
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organocatalytic synthesis of enantioenriched 1,2,4-triazolines containing a chiral quaternary carbon center†
Li-Jun Zhang,
Si-Xiang Zhang,
Li-Na Fu,
Xiao-Yun Li and
Qi-Xiang Guo *
*
Key Laboratory of Applied Chemistry of Chongqing Municipality, School of Chemistry and Chemical Engineering, Southwest University, Chongqing, 400715, China. E-mail: qxguo@swu.edu.cn
First published on 28th March 2017
Abstract
An efficient asymmetric cyclization reaction of amino-acid-derived isothiocyanates with azodicarboxylates is described. The chiral 1,2,4-triazolines are prepared in good yield (up to 87%) and enantioselectivities (up to 95% ee).
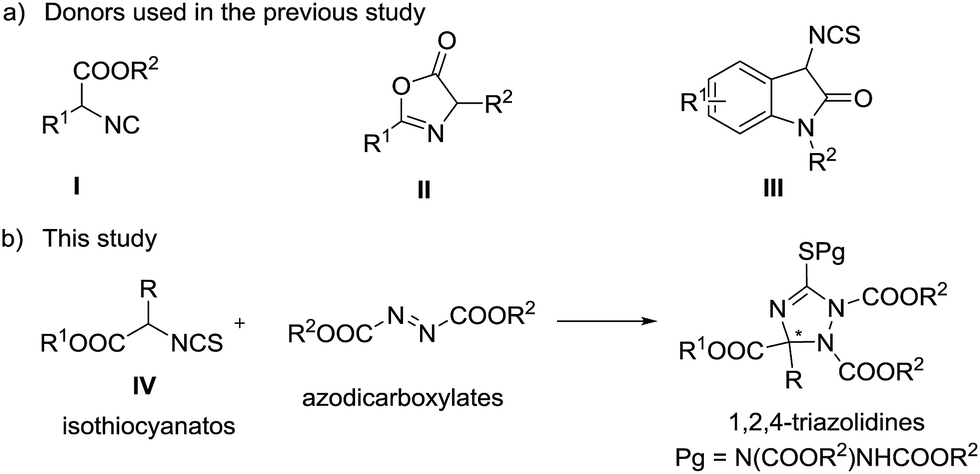
Triazoles are one of the most important classes of heterocycles because of their broad biological activities such as antiviral, antiepileptic, anticancer, and anti-HIV activities.1 Many triazoles are used in pharmaceuticals, agrochemicals, corrosion inhibitors, photographic materials, dyes, etc.2 Therefore, it is important to develop novel methods for the synthesis of structurally diverse triazoles. 1,2,4-Triazolines, one type of important triazole related compounds bearing a potential chiral carbon center at the C-3 position, exhibit good antiviral, anticancer, anti-inflammatory, and anticonvulsant properties.3 The racemic synthesis of 1,2,4-triazolines has been reported extensively;4 however, the asymmetric synthetic methods are very limited.5 In 2010, Jørgensen and co-workers reported preliminary results for the asymmetric synthesis of 1,2,4-triazolines by phase transfer catalysis.5a Then, Feng,5b Huang,5c and Shi5d reported the synthesis of chiral 1,2,4-triazolines independently. In these studies, α-isocyano esters (I) or azlactones (II) reacted with azadicarboxylates to generate the target chiral triazolines (Scheme 1). In 2013, Shi and co-workers reported an efficient [3 + 2] reaction of 3-aminooxindole derived isothiocyanates (III) with azodicarboxylates, producing spirooxindoles in high yields and enantioselectivities.6 α-Isothiocyanato esters (IV), which can be readily prepared from commercially available amino acids, are useful reactants in the catalytic asymmetric addition reactions with aldehydes,7 ketones,8 or imines,9 affording chiral cyclic thiocarbamates in good experimental results. Inspired by Shi's work,6 we assumed that the amino acid derived α-isothiocyanato esters would be good reactants in the catalytic asymmetric addition to azodicarboxylates. Such a reaction will produce chiral polysubstituted 1,2,4-triazolines bearing a carboxylic functional group at the C-3 position and sulfur substitution at the C-5 carbon (Scheme 1b), and provide an alternative pathway leading to chiral 1,2,4-triazolines. Also, the corresponding products can be converted into structural diversity chiral nitrogen containing heterocycles easily. Herein, we report this chiral tertiary amine catalyzed [3 + 2] reaction of α-isothiocyanato esters with azodicarboxylates, leading to polysubstituted 1,2,4-triazolines in good yields and enantioselectivities.
