
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Further insight into the photochemical behavior of 3-aryl-N-(arylsulfonyl)propiolamides: tunable synthetic route to phenanthrenes†
Ming Chen,
Xinxin Zhao,
Chao Yang,
Yanpei Wang and
Wujiong Xia *
*
State Key Lab of Urban Water Resource and Environment, Shenzhen Graduate School, Harbin Institute of Technology, Harbin 150080, P. R. China. E-mail: xiawj@hit.edu.cn
First published on 20th February 2017
Abstract
Reported herein is further insight into the photochemical behaviour of 3-aryl-N-(arylsulfonyl)-propiolamides, which provides a straightforward way to access meaningful phenanthrenes. Mechanistic investigation indicated that aryl migration, C–C coupling, 1,3-hydrogen shift, desulfonylation and elimination were involved in the process. Moreover, this protocol allowed for scale-up using a flow reactor.
Phenanthrenes represent an important class of the simplest polycyclic aromatic hydrocarbons (PAHs) and they have received much interest in materials science and technology due to their utility in functional materials, especially in photoelectronic devices.1 Moreover, compounds containing this structural core are often of great value in pharmaceutical chemistry because they generally display a broad spectrum of biological activities, such as anti-microbial, anti-tumor, anti-inflammatory, anti-malarial and others.2 Additionally, phenanthrene motifs can be found in a large class of natural products.3 Therefore, continuous effort has been devoted to the preparation of phenanthrene derivatives, and in the past decades, numerous relevant studies have been reported.4 Among all the available strategies, the Lewis acid/base/metal mediated intramolecular annulation of alkynylated biaryls5 and various metal induced intermolecular [4 + 2] cyclization of biaryls with alkynes6/bis(pinacolatoboryl) alkenes7 represented the typical approaches to construct the phenanthrene skeleton. In addition, the preparation of stilbene followed by photocyclization, radical or metal catalyzed oxidative C–C coupling reactions has been extensively studied as well.8 Besides, the construction of a phenanthrene ring could also be achieved via intramolecular ring-closing metathesis.9 Despite the above, most of these methods suffered from certain limitations, such as the use of complex and expensive metal catalysts which may be unfriendly to the environment, not-easily accessible precursors, air- or moisture-sensitive reagents, and harsh reaction conditions. Thus, development of an efficient and convenient method for the construction of phenanthrene scaffold from readily prepared substrates is of important significance.
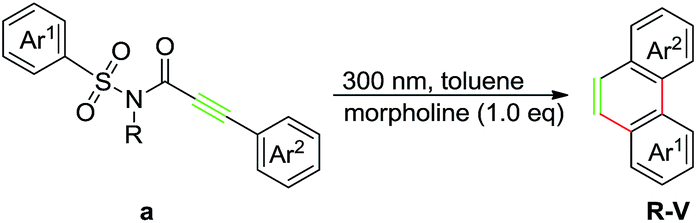
During our recent exploration of the ultraviolet light induced photochemical behavior of N-methyl-3-phenyl-N-(phenylsulfonyl)propiolamide in CH3CN, we disclosed that along with the main product 2-methylphenanthro[9,10-d]isothiazol-3(2H)-one 1,1-dioxide through the Smiles rearrangement/Mallory reaction, a trace amount of phenanthrene was also observed after removal of the sulfonylamide group (Scheme 1a).10 Such a result prompted us to explore suitable reaction conditions for the preferential preparation of phenanthrenes, and described in this paper are our continuous work on the photochemical behavior of 3-aryl-N-(arylsulfonyl)propiolamides to provide a straightforward way to a series of phenanthrenes tuned by base (Scheme 1b).
