. It is in agreement with Watanabe's group experiments.31 Fegan's group has also reported that crystals formed at high alkalinity are smaller.32 Except silicalite-1 particles, there is also existing amorphous silica around silicalite-1 particles. Fig. 2 also shows the XRD patterns of the three samples. The intensity of diffraction peaks of silicalite-1 increases when the NaOH/SiO2 previous work, we successfully synthesized the silicalite-1 with high crystallinity.28 Using the synthesized silicalite-1 as comparison, the crystallinity degree of SiO2@silicalite-1 is 68.95%, 82.71%, 77.19%, respectively. It indicates that when the NaOH/SiO2 mass ratio is 0.2:9, it is beneficial to synthesize the MFI-type structure. The result is in line with the observations reported in the literature.33
:9, it is composed of silica and silicalite-1 crystals. The particle size of silicalite-1 is about 40 μm (Fig. 2a). With the increase of the NaOH/SiO2 mass ratio (from 0.1:9 to 0.3:9), the particle size of silicalite-1 particles decreases (from 40 μm to 25 μm). It indicates that more nuclei are formed at the higher sodium hydroxide content and relative smaller crystals are produced. It is in agreement with Watanabe's group experiments.31 Fegan's group has also reported that crystals formed at high alkalinity are smaller.32 Except silicalite-1 particles, there is also existing amorphous silica around silicalite-1 particles. Fig. 2 also shows the XRD patterns of the three samples. The intensity of diffraction peaks of silicalite-1 increases when the NaOH/SiO2 previous work, we successfully synthesized the silicalite-1 with high crystallinity.28 Using the synthesized silicalite-1 as comparison, the crystallinity degree of SiO2@silicalite-1 is 68.95%, 82.71%, 77.19%, respectively. It indicates that when the NaOH/SiO2 mass ratio is 0.2:9, it is beneficial to synthesize the MFI-type structure. The result is in line with the observations reported in the literature.33
 | ||
| Fig. 2 SEM images of products prepared with NaOH/SiO2 mass ratios of (a) 0.1:9, (b) 0.2:9, (c) 0.3:9 and XRD patterns. | ||
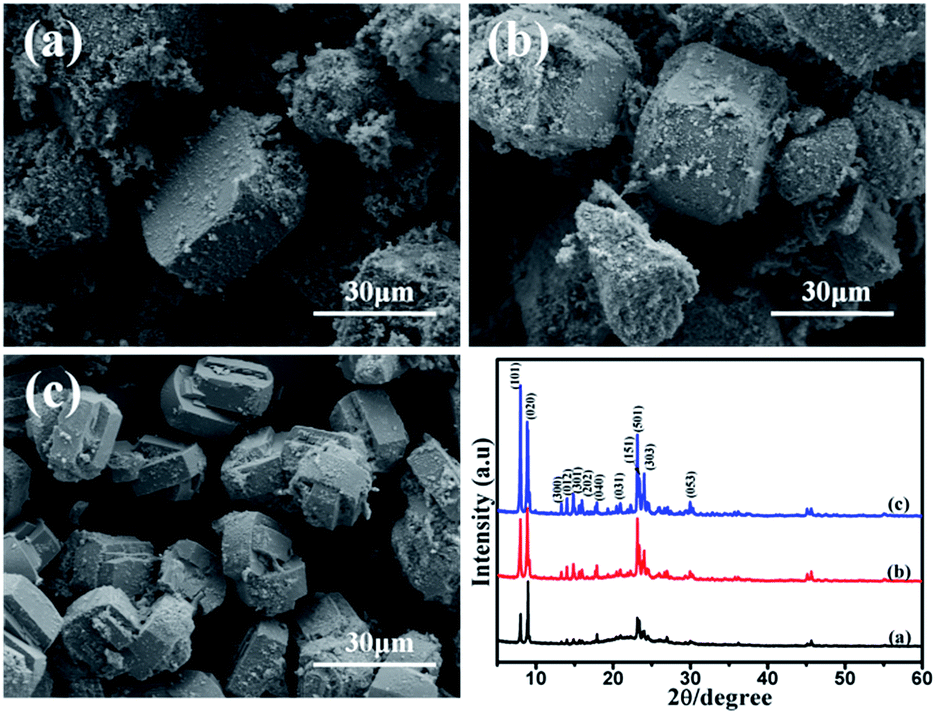 | ||
| Fig. 3 SEM images of products prepared with different amount of TPABr (a) 0.5 g, (b) 1.0 g, (c) 1.5 g and XRD patterns. | ||
 | ||
| Fig. 4 SEM images of products prepared with a crystallization time of (a) 3 h, (b) 12 h, (c) 24 h and XRD patterns. | ||
Fig. 4a is the SME image of the product which is crystallized for 3 h. It can be seen that silicalite-1 particles are not formed. The XRD patterns just shows weak characteristic peaks of silicalite-1 and exhibits a wide range diffraction from 20 to 25°, which is corresponding to amorphous SiO2. It can be inferred that SiO2 colloidal particles just aggregate and form gelation with the three-dimensional net structure after 3 h of