 | ||
| Scheme 1 Strategy for the synthesis of polysubstituted chiral 1,2,4-triazolines. | ||
In the initial study, the phenyl glycine derived isothiocyanato ester 1a was used as reactant to react with di-tert-butyl azodicarboxylate 2a in the promotion of cinchonine 3a. As expected, the desired product 4a was obtained in a high yield (96%), albeit with a low enantioselectivity (19% ee). We tried to improve the stereocontrol ability of the catalyst by modifying the 9-OH group of cinchonine and cinchonidine. As shown in Table 1, the modified catalysts 3b–d gave 4a in low yields and enantioselectivities (Table 1, entries 2–4). Quinidine 3e and its derivatives 3f–3j were then used as catalysts (Table 1, entries 5–10). Among them, 3f has the best enantioselective control ability (68% ee) (Table 1, entry 6). A comparison of the results gave by catalysts 3d and 3f showed that the methoxy group of 3f benefited the enantioselectivity of 4a significantly; therefore, modified catalysts 3k and 3l were synthesized and used in this reaction. However, no better result was obtained (Table 1, entries 11 and 12). Bifunctional catalysts 3m–3r have been extensively used as chiral organocatalysts, but they were not suitable promoters for this transformation (Table 1, entries 13–18). Therefore, catalyst 3f was selected as the optimal catalyst for further investigating the reaction conditions.
|
|||||
|---|---|---|---|---|---|
| Entry | 3 | T (°C) | Time (h) | Yielda (%) | eeb (%) |
| a Isolated yield.b Determined by chiral HPLC. | |||||
| 1 | 3a | 10 | 16 | 96 | 19 |
| 2 | 3b | 10 | 120 | 10 | <5 |
| 3 | 3c | 20 | 96 | 11 | <5 |
| 4 | 3d | 10 | 144 | 25 | −40 |
| 5 | 3e | 10 | 21 | 64 | <5 |
| 6 | 3f | 20 | 92 | 22 | −68 |
| 7 | 3g | 20 | 36 | 14 | −62 |
| 8 | 3h | 20 | 76 | 9 | −58 |
| 9 | 3i | 20 | 53 | 16 | 46 |
| 10 | 3j | 20 | 70 | 29 | <−5 |
| 11 | 3k | 20 | 104 | 19 | 58 |
| 12 | 3l | 20 | 104 | 14 | 69 |
| 13 | 3m | 10 | 66 | 29 | <5 |
| 14 | 3n | 10 | 66 | 29 | <5 |
| 15 | 3o | 10 | 53 | 29 | <5 |
| 16 | 3p | 10 | 120 | 20 | 29 |
| 17 | 3q | 10 | 65 | 10 | 19 |
| 18 | 3r | 20 | 144 | 20 | <5 |
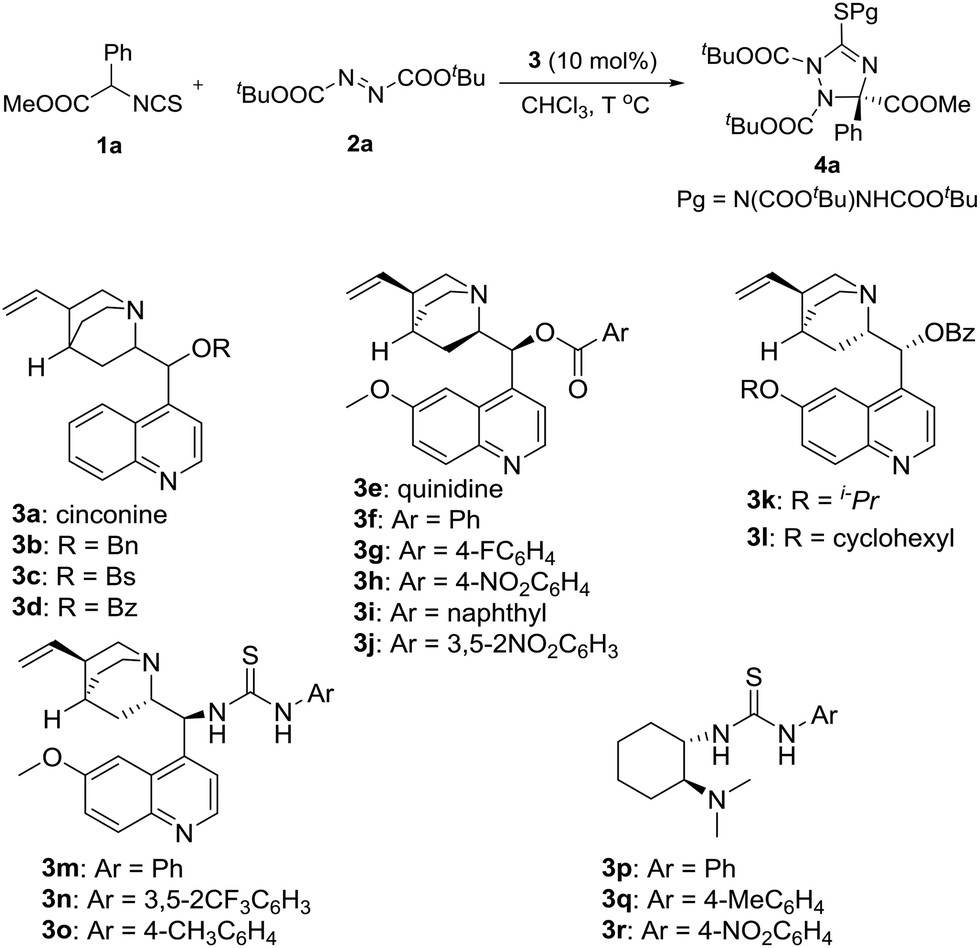
