
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Theoretical insight into effect of doping of transition metal M (M = Ni, Pd and Pt) on CO2 reduction pathways on Cu(111) and understanding of origin of electrocatalytic activity†
Lihui Ou *ab,
Wenqi Longa,
Jianxing Huanga,
Yuandao Chenab and
Junling Jina
*ab,
Wenqi Longa,
Jianxing Huanga,
Yuandao Chenab and
Junling Jina
aCollege of Chemistry and Materials Engineering, Hunan University of Arts and Science, Changde 415000, China
bHunan Province Cooperative Innovation Center for the Construction & Development of Dongting Lake Ecologic Economic Zone, Hunan University of Arts and Science, Changde 415000, China. E-mail: oulihui666@126.com; Tel: +86-736-7186115
First published on 17th February 2017
Abstract
The effect of the doped transition metal M (M = Ni, Pd and Pt) on CO2 reduction pathways and the origin of the electrocatalytic activity are investigated systematically by focusing on the CH4 and CH3OH formation pathways based on DFT calculations associated with the computational hydrogen electrode model. Our studies show that the doping of Ni, Pd and Pt can promote CO2 reduction into hydrocarbons and influence the selectivity of reduction pathways, in which the doping of Pt may be able to lead to the strongest catalytic activity. The adsorption behavior between reaction intermediates and surfaces is crucial and the interactions of intermediates with the catalysts should be moderate in order to efficiently catalyze CO2 reduction into CH4 and CH3OH, and avoid OH surface poisoning. The enhanced electrocatalytic activity of transition metal-doped Cu(111) surfaces may be owing to decreased overpotential and moderate electronic interactions between Cu and the doped transition metals. The doped Ni, Pd and Pt atoms can considerably decrease the overpotential and remove surface OH poisoning, in which the doped Pt can simultaneously reduce overpotential for CO formation and further reduction, and most easily remove OH, thus suggesting the best electrocatalytic activity. The moderate electron interaction between Cu and Pt and moderate upshift of the d-band center of Pt also explain why the Pt-doped Cu(111) surface has the best electrocatalytic activity for CO2 reduction. Two possible descriptors can be proposed in order to scale the electrocatalytic activity of Cu-based electrocatalysts for CO2 reduction, in which an ideal Cu-based electrocatalyst should be able to reduce barriers for CO formation and further reduction, and should have moderate electron interactions between Cu and the doped transition metals, and a moderate upshift of d-band center of the doped transition metals. In these ways, CO2 reduction pathways can be facilitated and the yield of hydrocarbons CH4 and CH3OH can be enhanced.
1. Introduction
Excessive consumption of fossil fuels has gradually led to the increasing anthropogenic emission of CO2 and depletion of finite natural resources.1 To mitigate over-production of CO2 and prepare for a fluctuating supply of fossil fuels, efforts are being made to convert CO2 into reusable hydrocarbons.2 Among various approaches for conversion, electrochemical reduction of CO2 is considered a potentially clean and promising technique to produce various value-added hydrocarbons at only the cost of a sustainable supply of electrical energy.3,4 However, the selectivity and faradic efficiency of the CO2 electroreduction process are dependent on many factors, such as the electrode materials, the surface structures of the electrode and the type of electrolytes,5–10 in which the major obstacle preventing the efficient reduction of CO2 is the lack of electrode materials that can readily couple with electric energy to achieve rapid and selective cleavage of C–O bonds in CO2 and formation of new bonds in the products. Towards this goal, various electrode materials and promoters have been screened experimentally and analyzed computationally to optimize their activity and selectivity for CO2 reduction,10–19 such as transition metals,15 ionic liquids,16 organometallic complexes17,18 and doped graphene.19 Unfortunately, present electrocatalysts for CO2 reduction suffer from high overpotentials, low current densities, low selectivity or poor durability.20 Among these electrocatalysts explored to date, Cu has received considerable attention because it has been demonstrated experimentally as a valid low-cost electrocatalyst for CO2 reduction21,22 and is the only pure elemental catalyst that produces hydrocarbons at significant faradic efficiency.20 Reduction products include formate, CH4, C2H4 and other higher hydrocarbons.23–26 Furthermore, theoretical study of CO2 reduction on transition metals Cu, Pt, Rh, Pd, Ni, Au and Ag by Nørskov and co-authors27 revealed the “volcano” type of the activity diagram, in which Cu has also the best catalytic activity and is located at the top of the diagram among these transition metals. Thus, experimental and theoretical studies all showed that Cu is the best known metal electrocatalyst for CO2 reduction. However, in order to achieve high faradic efficiency, significant overpotentials are required on Cu electrodes. For example, the theoretical reversible potentials for CH4 and C2H4 are 0.17 V and 0.08 V (vs. RHE), respectively, but experimentally it was observed that a very large potential of about −1.0 V is applied for CO2 reduction into these hydrocarbons.28 Simultaneously, the H2 evolution reaction (HER) at sufficiently negative potential can reduce the faradic efficiency for CO2 reduction by consuming protons and electrons. Thus, to make the CO2 reduction more active and selective, properly designing new electrocatalysts is extremely urgent under the current situation.In surface-catalyzed reactions, the surface selectivity, activity and stability can be dramatically improved through the doping of transition metals to the surface of a host, and some novel properties that are not present on the parent metal surfaces are often exhibited.29–32 Thus, bimetallic Cu-based alloys with Cu-rich composition experimentally have been extensively used to improve CO2 electroreduction owing to the high overpotential and low current density of the pure Cu surface, such as Cu–Ni, Cu–Zn, Cu–Cd, Cu–Sn, Cu–Pb, Cu–Au, Cu–Ag, Cu–Pd and Cu–Pt.33–40 Compared to pure Cu, doping of other transition metals into Cu catalysts can modify the activation barrier for different steps, thereby leading to a reduction of overpotential and a major change of faradic efficiency. For example, Cu–Au alloy has higher faradic efficiency for CO2 electroreduction than that of pure Cu,37 and the experimental onset potential on the Cu–Au alloy was positively shifted, indicating that the overpotential of CO2 reduction can be reduced through the doping of Au.38 The Ni-doped Cu surface also displayed experimentally a superior catalytic activity with respect to the CH3OH synthesis from a mixture of CO, CO2 and H2 in comparison with pure Cu.39,40 Such an improvement was ascribed to the capability of Ni to promote CH3OH production by activating CO2 and stabilizing the intermediates owing to the higher oxygen affinity of Ni.41 Recently, a Pd–Cu catalyst for CO2 electroreduction was investigated.42 A sharp increase of the reduction current and positive shift of potential were observed on the Pd–Cu electrode compared with those for pure Cu, indicating that the doping of Pd could effectively suppress the HER and enhance the CO2 electroreduction activity. Actually, in more previous studies, both Pd single-crystal43 and oxide-supported Pd catalysts44,45 have also been shown to be catalytically active toward CH3OH synthesis from CO2 reduction. Given that Pt is also a metal with high chemical stability and oxygen affinity, the doping of Pt could be a promising approach to improve CO2 electroreduction. Thus, a Cu–Pt alloy with high Cu concentration was developed by Xiong and co-authors. The greatly improved chemical stability and superior electrocatalytic activity towards CO2 reduction were exhibited owing to the presence of Pt.36 Moreover, the alloying of Cu with Pt can lower the decomposition activation energy of formate by up to 13%.46 In the recent theoretical studies on CO2 reduction, density functional theory (DFT) was employed by Liu and co-authors to investigate the CH3OH synthesis reaction from CO2 reduction on transition metals-doped Cu(111) surfaces.47 The overall CH3OH yield was increased by the doped Ni, Pd and Pt compared with pure Cu, suggesting that the doping of Ni, Pd and Pt is able to promote the CH3OH production of the Cu(111) surface. Using DFT calculations associated with the standard hydrogen electrode model,48,49 Hirunsit and co-authors performed a systematic thermodynamic investigation for CO2 electroreduction into CH4 and CH3OH on Cu-based electrocatalysts with a Cu-rich composition of Cu3X (X is Ag, Au, Co, Ni, Pd, Pt, Rh and Ir). The investigation exhibited that a considerably different electrocatalytic activity was produced and hydrocarbon selectivity was changed compared with those on the pure Cu surface, in which on the mostly Cu-based alloy catalysts CH4 is more energetically favorable to be yielded than CH3OH, and CH3OH was found to be more favorable than CH4 production on Cu3Pd and Cu3Pt surfaces. On most surfaces the potential-limiting step is the CO protonation with the exception of on Cu3Au and Cu3Co surfaces. Most recently, the activation of CO2 on transition metal TM (TM = Fe, Co, Ni, Ru, Rh, Pd, Ag, Os, Ir, Pt and Au)-doped Cu(111) and Cu(100) surfaces was investigated with a dopant coverage of 1/9 ML by Qiu and co-authors using first-principle DFT calculations combined with a slab model.50,51 The studies predicted that Co, Ru and Os may be potential dopants to enhance the chemisorption of CO2 on both TM-doped Cu surfaces. However, previous experimental and theoretical studies only showed that the doped transition metals could reduce the thermodynamic overpotential of CO2 reduction and activate CO2 molecules. The exact CO2 electroreduction mechanisms and origin of electrocatalytic activation on Cu-based electrocatalysts still remain unclear, especially kinetic analysis of the elementary reaction steps and the relationship between the kinetic barriers and adsorption ability of the reaction intermediates. Essentially, the doping of transition metals can change the CO2 reduction pathways and reduce the activation barriers, thereby leading to the reduced overpotential and enhanced catalytic activity. A general understanding of the adsorption behavior, reaction mechanism and atomic-level origin of the electrocatalytic activity of Cu-based catalyst bimetallic surfaces will help us discover more efficient catalysts for a given reaction.
Based on the above analysis, Ni, Pd and Pt atoms as potential dopants may be able to enhance CO2 reduction into hydrocarbons on the Cu surface. Furthermore, Cu, Ni, Pd and Pt have the same face-centered cubic structure, which gives them a crystallographic match. Thus, the activation barriers of key steps can be maneuvered through the miscible bimetallic combinations of Cu with Ni, Pd and Pt, thereby resulting in improved surface catalytic activity and selectivity. Focusing on the CH4 and CH3OH formation pathways, the effect of the doped transition metal M (M = Ni, Pd and Pt) on the CO2 reduction pathways and the origin of the electrocatalytic activity are investigated systematically based on DFT calculations associated with the computational hydrogen electrode model. The (111) facet is chosen owing to its high selectivity for CH4 production. The major goal of this work is to examine the effects of the doped Ni, Pd and Pt atoms on the CO2 reduction pathways, surface electrocatalytic activity and selectivity in comparison with pure Cu catalyst. Some key factors that influence surface electrocatalytic activity, such as OH surface poisoning, the limiting potential, potential-limiting step and electronic interactions between Cu and Ni, Pd and Pt, are also demonstrated. The development of more superior electrocatalysts requires an essential understanding of these effects.