hydrothermal treatment.34 The solvent and solute are immobilized in the three-dimensional net structure and loses its mobility. After being dried, water molecules are evaporated and SiO2 colloidal particles are left and bond together. When the product is synthesized by crystallizing for 12 h, it shows the obvious characteristic peaks of silicalite-1. And silicalite-1 particles exhibit the twinned-lozenge shape and the average crystal size of the harvested particles is about 20 μm (Fig. 4b). The crystallinity degree is about 117.76%. Further extension of the crystallization time (after 24 h), the particle size shape and crystal size of silicalite-1 are almost the same with the sample crystallizing for 12 h (Fig. 4c). But the crystallinity degree decreases to 88.11%. It indicates that 12 h is the suitable time to form silicalite-1 zeolite.
3.2. Preparation of TiO2@SiO2@silicalite-1 and its characterization
In order to draw a comparison, the traditional silicalite-1 was synthesized according to our previous report.28 In a typical process, 40 ml of silica sol (30 wt%, pH = 9.46) and 60 ml of distilled water were mixed together. Then 1.5 g of sodium hydroxide and 3 g of tetrapropylammonium bromide were added into above mixture. After the mixture was stirred at room temperature for 6 h, the obtained gel precursor was transformed into the Teflon-lined stainless-steel autoclave and heated at 180 °C for 24 h to obtain the silicalite-1. Fig. 5 illustrates the high magnification SEM images of silicalite-1 and SiO2@silicalite-1. It can be seen that the surface of the traditional silicalite-1 is smooth (Fig. 5a). Fig. 5b is the high magnification SEM images of SiO2@silicalite-1. It can be seen that the surface of SiO2@silicalite-1 coats with nanoparticles. The size of the nanoparticles is about 50 nm. Except these nanoparticles, there are also existing lots of grooves. | ||
| Fig. 5 High magnification SEM images of (a) silicalite-1 and (b) SiO2@silicalite-1. | ||
In order to investigate the physicochemical properties of the SiO2@silicalite-1 (30 ml silica sol, 0.3 g NaOH, 1.5 g TPABr, crystallized for 12 h), BET techniques was employed. As references, silicalite-1 (15 μm) and SiO2@silicalite-1 were characterized. The nitrogen sorption isotherms and pore size distributions are shown in Fig. 6. It can be seen that the nitrogen sorption isotherms of silicalite-1 and SiO2@silicalite-1 are both type I, which shows a steep increase at low relative pressure (P/P0 < 0.1) region corresponding to the presence of micropores in the samples.35 Silicalite-1 and SiO2@silicalite-1 almost keep the same micropore size distribution and have a very narrow micropore size distribution with a mean pore diameter of 0.5–0.55 nm, which is comparable to the pore diameter of the crystallographic five membered ring of silicalite-1.