 | ||
| Scheme 1 Previous work and this work. | ||
At the outset of further investigation, compound 1a was chosen as the model substrate and additives with different property such as acid/base/salt were employed to modify the reaction, respectively. The results turned out to be that, unlike the less effect of acid and salt, the addition of 1 equivalent of dimethylamine dramatically improved the yield of A in comparison to the reaction with no additives (Table 1, entries 1–4). Moreover, the increasing amount of dimethylamine to 3 equivalents totally yielded the product A with the retard of formation of 1b (Table 1, entry 5). Based on this observation, screening on the bases, such as triethylamine, methyl amine, cyclohexamine and morpholine, was conducted which led to comparable yields (Table 1, entries 6–9). Remarkably, the decrease in the amount of morpholine to 1 equivalent gave the best yield of 80% (Table 1, entry 10), whereas further decrease to 0.5 equivalent led to lower yield and poor selectivity (Table 1, entry 11). Gladly, we found that reactions run equally efficiently under air/inert gas conditions (Table 1, entries 10 and 12). Then a set of solvents was screened and the reaction performed in toluene provided the best result than in other medium (Table 1, entries 13–18). It is noteworthy that the solvent plays a critical role in the result in which most of the tested substrates have been decomposed in strong polar solvents, leading to a low yield of the desired product. The screening of light source indicated that the suitable wavelength was proved to be 300 nm (Table 1, entries 10, 19–21). Finally, we found there was a little loss on yield (in comparison with entry 10) when the irradiation time was increased to 1.8 hours (Table 1, entries 22).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Additive (equiv.) | Light | t (h) | Yieldb (%) | |
| A | 1b | |||||
| a Reaction conditions: 1a (0.2 mmol), solvent (anhydrous 40 ml), under air atmosphere, irradiation at room temperature.b Isolated yield.c Under N2 atmosphere. | ||||||
| 1 | Toluene | No additive | 300 nm | 1.5 | <5 | 71 |
| 2 | Toluene | Acetic acid (1.0) | 300 nm | 1.5 | <5 | 68 |
| 3 | Toluene | Dimethylamine (1.0) | 300 nm | 1.0 | 40 | 45 |
| 4 | Toluene | NaCl (1.0) | 300 nm | 1.3 | <5 | 65 |
| 5 | Toluene | Dimethylamine (3.0) | 300 nm | 1.0 | 70 | 0 |
| 6 | Toluene | Et3N (3.0) | 300 nm | 1.0 | 68 | 0 |
| 7 | Toluene | Methylamine (3.0) | 300 nm | 1.2 | 63 | 0 |
| 8 | Toluene | Cyclohexamine (3.0) | 300 nm | 0.9 | 65 | 0 |
| 9 | Toluene | Morpholine (3.0) | 300 nm | 0.7 | 72 | 0 |
| 10 | Toluene | Morpholine (1.0) | 300 nm | 0.9 | 80 | 0 |
| 11 | Toluene | Morpholine (0.5) | 300 nm | 1.2 | 55 | 20 |
| 12c | Toluene | Morpholine (1.0) | 300 nm | 0.9 | 80 | 0 |
| 13 | MeCN | Morpholine (1.0) | 300 nm | 1.2 | 15 | 60 |
| 14 | MeOH | Morpholine (1.0) | 300 nm | 1.2 | 23 | 55 |
| 15 | DMF | Morpholine (1.0) | 300 nm | 1.0 | 20 | 53 |
| 16 | THF | Morpholine (1.0) | 300 nm | 1.3 | 72 | 0 |
| 17 | Benzene | Morpholine (1.0) | 300 nm | 1.0 | 70 | 0 |
| 18 | DCM | Morpholine (1.0) | 300 nm | 1.5 | 65 | Trace |
| 19 | Toluene | Morpholine (1.0) | 350 nm | 12.0 | 58 | Trace |
| 20 | Toluene | Morpholine (1.0) | HPML | 4.0 | 60 | Trace |
| 21 | Toluene | Morpholine (1.0) | MPML | 6.0 | 62 | Trace |
| 22 | Toluene | Morpholine (1.0) | 300 nm | 1.8 | 78 | 0 |
Subsequently, a series of substrates were subjected to the optimal conditions to investigate the reaction scope, and the results were summarized in Tables 2 and 3. As expected, substrate with the same aryl groups tethered to the sulfonyl or the alkynyl resulted in the same adduct regardless of divergent R3 substituent on nitrogen atom (Table 2, A, B, C and Table 3, R), it should be noted that the non-substituted phenanthrene A, 3-methylphenanthrene B, 3-methoxyphenanthrene C and the heterocyclic aromatic compound R could be readily prepared in moderate to excellent yields. Substrates with electrophilic groups such as the halogen atom, trifluoromethyl and cyano on the para-position of aryl ring were perfectly tolerant with the reaction conditions, generating the corresponding products in 30–72% yields (Table 2, D–G, J). Sterically bulkier group such as butyl- or phenyl-substituted substrate 15a and 16a could furnish the corresponding products as well in the yield of 68% and 56%, respectively (Table 2, H and I). Substrate 18a with p-methyl on both benzene rings afforded the single product K in 67% yield (Table 2, K). The reaction of compound 19a bearing meta-methyl on aryl ring delivered a mixture of regioisomers in a ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (Table 2, L and L′), while the meta-fluoro substituted substrate 20a gave two regioselective products M and M′ in a ratio of 8:1.4c As for the photoreaction of ortho-methyl substituted substrates 21a, this protocol allowed access to both the desired 1-methylphenanthrene N and the unexpected 1-methyl-2-(phenylethynyl)benzene N′ in a ratio of 3:5. In view of our previous work, the arising of N′ was supposed possibly from the Smiles rearrangement of 21a followed by the elimination of sulfonylamide group. Replacing the methyl group with F, Cl atom and trifluoromethyl group equally afforded corresponding structural isomers in different ratios (Table 2, N–Q). More importantly, other polycyclic compounds, such as benzo[c]phenanthrene and chrysene, could also be prepared via this synthetic protocol (Table 3, T and U). In addition, the reaction was also applicable for the synthesis of heteroaromatic compounds and poly-substituted phenanthrene (Table 3, R, S and V). Particularly, the yield of compound V reached to 50% even the amount of 30a increased to 0.31 gram.
:3 (Table 2, L and L′), while the meta-fluoro substituted substrate 20a gave two regioselective products M and M′ in a ratio of 8:1.4c As for the photoreaction of ortho-methyl substituted substrates 21a, this protocol allowed access to both the desired 1-methylphenanthrene N and the unexpected 1-methyl-2-(phenylethynyl)benzene N′ in a ratio of 3:5. In view of our previous work, the arising of N′ was supposed possibly from the Smiles rearrangement of 21a followed by the elimination of sulfonylamide group. Replacing the methyl group with F, Cl atom and trifluoromethyl group equally afforded corresponding structural isomers in different ratios (Table 2, N–Q). More importantly, other polycyclic compounds, such as benzo[c]phenanthrene and chrysene, could also be prepared via this synthetic protocol (Table 3, T and U). In addition, the reaction was also applicable for the synthesis of heteroaromatic compounds and poly-substituted phenanthrene (Table 3, R, S and V). Particularly, the yield of compound V reached to 50% even the amount of 30a increased to 0.31 gram.
|
||||||
|---|---|---|---|---|---|---|
|
||||||
| Subs | R1 | R2 | R3 | t (h) | Product | Yieldb (%) |
| a All reactions are carried out under the standard conditions.b Isolated yield.c The ratio was determinated by GC.d The total yield of both isomers. | ||||||
| 1a | H | H | Me | 0.9 | A | 80 |
| 2a | H | H | Bn | 1.2 | A | 75 |
| 3a | H | H | Propargyl | 1.0 | A | 70 |
| 4a | H | H | Allyl | 1.1 | A | 75 |
| 5a | H | H | 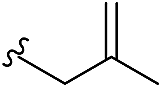 |
1.0 | A | 87 |
| 6a | Me | H | Me | 1.0 | B | 85 |
| 7a | H | Me | Me | 1.2 | B | 90 |
| 8a | Me | H | Allyl | 1.2 | B | 80 |
| 9a | OMe | H | Me | 2.5 | C | 55 |
| 10a | H | OMe | Me | 2.5 | C | 65 |
| 11a | F | H | Me | 1.2 | D | 55 |
| 12a | CI | H | Me | 1.2 | E | 72 |
| 13a | Br | H | Me | 2.5 | F | 30 |
| 14a | CF3 | H | Me | 1.5 | G | 70 |
| 15a | Ph | H | Me | 2.5 | H | 56 |
| 16a | t-Butyl | H | Me | 1.2 | I | 84 |
| 17a | CN | H | Me | 2.0 | J | 55 |
| 18a | Me | Me | Me | 1.2 | K | 67 |
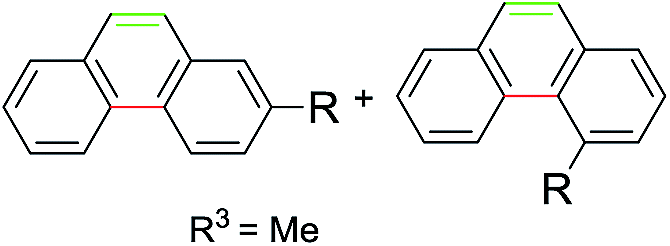
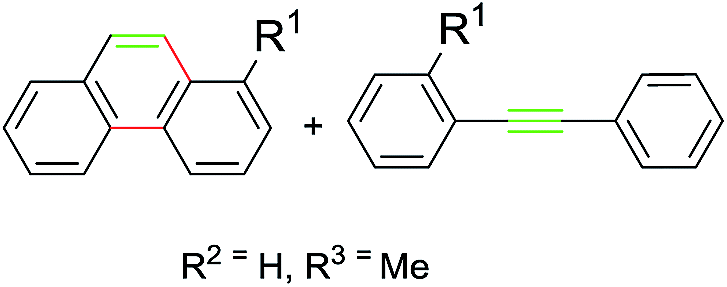
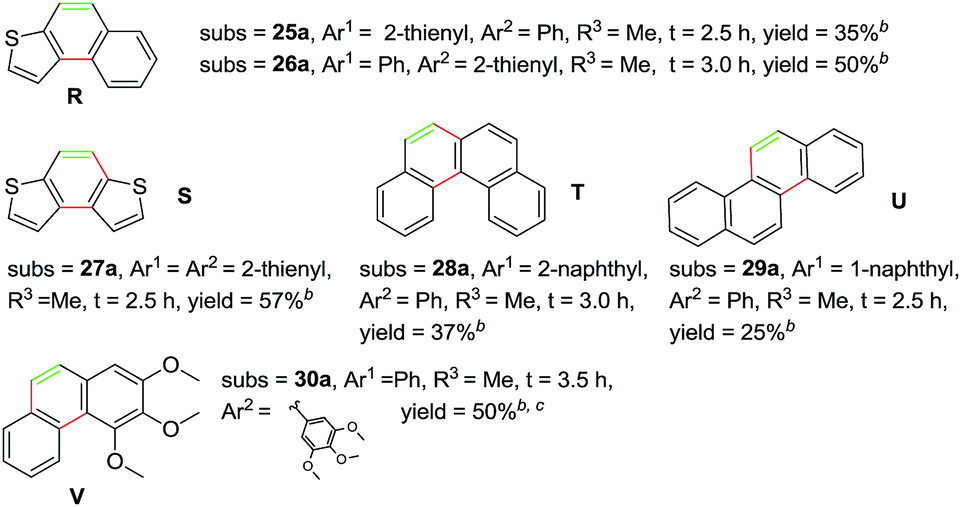
Notably, among all the prepared products, compounds A, B, L′, N, K, U and V were identified as natural products and have been reported in previous literatures,11–17 which indicated the inherent synthetic utility of this novel protocol.
Before the reaction mechanism was proposed, some control experiments were conducted using 7a as representative compound. First, 7a was irradiated under the standard conditions for 0.2 h, fortunately, except for the product B, both the rearrangement product 7c and a new compound 7d which was quite labile and could be rapidly converted into B was isolated (Scheme 2, eqn (1)). Further exploration disclosed that 7d was derived from compound 7c, thus suggesting that product B was probably resulted from a tandem three-step photo transformation (Scheme 2, eqn (2)), and all these transformations only took place under the irradiation of UV light.
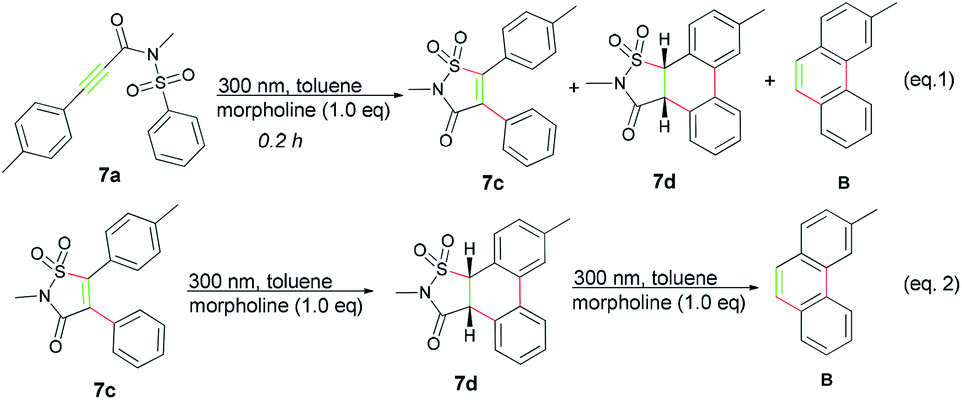 | ||
| Scheme 2 Control experiments. | ||
Based upon the above results, a tentative mechanism for this photoreaction was proposed as shown in Scheme 3. The alkyne group of substrate a was first excited to 1,2-biradicals which initiated radical Smiles rearrangement/C–S bonding cascade reaction to form the isolatable intermediate c,10 then subsequently oxidative cyclization delivered product b (Scheme 3, path a).10 And cyclization reaction occurred to form the intermediate II which underwent 1,3-H shift process with the morpholine served as an assistant agent18 to form compound d that was identified by the isolation of 7d as shown in Scheme 2. Then a ring opening reaction of d occurred and afforded the 1,4-biradical intermediate III after exclusion of sulfur dioxide. The final phenanthrene product was formed from intermediate III after elimination of isocyanic intermediate e, which could be trapped by morpholine to produce morpholine-4-carboxamide f (Scheme 3, path c). Furthermore, to add more credence to the existence of compound f, N-benzylmorpholine-4-carboxamide (R3 = Bn) was isolated in 40% yield during the reaction of substrate 2a.19 It was reasonable to assume that some of the intermediate c, which was arisen from the substrate containing ortho-substituent on aryl ring, could not effectively undergo the C–C coupling due to the steric hindrance, but be converted into the diphenylethyne product instead after removing the sulfonylamide group with the aid of morpholine in the manner as described above (Scheme 3, path b).