2. Computational method and modeling
All DFT calculations were implemented through the PWSCF codes included in the Quantum ESPRESSO distribution,52 while figures of the chemical structures were produced by the XCRYSDEN graphical package.53–55 The exchange–correction interactions were treated by using the generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE).56 The nuclei and core electrons were described by ultrasoft pseudopotentials.57 The transition metal M (M = Ni, Pd and Pt)-doped Cu electrodes modeled by three-layer (111) slabs with a 2 × 3 supercell were used to perform the calculations of the all stable geometry structure of various adsorption and co-adsorption intermediates, which has been shown to be thick enough to well describe the CO2 reduction. The calculated equilibrium lattice constant of Cu was 3.66 Å, which was in good agreement with experimental and theoretical values (3.62 and 3.66 Å, respectively).58,59 During the geometry optimization, the bottom two atomic layers in the transition metal M (M = Ni, Pd and Pt)-doped Cu(111) slabs were kept fixed, whereas the top one layer and the adsorbates were relaxed to geometry configurations with the lowest energy. A vacuum region of 16 Å was used along the z-direction to avoid any periodic interactions. For the 2 × 3 supercell, the surface Brillouin zone was sampled by using a 4 × 3 × 1 uniformly shifted k-mesh. The Kohn–Sham orbitals were expanded in a plane-wave basis set, and the plane-wave cutoff energy was optimized at 26 Ry. The smearing technique of Methfessel and Paxton was used to treat the Fermi-surface effects with a value of smearing parameter of 0.02 Ry.60 The convergence criteria for the total energy and Cartesian force components acting on each atom were set to within 10−5 Ry and below 10−3 Ry Bohr−1, respectively. Additionally, it has been shown that dopants Pd and Pt prefer to stay in the surface of Cu(111), while Ni is in favor of the bulk.61 However, based on the experiments conducted by Chorkendorff and coauthors,39,40 the active adsorbents are able to pull the Ni atom out to the surface under hydrocarbon synthesis conditions, such as CO. Thus, our present study did not include the segregation of Ni into the bulk. The doping range of dopants on the Cu-based electrocatalyst surfaces from ca. 1/9 to 1/4 has been studied in the previous experimental and theoretical studies,35,36,47,50,51,62 and the surface with dopant coverage of ca. 1/6 ML has superior electrocatalytic activity towards CO2 reduction. Thus, only one surface Cu atom was substituted by a dopant (Ni, Pd and Pt) atom in each periodic supercell in the present study, corresponding to a dopant coverage of 1/6 ML. Selecting dopant coverage of 1/6 ML is also in order to compare with our previous studies on a pure 2 × 3 Cu(111) surface.63The minimum energy paths (MEPs) for each step of the CO2 reduction into hydrocarbons were determined by the climbing-image nudged elastic band (CI-NEB) method.64,65 The image of highest energy approximated the transition state of the optimized reaction coordinate, and the transition state images from the CI-NEB calculations were optimized by the quasi-Newton method, which minimizes the forces to find the saddle point. For each intermediate point in the MEPs, geometry optimization was also performed.
3. Results and discussion
3.1 The optimal reaction pathways of CO2 reduction
The calculated results of CO2 reduction into CH4 and CH3OH on the pure Cu(111) surface were reported in our previous work63 and are summarized in the ESI† again to complete the systematic comparison. Various possible pathways obtained by minimum energy pathway (MEP) analysis during the course of CO2 reduction on transition metal M (M = Ni, Pd and Pt)-doped Cu(111) are also demonstrated in the ESI.† Based on the MEP analyses of the elementary reaction steps, it can be concluded that CO, CHO and CH2O are key reaction intermediates during the course of CO2 reduction into hydrocarbons on Ni-, Pd- and Pt-doped Cu(111) surfaces, in which CO is firstly formed through CO2 dissociative hydrogenation, and the COOH intermediate is involved during the course of CO formation. On the Ni-doped Cu(111) surface, formation of intermediates CH3O, CH2OH and CH2 through further CH2O hydrogenation and hydrogenative dissociation are all favorable reaction pathways, which may be parallel pathways in the CO2 reduction mechanism based on the MEP analyses. CH3OH formation more easily occurs through CH2OH hydrogenation in CH3O and CH2OH further reduction on the Ni-doped Cu(111) surface. Simultaneously, CH2 that is produced through CH2O hydrogenative dissociation also leads to CH4 formation. Notably, CH2OH hydrogenation into CH3OH formation and CH2 serial hydrogenation into CH4 require very low activation barriers on the Ni-doped Cu(111) surface, which can be all overcome by the thermoactive process at ambient temperature. Thus, we conclude that CH4 and CH3OH may be able to be formed during CO2 reduction simultaneously on the Ni-doped Cu(111) surface. The MEP analyses on the Pd-doped Cu(111) surface show that CH2O direct hydrogenation into CH3O is the most favorable pathway among all possibilities of further CH2O reduction. The most preferred reaction pathway among further CH3O reduction is CH3O hydrogenative dissociation into CH3 on the Pd-doped Cu(111) surface. Thus, CH4 can be formed through CH3 hydrogenation. On the Pt-doped Cu(111) surface, direct hydrogenation of CH2O to form CH3O and CH2OH intermediates are the most favorable pathways. Thus, CH3O and CH2OH may be reaction intermediates in CO2 reduction on the Pt-doped Cu(111) surface. The MEP analyses show that CH3 and CH3OH can be formed easily through CH3O hydrogenative dissociation and CH2OH hydrogenation, respectively. Simultaneously, a CH2 intermediate can be also formed through CH2OH hydrogenative dissociation. Intermediates CH2 serial hydrogenation and CH3 hydrogenation possibly result in the formation of the final CH4 product. Thus, we can conclude that CH4 and CH3OH in CO2 reduction on the Pt-doped Cu(111) surface are also possible products. Based on thermodynamic and kinetic analyses, (CO + H)* → CHO* is the slowest step on these three surfaces. The optimal reaction pathways of CO2 reduction on the pure and transition metal M (M = Ni, Pd and Pt)-doped Cu(111) surfaces are summarized in Fig. 1.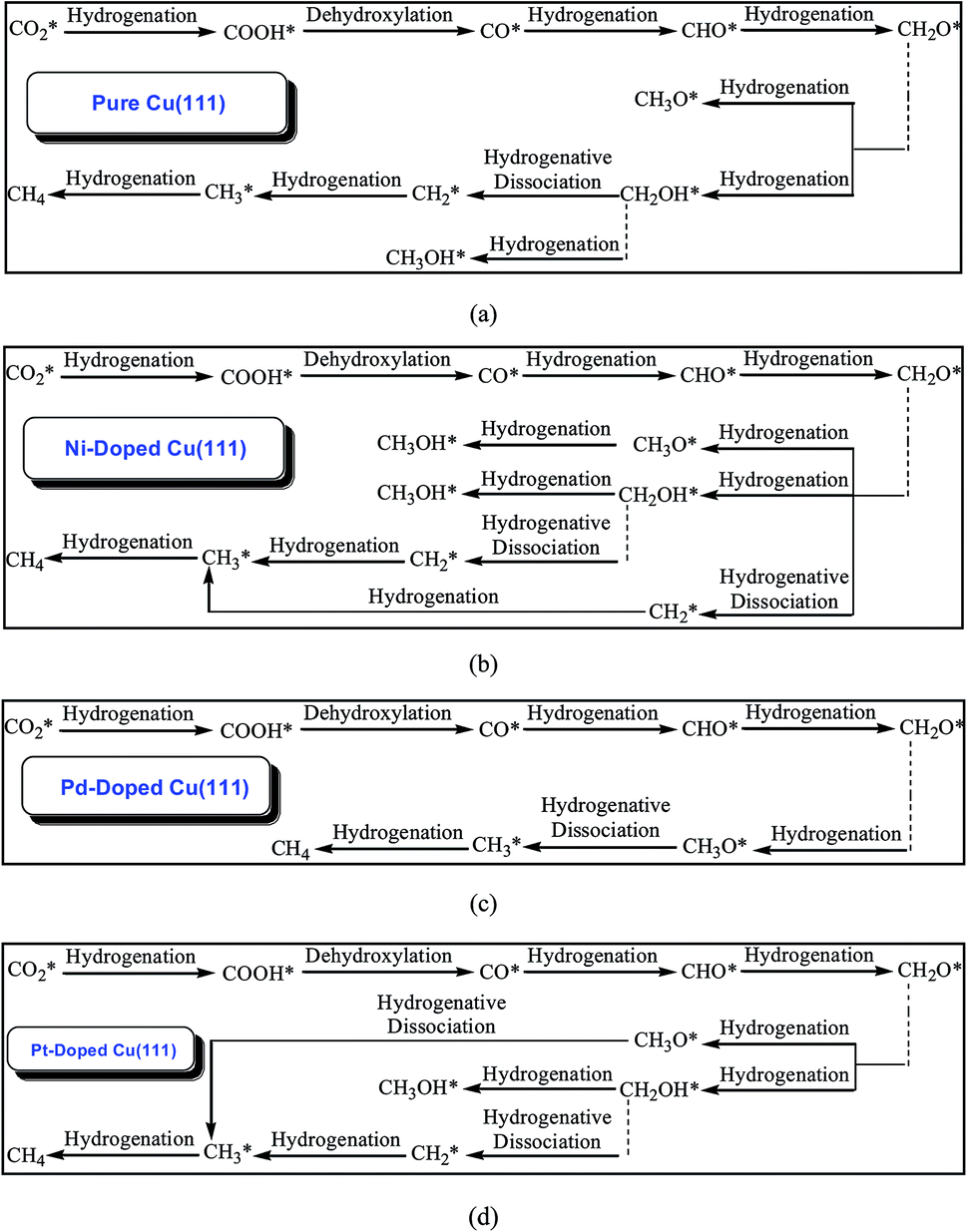 | ||
| Fig. 1 The optimal reaction pathways of CO2 reduction into CH4 and CH3OH on: (a) pure Cu(111); (b) Ni-doped Cu(111); (c) Pd-doped Cu(111); and (d) Pt-doped Cu(111). | ||
3.2 Comparison of CO2 reduction pathways among pure and Ni-, Pd- and Pt-doped Cu(111)
The reaction free energies and activation barriers for the optimal CO2 reduction pathways into CH4 and CH3OH production are listed in Tables 1 and 2 on the pure and transition metal Ni-, Pd- and Pt-doped Cu(111) surfaces.| Reaction paths | Pure Cu(111) | Ni-doped Cu(111) | ||
|---|---|---|---|---|
| ΔGreac (eV) | Eact (eV) | ΔGreac (eV) | Eact (eV) | |
| a The asterisk (*) indicates that the species is adsorbed on the surface. The zero point energies (ZPE) for all species, which are obtained by the present DFT calculations, are included in the reaction free energy. For instance, for the reaction step of (CO + H)* → CHO*, the reaction free energy is calculated according to “E(CHO*) − E(CO + H)* + ZPE(CHO*) − ZPE(CO*) − ZPE(H*)”. | ||||