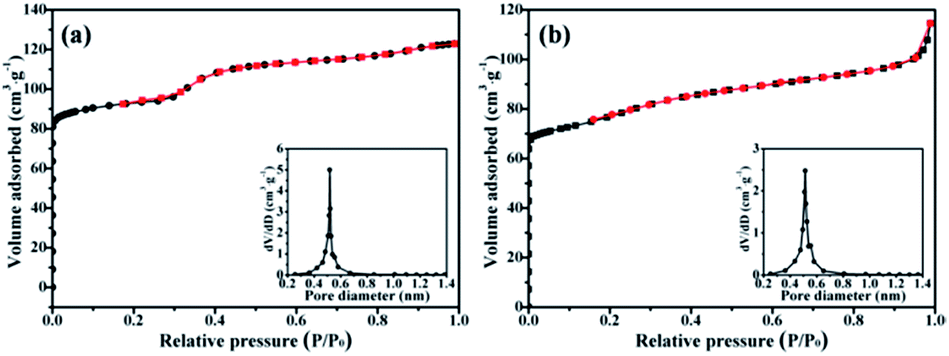 | ||
| Fig. 6 Nitrogen sorption isotherms and pore size distribution of (a) silicalite-1 and (b) SiO2@silicalite-1. | ||
The textural properties of the synthesized silicalite-1 and SiO2@silicalite-1 are shown in Table 1. The particle size of silicalite-1 and SiO2@silicalite-1 are 15 and 20 μm, respectively. The BET surface area and external surface area of silicalite-1 are 334 m2 g−1 and 195 m2 g−1, respectively. When the sample is formed into SiO2@silicalite-1, it keeps the high BET surface area (279 m2 g−1).
Fig. 7 shows the TEM images of SiO2@silicalite-1 and TiO2@SiO2@silicalite-1. The SiO2@silicalite-1 was synthesized with the sol composition of 30 ml silica sol, 0.2 g NaOH, and 1.5 g TPABr. The high magnification SEM image of SiO2@silicalite-1 (Fig. 5a) shows that the surface of SiO2@silicalite-1 is coated with nanoparticles. From Fig. 7a, it can be seen that these nanoparticles are amorphous and aggregate together. It can be inferred that silicalite-1 grains are formed in the three-dimensional net structure of silica and grow in situ by transforming amorphous SiO2 into MFI-type structure. Fig. 7b is the high magnification TEM image of TiO2@SiO2@silicalite-1. It can be seen that there are existing diffractions with lattice constant of 0.352. It indicates that TiO2 is well loaded in the pores of amorphous SiO2.
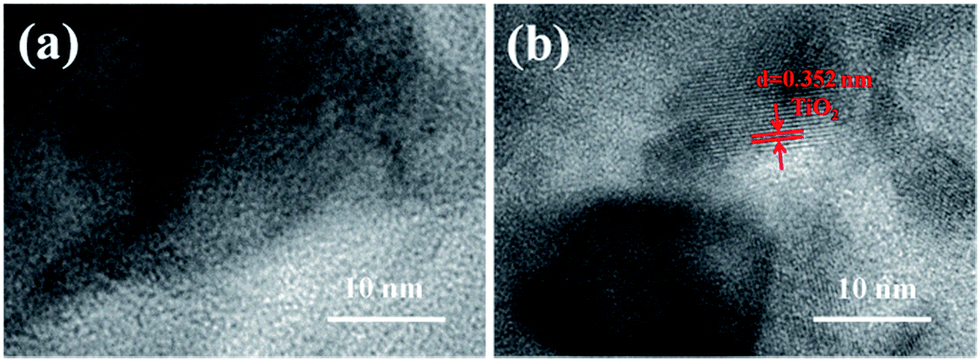 | ||
| Fig. 7 TEM images of (a) SiO2@silicalite-1 and (b) TiO2@SiO2@silicalite-1. | ||
In the preparation of TiO2-loaded SiO2@silicalite-1 (TiO2@SiO2@silicalite-1), we used the SiO2@silicalite-1 as support, which was synthesized with 30 ml silica sol, 0.2 g NaOH, 1.5 g TPABr and crystallized for 12 h at 180 °C. The synthetic procedure is given as above. Fig. 8 shows that diffuse reflectance UV-vis spectra of SiO2@silicalite-1, TiO2@silicalite-1, and TiO2@SiO2@silicalite-1. It can be seen that SiO2@silicalite-1 just shows a gentle absorption band at about 250–280 nm. When silicalite-1 and SiO2@silicalite-1 are loaded with TiO2 nanoparticles, they all exhibit a highly intense band at about 388 nm. According to the Kubelka–Munk radiative transfer model, the band gap (Eg) of TiO2@silicalite-1 and TiO2@SiO2@silicalite-1 are about 3.2 eV.36 It indicates that TiO2 nanoparticles are successfully loaded silicalite-1 and SiO2@silicalite-1. However, from Fig. 8, it can be found that the absorption peak of TiO2@SiO2@silicalite-1 corresponding to TiO2 is higher than TiO2@silicalite-1. It can be inferred that content of TiO2 in TiO2@SiO2@silicalite-1 may be higher than in TiO2@silicalite-1.