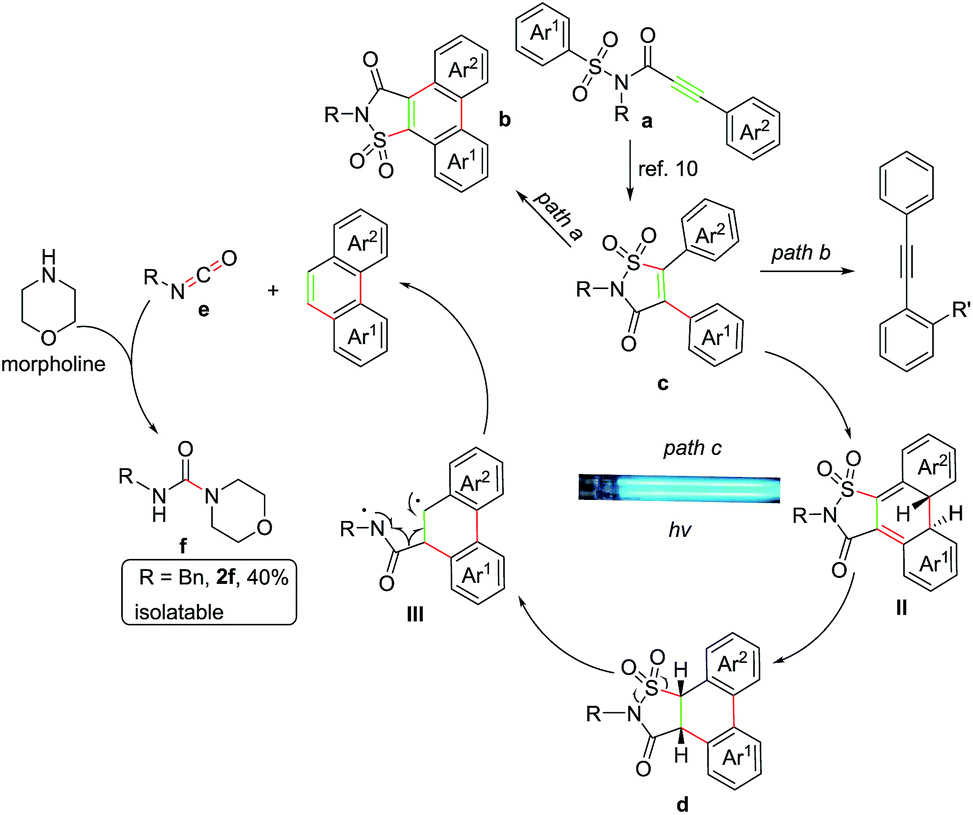 | ||
| Scheme 3 The proposed reaction mechanism. | ||
In order to demonstrate the synthetic value and utility of this protocol, we turned our attention toward the preparation of phenanthrenes on a large scale.20 With this aim in mind, we carried out the photoreaction in a continuous flow reactor with compound 1a, of which the loading was increased to 1.0 gram and concentration to 10 mM, as the subject substrate. Then the reaction was run for 4 hours to afford the target product A in 75% with little loss on yield (Scheme 4).
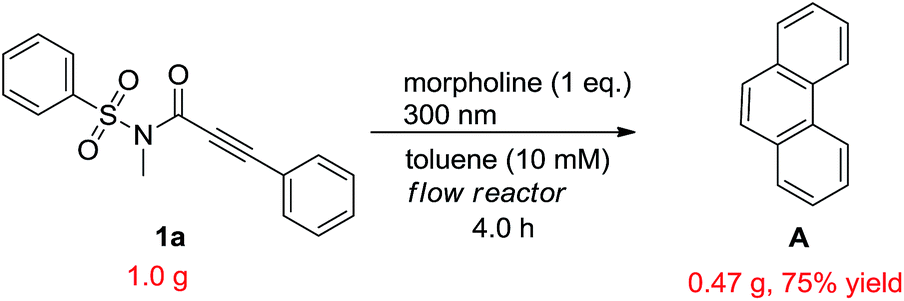 | ||
| Scheme 4 Gram scale study with a flow reactor. | ||
In conclusion, the photoreaction of easily accessible 3-aryl-N-(arylsulfonyl)propiolamides to prepare phenanthrene derivatives via radical Smiles rearrangement/elimination has been developed. The addition of morpholine enables the 1,3-H shift which is crucial to the regioselective formation of phenanthrenes. Moreover, reasonable mechanism was elaborated in detail based on the isolated intermediates. It's worthy to point out that, using our established protocol, the desired substituents on phenanthrene rings could be simply introduced in advance on the aromatic groups of the starting material. Besides, the availability and efficiency of this approach makes it appealing for the syntheses of certain natural products.
Acknowledgements
We are grateful for the financial support from China NSFC (No. 21372055, 21472030 and 21672047), SKLUWRE (No. 2015DX01), the Fundamental Research Funds for the Central Universities (Grant No. HIT.BRETIV.201310) and HLJNSF (B201406).Notes and references
- (a) A. C. Grimsdale and K. Mullen, Angew. Chem., Int. Ed., 2005, 44, 5592 CrossRef CAS PubMed; (b) J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452 CrossRef CAS PubMed; (c) G. Marcelo, T. J. V Prazeres, M. T. Charreyre, J. M. Martinho and J. P. S. Farinha, Macromolecules, 2009, 43, 501 CrossRef; (d) M. P. Robin and R. K. O'Reill, Polym. Int., 2015, 64, 174 CrossRef CAS; (e) T. Kojima, I. Kawajiri, J. I. Nishida, C. Kitamura, H. Kurata, M. Tanaka, H. Ikeda and T. Kawase, Bull. Chem. Soc. Jpn., 2016, 89, 931 CrossRef CAS.
- Selected examples for anti-microbial: (a) H. Shimura, M. Matsuura, N. Takada and Y. Koda, Phytochemistry, 2007, 68, 1442 CrossRef CAS PubMed; (b) A. Kovács, A. Vasas and J. Hohmann, Phytochemistry, 2008, 69, 1084 CrossRef PubMed; (c) K. Yoshikawa, C. Baba, K. Iseki, T. Ito, Y. Asakawa, S. Kawano and T. Hashimoto, J. Nat. Med., 2014, 68, 743 CrossRef CAS PubMed. For anti-tumor: (d) X. Yang, Q. Shi, Y. N. Liu, G. Zhao, K. F. Bastow, J. C. Lin, S. C. Yang, P. C. Yang and K. H. Lee, J. Med. Chem., 2009, 52, 5262 CrossRef CAS PubMed; (e) A. R. Kirtane, H. L. Wong, B. R. Guru, L. G. Lis, G. I. Georg, V. J. Gurvich and J. Panyam, Mol. Pharmaceutics, 2015, 12, 2912 CrossRef CAS PubMed; (f) Y. Kwon, J. Song, H. Lee, E. Y. Kim, K. Lee, S. K. Lee and S. Kim, J. Med. Chem., 2015, 58, 7749 CrossRef CAS PubMed. For anti-inflammatory: (g) R. W. Friesen and J. A. Mancini, J. Med. Chem., 2008, 51, 4059 CrossRef CAS PubMed; (h) Y. Kanekar, K. Basha, S. Duche, R. Gupte and A. Kapat, Eur. J. Med. Chem., 2013, 67, 454 CrossRef CAS PubMed; (i) Y. Lin, F. Wang, L. Yang, Z. Chun, J. Bao and G. Zhang, Phytochemistry, 2013, 95, 242 CrossRef CAS PubMed. For anti-malarial: (j) L. Krbechek, R. R. Riter, R. G. Wagner and C. W. Huffman, J. Med. Chem., 1970, 13, 234 CrossRef CAS PubMed; (k) E. A. Nodiff, A. J. Saggiomo, M. Shinbo, E. H. Chen, H. Otomasu, Y. Kondo, T. Kikuchi, B. L. Verma and S. Matsuura, J. Med. Chem., 1972, 15, 775 CrossRef CAS PubMed; (l) M. Leven, J. Held, S. Duffy, S. Tschan, S. Sax, J. Kamber, W. Frank, K. Kuna, D. Geffken, C. Siethoff, S. Barth, V. M. Avery, S. Wittlin, B. Mordmüller and T. Kurz, J. Med. Chem., 2014, 57, 7971 CrossRef CAS PubMed.
- (a) S. Estrada, R. A. Toscano and R. Mata, J. Nat. Prod., 1999, 62, 1175 CrossRef CAS PubMed; (b) G. M. Cragg and D. J. Newman, J. Nat. Prod., 2004, 67, 232 CrossRef CAS PubMed; (c) E. R. Walker, S. Y. Leung and A. G. M. Barrett, Tetrahedron Lett., 2005, 46, 6537 CrossRef CAS; (d) K. Yoshikawa, T. Ito, K. Iseki, C. Baba, H. Imagawa, Y. Yagi, H. Morita, Y. Asakawa, S. Kawano and T. Hashimoto, J. Nat. Prod., 2012, 75, 605 CrossRef CAS PubMed; (e) J. Ren, X. P. Qian, Y. G. Guo, T. Li, S. K. Yan, H. Z. Jin and W. D. Zhang, Phytochem. Lett., 2016, 18, 64 CrossRef CAS; (f) Q. Ye, Y. Mei, P. Yang, L. Cheng and D. Kong, Chem. Nat. Compd., 2016, 52, 381 CrossRef CAS; (g) X. M. Zhou, C. J. Zheng, L. S. Gan, G. Y. Chen, X. P. Zhang, X. P. Song, G. N. Li and C. G. Sun, J. Nat. Prod., 2016, 79, 1791 CrossRef CAS PubMed.
- For reviews, see: (a) A. J. Floyd, S. F. Dyke and S. E. Ward, Chem. Rev., 1976, 76, 509 CrossRef CAS; (b) S. Kotha, S. Misra and S. Halde, Tetrahedron, 2008, 64, 10775 CrossRef CAS; (c) K. B. Jørgensen, Molecules, 2010, 15, 4334 CrossRef PubMed; (d) E. Aguilar, R. Sanz, M. A. F. Fernández-Rodríguez and P. García-García, Chem. Rev., 2016, 116, 8256 CrossRef CAS PubMed.
- Lewis acid, see: (a) M. B. Goldfinger and T. M. Swager, J. Am. Chem. Soc., 1994, 116, 7895 CrossRef CAS; (b) J. D. Tovar and T. M. Swager, J. Organomet. Chem., 2002, 653, 215 CrossRef CAS; (c) T. Yao, M. A. Campo and R. C. Larock, Org. Lett., 2004, 6, 2677 CrossRef CAS PubMed; (d) T. Yao, M. A. Campo and R. C. Larock, J. Org. Chem., 2005, 70, 3511 CrossRef CAS PubMed. Base: (e) Y. Wang, J. Xu and D. J. Burton, J. Org. Chem., 2006, 71, 7780 CrossRef CAS PubMed; (f) A. Kondoh, T. Aoki and M. Terada, Chem.–Eur. J., 2015, 21, 12577 CrossRef CAS PubMed. Metal: (g) A. Fürstner and V. Mamane, J. Org. Chem., 2002, 67, 6264 CrossRef; (h) V. Mamane, P. Hannen and A. Fürstner, Chem.–Eur. J., 2004, 10, 4556 CrossRef CAS PubMed; (i) M. L. Hossain, F. Ye, Z. Liu, Y. Xia, Y. Shi, L. Zhou, Y. Zhang and J. Wang, J. Org. Chem., 2014, 79, 8699 CrossRef PubMed; (j) K. Bera, S. Sarkar and U. Jana, Tetrahedron Lett., 2015, 56, 312 CrossRef CAS.
- (a) Z. Shi, S. Ding, Y. Cui and N. Jiao, Angew. Chem., Int. Ed., 2009, 48, 7895 CrossRef CAS PubMed; (b) C. Wang, S. Rakshit and F. Glorius, J. Am. Chem. Soc., 2010, 132, 14006 CrossRef CAS PubMed; (c) A. Matsumoto, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 6557 CrossRef CAS PubMed; (d) T. Xiao, X. Dong, Y. Tang and L. Zhou, Adv. Synth. Catal., 2012, 354, 3195 CrossRef CAS; (e) Y. D. Lin, C. L. Cho, C. W. Ko, A. Pulte and Y. T. Wu, J. Org. Chem., 2012, 77, 9979 CrossRef CAS PubMed; (f) T. Nagata, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2014, 79, 8960 CrossRef CAS PubMed; (g) M. Bu, G. Lu and C. Cai, Org. Chem. Front., 2016, 3, 630 RSC.
- (a) M. Shimizu, I. Nagao, Y. Tomioka and T. Hiyama, Angew. Chem., Int. Ed., 2008, 47, 8096 CrossRef CAS PubMed; (b) M. Shimizu, I. Nagao, Y. Tomioka, T. Kadowaki and T. Hiyama, Tetrahedron, 2011, 67, 8014 CrossRef CAS.
- Selected example for photocyclization: (a) F. B. Mallory, K. E. Butler, A. C. Evans, E. J. Brondyke, C. W. Mallory, C. Yang and A. Ellenstein, J. Am. Chem. Soc., 1997, 119, 2119 CrossRef CAS; (b) Q. Lefebvre, M. Jentsch and M. Rueping, Beilstein J. Org. Chem., 2013, 9, 1883 CrossRef PubMed; (c) M. Y. Chang, Y. C. Chen, S. Y. Lin and C. K. Chan, Tetrahedron, 2014, 70, 1740 CrossRef CAS; (d) Z. Li and R. J. Twieg, Chem.–Eur. J., 2015, 21, 15534 CrossRef CAS PubMed. For radical: (e) D. C. Harrowvena, M. I. T. Nunna and D. R. Fenwick, Tetrahedron Lett., 2002, 43, 3185 CrossRef; (f) D. C. Harrowven, I. L. Guy and L. Nanson, Angew. Chem., Int. Ed., 2006, 45, 2242 CrossRef CAS PubMed; (g) S. Radix and R. Barret, Tetrahedron, 2007, 63, 12379 CrossRef CAS. For metal: (h) K. L. Wang, M. Y. Lü, Q. M. Wang and R. Q. Huang, Tetrahedron, 2008, 64, 7504 CrossRef CAS; (i) A. K. Yadav, H. Ila and H. Junjappa, Eur. J. Org. Chem., 2010, 2010, 338 CrossRef; (j) V. Gupta, V. U. B. Rao, T. Das, K. Vanka and R. P. Singh, J. Org. Chem., 2016, 81, 5663 CrossRef CAS PubMed.
- (a) T. J. Katz and R. Rothchild, J. Am. Chem. Soc., 1976, 98, 2519 CrossRef CAS; (b) A. Iuliano, P. Piccioli and D. Fabbri, Org. Lett., 2004, 6, 3711 CrossRef CAS PubMed; (c) T. J. Katz, Angew. Chem., Int. Ed., 2005, 44, 3010 CrossRef CAS PubMed; (d) Y. Xia, Z. Liu, Q. Xiao, P. Qu, R. Ge, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 5714 CrossRef CAS PubMed; (e) K. Bera, S. Sarkar, S. Jalal and U. Jana, J. Org. Chem., 2012, 77, 8780 CrossRef CAS PubMed.
- M. Chen, C. Yang, Y. Wang, D. Li and W. Xia, Org. Lett., 2016, 18, 2280 CrossRef CAS PubMed.
- M. Scholz, H. Ulbrich, A. Mattern, J. P. Kramb, W. Kiefer and G. Dannhardt, Arch. Pharm., 2008, 341, 281 CrossRef CAS PubMed.
- R. J. Kumar, D. Jyostna, G. D. Krupadanam and G. Srimannarayana, Phytochemistry, 1988, 27, 3625 CrossRef CAS.
- O. Kruber and A. Marx, Chem. Ber., 1938, 71, 2478 CrossRef.
- O. Kruber and A. Raeithel, Chem. Ber., 1954, 87, 1469 CrossRef CAS.
- O. Kruber and A. Marx, Chem. Ber., 1938, 71, 2478 CrossRef.
- W. Kern, Helv. Chim. Acta, 1947, 30, 1595 CrossRef CAS PubMed.
- M. Tori, A. Hashimoto, K. Hirose and Y. Asakawa, Phytochemistry, 1995, 40, 1263 CrossRef CAS.
- J. B. M. Somers, A. Couture, A. Lablache-Combier and W. H. Laarhoven, J. Am. Chem. Soc., 1985, 107, 1387 CrossRef CAS.
- Relative compounds: see the ESI.†.
- H. Okamoto, T. Takane, S. Gohda, Y. Kubozono, K. Sato, M. Yamaji and K. Satake, Chem. Lett., 2014, 43, 994 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, product characterization and NMR spectral data. See DOI: 10.1039/c7ra00193b |
| This journal is © The Royal Society of Chemistry 2017 |