| CO2(g) + H* → (CO + OH)* | 0.27 | 1.18 | −0.23 | 0.40 |
| (CO + H)* → CHO* | 0.90 | 1.06 | 1.03 | 1.08 |
| (CHO + H)* → CH2O* | −0.20 | 0.72 | −0.04 | 0.28 |
| (CH2O + H)* → (CH2 + OH)* | 0.01 | 1.12 | 0.06 | 0.24 |
| (CH2O + H)* → CH2OH* | 0.13 | 0.95 | −0.60 | 0.27 |
| (CH2O + H)* → CH3O* | −1.05 | 1.86 | −0.08 | 0.24 |
| (CH2OH + H)* → CH2* + H2O (l) | −0.19 | 0.66 | −0.13 | 0.58 |
| (CH2OH + H)* → CH3OH (l) | −0.95 | 0.68 | −0.61 | 0.16 |
| (CH2 + H)* → CH3* | −0.83 | 0.63 | −0.55 | 0.30 |
| (CH3 + H)* → CH4* | −0.80 | 1.03 | −0.59 | 0.21 |
| Reaction paths | Pd-doped Cu(111) | Pt-doped Cu(111) | ||
|---|---|---|---|---|
| ΔGreac (eV) | Eact (eV) | Ereac (eV) | ΔGreac (eV) | |
| CO2(g) + H* → (CO + OH)* | 0.31 | 0.83 | 0.07 | 0.65 |
| (CO + H)* → CHO* | 0.51 | 0.87 | 0.41 | 0.84 |
| (CHO + H)* → CH2O* | −0.25 | 0.10 | −0.02 | 0.35 |
| (CH2O + H)* → (CH2 + OH)* | 0.24 | 0.52 | 0.26 | 0.82 |
| (CH2O + H)* → CH2OH* | −0.12 | 0.68 | −0.35 | 0.59 |
| (CH2O + H)* → CH3O* | −0.80 | 0.06 | −0.62 | 0.17 |
| (CH2OH + H)* → CH2* + H2O (l) | −0.01 | 0.33 | 0.10 | 0.60 |
| (CH2OH + H)* → CH3OH (l) | −0.86 | 0.30 | −0.53 | 0.34 |
| (CH3O + H)* → CH3OH (l) | −0.10 | 1.86 | −0.17 | 1.36 |
| (CH3O + H)* → (CH3 + OH)* | 0.01 | 0.29 | −0.12 | 0.63 |
| (CH2 + H)* → CH3* | −0.83 | 0.35 | −0.95 | 0.33 |
| (CH3 + H)* → CH4* | −1.01 | 0.23 | −0.82 | 0.70 |
Based on the data in Tables 1 and 2, potential energy diagrams for formation of CH4 and CH3OH through CO2 reduction on the pure and transition metal Ni-, Pd- and Pt-doped Cu(111) surfaces are given in Fig. 2–5, respectively. We observed that the activation barriers of CO formation through initial CO2 reduction are considerably decreased by the doping of Ni, Pd and Pt, which is the rate-determining step of CO2 reduction on the pure Cu(111) surface (ca. 1.18 eV). Thus, the doping of Ni, Pd and Pt can activate CO2 and enhance the surface activity of Cu catalysts for CO2 reduction. Therefore, CO formation requires significantly lower activation barriers on Ni and Pt-doped Cu(111) surfaces (ca. 0.40 and 0.65 eV), whereas an activation barrier of 0.83 V is required after doping of Pd (see Fig. S1, S19 and S16†), which may be attributed to chemisorbed CO2 molecule being observed after doping of Ni and Pt by binding C atom at Ni and Pt sites, whereas only physisorbed CO2 was observed on the Pd-doped Cu(111) surface. For further CO reduction, CO hydrogenation into CHO was shown to be the preferred reaction pathway on the Ni-, Pd- and Pt-doped Cu(111) surfaces, as obtained on the pure Cu(111) surface.63 Our previous study and the work from Mei and co-authors63,66 have shown that CO prefers to desorb from Cu rather than to undergo further hydrogenation into CHO, which is in favor of dissociation back to CO and H. For example, CO hydrogenation to form CHO needs to overcome an activation barrier of 1.06 eV on the pure Cu(111) surface, whereas the inverse process only requires an activation barrier of 0.16 eV. The corresponding pathway has nearly equal activation barriers on the Ni-doped Cu(111) surface and the pure Cu(111) surface (1.06 eV vs. 1.08 eV), and a very low activation barrier for CHO dissociation back to CO was required (ca. 0.10 eV). Both steps may hinder CH4 and CH3OH formation in CO2 reduction on the pure and Ni-doped Cu(111) surfaces. In fact, a large amount of CO was observed experimentally on Cu for CH3OH synthesis.67 Based on our present calculations, the doping of Pd and Pt helps to improve the corresponding hydrogenation process. The activation barriers are 0.87 and 0.84 eV for CO hydrogenation after the doping of Pd and Pt, which are 0.19 and 0.22 eV lower than the corresponding barrier on the pure Cu(111) surface, respectively. Moreover, the activation barriers for CHO dissociation back to CO are increased on Pd- and Pt-doped Cu(111) surfaces (see Fig. S11 and S18†), which are 0.10 and 0.27 eV higher than the corresponding barriers on the pure Cu(111) surface, respectively. Improvement of further CO reduction may be attributed to the stronger CO adsorption. The adsorption energy of CO is 0.13 and 0.48 eV stronger after doping of Pd and Pt than that on the pure Cu(111) surface, respectively, i.e. the CO intermediate can be stabilized. Our present studies also show that the CO molecule can be stabilized by the doping of Ni. However, CO adsorption is stronger than that on the pure Cu(111) surface by 0.80 eV after doping of Ni, which is significantly higher than that on the Pd- and Pt-doped Cu(111) surfaces, thereby leading to a relatively poor improvement for further CO reduction into CHO. Although the doped Ni atom cannot significantly improve the further CO reduction into CHO, it makes CO formation the most facile to occur. According to our calculations, CO prefers to adsorb on the top sites of Ni, Pd and Pt. In comparison, the energetically most favorable site for CO adsorption is the fcc-hollow site on the pure Cu(111) surface.63
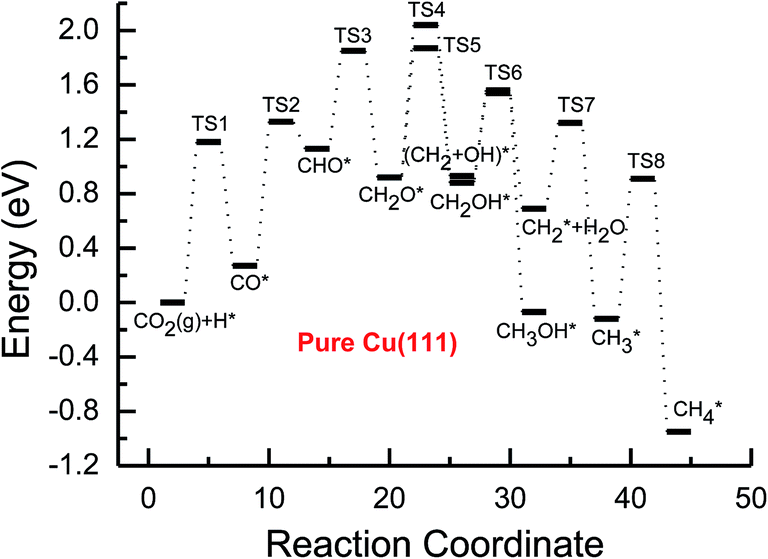 | ||
| Fig. 2 Potential energy diagrams for the CH4 and CH3OH formation through CO2 reduction on the pure Cu(111) surface. TS stands for transition state. | ||
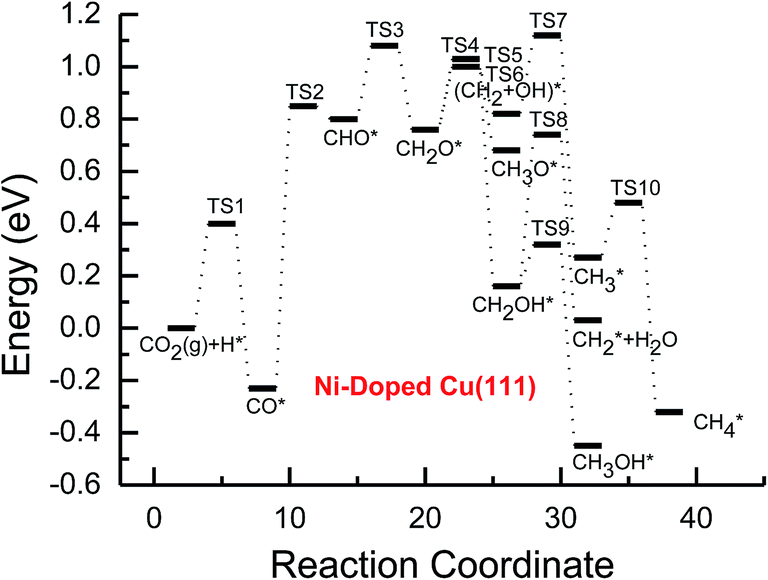 | ||
| Fig. 3 Potential energy diagrams for CH4 and CH3OH formation through CO2 reduction on the transition metal Ni-doped Cu(111) surface. | ||
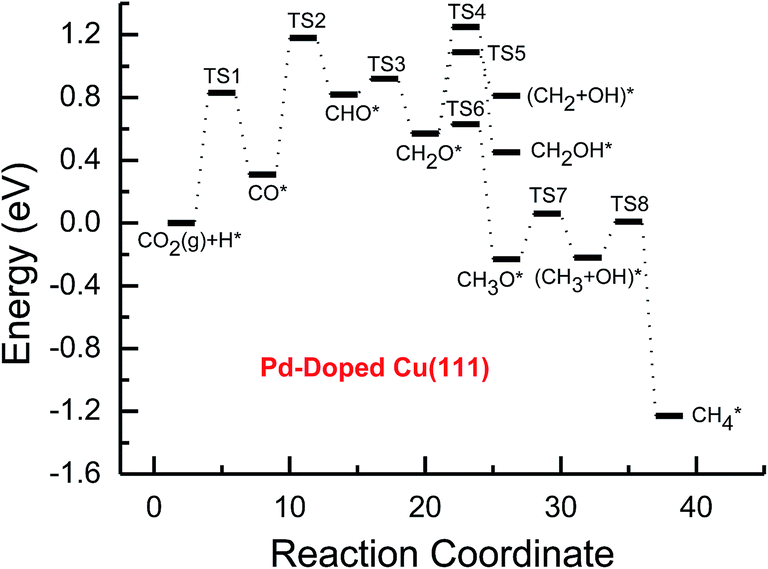 | ||
| Fig. 4 Potential energy diagrams for CH4 and CH3OH formation through CO2 reduction on the transition metal Pd-doped Cu(111) surface. | ||
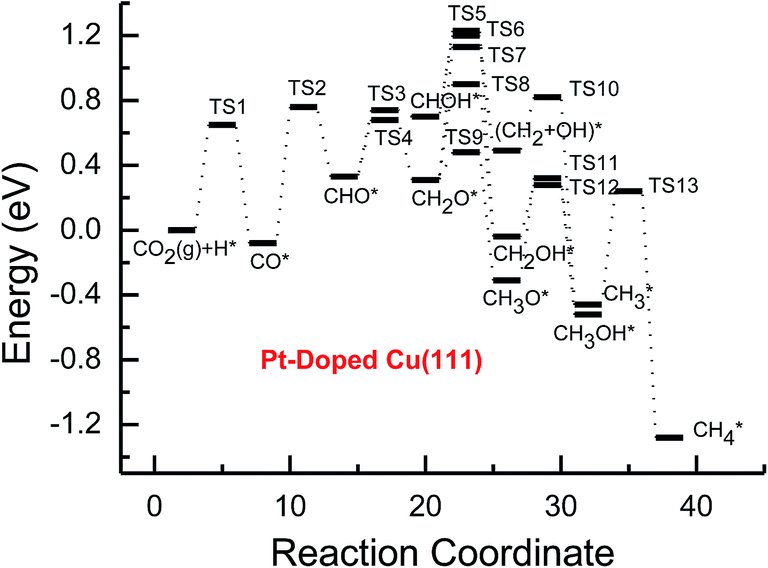 | ||
| Fig. 5 Potential energy diagrams for CH4 and CH3OH formation through CO2 reduction on the transition metal Pt-doped Cu(111) surface. | ||
Subsequent further CHO reduction into the CH2O intermediate is the most favorable pathway among various possibilities on the pure and Ni-, Pd- and Pt-doped Cu(111) surfaces (see Fig. S4, S12 and S19†). The activation barriers of 0.28, 0.10 and 0.35 eV are required for this pathway, respectively, which are significantly lower than that on the pure Cu(111) surface (ca. 0.72 eV). Simultaneously, it can be found that the CHOH intermediate may be formed on the Pt-doped Cu(111) surface since CH2O and CHOH formation through CHO hydrogenation have nearly equal activation barriers (ca. 0.35 and 0.40 eV). We also observed that the further reduction of CHO is energetically comparable to CHO dissociation back to CO after the doping of Ni, Pd and Pt. Thus, although the doping of Ni has a poor improvement for further CO reduction, it considerably improves the further CHO reduction into key intermediate CH2O. The significantly lower activation barriers after the doping of Ni, Pd and Pt may be owing to the stronger CHO adsorption. CHO prefers to bind at Ni, Pd and Pt sites through a C atom after the doping of Ni, Pd and Pt, which is 0.67, 0.29 and 0.68 eV more stable than that on the pure Cu(111) surface, respectively. Thus, the CHO intermediate is able to be stabilized and further CHO reduction can be promoted by the doping of Ni, Pd and Pt atoms.