 | ||
| Fig. 8 UV-vis spectra of silicalite-1, TiO2@silicalite-1 and TiO2@SiO2@silicalite-1. | ||
The catalytic performance of all samples was examined by degradation of RhB. Fig. 9a shows the results of RhB removal by the catalysts. In order to ensure the adsorption–desorption equilibrium between RhB and the catalyst, all samples were mixed with RhB solution and stirred in the dark. After 30 min of continuously stirring in the dark, 5.24% of RhB is absorbed by SiO2@silicalite-1. When SiO2@silicalite-1 is loaded with TiO2, it shows 4.75% absorption. The adsorptive property of the catalyst is reduced, which may be contributed to the loading of TiO2 in the pores of SiO2@silicalite-1. The absorption of TiO2@silicalite-1 is 5.22%, which is higher than TiO2@SiO2@silicalite-1. It may be the reason that the surface area of TiO2@silicalite-1 is higher than the surface of TiO2@SiO2@silicalite-1. Then, all experiments are exposed under the UV light for 120 min. From Fig. 9a, we can see that 4.68% of RhB is degraded by itself under the UV light. The existing of SiO2@silicalite-1 powders cannot accelerate the degradation of RhB. When SiO2@silicate-1 is loaded with TiO2 nanoparticles, it can degrade 89.19% of RhB. It shows high photocatalytic activity. And the traditional silicalite-1 was used as support to load TiO2 at the same process to prepare the TiO2@silicalite-1 catalyst. Only 34.58% of RhB is degraded by TiO2@silicalite-1 catalyst. The catalytic activity of TiO2@SiO2@silicalite-1 is higher than TiO2@silicalite-1. It may be the reason that SiO2@silicalite-1 can load more TiO2 than the traditional silicalite-1. The kinetics of all samples for degradation of RhB under UV light irradiation is illustrated in Fig. 9b. The photocatalysis degradation kinetic reaction can be described by pseudo-first-rate order kinetics, In(C0/C) = kt, where k is a pseudo-first-rate kinetic constant and t is the irradiation time. The calculated k value of TiO2@silicalite-1 is 0.00384 min−1. For SiO2@TiO2@silicalite-1, the calculated k value is 0.0165 min−1, which is 4.3 times the k value of TiO2@silicalite-1. The photocatalysis activity of the composite is improved greatly. Accordingly, the change of the photocatalytic activity of TiO2@SiO2@silicalite-1 catalyst is probably due to the increase of TiO2 loading in the catalyst. More TiO2 nanoparticles can be loaded in the pores of amorphous SiO2 and the grooves of silicalite-1.
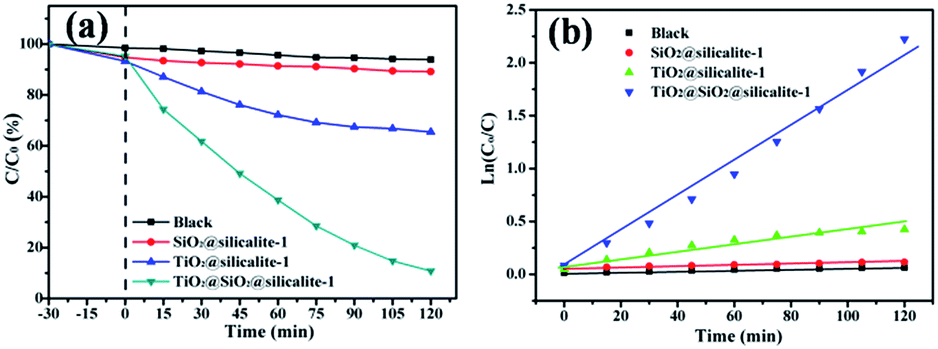 | ||
| Fig. 9 Photocatalytic degradation (a) and kinetics (b) of blank, silicalite-1, TiO2@silicalite-1 and TiO2@SiO2@silicalite-1 for degradation of RhB in the dark and under UV light irradiation with a catalyst content of 1 g l−1, C0 = 5 mg l−1. | ||
The energy dispersive spectrometer of TiO2@silicalite-1 and TiO2@SiO2@silicalite-1 was measured by a Hitachi SU70 microscope and the element proportion was shown in Fig. 10. It can be seen that the atomic ratio of O/Si of TiO2@silicalite-1 and TiO2@SiO2@silicalite-1 is about 2. After five times of loading, the Ti element in TiO2@silicalite-1 is just 0.13%. However, the Ti element in TiO2@SiO2@silicalite-1 is 0.44%, which is almost 3.48 times the Ti content of TiO2@silicalite-1.