With the optimal catalyst in hand, other reaction conditions were investigated for further improving the yield and enantioselectivity. First, solvents were screened. As shown in Table 2, m-xylene was the best solvent for this transformation. Not only the yield increased to 70%, but the enantioselectivity excess was enhanced to 78% (Table 2, entry 7). Then, this reaction was carried out at a low temperature (−20 °C). The enantioselectivity was further improved to 86% ee (Table 2, entry 8). Additives also affected the reaction outcomes. The yield and enantioselectivity of 4a increased to 77% and 89% ee when Na2SO4 was added to the reaction (Table 2, entry 9).
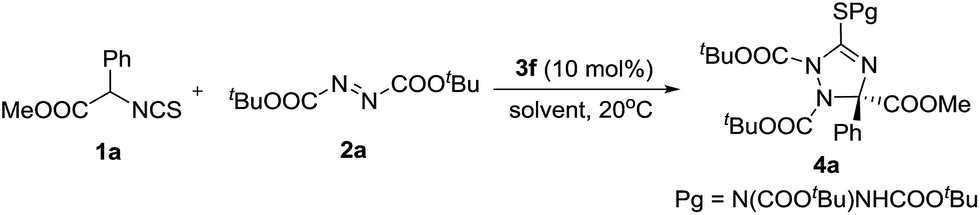
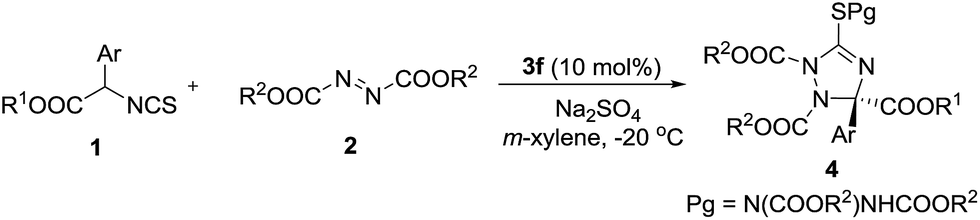
The substrate scope of the reaction was investigated under the optimal reaction conditions. First, the substituent effects of isothiocyanato esters 1 were studied. The results are shown in Table 3. Electron-withdrawing group substituted isothiocyanates were good reaction partners in this reaction, leading to the chiral 1,2,4-triazolines 4b–4e in good yields and enantioselectivities (Table 3, entries 2–5). The low yield of product 4f (15%) was partially caused by the steric influence of the o-substituent (Table 3, entry 6). We found the electron-donating groups of compounds 1 could give the products 4h–4j in high enantioselectivities, albeit the yield decreased slightly (Table 3, entries 8–10). The 2,5-dimethyl-phenyl-substituted isothiocyanate did not participate in this reaction (Table 3, entry 11). This was probably caused by the steric effect of the bulky o-methyl group. The alkyl groups of compounds 2 and 1 were then investigated. We found the yields increased greatly when the tert-butyl group of 2 was replaced by iso-propyl and ethyl groups, albeit the enantioselectivities decreased (Table 3, entries 12 and 13). These results indicated this transformation would be speeded if an azodicarboxylate bearing small alkyl group was introduced as acceptor. So, compounds 1 having o-substituted phenyls, which could not react with di-tert-butyl azodicarboxylate efficiently (Table 3, entries 6 and 11), were used as donors to react with di-iso-propyl and ethyl azodicarboxylates, respectively (Table 3, entries 17–20). As expected, these reactions took place smoothly, giving the products in moderate to good yields and high to excellent enantioselectivities. Particularly, the highest enantioselectivity (95% ee) in this work was obtained in the reaction of 2-Cl phenyl substituted isothiocyanate with diisopropyl azodicarboxylate (Table 3, entry 17). The damage of enantioselectivities of products 4l and 4m could be complemented through increasing the bulk of alkyl group of isothiocyanate 1. For example, the enantioselectivities increased greatly when we used ethyl and isopropyl isothiocyanato esters as donors (Table 3, entries 14–16). The absolute configuration (S) of compound 4q was determined by single crystal X-ray analysis (see ESI†).10 The absolute configuration of compounds 4 and 5 were assigned accordingly.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 4 | Ar | R1 | R2 | Time (h) | Yielda (%) | eeb (%) |
| a Isolated yield.b Determined by chiral HPLC.c Not determined.d At 20 °C.e At 50 °C. | |||||||
| 1 | 4a | Ph | Me | tBu | 96 | 77 | 89 |
| 2 | 4b | 4-ClC6H4 | Me | tBu | 91 | 75 | 88 |
| 3 | 4c | 4-FC6H4 | Me | tBu | 120 | 68 | 88 |
| 4 | 4d | 4-CF3C6H4 | Me | tBu | 64 | 65 | 82 |
| 5 | 4e | 3-CF3C6H4 | Me | tBu | 70 | 70 | 71 |
| 6 | 4f | 2-ClC6H4 | Me | tBu | 120 | 15 | Ndc |
| 7 | 4g | 4-F-3-MeC6H3 | Me | tBu | 88 | 70 | 85 |
| 8 | 4h | 4-MeOC6H4 | Me | tBu | 120 | 54 | 85d |
| 9 | 4i | 4-VillylOC6H4 | Me | tBu | 120 | 53 | 84d |
| 10 | 4j | 4-BnOC6H4 | Me | tBu | 120 | 56 | 84d |
| 11 | 4k | 2,5-2MeC6H3 | Me | tBu | 24 | Trace | Nd |
| 12 | 4l | Ph | Me | iPr | 79 | 72 | 38 |
| 13 | 4m | Ph | Me | Et | 26 | 76 | 14 |
| 14 | 4n | Ph | Et | iPr | 24 | 49 | 49 |
| 15 | 4o | Ph | iPr | Et | 57 | 78 | 62 |
| 16 | 4p | Ph | iPr | iPr | 108 | 51 | 76 |
| 17 | 4q | 2-ClC6H4 | Me | iPr | 64 | 58 | 95 |
| 18 | 4r | 2-ClC6H4 | Me | Et | 94 | 87 | 83 |
| 19 | 4s | 2,5-2MeC6H3 | Me | iPr | 216 | 41 | 77e |
| 20 | 4t | 2,5-2MeC6H3 | Me | Et | 72 | 44 | 72e |
Enantioenriched compounds 4 can be readily converted into other 1,2,4-triazolines under mild reaction conditions. As described in Scheme 2, the alkylation of compound 4q with benzyl bromide or allylic bromide afforded novel triazolines 5a and 5b in good yields (Scheme 1, eqn (1) and (2)). Furthermore, compound 4q could couple with phenylboronic acid and give product 5c in a high yield (Scheme 1, eqn (3)). Importantly, the enantioselectivity of 4q was transformed into the corresponding products completely.
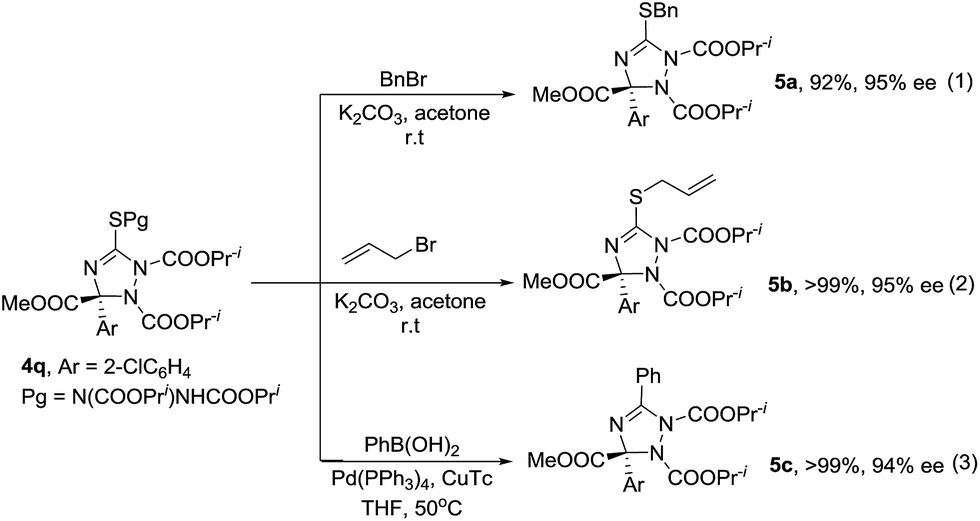 | ||
| Scheme 2 Transformation of product 4q. | ||
Conclusion
In conclusion, an efficient organocatalytic method was developed for the synthesis of chiral 1,2,4-triazolines, in which, the amino-acid-derived isothiocyanato esters were used as the donors for the first time. Triazoline products were obtained in good yields and good to excellent enantioselectivities, and could be converted into other compounds without the loss of enantioselectivities.Acknowledgements
We are grateful for financial support from NSFC (21472150, 21272002), the Fundamental Research Funds for the Central Universities (XDJK2013B028) and the Program for New Century Excellent Talents in Universities (NCET-12-0929).Notes and references
- (a) K. T. Potts, Chem. Rev., 1961, 61, 87 CrossRef CAS; (b) A. Moulin, M. Bibian, A.-L. Blayo, S. EI Habnouni, J. Martinez and J.-A. Fehrentz, Chem. Rev., 2010, 110, 1809 CrossRef CAS PubMed; (c) D. Astruc, L. Liang, A. Rapakousiou and J. Ruiz, Acc. Chem. Res., 2012, 45, 630 CrossRef CAS PubMed.
- (a) S. Maddila, R. Pagadala and S. B. Jonnalagadda, Lett. Org. Chem., 2013, 10, 693 CrossRef CAS; (b) C. H. Zhou and Y. Wang, Curr. Med. Chem., 2012, 19, 239 CrossRef CAS PubMed.
- (a) L. Yet, Prog. Heterocycl. Chem., 2011, 23, 231 CAS; (b) K. Sharma and B. Lal, J. Steroid Biochem. Mol. Biol., 2007, 107, 270 CrossRef CAS PubMed; (c) S. D. Barchéchath, R. I. Tawatao, M. Corr, D. A. Carson and H. B. Cottam, J. Med. Chem., 2005, 48, 6409 CrossRef PubMed; (d) O. G. Todoulou, A. E. Papadaki-Valiraki, S. Ikeda and E. D. Clercq, Eur. J. Med. Chem., 1994, 29, 611 CrossRef CAS.
- Selected examples see: (a) T. Kolasa and M. Miller, Tetrahedron Lett., 1988, 29, 4661 CrossRef CAS; (b) T. Ibata, Y. Isogami and H. Tamura, Chem. Lett., 1988, 1551 CrossRef CAS; (c) A. Hassner and B. Fischer, Tetrahedron, 1989, 45, 3535 CrossRef CAS; (d) D. J. Anderson and W. Watt, J. Heterocycl. Chem., 1995, 32, 1525 CrossRef CAS; (e) O. Tsuge, T. Hatta, H. Tashiro and H. Maeda, Heterocycles, 2001, 55, 243 CrossRef CAS; (f) R. S. Z. Saleem and J. J. Tepe, J. Org. Chem., 2010, 75, 4330 CrossRef CAS PubMed.
- (a) D. Monge, K. L. Jensen, R. Marín and K. A. Jørgensen, Org. Lett., 2011, 13, 328 CrossRef CAS PubMed; (b) M. Wang, X. Liu, P. He, L. Lin and X. Feng, Chem. Commun., 2013, 49, 2572 RSC; (c) Q. Shao, J. Chen, M. Tu, D. W. Piotrowski and Y. Huang, Chem. Commun., 2013, 49, 11098 RSC; (d) M.-X. Zhao, H.-L. Bi, H. Zhou, H. Yang and M. Shi, J. Org. Chem., 2013, 78, 9377 CrossRef CAS PubMed.
- Y. Jiang, C.-K. Pei, D. Du, X.-G. Li, Y.-N. He, Q. Xu and M. Shi, Eur. J. Org. Chem., 2013, 7895 CrossRef CAS.
- Selected examples: (a) L. Li, E. G. Klauber and D. Seidel, J. Am. Chem. Soc., 2008, 130, 12248 CrossRef CAS PubMed; (b) M. C. Willis, G. A. Cutting, V. J.-D. Piccio, M. J. Durbin and M. P. John, Angew. Chem., Int. Ed., 2005, 44, 1543 CrossRef CAS PubMed; (c) X. Chen, Y. Zhu, Z. Qiao, M. Xie, L. Lin, X. Liu and X. Feng, Chem.–Eur. J., 2010, 16, 10124 CrossRef CAS PubMed; (d) W.-Y. Han, J.-Q. Zhao, Z.-J. Wu, X.-M. Zhang and W.-C. Yuan, J. Org. Chem., 2013, 78, 10541 CrossRef CAS PubMed; (e) Y.-M. Cao, F.-F. Shen, F.-T. Zhang, J.-L. Zhang and R. Wang, Angew. Chem., Int. Ed., 2014, 53, 1862 CrossRef CAS PubMed; (f) W.-B. Chen, W.-Y. Han, Y.-Y. Han, X.-M. Zhang and W.-C. Yuan, Tetrahedron, 2013, 69, 5281 CrossRef CAS.
- Selected examples: (a) X. Jiang, Y. Cao, Y. Wang, L. Liu, F. Shen and R. Wang, J. Am. Chem. Soc., 2010, 132, 15328 CrossRef CAS PubMed; (b) W.-B. Chen, Z.-J. Wu, J. Hu, L.-F. Cun, X.-M. Zhang and W.-C. Yuan, Org. Lett., 2011, 13, 2472 CrossRef CAS PubMed; (c) T. Yoshino, H. Morinoto, G. Lu, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 17082 CrossRef CAS PubMed; (d) M. K. Vecchione and D. Seidel, Chem. Commun., 2010, 46, 4604 RSC; (e) X. Jiang, G. Zhang, D. Fu, Y. Cao, F. Shen and R. Wang, Org. Lett., 2010, 12, 1544 CrossRef CAS PubMed.
- Selected examples: (a) L. Li, M. Ganesh and D. Seidel, J. Am. Chem. Soc., 2009, 131, 11648 CrossRef CAS PubMed; (b) Z. Shi, P. Yu, P. J. Chua and G. Zhong, Adv. Synth. Catal., 2009, 351, 2797 CrossRef CAS; (c) G. A. Cutting, M. E. Stainforth, M. P. John, C. Kociok-Kohn and M. C. Willis, J. Am. Chem. Soc., 2007, 129, 10632 CrossRef CAS PubMed; (d) X. Chen, S. Dong, Z. Qiao, M. Xie, L. Lin, X. Liu and X. Feng, Chem.–Eur. J., 2011, 17, 2583 CrossRef CAS PubMed; (e) S. Kato, T. Yoshino, M. Shibasaki, M. Kanai and S. Matsunaga, Angew. Chem., Int. Ed., 2012, 51, 7007 CrossRef CAS PubMed.
- CCDC 1043418 (4q) contains the supplementary crystallographic data for this paper.†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1043418. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra28800f |
| This journal is © The Royal Society of Chemistry 2017 |