On the Ni-doped Cu(111) surface, CH2O hydrogenation into CH3O and CH2OH, and hydrogenative dissociation into CH2 may be parallel pathways since they have very low and almost equal activation barriers. The barriers for the corresponding pathways are 0.24, 0.27 and 0.24 eV, respectively. Only CH3O formation is the most preferred pathway in further CH2O reduction on the Pd-doped Cu(111) surface with a very low activation barrier of 0.06 eV, which is a non-activated process, as shown in Fig. S13.† On the Pt-doped Cu(111) surface, CH3O and CH2OH formations through CH2O hydrogenation are the most favorable pathways among four possibilities owing to the relatively lower activation barriers (0.21 and 0.59 eV), as shown in Fig. S20.† However, considerably higher activation barriers are required on the pure Cu(111) surface for further CH2O reduction compared with that on Ni-, Pd- and Pt-doped Cu(111), and the most favorable reduction pathway is hydrogenation of CH2O into CH2OH with an activation barrier of 0.95 eV. The significantly lower barriers may be owing to the doping of Ni, Pd and Pt stabilizing the CH2O intermediate, which adsorbs on the Ni-, Pd- and Pt-doped Cu(111) surfaces through the C atom directly interacting with Ni, Pd and Pt and O atoms interacting with Cu and transition metal M (M = Ni, Pd and Pt). In comparison, the adsorption of CH2O intermediate on the Ni-, Pd- and Pt-doped Cu(111) surfaces is 0.55, 0.19 and 0.31 eV more stable than that on pure Cu(111), respectively. On the Pt-doped Cu(111) surface, CH2 and CH2OH may be able to be formed through CHOH hydrogenative dissociation and direct hydrogenation owing to almost identical activation barriers (ca. 0.53 and 0.54 eV). However, CHOH dissociation back into CHO only requires a very low activation barrier of ca. 0.04 eV, as shown in Fig. S4 and S19,† which is a non-activated process. Thus, although CHOH can be formed on the Pt-doped Cu(111) surface, its further reduction may not be able to occur.
Further reduction of CH2OH intermediate into CH3OH is more favorable to occur with an activation barrier as low as 0.16 eV on the Ni-doped Cu(111) surface, which is significantly more facile in contrast with CH3O further reduction, as shown in Fig. S6 and S7.† Thus, CH3OH can be formed easily by CH2OH hydrogenation after the doping of Ni atom. CH3O hydrogenative dissociation into the CH3 intermediate is the most preferred pathway in further CH3O reduction on the Pd- and Pt-doped Cu(111) surfaces with relatively lower activation barriers of 0.29 and 0.63 eV, respectively. On the Pt-doped Cu(111) surface, CH3OH and CH2 can be also formed easily through hydrogenation and hydrogenative dissociation in further reduction of CH2OH with relatively lower activation barriers of 0.34 and 0.60 eV, respectively. Thus, the doping of Pt may lead to simultaneous formation of CH2, CH3 and CH3OH, as shown in Fig. S22 and S23.† Our previous studies also suggested that CH3OH and CH2 were formed on the pure Cu(111) surface through further reduction of CH2OH with activation barriers of 0.68 and 0.66 eV, respectively.63 However, the doping of Ni and Pt leads to a significant decrease of the activation barriers for CH3OH formation. Thus, further CH2OH reduction can be improved through the doped Ni and Pt atoms, whereas the doping of Pd promotes CH3O formation and further reduction, leading to enhanced CH4 and CH3OH yields. Further reduction of CH3O was not considered on the pure Cu(111) surface owing to the significantly higher activation barrier of CH3O formation. The significant promotion effect on further CH2OH reduction may be owing to the stronger CH2OH adsorption after doping of Ni and Pt atoms. The binding of CH2OH is 0.39 and 0.51 eV more stable than that on the pure Cu(111) surface, and it prefers to bind at the top site of the Ni and Pt atoms by C atom on the Ni- and Pt-doped Cu(111) surfaces. Compared with that on the pure Cu(111) surface, CH3O on the Ni-doped Cu(111) surface is 0.05 eV more stable. However, CH3O is 0.09 and 0.25 eV less stable after the doping of Pd and Pt than that on the pure Cu(111) surface, which may explain why the CH3 intermediate can be formed through hydrogenative dissociation of CH3O after doping of Pd and Pt.
Based on the above analyses, CH4 production possibly occurs through serial CH2 hydrogenation on the Ni- and Pt-doped Cu(111) surfaces or direct CH3 hydrogenation on the Pd- and Pt-doped Cu(111) surfaces. The activation barriers for CH2 hydrogenation into CH3 and CH3 hydrogenation into CH4 are 0.30 and 0.21 eV, 0.35 and 0.23 eV, 0.33 and 0.70 eV on Ni, Pd- and Pt-doped Cu(111) surfaces (see Fig. S8, S15 and S24†), respectively, which are notable lower than that for the corresponding pathways on the pure Cu(111) surface (0.63 eV and 1.03 eV, respectively). Based on our present calculations, the doping of Ni and Pt helps to stabilize CH2 and CH3 intermediates and enhance the corresponding hydrogenation processes, in which CH2 and CH3 are 0.43 and 0.22 eV, 0.18 and 0.23 eV more stable after doping of Ni and Pt, respectively. Although CH3 is 0.07 eV less stable on the Pd-doped Cu(111) surface, the doping of Pd still can improve further CH3 reduction into CH4. This may be attributed to different electronic interactions among Ni and Pd, Ni and Pt, which will be confirmed by subsequent electronic structure analysis. Additionally, OH can be formed during the course of CO2 reduction into CH4 and CH3OH on the pure and Ni-, Pd- and Pt-doped Cu(111) surfaces, which may lead to poisoning of surface active sites. Thus, the OH removal to form H2O is an important step for CO2 reduction. The activation barriers for OH removal are 1.09, 0.68, 0.56 and 0.21 eV on pure and Ni-, Pd- and Pt-doped Cu(111) surfaces, respectively, as shown in Fig. 6. We observe that the doping of Ni, Pd and Pt atoms significantly decreases the activation barriers of OH removal, which is 0.41, 0.53 and 0.88 eV lower than the corresponding barrier on pure Cu(111), respectively. Therefore, the reduced value of the activation barrier after the doping of Ni atom is lower than that on the Pd- and Pt-doped Cu(111) surfaces. The significant improvement of OH removal on the Pd- and Pt-doped Cu(111) may be owing to the weaker OH adsorption, in which the adsorption of OH is 0.16 and 0.26 eV less stable than that on the pure Cu(111) surface, respectively, whereas the doping of Ni makes OH adsorption 0.08 eV stronger. Thus, the doping of metals with less affinity toward OH, such as Pd and Pt, improves the OH removal step and proves to be more efficient than pure Cu and high OH affinity metal Ni.
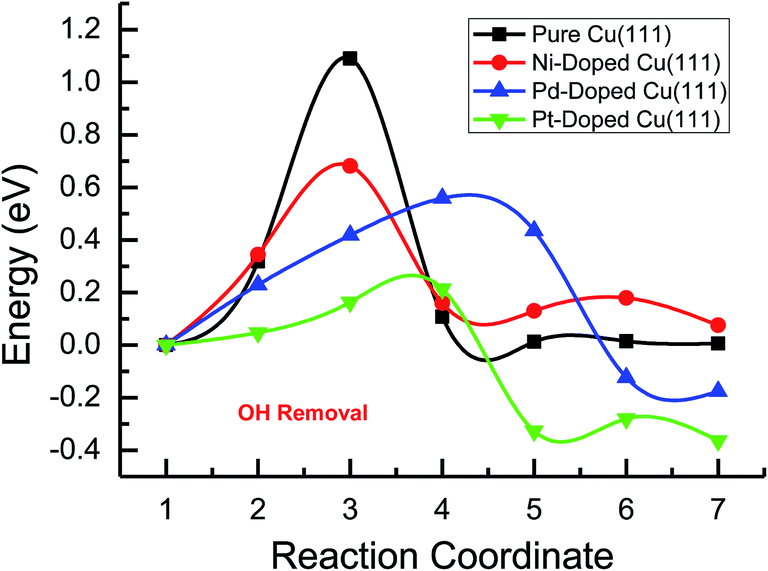 | ||
| Fig. 6 The minimum energy pathways of OH removal to form H2O on pure and Ni-, Pd- and Pt-doped Cu(111) surfaces. | ||
On this basis, the effect of the doped Ni, Pd and Pt atoms on the selectivity of the reaction pathways and reduction production can be revealed. The doped Ni, Pd and Pt atoms changed the rate-determining step of CO2 reduction, which is (CO2 + H)* → (CO + OH)* on the pure Cu(111) surface, whereas it is changed into (CO + H)* → CHO* after the doping of Ni, Pd and Pt. The activation barriers of rate-determining steps are also reduced, which are 0.10, 0.31 and 0.34 eV lower than that of the corresponding process on the pure Cu(111) surface, respectively. Simultaneously, we observed that the activation barriers in overall optimal pathways for CO2 reduction into CH4 and CH3OH are 1.88, 1.12, 1.18 and 0.90 eV on the pure and Ni-, Pd- and Pt-doped Cu(111) surfaces, respectively, as shown in Fig. 2–5, in which the barriers are considerably decreased compared with that on pure Cu(111) and the doping of Pt may be able to lead to the strongest catalytic activity owing to the most reduced value. Therefore, higher CH4 and CH3OH yields can be expected on the Ni-, Pd- and Pt-doped Cu(111) surfaces. The higher reactivity of Ni-, Pd- and Pt-doped Cu(111) surfaces is also consistent with the previous experimental observations.36,39–42 The adsorption behavior of intermediates may be able to determine CO2 reduction activity based on the above discussion. The most stable adsorption sites and adsorption energies for the possible intermediates involved in CO2 reduction into CH4 and CH3OH on the pure and Ni-, Pd- and Pt-doped Cu(111) surfaces are given in Table 3. It is clear that the doping of Ni, Pd and Pt atoms can stabilize the relatively weaker adsorbed C- and O-containing intermediates on the pure Cu(111) surface, such as CO, CHO and CH2O, where C and O atoms directly interact with the doped Ni, Pd and Pt atoms, thereby resulting in easier formation and reduction of CO, CHO and CH2O in spite of the doped Ni atom not notably improving further CO reduction into CHO owing to excessively strongly adsorbed CO. However, the stronger adsorbed intermediates, such as CH3O and OH, are destabilized by the doped Pd and Pt atoms in comparison with pure Cu(111), which make CH3O formation and further reduction able to occur on the Pd- and Pt-doped Cu(111) surfaces and OH removal be easier. Owing to the high carbon and oxygen affinity of Ni, all intermediates can be stabilized by the doped Ni atom in CO2 reduction and the Ni-doped Cu(111) surface has the strongest adsorption ability among the Ni-, Pd- and Pt-doped Cu(111) surfaces, whereas the doping of Pd and Pt leads to the weakest and moderate adsorption of most intermediates, respectively, which may explain why Pt-doped Cu(111) surfaces have the best catalytic activity for CO2 reduction. Thus, it is concluded that moderate adsorbed intermediates on the Cu-based electrocatalyst surfaces will favor CO2 reduction.
| Species | Cu(111) | Ni-doped Cu(111) | Pd-doped Cu(111) | Pt-doped Cu(111) | ||||
|---|---|---|---|---|---|---|---|---|
| Sites | Eads (eV) | Sites | Eads (eV) | Sites | Eads (eV) | Sites | Eads (eV) | |
| COOH | Bridge | −1.56 | Bridge | −1.95 | Bridge | −1.69 | Bridge | −2.03 |
| CO | fcc | −0.80 | Top | −1.59 | Top | −0.93 | Top | −1.28 |
| CHO | fcc | −1.25 | Top | −1.93 | Top | −1.54 | Top | −1.93 |
| CH2O | fcc | −0.05 | fcc | −0.60 | fcc | −0.24 | fcc | −0.36 |
| CHOH | fcc | −2.50 | Top | −3.02 | fcc | −2.60 | Top | −2.95 |
| CH2OH | fcc | −0.90 | fcc | −1.29 | fcc | −1.11 | fcc | −1.41 |
| CH3O | fcc | −2.28 | fcc | −2.33 | fcc | −2.19 | hcp | −2.03 |
| CH2 | fcc | −2.94 | fcc | −3.37 | fcc | −2.90 | fcc | −3.12 |
| CH3 | fcc | −1.36 | fcc | −1.58 | Top | −1.29 | Top | −1.59 |
| OH | fcc | −3.16 | fcc | −3.24 | hcp | −3.00 | hcp | −2.90 |
3.3 Origin of catalytic activity
The calculated limiting potentials (E) and reaction free energies (ΔGreac) at 0.17 V vs. RHE in CO2 electroreduction to CH4 and CH3OH on pure and Ni-, Pd-, and Pt-doped Cu(111) surfaces are listed in Tables 4 and 5. We found that the potential-limiting step is CHO formation (CO* + H+ + e− → CHO*) with the most positive reaction free energy at 0.17 V vs. RHE on the pure and Ni-, Pd- and Pt-doped Cu(111) surfaces, requiring limiting potentials of −0.60, −0.72, −0.44 and −0.40 V vs. RHE by the CHE model, respectively. It can be observed that the limiting potentials are positively shifted after the doping of Pd and Pt atoms, whereas it is more negative after the doping of Ni atom, implying that the doped Pd and Pt atoms can decrease the overpotential of this pathway. The strongest CO adsorption on the Ni-doped Cu(111) surface may induce the protonation to be potentially limited, resulting in a relatively high overpotential for CHO formation. The formation of CHO is also the rate-determining step in CO2 reduction on the Ni-, Pd- and Pt-doped Cu(111) surfaces based on the above kinetic analyses, and its activation barriers are lower after the doping of Pd and Pt atoms, and slightly higher after the doping of Ni atom, compared with that on the pure Cu(111) surface. Although the doped Ni atom leads to more negative limiting potential (−0.72 V) and more positive reaction free energy (0.89 eV) for CHO formation, it can notably shift the limiting potential and reaction free energy to more positive (1.11 V) and more negative values (−0.94 eV) for CO formation (CO2* + H+ + e− → (CO + OH)*), respectively, in which the formation of CO has a slightly negative limiting potential of −0.07 V and a positive reaction free energy of 0.24 eV on the pure Cu(111) surface. The above kinetic studies also show that the activation barrier of CO formation is significantly decreased by the doping of Ni atom. Thus, the decreased overpotential for CO formation can be concluded after the doping of Ni. As shown in Table 5, the doping of Pt atom also can significantly shift the limiting potential of CO formation to a more positive value and make the reaction free energy more negative, thus leading to decrease of the overpotential. However, the limiting potential and reaction free energy of CO formation after the doping of Pd are almost unchanged compared with that on the pure Cu(111) surface. These may be owing to chemisorbed CO2 being observed on Ni- and Pt-doped Cu(111) surfaces and only physisorbed CO2 is formed on the Pd-doped Cu(111) surface. Simultaneously, the limiting potentials of OH removal are also shifted positively after the doping of Ni, Pd and Pt, which are 0.06, 0.24 and 0.36 V more positive than that on the pure Cu(111) surface, respectively, leading to the reduction of the overpotential. Therefore, the Pt-doped Cu(111) surface with the weakest OH adsorption has the most positive limiting potential and the most negative reaction free energy, which is in agreement with the above kinetic analyses, suggesting that the doped metals with less affinity toward OH could decrease surface OH poisoning and enhance the catalytic activity. Thus, the best electrocatalytic activity of the Pt-doped Cu(111) surface for CO2 reduction may be able to be attributed to the simultaneous reduction of overpotential for CO formation and further reduction, and the easiest OH removal.
| Possible reduction steps | Pure Cu(111) | Ni-doped Cu(111) | ||
|---|---|---|---|---|
| E (V) | ΔGreac (eV) | E (V) | ΔGreac (eV) | |
| (a) CO2* + H+ + e− → (CO + OH)* | −0.07 | 0.24 | 1.11 | −0.94 |
| (b) CO* + H+ + e− → CHO* | −0.60 | 0.77 | −0.72 | 0.89 |
| (c) CHO* + H+ + e− → CH2O* | 0.45 | −0.28 | 0.32 | −0.15 |
| (d) CH2O* + H+ + e− → (CH2 + OH)* | 0.38 | −0.21 | 0.10 | 0.07 |
| (e) CH2O* + H+ + e− → CH3O* | 1.26 | −1.09 | 0.76 | −0.59 |
| (f) CH2O* + H+ + e− → CH2OH* | 0.23 | −0.06 | 0.07 | 0.10 |
| (g) CH2OH* + H+ + e− → CH3OH (l) | 1.19 | −1.02 | 0.76 | −0.59 |
| (h) CH2OH* + H+ + e− → CH2* + H2O (l) | 0.43 | −0.26 | 0.40 | −0.23 |
| (i) CH3O* + H+ + e− → CH3OH (l) | 0.17 | 0.00 | 0.07 | 0.10 |
| (j) CH3O* + H+ + e− → (CH3 + OH)* | 0.24 | −0.07 | 0.22 | −0.05 |
| (k) CH2* + H+ + e− → CH3* | 1.05 | −0.88 | 0.84 | −0.67 |
| (m) CH3* + H+ + e− → CH4* | 1.07 | −0.90 | 0.86 | −0.69 |
| (n) OH* + H+ + e− → H2O (l) | 0.12 | 0.05 | 0.18 | −0.01 |
| Possible reduction steps | Pd-doped Cu(111) | Pt-doped Cu(111) | ||
|---|---|---|---|---|
| E (V) | ΔGreac (eV) | E (V) | ΔGreac (eV) | |
| (a) CO2* + H+ + e− → (CO + OH)* | −0.11 | 0.28 | 0.82 | −0.65 |
| (b) CO* + H+ + e− → CHO* | −0.44 | 0.61 | −0.40 | 0.57 |
| (c) CHO* + H+ + e− → CH2O* | 0.35 | −0.18 | 0.08 | 0.09 |
| (d) CH2O* + H+ + e− → (CH2 + OH)* | −0.08 | 0.25 | −0.16 | 0.33 |
| (e) CH2O* + H+ + e− → CH3O* | 0.97 | −0.80 | 0.69 | −0.52 |
| (f) CH2O* + H+ + e− → CH2OH* | 0.25 | −0.08 | 0.43 | −0.26 |
| (g) CH2OH* + H+ + e− → CH3OH (l) | 0.93 | −0.76 | 0.62 | −0.45 |
| (h) CH2OH* + H+ + e− → CH2* + H2O (l) | 0.11 | 0.06 | 0.00 | 0.17 |
| (i) CH3O* + H+ + e− → CH3OH (l) | 0.21 | −0.04 | 0.36 | −0.19 |
| (j) CH3O* + H+ + e− → (CH3 + OH)* | 0.01 | 0.16 | 0.32 | −0.15 |
| (k) CH2* + H+ + e− → CH3* | 1.03 | −0.86 | 1.11 | −0.94 |
| (m) CH3* + H+ + e− → CH4* | 1.14 | −0.97 | 0.84 | −0.67 |
| (n) OH* + H+ + e− → H2O (l) | 0.36 | −0.19 | 0.48 | −0.31 |
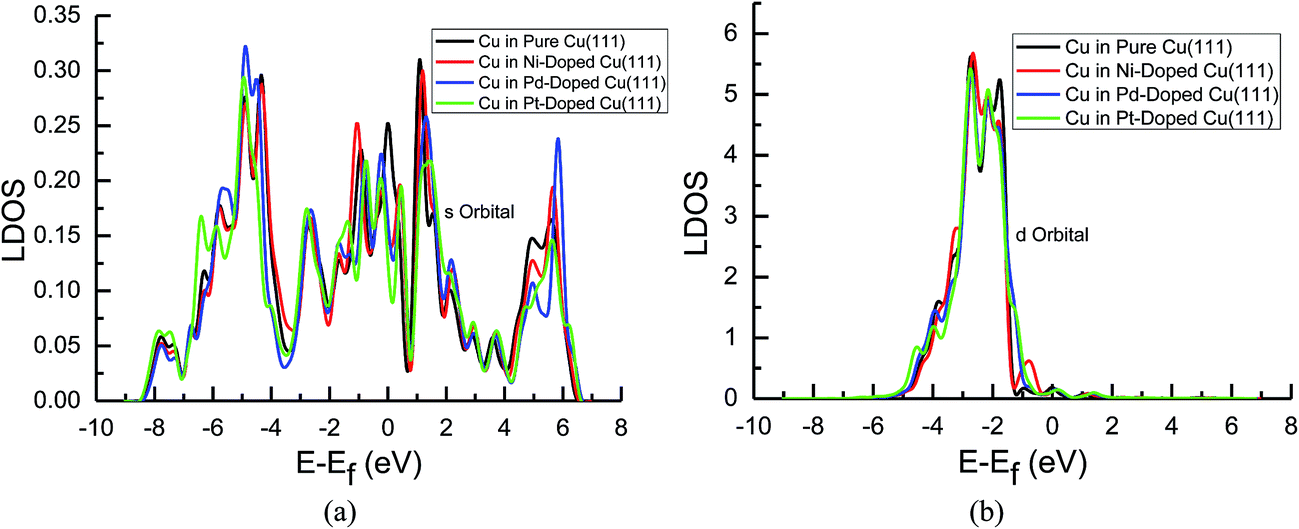 | ||
| Fig. 7 Local density of states of pure and transition metals Ni-, Pd- and Pt-doped Cu(111) surfaces: (a) s orbital of Cu and (b) d orbital of Cu. | ||
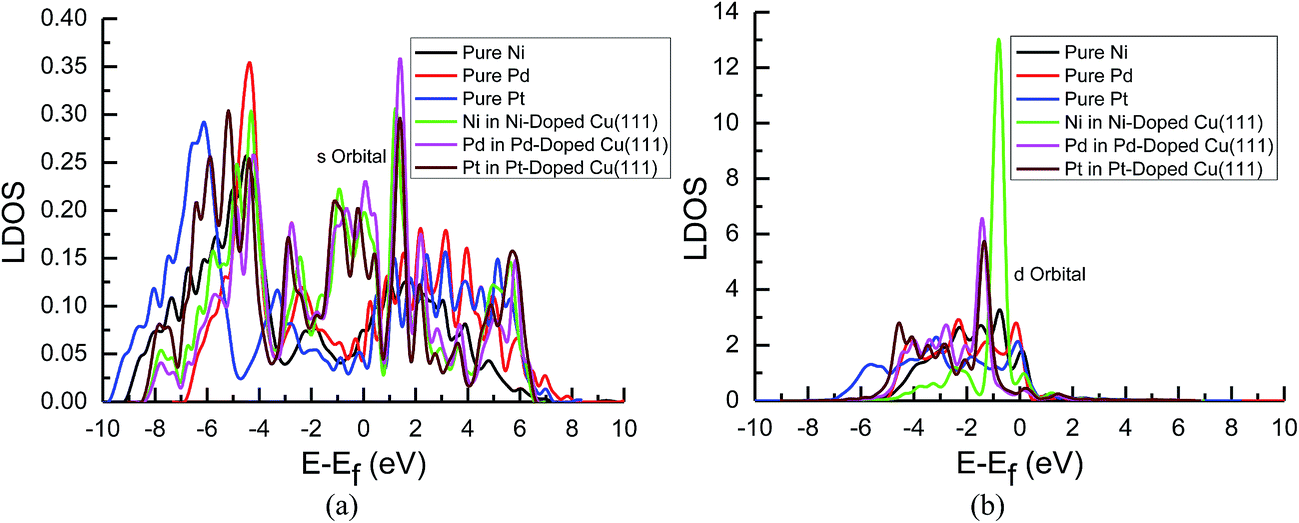 | ||
| Fig. 8 Local density of states of pure and transition metals Ni-, Pd- and Pt-doped Cu(111) surfaces: (a) s orbitals of Ni, Pd and Pt; and (b) d orbitals of Ni, Pd and Pt. | ||
In the mean time, we also observed that the s and d states of Ni, Pd and Pt in Ni-, Pd- and Pt-doped Cu(111) are changed significantly near the Fermi energy level compared with those of pure Ni, Pd and Pt, as shown in Fig. 8(a) and (b). The considerably higher LDOS near the Fermi energy level shows that electrons in the s state of Cu are transferred into the s and d states of Ni, Pd and Pt. The highest LDOS in the d states of Ni in Ni-doped Cu(111) may imply that the most electron transfer and strongest interactions occur between Cu and Ni. Thus, the present studies explain why the reaction intermediates have the strongest adsorption on the Ni-doped Cu(111) surface. The relatively weaker interaction between Cu and transition metals Pd and Pt may result in weaker adsorption of intermediates.
The d-band center of surface atoms, εd, is a key parameter that influences surface adsorption characteristics,69,70 and its upshift and downshift can be used to judge the catalytic activity of the electrocatalysts.71–73 The shift of the d-band center into a lower energy level (downshift) corresponds to the weaker adsorption of reaction intermediates, whereas the shift into a higher energy level (upshift) corresponds to the stronger adsorption. Thus, the d-band centers of surface atoms were calculated in pure and Ni-, Pd- and Pt-doped Cu(111), which is the first moment of the projected d-band density of states on the surface atoms referenced to the Fermi energy level. The corresponding values for surface atoms are listed in Table 6. It was found that there is a slight and considerably upshift of the d-band center of surface Cu and Ni atoms (ca. 0.02 and 0.74 eV) in Ni-doped Cu(111) compared with that of the pure metal surfaces, respectively, whereas that of surface Cu atoms is not changed after the doping of Pd and Pt atoms. The relatively lower upshift of the d-band center of the surface Pt atom in Pt-doped Cu(111) is also observed (ca. 0.20 eV). However, the d-band center of the surface Pd atom is downshifted significantly in Pd-doped Cu(111) (ca. −0.34 eV) compared with that of pure Pd. Thus, we conclude that the doping of Ni and Pd can lead to the strongest and weakest adsorption of reaction intermediates on the Ni and Pd-doped Cu(111) surfaces, respectively. Furthermore, the significant upshift of the d-band center of the surface Ni atom and the strongest interaction between CO and the Ni-doped Cu(111) surface may also be able to explain why further CO reduction has the most positive activation barrier and the most negative limiting potential on the Ni-doped Cu(111) surface among the Ni-, Pd- and Pt-doped Cu(111) surfaces, whereas the significant downshift of the d-band center of the surface Pd atom in Pd-doped Cu(111) explains why only physisorbed CO2 is observed, which even leads to slightly more negative limiting potential for CO formation in comparison with that on pure Cu(111). Accordingly, we infer that the dopant Pt with moderate upshift of the d-band center is the most capable of enhancing the electrocatalytic activity of Cu catalysts for CO2 reduction, which confirms the above thermodynamic and kinetic studies.
| Surface atoms | εd (eV) | Δεda (eV) | |
|---|---|---|---|
| a Δεd represents difference of the d-band center, εd of surface atoms between surface atoms in Ni-, Pd- and Pt-doped Cu(111) and pure surface atoms. | |||
| Pure Cu | −2.52 | — | |
| Pure Ni | −1.79 | — | |
| Pure Pd | −2.02 | — | |
| Pure Pt | −2.69 | — | |
| Ni-doped Cu(111) | Cu | −2.50 | 0.02 |
| Ni | −1.05 | 0.74 | |
| Pd-doped Cu(111) | Cu | −2.52 | 0.00 |
| Pd | −2.36 | −0.34 | |
| Pt-doped Cu(111) | Cu | −2.52 | 0.00 |
| Pt | −2.49 | 0.20 | |
Based on our present calculations, CO formation and further reduction are key reaction steps during the course of CO2 reduction into hydrocarbons. CO2 reduction on the Cu catalyst can be promoted and higher hydrocarbons yield may be able to be expected by the doping of transition metals. On the basis of the above discussion, two possible descriptors can be proposed in order to scale the electrocatalytic activity of Cu-based alloy catalysts for CO2 reduction. One is activation barriers of CO formation and further reduction. The transition metal-doped Cu-based alloy electrocatalysts with lower activation barriers for CO formation and further reduction will exhibit better catalytic activity for CO2 reduction. For example, the barriers of CO formation are decreased by the doped Ni, Pd and Pt atoms, and those of further CO reduction on the Pd- and Pt-doped Cu(111) surfaces are also reduced. The chemisorbed CO2 on Cu-based electrocatalyst surfaces will favor the formation of the key intermediate CO, i.e. the doping of Ni and Pt lead to the formation of chemisorbed CO2 and make CO formation easier. The moderately adsorbed CO on Cu-based electrocatalyst surfaces can avoid CO desorption and poisoning on surface active sites, thereby being in favor of further CO reduction, i.e. the Ni-doped Cu(111) surface has excessively strong CO adsorption, resulting in the highest barrier and the most negative limiting potential for further CO reduction among pure, Ni-, Pd- and Pt-doped Cu(111) surfaces. Another is the electronic structure of the transition metals-doped Cu-based electrocatalysts. The moderate interactions between Cu and transition metals and the moderate upshift of the d-band center of the doped transition metals in transition metal-doped Cu(111) will favor CO2 reduction, which can lead to moderate adsorption of intermediates. For example, the Ni-doped Cu(111) surface with the strongest interaction between Cu and Ni and the most upshift of the d-band center of the Ni atom, and Pd-doped Cu(111) surface with the downshift of the d-band center of Pd atom have relatively poorer electrocatalytic activity for CO2 reduction than the Pt-doped Cu(111) surface. Thus, an ideal Cu-based alloy electrocatalyst toward CO2 reduction should be able reduce activation barriers for CO formation and further reduction, and should have moderate electron interactions between Cu and the doped transition metals, and a moderate upshift of the d-band center of the doped transition metals. In these ways, CO2 reduction pathways can be facilitated and the yield of hydrocarbons CH4 and CH3OH can be enhanced.
4. Conclusions
In our present paper, the effect of the doped transition metal M (M = Ni, Pd and Pt) on CO2 reduction pathways and the origin of the electrocatalytic activity are investigated systematically by focusing on the CH4 and CH3OH formation pathways based on DFT calculations of the key adsorbates associated with the computational hydrogen electrode model. Our studies show that the doping of Ni, Pd and Pt can promote CO2 reduction and influence the selectivity of the reduction pathways and products, in which the doping of Pt may be able to lead to the strongest catalytic activity. The adsorption behavior of intermediates may be able to determine the CO2 reduction activity. The destabilization of the stronger adsorbed intermediates to CH3O by the doped Pd and Pt atoms make CH3O formation and further reduction occur, thus the selectivity of reduction pathways is revealed. Owing to high carbon and oxygen affinity of Ni, all intermediates can be stabilized by the doped Ni atom. The Ni-doped Cu(111) surface has the strongest adsorption ability among the Ni-, Pd- and Pt-doped Cu(111) surfaces, whereas the doping of Pd and Pt leads to the weakest and moderate adsorption of most intermediates, respectively, which may explain why Pt-doped Cu(111) surfaces have the best catalytic activity for CO2 reduction. Thus, it is concluded that moderate adsorbed intermediates on the Cu-based electrocatalyst surfaces will favor CO2 reduction.The enhanced electrocatalytic activity of the transition metal-doped Cu(111) surfaces may be owing to the decrease of the overpotential and electronic interactions between Cu and the transition metals. Further reduction of CO into CHO is the potential-limiting step on the pure and Ni-, Pd- and Pt-doped Cu(111) surfaces. The doping of Pd and Pt atoms can reduce the overpotential of CHO formation, whereas the doped Ni makes it slightly higher in comparison with pure Cu(111). The overpotential of CO formation is notably decreased after the doping of Ni, which is the rate-determining step on pure Cu(111). The significantly reduced overpotential for CO formation is also observed on the Pt-doped Cu(111). However, a slightly increased overpotential is obtained on the Pd-doped Cu(111) for CO formation. Simultaneously, the Pt-doped Cu(111) surface has the lowest overpotential for OH removal, suggesting that the doped metals with less affinity toward OH could enhance the catalytic activity. Thus, the doped Pt can simultaneously reduce the overpotential for CO formation and further reduction and most easily remove OH. The electronic structures analyses show that there is a moderate interaction between Cu and Pt and a moderate upshift of the d-band center of Pt. These analyses confirm why the Pt-doped Cu(111) surface has the best electrocatalytic activity for CO2 reduction.
Two possible descriptors can be proposed in order to scale the electrocatalytic activity of Cu-based electrocatalysts for CO2 reduction. One is activation barriers of CO formation and further reduction. The transition metal-doped Cu-based electrocatalysts with lower barriers will exhibit better catalytic activity for CO2 reduction. The chemisorbed CO2 on the Cu-based electrocatalyst will favor the formation of the key intermediate CO. The moderate adsorbed CO on the Cu-based electrocatalyst can avoid CO desorption and poisoning on surface active sites, thereby being in favor of further reduction of CO. Another is the electronic structure of the Cu-based electrocatalysts. The moderate interactions between Cu and the doped transition metals and the moderate upshift of the d-band center of the doped transition metals will be in favor of CO2 reduction. Thus, an ideal Cu-based electrocatalyst should be able to reduce barriers for CO formation and further reduction, and should have moderate interactions between Cu and the doped transition metals, and a moderate upshift of d-band center of the doped transition metals. In these ways, CO2 reduction pathways can be facilitated and the yield of hydrocarbons CH4 and CH3OH can be enhanced.
Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (Grant No. 21303048), the Outstanding Youth Foundation of the Education Department of Hunan Province (Grant No. 16B178), the Hunan Provincial Natural Science Foundation of China (Grant No. 13JJ4101), the Construct Program of the Key Discipline in Hunan Province (Applied Chemistry), the Doctoral Start-up Fund of Hunan University of Arts and Science, the Hunan Provincial College Students Inquiry Learning and Innovative Pilot Projects (Grant No. 201510549007) and the Innovation Project of Hunan University of Arts and Science (Grant No. YB1509 and YB1612).References
- A. Goeppert, M. Czaun, R. B. May, G. K. S. Prakash, G. A. Olah and S. R. Narayanan, Carbon Dioxide Capture from the Air Using a Polyamine Based Regenerable Solid Adsorbent, J. Am. Chem. Soc., 2011, 133, 20164–20167 CrossRef CAS PubMed.
- C. Graves, S. D. Ebbesen, M. Mogensen and K. S. Lackner, Sustainable Hydrocarbon Fuels by Recycling CO2 and H2O with Renewable or Nuclear Energy, Renewable Sustainable Energy Rev., 2011, 15, 1–23 CrossRef CAS.
- D. T. Whipple and P. J. A. Kenis, Prospects of CO2 Utilization via Direct Heterogeneous Electrochemical Reduction, J. Phys. Chem. Lett., 2011, 1, 3451–3458 CrossRef.
- W. L. Zhu, R. Michalsky, O. Metin, H. F. Lv, S. J. Guo, C. J. Wright, X. L. Sun, A. A. Peterson and S. H. Sun, Monodisperse Au Nanoparticles for Selective Electrocatalytic Reduction of CO2 to CO, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, New Insights into the Electrochemical Reduction of Carbon Dioxide on Metallic Copper Surfaces, Energy Environ. Sci., 2012, 5, 7050–7059 CAS.
- K. J. P. Schouten, E. Pérez Gallent and M. T. M. Koper, Structure Sensitivity of the Electrochemical Reduction of Carbon Monoxide on Copper Single Crystals, ACS Catal., 2013, 3, 1292–1295 CrossRef CAS.
- M. Le, M. Ren, Z. Zhang, P. T. Sprunger, R. L. Kurtz and J. C. Flake, Electrochemical Reduction of CO2 to CH3OH at Copper Oxide Surfaces, J. Electrochem. Soc., 2011, 158, E45–E49 CrossRef CAS.
- X. Nie, G. L. Griffin, M. J. Janik and A. Asthagiri, Surface Phases of Cu2O(111) under CO2 Electrochemical Reduction Conditions, Catal. Commun., 2014, 52, 88–91 CrossRef CAS.
- C. W. Li and M. W. Kanan, CO2 Reduction at Low Overpotential on Cu Electrodes Resulting from the Reduction of Thick Cu2O Films, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS PubMed.
- B. Innocent, D. Liaigre, D. Pasquier, F. Ropital, J. M. Legerand and K. B. Kokoh, Electro-Reduction of Carbon Dioxide to Formate on Lead Electrode in Aqueous Medium, J. Appl. Electrochem., 2009, 39, 227–232 CrossRef CAS.
- Y. Chen and M. W. Kanan, Tin Oxide Dependence of the CO2 Reduction Efficiency on Tin Electrodes and Enhanced Activity for Tin/Tin Oxide Thin-Film Catalysts, J. Am. Chem. Soc., 2012, 134, 1986–1989 CrossRef CAS PubMed.
- M. Jitaru, D. A. Lowy, M. Toma, B. C. Toma and L. Oniciu, Electrochemical Reduction of Carbon Dioxide on Flat Metallic Cathodes, J. Appl. Electrochem., 1997, 27, 875–889 CrossRef CAS.
- H. Noda, S. Ikeda, Y. Oda, K. Imai, M. Maeda and K. Ito, Electrochemical Reduction of Carbon Dioxide at Various Metal Electrodes in Aqueous Potassium Hydrogen Carbonate Solution, Bull. Chem. Soc. Jpn., 1990, 63, 2459–2462 CrossRef CAS.
- A. A. Peterson and J. K. Nørskov, Activity Descriptors for CO2 Electroreduction to Methane on Transition-Metal Catalysts, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, Electrocatalytic Conversion of Carbon Dioxide to Methane and Methanol on Transition Metal Surfaces, J. Am. Chem. Soc., 2014, 136, 14107–14113 CrossRef CAS PubMed.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Ionic Liquid-Mediated Selective Conversion of CO2 to CO at Low Overpotentials, Science, 2011, 334, 643–644 CrossRef CAS PubMed.
- J. F. Hull, Y. Himeda, W. H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Reversible Hydrogen Storage Using CO2 and a Proton-Switchable Iridium Catalyst in Aqueous Media under Mild Temperatures and Pressures, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed.
- R. Angamuthu, P. Byers, M. Lutz, A. L. Spek and E. Bouwman, Electrocatalytic CO2 Conversion to Oxalate by a Copper Complex, Science, 2010, 327, 313–315 CrossRef CAS PubMed.
- V. Tripkovic, M. Vanin, M. Karamad, M. E. Björketun, K. W. Jacobsen, K. S. Thygesen and J. Rossmeisl, Electrochemical CO2 and CO Reduction on Metal-Functionalized Porphyrin-Like Graphene, J. Phys. Chem. C, 2013, 117, 9187–9195 CAS.
- Y. Hori, Electrochemical CO2 Reduction on Metal Electrodes, in Modern Aspects of Electrochemistry, ed. G. Constantinos, C. Vayenas, R. E. White and M. E. Gamboa-Aldeco, Springer, New York, 2008, vol. 42, pp. 89–189 Search PubMed.
- N. Yang, F. Gao and C. E. Nebel, Diamond Decorated with Copper Nanoparticles for Electrochemical; Reduction of Carbon Dioxide, Anal. Chem., 2013, 85, 5764–5769 CrossRef CAS PubMed.
- W. Tang, A. A. Peterson, A. S. Varela, Z. P. Jovanov, L. Bech, W. J. Durand, S. Dahl, J. K. Nørskov and I. Chorkendorff, The Importance of Surface Morphology in Controlling the Selectivity of Polycrystalline Copper for CO2 Electroreduction, Phys. Chem. Chem. Phys., 2012, 14, 76–81 RSC.
- Y. Hori, K. Kikuchi, A. Murata and S. Suzuki, Production of Methane and Ethylene in Electrochemical Reduction of Carbon Dioxide at Copper Electrode in Aqueous Hydrogen Carbonate Solution, Chem. Lett., 1986, 15, 897–898 CrossRef.
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, Electroreduction of CO to CH4 and C2H4 at a Copper Electrode in Aqueous Solutions at Ambient Temperature and Pressure, J. Am. Chem. Soc., 1987, 109, 5022–5023 CrossRef CAS.
- R. L. Cook, R. C. MacDuff and A. F. Sammells, Evidence for Formaldehyde, Formic Acid, and Acetaldehyde as Possible Intermediates during Electrochemical Carbon Dioxide Reduction at Copper, J. Electrochem. Soc., 1989, 136, 1982–1984 CrossRef CAS.
- Y. Hori, A. Murata and R. Takahashi, Formation of Hydrocarbons in the Electrochemical Reduction of Carbon Dioxide at a Copper Electrode in Aqueous Solution, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- A. A. Peterson and J. K. Nørskov, Activity Descriptors for CO2 Electroreduction to Methane on Transition-Metal Catalysts, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- K. P. Kuhl, E. Cave, D. N. Abram and T. F. Jaramillo, New Insights into the Electrochemical Reduction of Carbon Dioxide on Metallic Copper Surfaces, Energy Environ. Sci., 2012, 5, 7050–7059 CAS.
- J. G. Chen, C. A. Menning and M. B. Zellner, Monolayer Bimetallic Surfaces: Experimental and Theoretical Studies of Trends in Electronic and Chemical Properties, Surf. Sci. Rep., 2008, 63, 201–254 CrossRef CAS.
- W. Yu, M. D. Porosoff and J. G. Chen, Review of Pt-Based Bimetallic Catalysis: From model Surfaces to Supported Catalysts, Chem. Rev., 2012, 112, 5780–5817 CrossRef CAS PubMed.
- D. A. Hansgen, D. G. Vlachos and J. G. Chen, Using First Principles to Predict Bimetallic Catalysts for the Ammonia Decomposition Reaction, Nat. Chem., 2010, 2, 484–489 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Towards the Computational Design of Solid Catalysts, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- N. D. Subramanian, G. Balaji, C. S. S. R. Kumar and J. J. Spivey, Development of Cobalt–Copper Nanoparticles as Catalysts for Higher Alcohol Synthesis from Syngas, Catal. Today, 2009, 147, 100–106 CrossRef CAS.
- N. Schumacher, K. Andersson, L. C. Grabow, M. Mavrikakis, J. Nerlov and I. Chorkendorff, Interaction of Carbon Dioxide with Cu Overlayers on Pt(111), Surf. Sci., 2008, 602, 702–711 CrossRef CAS.
- M. Watanabe, M. Shibata, A. Kato, M. Azuma and T. Sakata, Design of Alloy Electrocatalysts for CO2 Reduction III. The Selective and Reversible Reduction of CO2 on Cu Alloy Electrodes, J. Electrochem. Soc., 1991, 138, 3382–3389 CrossRef CAS.
- X. Y. Zhao, B. B. Luo, R. Long, C. M. Wang and Y. J. Xiong, Composition-Dependent Activity of Cu-Pt Alloy Nanocubes for Electrocatalytic CO2 Reduction, J. Mater. Chem. A, 2015, 3, 4134–4138 CAS.
- J. Christophe, T. Doneux and C. Buess-Herman, Electroreduction of Carbon Dioxide on Copper-Based Electrodes: Activity of Copper Single Crystals and Copper–Gold Alloys, Electrocatalysis, 2012, 3, 139–146 CrossRef CAS.
- Z. Xu, E. Lai, Y. Shao-Horn and K. Hamad-Schifferli, Compositional Dependence of the Stability of AuCu Alloy Nanoparticles, Chem. Commun., 2012, 48, 5626–5628 RSC.
- J. Nerlov and I. Chorkendorff, Methanol Synthesis from CO2, CO, and H2 over Cu(100) and Ni/Cu(100), J. Catal., 1999, 181, 271–279 CrossRef CAS.
- J. Nerlov, S. Sckerl, J. Wambach and I. Chorkendorff, Methanol Synthesis from CO2, CO and H2 over Cu(100) and Cu(100) Modified by Ni and Co, Appl. Catal., A, 2000, 191, 97–109 CrossRef CAS.
- E. Vesselli, L. D. Rogatis, X. Ding, A. Baraldi, L. Savio, L. Vattuone, M. Rocca, P. Fornasiero, M. Peressi, A. Baldereschi, R. Rosei and G. Comelli, Carbon Dioxide Hydrogenation on Ni(110), J. Am. Chem. Soc., 2008, 130, 11417–11422 CrossRef CAS PubMed.
- X. Liu, L. S. Zhu, H. Wang, G. Y. He and Z. Y. Biao, Catalysis Performance Comparison for Electrochemical Reduction of CO2 on Pd–Cu/Graphene Catalyst, RSC Adv., 2016, 6, 38380–38387 RSC.
- P. J. Berlowitz and D. W. Goodman, Kinetics of Methanol and Methane Synthesis over Pd/SiO2 and Pd/La2O3, J. Catal., 1987, 108, 364–368 CrossRef CAS.
- M. L. Poutsma, L. F. Elek, P. A. Ibarbia, A. P. Risch and J. A. Rabo, Selective Formation of Methanol from Synthesis Gas over Palladium Catalysts, J. Catal., 1978, 52, 157–168 CrossRef CAS.
- Y. A. Ryndin, R. F. Hicks, A. T. Bell and Y. I. Yermakov, Effects of Metal-Support Interactions on the Synthesis of Methanol over Palladium, J. Catal., 1981, 70, 287–297 CrossRef CAS.
- J. P. Reilly, D. O'Connell and C. J. Barnes, Modification of Formate Stability by Alloying: the Cu(100)-c(2 × 2)-Pt System, J. Phys.: Condens. Matter, 1999, 11, 8417–8430 CrossRef CAS.
- Y. X. Yang, M. G. White and P. Liu, Theoretical Study of Methanol Synthesis from CO2 Hydrogenation on Metal-Doped Cu(111) Surfaces, J. Phys. Chem. C, 2012, 116, 248–256 CAS.
- P. Hirunsit, Electroreduction of Carbon Dioxide to Methane on Copper, Copper–Silver, and Copper–Gold Catalysts: A DFT Study, J. Phys. Chem. C, 2013, 117, 8262–8268 CAS.
- P. Hirunsit, W. Soodsawang and J. Limtrakul, CO2 Electrochemical Reduction to Methane and Methanol on Copper-Based Alloys: Theoretical Insight, J. Phys. Chem. C, 2015, 119, 8238–8249 CAS.
- M. Qiu, Z. X. Fang, Y. Li, J. Zhu, X. Huang, K. N. Ding and W. K. Chen, First-Principles Investigation of the Activation of CO2 Molecule on TM/Cu (TM = Fe, Co and Ni) Surface Alloys, Appl. Surf. Sci., 2015, 353, 902–912 CrossRef CAS.
- M. Qiu, Y. Liu, J. Wu, Y. Li, X. Huang, W. K. Chen and Y. F. Zhang, Theoretical Investigations of the Activation of CO2 on the Transition Metal-Doped Cu(100) and Cu(111) Surfaces, Chin. J. Struct. Chem., 2016, 35, 669–678 CAS.
- S. Baroni, A. Dal Corso, S. de Gironcoli and P. Giannozzi, PWSCF and PHONON: Plane-Wave Pseudo-Potential Codes, 2001, http://www.pwscf.org Search PubMed.
- A. Kokalj, XCrySDen—A New Program for Displaying Crystalline Structures and Electron Densities, J. Mol. Graphics Modell., 1999, 17, 176–179 CrossRef CAS PubMed.
- A. Kokalj and M. Causà, Scientific Visualization in Computational Quantum Chemistry, in Proceedings of High Performance Graphics Systems and Applications European Workshop, CINECA-Interuniversity Consortium, Bologna, Italy, 2000 Search PubMed.
- A. Kokalj and M. Causà, XCrySDen: (X-Window) CRYstalline Structures and DENsities, 2001, http://www-k3.ijs.si/kokalj/xc/XCrySDen.html Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- D. Vanderbilt, Soft Self-Consistent Pseudopotentials in a Generalized Eigenvalue Formalism, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef.
- J. Greeley, A. A. Gokhale, J. Kreuser, J. A. Dumesic, H. Topsøe, N. Y. Topsøe and M. Mavrikakis, CO Vibrational Frequencies on Methanol Synthesis Catalysts: A DFT study, J. Catal., 2003, 213, 63–72 CrossRef CAS.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC Press/Taylor and Francis, Boca Raton, FL, 93rd edn, Internet Version 2012, 2012 Search PubMed.
- M. Methfessel and A. T. Paxton, High-Precision Sampling for Brillouin-zone Integration in Metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 3616–3621 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Theoretical Surface Science and Catalysis-Calculations and Concepts, Adv. Catal., 2000, 45, 71–129 CAS.
- H. A. Hansen, C. Shi, A. C. Lausche, A. A. Peterson and J. K. Nørskov, Bifunctional Alloys for the Electroreduction of CO2 and CO, Phys. Chem. Chem. Phys., 2016, 18, 9194–9201 RSC.
- L. H. Ou, Chemical and Electrochemical Hydrogenation of CO2 to Hydrocarbons on Cu Single Crystal Surfaces: Insights into the Mechanism and Selectivity from DFT Calculations, RSC Adv., 2015, 5, 57361–57371 RSC.
- G. Henkelman and H. Jonsson, Improved Tangent Estimate in the Nudged Elastic Band Method for Finding Minimum Energy Paths and Saddle Points, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jonsson, A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- Y. F. Zhao, Y. Yang, C. Mims, C. H. F. Peden, J. Li and D. Mei, Insight into Methanol Synthesis from CO2 Hydrogenation on Cu(111): Complex Reaction Network and the Effects of H2O, J. Catal., 2011, 281, 199–211 CrossRef CAS.
- Y. X. Yang, J. Evans, J. A. Rodriguez, M. G. White and P. Liu, Fundamental Studies of Methanol Synthesis from CO2 Hydrogenation on Cu(111), Cu Clusters, and Cu/ZnO(0001), Phys. Chem. Chem. Phys., 2010, 12, 9909–9917 RSC.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, Origin of Overpotential for Oxygen Reduction at a Fuel-Cell Cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- B. Hammer and J. K. Nørskov, Electronic Factors Determining the Reactivity of Metal Surfaces, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Why Gold is the Noblest of All the Metals, Nature, 1995, 376, 238–240 CrossRef CAS.
- J. Greeley, J. K. Nørskov and M. Mavrikakis, Electronic Structure and Catalysis on Metal Surfaces, Annu. Rev. Phys. Chem., 2002, 53, 319–348 CrossRef CAS PubMed.
- B. Hammer, Y. Morikawa and J. K. Nørskov, CO Chemisorption at Metal Surfaces and Overlayers, Phys. Rev. Lett., 1996, 76, 2141–2144 CrossRef CAS PubMed.
- M. Gajdoš, A. Eichler and J. Hafner, CO Adsorption on Close-Packed Transition and Noble Metal Surfaces: Trends from ab initio Calculations, J. Phys.: Condens. Matter, 2004, 16, 1141–1164 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28815d |
| This journal is © The Royal Society of Chemistry 2017 |