 | ||
| Fig. 10 The content of Ti, Si, O element in TiO2@silicalite-1 and TiO2@SiO2@silicalite-1. | ||
More TiO2 nanoparticles can be loaded in the pores of amorphous SiO2 and grooves on silicalite-1 surface.
In order to investigate the stability of photocatalytic performance under UV light, TiO2@SiO2@silicalite-1 was used as catalyst to degrade RhB in five repeated cycles. The results are shown in Fig. 11. From Fig. 11, it can be seen that the photocatalytic performance of SiO2@TiO2@silicalite-1 exhibits effective photostability under the UV light. After five repeated cycles, the photocatalytic efficiency of TiO2@SiO2@silicalite-1 is 88.09%, which reduces only by 1.1%. It may be the reason that TiO2 nanoparticles load in the pores of amorphous of SiO2 and tightly adhere on the surface of silicalite-1.
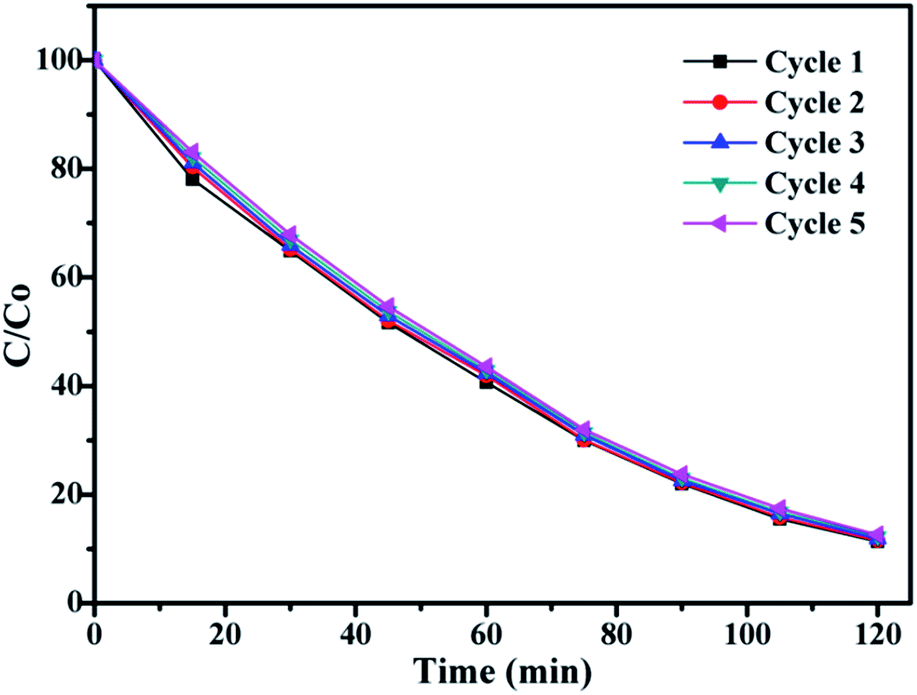 | ||
| Fig. 11 Five photocatalytic degradation cycles of RhB using TiO2@SiO2@silicalite-1 under UV light. | ||
Fig. 12 shows the schematic diagram for the adsorption and photocatalytic degradation of RhB molecules on catalyst. As we know silicalite-1 zeolite has high adsorptive property. It can keep a high RhB concentration on its surface for its adsorption. After a period of contact, there is an adsorption–desorption equilibrium between the catalyst and the solution. When TiO2 particles are loaded on SiO2@silicalite-1 and irradiated by UV light, it can generate the hydroxyl radicals and valence band holes to oxidize RhB molecules into inorganic compounds. The adsorption of the zeolites and photocatalysis of the deposited TiO2 have been combined, resulting in synergistic effects in improving the photocatalytic efficiency of TiO2. When SiO2@silicalite-1 monolith is loaded with TiO2 particles, it can degrade RhB molecular effectively. From Fig. 9a, it can be seen that after 120 min of UV light irradiation, 89.19% of RhB is degraded. The catalyst shows high photocatalytic activity.
 | ||
| Fig. 12 The schematic diagram for the adsorption and photocatalytic degradation of RhB molecules on catalyst. | ||
4. Conclusions
A series of SiO2@silicalite-1 with different silicalite-1 particle size and crystallinity degree were synthesized according to the hydrothermal method using silica sol as Si source, TPABr as template. By adding an appropriate amount of tetrapropyl ammonium bromide and sodium hydroxide in the silica sol and controlling the crystallization time, the crystallinity degree of silicalite-1 monolith can be 117.76% comparing to the synthesized silicalite-1. SiO2@silicalite-1 containing TiO2 (TiO2@SiO2@silicalite-1) was also prepared by a sol–gel method. TiO2 was well loaded on the pores of amorphous SiO2 and the grooves of silicalite-1 surface. And it shows high catalytic performance for degradation of RhB and keeps high stability of photocatalytic performance. SiO2@silicalite-1 is potential to be used as support to load same materials using in different catalysis.Acknowledgements
The authors grateful acknowledge financial supported by the Scientific Technological Innovation Platform of Fujian Province (2006L2003).References
- L. A. Truter, V. Ordomsky, J. C. Schouten and T. A. Nijhuis, Appl. Catal., A, 2016, 515, 72–82 CrossRef CAS.
- Y. Jiao, X. Yang, C. Jiang, C. Tian, Z. Yang and J. Zhang, J. Catal., 2015, 332, 70–76 CrossRef CAS.
- J. Čejka, G. Centi, J. Perez-Pariente and W. J. Roth, Catal. Today, 2012, 179, 2–15 CrossRef.
- S. L. Hailu, B. U. Nair, M. Redi-Abshiro, R. Aravindhan, I. Diaz and M. Tessema, J. Porous Mater., 2015, 22, 1363–1373 CrossRef CAS.
- H. Liu, S. Peng, L. Shu, T. Chen, T. Bao and R. L. Frost, Chemosphere, 2013, 91, 1539–1546 CrossRef CAS PubMed.
- M. A. S. D. Barros, E. A. Silva, P. A. Arroyo, C. R. G. Tavares, R. M. Schneider, M. Suszek and E. F. Sousa-Aguiar, Chem. Eng. Sci., 2004, 59, 5959–5966 CrossRef CAS.
- Q. Guan, F. Wan, F. Han, Z. Liu and W. Li, Catal. Today, 2016, 259, 467–473 CrossRef CAS.
- Y. H. Zhuonan Song, L. Wang, S. Li and M. Yu, Chem. Commun., 2015, 51, 373–375 RSC.
- J. García-Martínez, M. Johnson, J. Valla, K. Li and J. Y. Ying, Catal. Sci. Technol., 2012, 2, 987 Search PubMed.
- Y.-T. Cheng, J. Jae, J. Shi, W. Fan and G. W. Huber, Angew. Chem., Int. Ed., 2012, 51, 1387–1390 CrossRef CAS PubMed.
- J. A. Thompson, N. A. Brunelli, R. P. Lively, J. R. Johnson, C. W. Jones and S. Nair, J. Phys. Chem. C, 2013, 117, 8198–8207 CAS.
- A. Nanoti, S. Dasgupta, Aarti, N. Biswas, A. N. Goswami, M. O. Garg, S. Divekar and C. Pendem, Ind. Eng. Chem. Res., 2012, 51, 13765–13772 CrossRef CAS.
- L. R. Rad, A. Momeni, B. F. Ghazani, M. Irani, M. Mahmoudi and B. Noghreh, Chem. Eng. J., 2014, 256, 119–127 CrossRef CAS.
- Z. Zhang, M. Sadakane, T. Murayama, N. Sakaguchi and W. Ueda, Inorg. Chem., 2014, 53, 7309–7318 CrossRef CAS PubMed.
- A. M. Abdel-Mageed, S. Eckle, H. G. Anfang and R. J. Behm, J. Catal., 2013, 298, 148–160 CrossRef CAS.
- J. Yang, J. Zhang, L. Zhu, S. Chen, Y. Zhang, Y. Tang, Y. Zhu and Y. Li, J. Hazard. Mater., 2006, 137, 952–958 CrossRef CAS PubMed.
- J. Panpranot, J. G. Goodwin Jr and A. Sayari, J. Catal., 2002, 211, 530–539 CrossRef CAS.
- D. Liu, W. Zhou and J. Wu, Fuel, 2016, 177, 251–259 CrossRef CAS.
- D. Liu, W. Zhou and J. Wu, Adsorption, 2016, 22, 327–334 CrossRef CAS.
- D. Liu, W. Zhou and J. Wu, Chem. Eng. J., 2016, 284, 862–871 CrossRef CAS.
- R. Zhang, D. Shi, N. Liu, B. Chen, L. Wu, L. Wu and W. Yang, Catal. Today, 2015, 258, 17–27 CrossRef CAS.
- J. Li, Y. Yu, X. Li, W. Wang, G. Yu, S. Deng, J. Huang, B. Wang and Y. Wang, Appl. Catal., B, 2015, 172–173, 154–164 CrossRef CAS.
- X. Li, J. Li, G. Zhou, Y. Feng, Y. Wang, G. Yu, S. Deng, J. Huang and B. Wang, Appl. Catal., A, 2014, 481, 173–182 CrossRef CAS.
- L. Zhao, J. Gao, C. Xu and B. Shen, Fuel Process. Technol., 2011, 92, 414–420 CrossRef CAS.
- W. Z. Johan, C. Groen, S. Brouwer, S. J. Huynink, F. Kapteijn, J. A. Moulijn and J. Perez-Ramırez, J. Am. Chem. Soc., 2007, 129, 355–360 CrossRef PubMed.
- A. Micke, M. Bulow and M. Kocirik, J. Phys. Chem., 1994, 98, 924–929 CrossRef CAS.
- Z. Zhu, G. Lu, Z. Zhang, Y. Guo, Y. Guo and Y. Wang, ACS Catal., 2013, 3, 1154–1164 CrossRef CAS.
- X. Yang, C. Gan, H. Xiong, L. Huang and X. Luo, RSC Adv., 2016, 6, 105737–105743 RSC.
- Y.-Q. Deng, S.-F. Yin and C.-T. Au, Ind. Eng. Chem. Res., 2012, 51, 9492–9499 CrossRef CAS.
- Y. F. Yeong, A. Z. Abdullah, A. L. Ahmad and S. Bhatia, J. Membr. Sci., 2009, 341, 96–108 CrossRef CAS.
- R. Watanabe, T. Yokoi and T. Tatsumi, J. Colloid Interface Sci., 2011, 356, 434–441 CrossRef CAS PubMed.
- S. G. Fegan and B. M. Lowe, J. Chem. Soc., 1986, 82, 785–799 CAS.
- N. Ren, J. Bronić, B. Subotić, X.-C. Lv, Z.-J. Yang and Y. Tang, Microporous Mesoporous Mater., 2011, 139, 197–206 CrossRef CAS.
- M. L. A. Catherine, A. Morris, R. M. Stroud, C. I. Merzbacher and D. R. Rolison, Science, 1999, 284, 622–624 CrossRef.
- N. Tsunoji, S. Yuki, Y. Oumi, M. Sekikawa, Y. Sasaki, M. Sadakane and T. Sano, ACS Appl. Mater. Interfaces, 2015, 7, 24360–24369 CAS.
- A. Murphy, Sol. Energy Mater. Sol. Cells, 2007, 91, 1326–1337 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |