
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regio- and stereoselective synthesis of novel isoxazolidine heterocycles by 1,3-dipolar cycloaddition between C-phenyl-N-methylnitrone and substituted alkenes. Experimental and DFT investigation of selectivity and mechanism†
Djamila Hellela,
Fouad Chafaaa,
Abdelmalek Khorief Nacereddine *ab,
Abdelhafid Djerouroua and
Emmanuel Vranckenc
*ab,
Abdelhafid Djerouroua and
Emmanuel Vranckenc
aLaboratoire de Synthèse et Biocatalyse Organique, Département de Chimie, Faculté des Sciences, Université Badji Mokhtar Annaba, BP 12, 23000 Annaba, Algeria. E-mail: khorief.abdelmalek@univ-annaba.org
bDépartement de Physique et Chimie, Ecole Normale Supérieure d’Enseignement Technologique de Skikda, Cité des frères Boucetta, Azzaba, Skikda, Algeria
cInstitut Charles Gerhardt, UMR 5253 CNRS-UM2-UM1 ENSCM, Laboratoire Architectures Moléculaires et Matériaux Nanostructurés (AM2N), Ecole Nationale Supérieure de Chimie de Montpellier, 8 Rue de l'Ecole Normale, 34296 Montpellier, France
First published on 12th June 2017
Abstract
A series of isoxazolidine heterocycles was synthesized through the 1,3-dipolar cycloaddition (13DC) reaction of C-phenyl-N-methylnitrone with different substituted alkenes. The structures and stereochemistry of the cycloadducts were determined by spectroscopic methods. These 13DC reactions are characterized by complete regioselectivity and high stereoselectivity. The molecular mechanism, reactivity and selectivity of these 13DC reactions have been investigated by means of transition state theory and reactivity indices derived from conceptual DFT using DFT methods at the B3LYP/6-31G(d,p) level of theory. The obtained results indicate that these cycloaddition reactions take place through a one-step synchronous mechanism with a non-polar mechanism and high activation energies. The theoretical results are in agreement with the experimental findings.
1. Introduction
The 1,3-dipolar cycloaddition (13DC) reaction is one of the most useful reactions in organic synthesis.1 Many dipoles and dipolarophiles have been employed to obtain either carbocyclic or heterocycles compounds with both oxygen and nitrogen atoms.2 Nitrones are a class of powerful reagents, which readily undergo 13DC with various dipolarophiles, in particular with alkenes to afford in high yields important isoxazolidines (Scheme 1).3 These cycloadducts have attracted considerable attention due to their potential biological activities, have been used as precursors for β-amino alcohols through reductive cleavage of the N–O bond, and have been considered as potential precursors for the synthesis of many natural products such as β-lactam antibiotics, alkaloids,4 and especially sugar and nucleoside analogues.5 Isoxazolidines possess medicinal activities such as antibacterial, anticonvulsant, antibiotic, antitubercular and antifungal activities5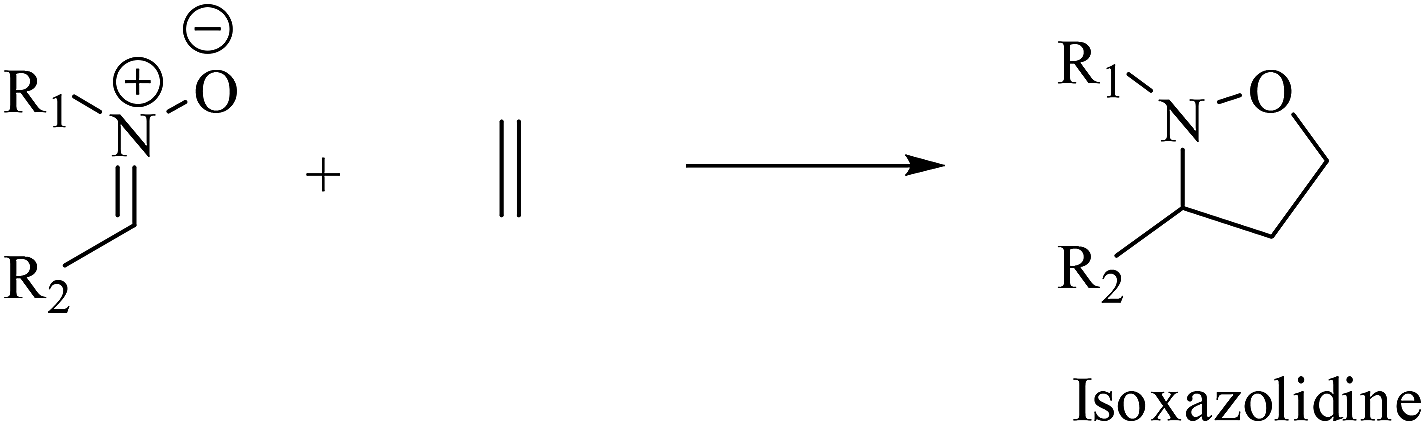 | ||
| Scheme 1 Synthesis of isoxazolidines by 13DC reaction of nitrones with simplest ethylene. | ||
A tremendous number of experimental and theoretical works can be found in the literature devoted to the study of the 13DC reaction of nitrones with alkenes for the synthesis of isoxazolidines.6 Tran et al.7 experimentally studied the 13DC reaction between C-phenyl-N-methylnitrone and dimethyl 2-benzylidenecyclopropane-1,1-dicarboxylate finding that this reaction is regio- and stereoselective yielding the corresponding ortho/endo cycloadduct as a single regio- and stereocycloadduct (Scheme 2). In order to explain the observed regio- and stereoselectivities, we have studied recently this 13DC reaction finding the formation of a non-classical hydrogen bond (HB) between the carbonyl oxygen atom of one of the two carboxylate groups of alkene and the nitrone C3–H hydrogen atom (Fig. 1). Formation of this non-classical HB, provokes a stabilization by 7 kcal mol−1 and can overcome the low meta regioselectivity.8
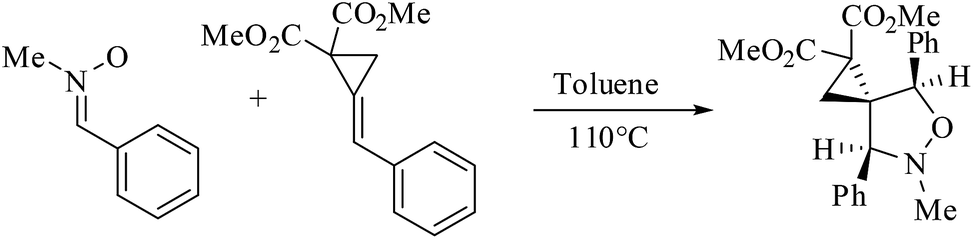 | ||
| Scheme 2 13DC reaction between C-phenyl-N-methylnitrone and dimethyl 2-benzylidenecyclopropane-1,1-dicarboxylate. | ||
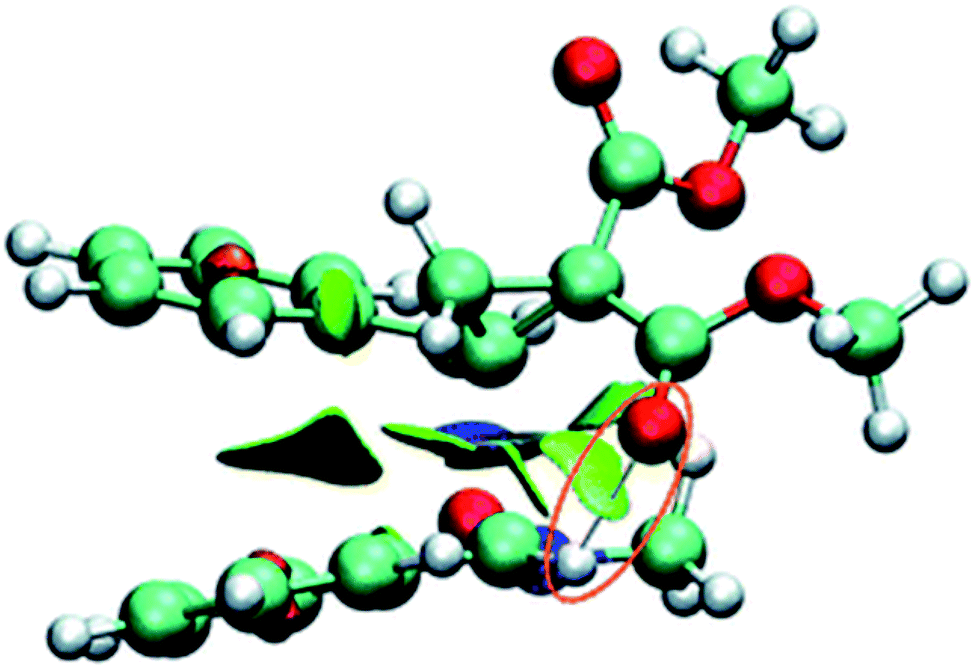 | ||
| Fig. 1 Non-classical HB at the meta–endo approach. | ||
Recently, Falkowska and co-workers9 reported the synthesis of SF5-substituted isoxazolidines which prepared in moderate yields by 13DC reactions between pentafluorosulfanyl-substituted acrylic ester or amides with nitrones as dipoles (Scheme 3). The authors have founds from his theoretical study that the selectivity of this 13DC reaction is a result of the balance between two sterical interactions, in which the sterical bulkiness of substituent Z is predominant, SF5-endo-approaches will be favored conducting to a mixture of diastereoisomers. On the contrary, minimizing the steric effect with SF5 will conduct to 3,4-trans diastereoisomer.
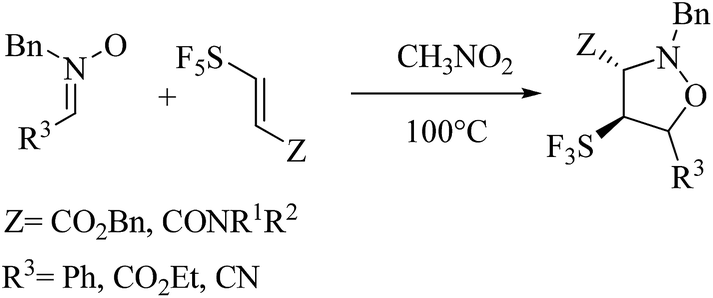 | ||
| Scheme 3 13DC reaction between pentafluorosulfanyl-substituted acrylic ester and amides with nitrones reported by Falkowska.9 | ||
From the literature, we can note that the regio- and stereoselectives of cycloaddition reaction and namely 13DC are mainly controlled by the electronic and steric factors of the substituent at the dipole or at the dipolarophile. Despite that there are many theoretical approaches that can predict the regioselectivity; such as the Houk's rule based on FMO analysis10 and the DFT-based reactivity indices,11 the stereoselectivity is often generally reliant to the steric effects of the substituent, alike, in many cases, when the reactants possess a voluminous substituent's, the regioselectivity cannot be predicted using these theoretical models. Thereby, it is necessary to study each reaction as a separate system.
Herein, we focused to perform an experimental as well as theoretical studies of the 13DC reactions between nitrone 1 prepared previously as dipole with various substituted alkenes 2a–e as a dipolarophiles leading to the formation of the corresponding isoxazolidines (Scheme 4) in order to shed light on the factors that controlled the regio- and stereoselectivities, as well as the molecular mechanism of these 13DC reactions.
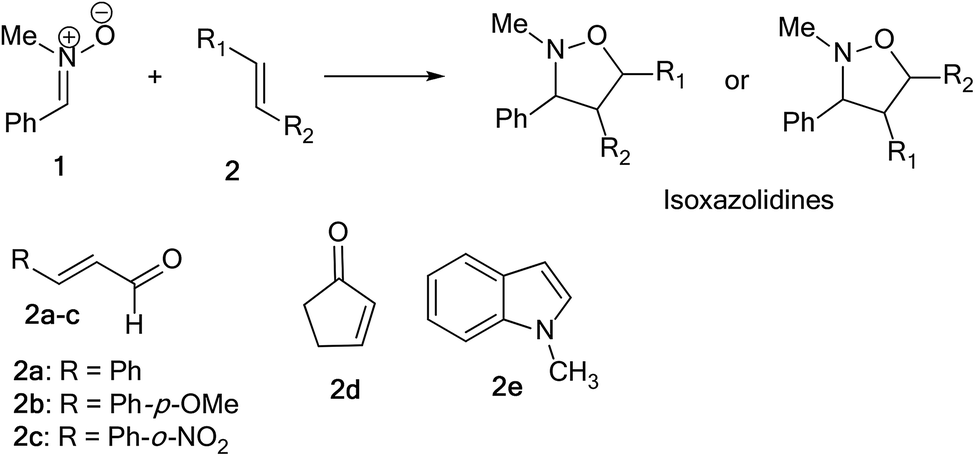 | ||
| Scheme 4 13DC reaction between nitrone 1 and dipolarophiles 2a–e. | ||
2. Results and discussion
The present study has been divided into two dependent parts; in the first part, the synthesis of isoxazolidines heterocycles using 13DC reactions between nitrone 1 and a series of alkenes 2a–e were performed and the structure of cycloadducts was characterized using spectroscopic usual methods (IR, NMR and MS). In the second part, we have performed a theoretical analysis using DFT methods of the regio- and stereoselectivities observed in the Experimental part as well as the molecular mechanism using theoretical approaches such as transition state theorem12 and reactivity indices defined within the context of the conceptual DFT.132.1. Experimental part
In order to synthesize the isoxazolidines heterocycles, we envisaged to prepare in first place the nitrone 1 which is easily accessible from the condensation reaction between benzaldehyde and the N-methyl-hydroxylamine-hydrochloride (Scheme 5). This nitrone has been obtained according to the procedure reported in literature,14 in which its structure was determined by spectroscopic methods (IR, NMR and mass). The 13DC reaction of nitrone 1 with the set of alkenes 2a–e were performed under refluxing polar solvent (toluene) at 110 °C in order to enhance the reactivity of both nitrone and alkenes. In the case of alkenes 2a, 2b and 2e, the corresponding isoxazolidines were obtained in a low yields 10, 12 and 23%, respectively (Table 1: entries 1, 2 and 5). Increasing the time of reaction or heating at 130 °C for 20 hours did not improve the yield. On the other hand, the 13DC reaction of nitrone 1 with both alkenes 2c and 2d leading to the formation of the corresponding isoxazolidines cycloadducts in a moderate (49%) and good yields (90%), respectively (see Table 1: entries 3 and 4). The slight increase in the yield can be explained by the presence of an electron-withdrawing group (NO2) at the alkene 2c which enhance the reactivity of the alkene to become a good electrophile.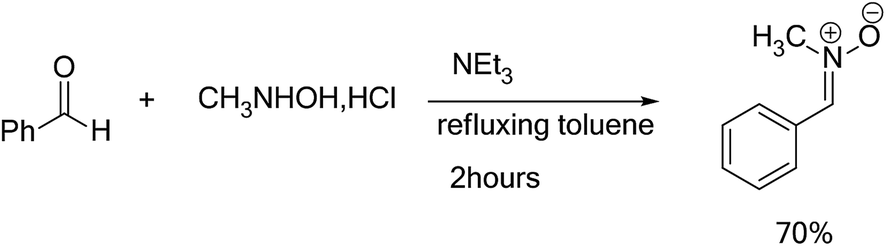 | ||
| Scheme 5 Synthesis of nitrone 1. | ||
| Alkene | Time (h) | Yield (%) | Cycloadduct |
|---|---|---|---|
| 2a | 24 | 10 | CA-a-mn (100%) |
| 2b | 24 | 12 | CA-b-mn (100%) |
| 2c | 24 | 49 | CA-c-mn (80%) |
| CA-c-mx (20%) | |||
| 2d | 08 | 90 | CA-d-mx (100%) |
| 2e | 12 | 23 | CA-e-ox (100%) |
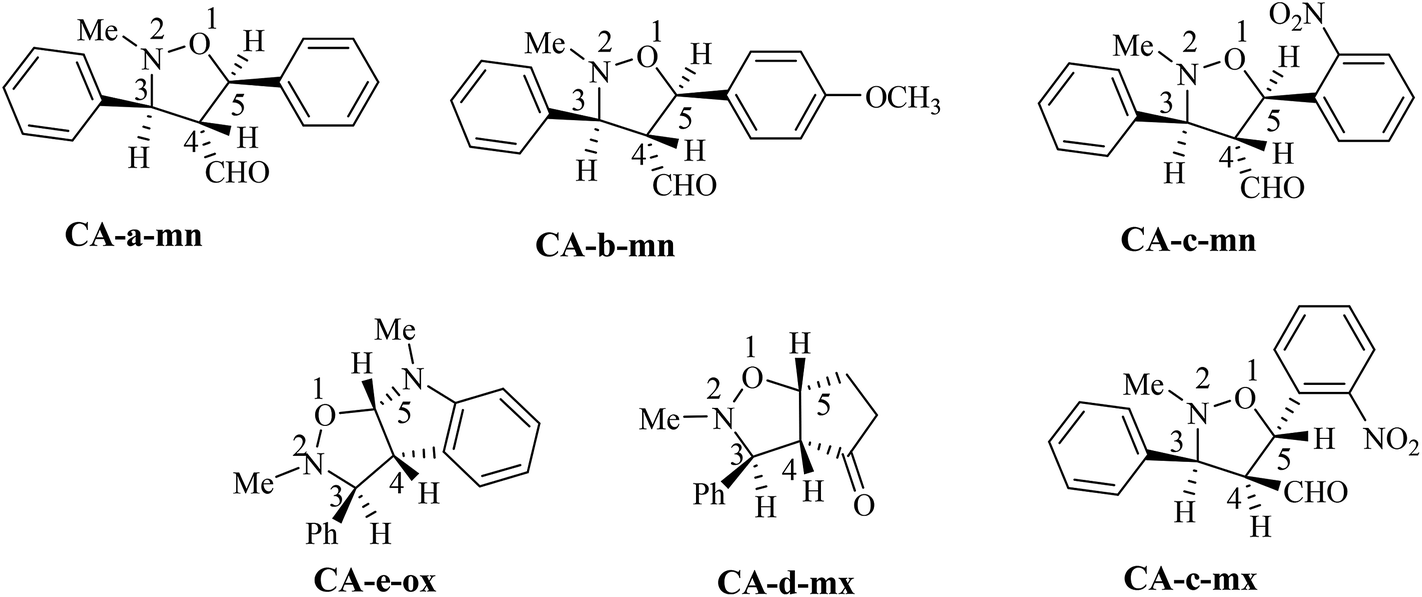
The structural determination of isoxazolidines 3a–e has been performed spectroscopically using usual methods such as IR, NMR and MS spectrums. In the case of isoxazolidines 3a and 3b, only one regioisomer (meta) and one diastereoisomer (trans) is observed. The assigned structures are again based on rigorous spectroscopic analysis for NMR data. The structure of different stereoisomers has been determined from coupling constant of selected hydrogens at the 1H NMR spectrum according to the data reported in the literature.15 Thus, the value of coupling constant of the hydrogens in position cis (J > 6 Hz) is always higher than that of hydrogens trans (J < 6 Hz).
The 13DC reaction between nitrone 1 and alkene 2c processed with completely meta regioselectivity and leads to the formation of a mixture of two diastereoisomers (trans/cis) in a ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 (see Table 1, entry 3). Note that the diastereoisomeric ratio was determined by integration of the corresponding 1H NMR signals of the crude mixture spectrum.
:20 (see Table 1, entry 3). Note that the diastereoisomeric ratio was determined by integration of the corresponding 1H NMR signals of the crude mixture spectrum.
The two-dimensional NMR (1H–1H and 1H–13C) and HRMS analysis has been also used to confirm the structures of the obtained compounds.
Using the same reaction conditions (Scheme 6), the alkene 2d reacts with the nitrone 1 during 8 hours to give a single regio- and diastereoisomer cycloadduct in a good yield (90%). However, in the case of the alkene 2e (Scheme 6), this 13DC reaction leads to the formation of a single regio- and stereoisomer in a poor yield (23%), for 12 hours of time (see Table 1, entry 5). This fact may be explained by the effects electronic donating of the conjugate nitrogen atom witch make the alkene 2e electron-rich ethylene, and thereby, it reacts as a nucleophile with nitrone 1 that it is naturally classified as a nucleophile species. Therefore, this 13DC become requiring strong experimental reaction conditions such as hard heating or high pressure, which explain the obtained low yield.
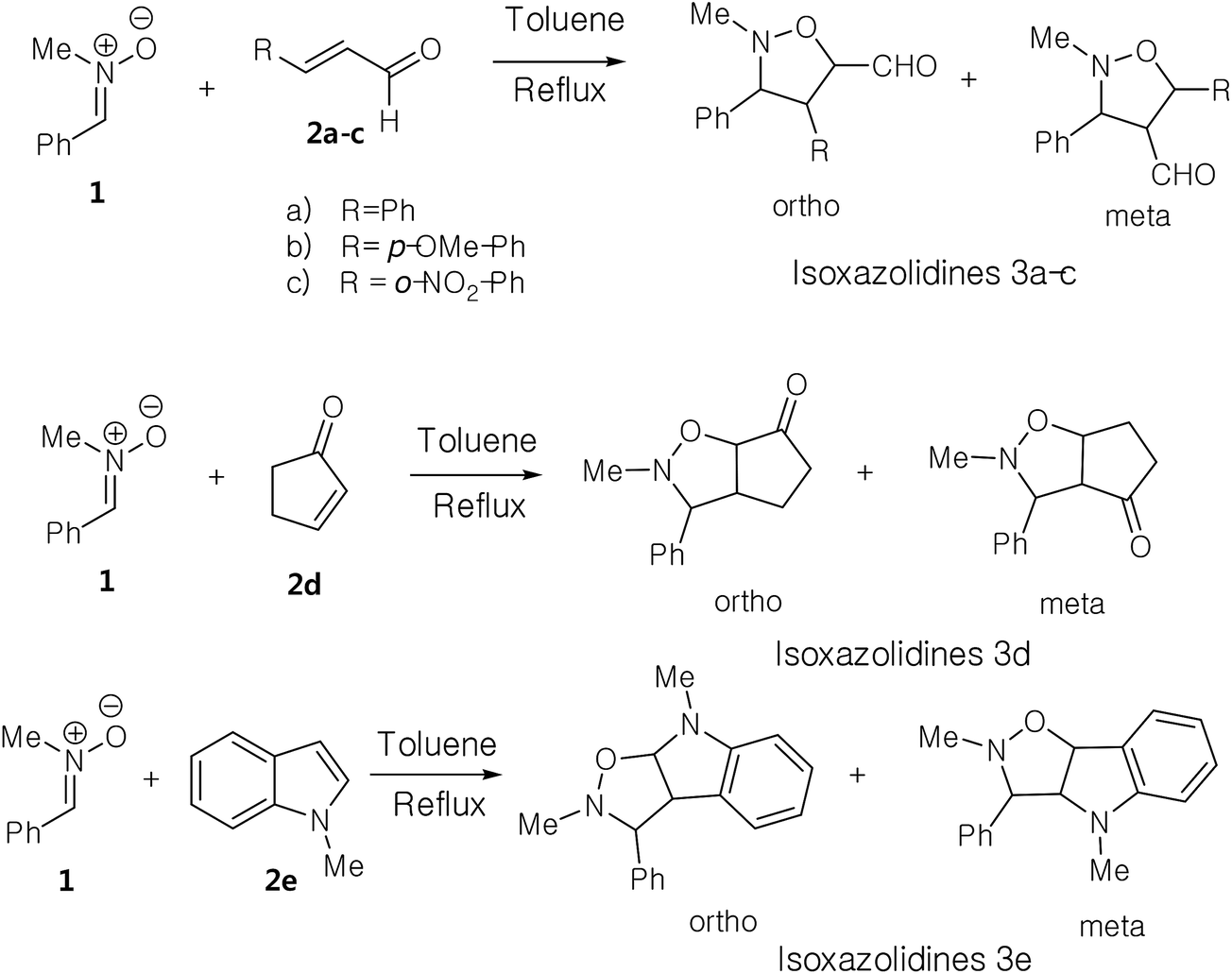 | ||
| Scheme 6 Synthesis of isoxazolidines via 13DC reaction of nitrone 1 with alkenes 2a–e. | ||
The obtained regioisomers (ortho/meta) were determined from cosy 1H/1H spectrum. On the other hand, the stereoisomers (trans/cis) determination has been performed using hydrogen coupling constants (J), which were extracted from the 1H NMR spectrum. The assigned regiochemistry of isoxazolidines 2a, 2b and 2c is based on the chemical shift value and multiplicity of C4–H. The trans/cis or endo/exo determination between C3–H and C4–H and between C4–H and C5–H are based on the value of coupling constant 1H NMR, using the rule that cis vicinal 1H coupling constant are always higher (5–9 Hz) than that of trans (0–5 Hz) in case of isoxazolidines and related heterocycles.15
The analysis of NMR cosy spectra of isoxazolidines 3a, 3b and 3c shows that the signal of the proton C4–H at δ 3.38 ppm appear as multiplet which is coupled with the protons C3–H and C5–H and is also coupled with the proton of the aldehyde function (CHO) which is appear in the range from δ 8–10 ppm (Scheme 7). Therefore, the isoxazolidines cycloadducts 3a, 3b and 3c, are a regioisomers meta. For the isoxazolidines 3d, the signal of C4–H appear as a multiplet at δ 2.90 ppm is also coupled with the C3–H that is appear as a doublet at δ 3.42 ppm and with C5–H that is appear as multiplet at δ 4.86 ppm. Thereby, the obtained isoxazolidines is the meta regioisomer.
 | ||
| Scheme 7 Stereochemistry of the obtained cycloadducts of the 13DC reaction of nitrone 1 with alkenes 2a–e. | ||
For the isoxazolidine 3e, the obtained cycloadduct is an ortho regioisomer, because the proton C5–H appears in a low intensity, this fact is due to the electron withdrawing effects of two electro-attractor atoms; oxygen and nitrogen. Thereby, the signal of C5 and C5–H atoms appear in low values of chemical shifts. Furthermore, this hydrogen appears as a doublet because it coupled only with the C4–H. The assigned 13C chemical shift values are in consonance with the assigned structures of isoxazolidines 3a–e.
For the isoxazolidines 3e, the low coupling constant of the C3–H with C4–H (J ≈ 0 Hz), being indicative that the C3–H and the C4–H are in trans relationship, based on the value obtained in the isoxazolidines heterocycles.15 Consequently, the stereochemistry of isoxazolidines 3e is ortho–trans or ortho–exo (see Scheme 7).
For, isoxazolidine 3a, the coupling constant J3,4 = 3.68 Hz account for a trans relationship between C3–H and C4–H. In addition the coupling constant J5,4 = 5.8 Hz indicated that the C4–H and C5–H are in trans position (see Scheme 7). The same reasoning in the case of isoxazolidine 3b, in which the low values of coupling constants J3,4 = 3.68 Hz and J5,4 = 5.8 Hz indicative for the trans relationship between them and leading to predict the structure shown in Scheme 7.
For the isoxazolidine 3c, the apparition of two signals adjacent with different integrations in the 1H NMR spectrum of the crude product for the C5–H indicative for the formation of a mixture of two diastereoisomers, in which one appears at δ 5.90 as doublet (J = 12 Hz) and the second also as a doublet at δ 5.95 (J = 4 Hz). From the value of coupling constants, these values indicative for a cis-diastereoisomer for signal at 5.90 ppm and a trans-diastereoisomer for a signal at δ 5.95 ppm. The proportion calculation shown that the trans-diastereoisomer formed in a major amount (80%), whereas, the cis-diastereoisomer is the minor (20%).
From these results, we can note that the presence of a electron withdrawing group at the alkene favour the formation of a meta regioisomers, however, the electron-donating group favor the ortho regioisomers. The exo stereoselectivity observed in these 13DC reactions may be attributed to steric factors.
2.2. Computational part
In this part, we have planned to study theoretically the effects of electronic and steric factors on the determination of the regio- and stereoselectivities as well as the nature of molecular mechanism. Therefore, we have chosen the alkenes 2a, 2d and 2e, which the first has a planner structure and the others having an electron withdrawing and donating group, respectively.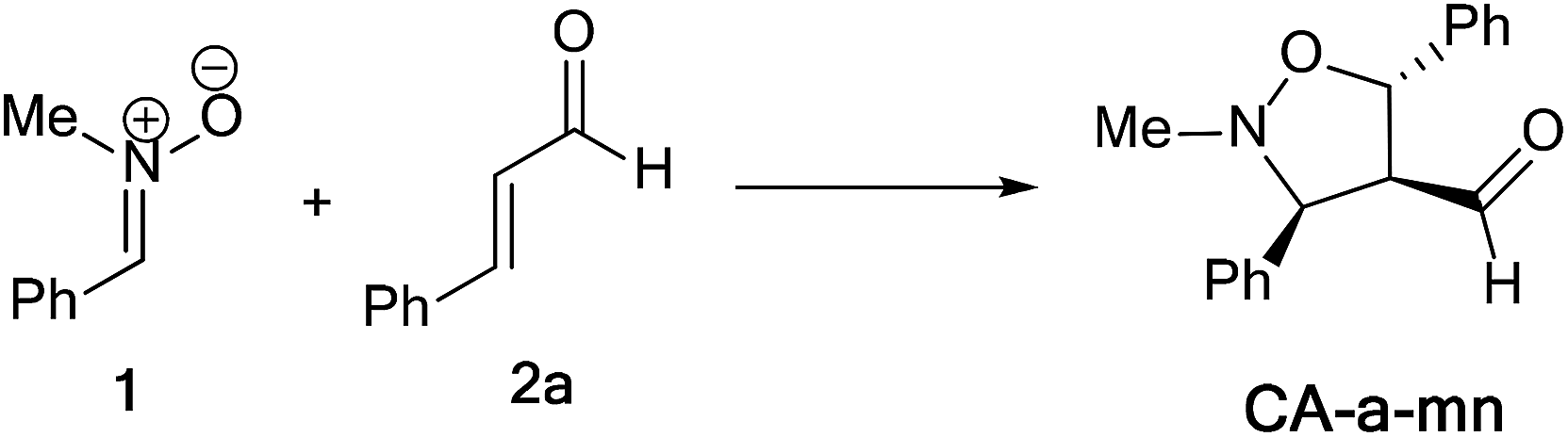 | ||
| Scheme 8 13DC reaction between nitrones 1 and alkene 2a. | ||
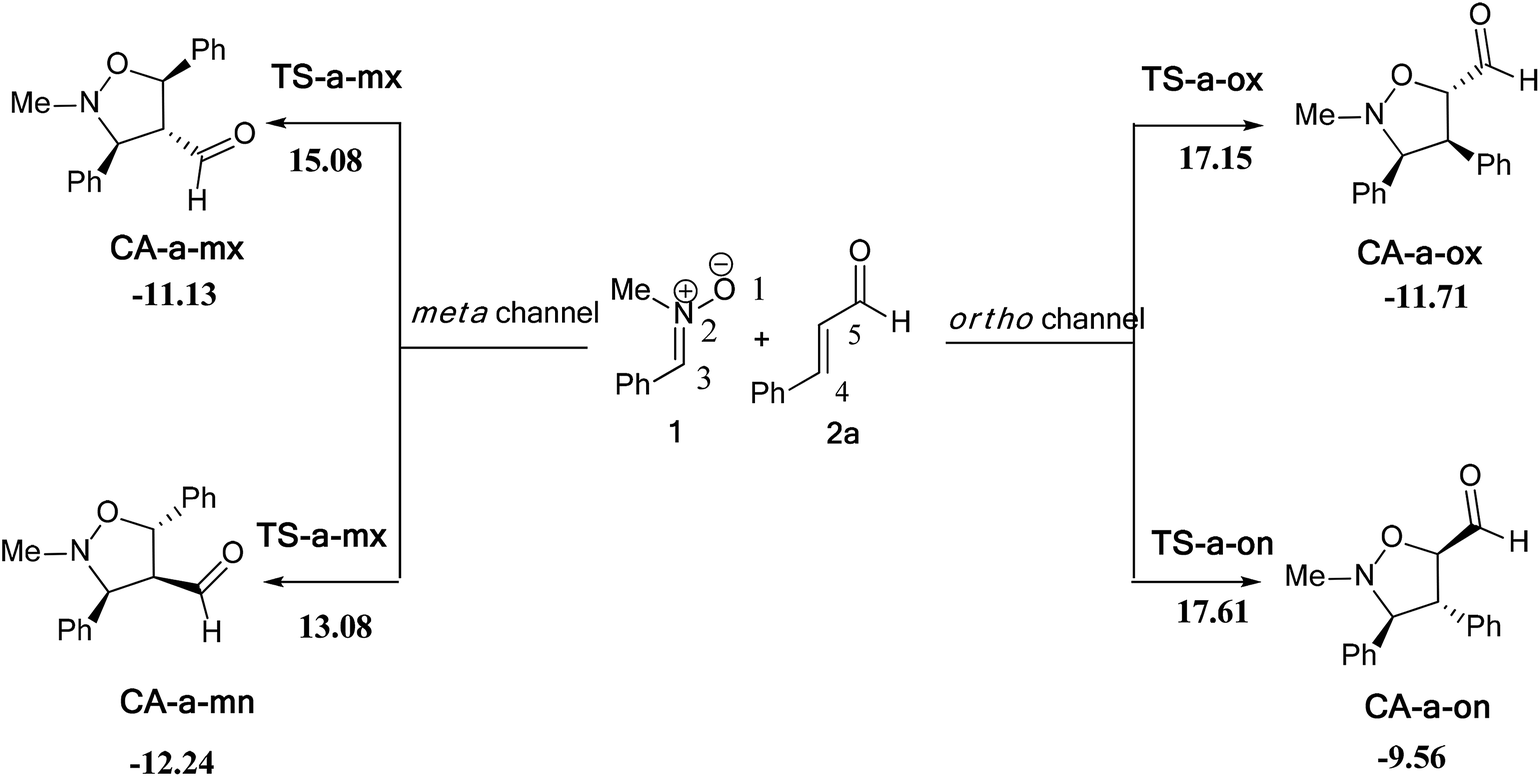 | ||
| Scheme 9 The possible regio- and stereoisomeric pathways for the 13DC reaction of nitrone 1 with alkene 2a. | ||
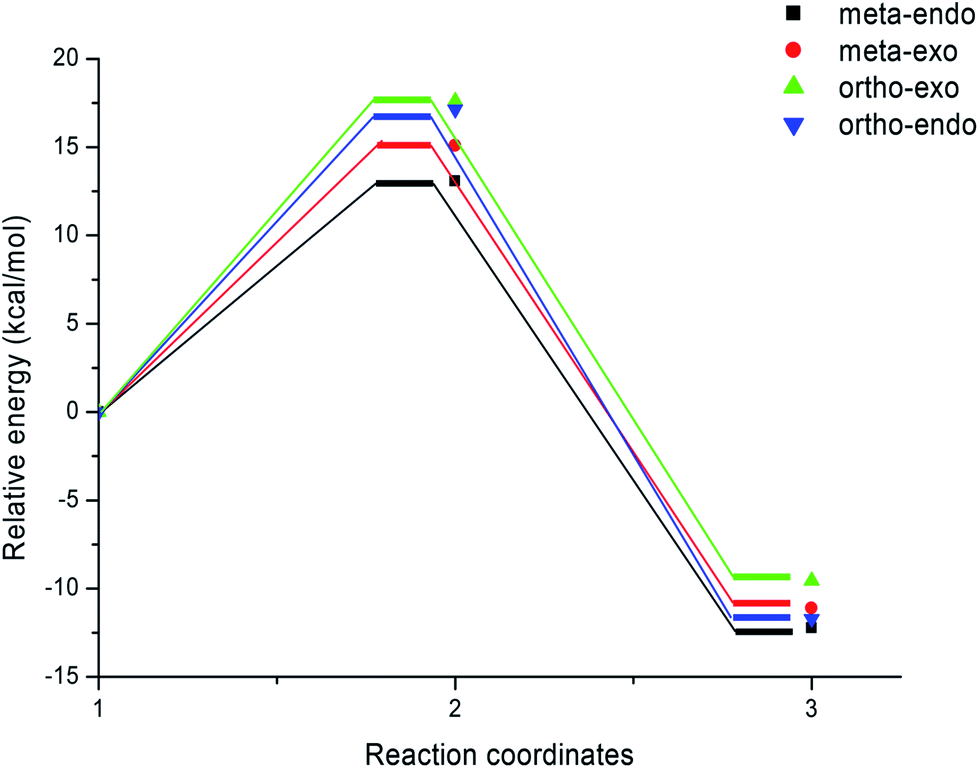 | ||
| Fig. 2 Energy profiles for the competitive reactive pathways associated with the 13DC reaction between nitrone 1 and alkene 2a. | ||
The activation energies associated with this 13DC reaction are 13.08 (TS-a-mn), 15.08 (TS-a-mx), 17.61 (TS-a-on) and 17.15 kcal mol−1 (TS-a-ox). These values indicates that TS-a-mn is more favored by 2.00 kcal mol−1 than the second favored approach (TS-a-mx) and by 4.06 kcal mol−1 than the more favoured ortho approach (TS-a-ox), account for the complete meta regioselectivity and endo stereoselectivity. Moreover, the former cycloadducts are slightly stables between −9.56 and −12.24 kcal mol−1, which account for the reversibility of this 13DC reaction, where, the CA-a-mn is the more stable one (ΔE = −12.24 kcal mol−1). Therefore, the 13DC reaction between nitrone 1 and alkene 2a favors kinetically and thermodynamically the formation of a single stereoisomer (CA-a-mn) generated from meta–exo approach, in great agreement with our experimental observation.
Values of relative enthalpies, entropies, and Gibbs free energies of the TSs and CAs involved in the 13DC reaction of nitrone 1 with alkene 2a are displayed in Table 2, while total ones are collected in Table S2 in ESI.†
| ΔH | ΔS | ΔG | |
|---|---|---|---|
| TS-a-mn | 15.08 | −46.921 | 33.06 |
| TS-a-mx | 17.17 | −46.257 | 34.89 |
| TS-a-on | 19.21 | −47.614 | 37.45 |
| TS-a-ox | 19.18 | −47.426 | 37.35 |
| CA-a-mn | −7.40 | −46.816 | 10.53 |
| CA-a-mx | −6.27 | −46.538 | 11.56 |
| CA-a-on | −4.61 | −48.899 | 14.13 |
| CA-a-ox | −6.69 | −46.947 | 11.30 |
Inclusion of thermal corrections to the electronic energies increases the relative enthalpies of the TSs and CAs in the borderline of 2 kcal mol−1 (see Table 2), which remaining TS-a-mn (ΔH = 15.08 kcal mol−1) the more favoured approach and the corresponding cycloadduct CA-a-mn (ΔH = −7.40 kcal mol−1) the more stable one. Addition of the entropic contribution to the enthalpy increases dramatically the activation Gibbs free energy to 33.06, 34.89, 37.45 and 37.35 kcal mol−1 for TS-a-mn, TS-a-mx, TS-a-on and TS-a-ox, respectively, but does not changes the regio- and stereoselectivities. This fact is a consequence of the of the unfavorable activation entropy associated with these bimolecular processes. In addition, the process becomes slightly endothermic, account for the reversibility of this 13DC reaction, which explains the experimental low yield. The reversibility of this 13DC reaction indicates that it is also under thermodynamic control, in which CA-a-mn is favoured both kinetically and thermodynamically (more stable one).
The structures of the TSs involved in the 13DC reaction between nitrone 1 and alkene 2a are given in Fig. 3. For the meta regioisomeric pathways, the lengths of the newly formed bonds O–C and C–C are 1.965 and 2.194 Å at TS-d-mn, 1.941 and 2.217 Å at TS-d-mx. For the ortho pathways, the lengths of the O–C and C–C new forming bonds are 2.036 and 2.142 Å at TS-d-on and 2.033 and 2.084 Å at TS-d-ox. These values indicate that at the ortho pathways the formation of O–C and C–C were performed synchronously. However, for the meta pathways, the formation of O–C and C–C were performed asynchronously; where, the new O–C σ bond is more advanced than the C–C one.
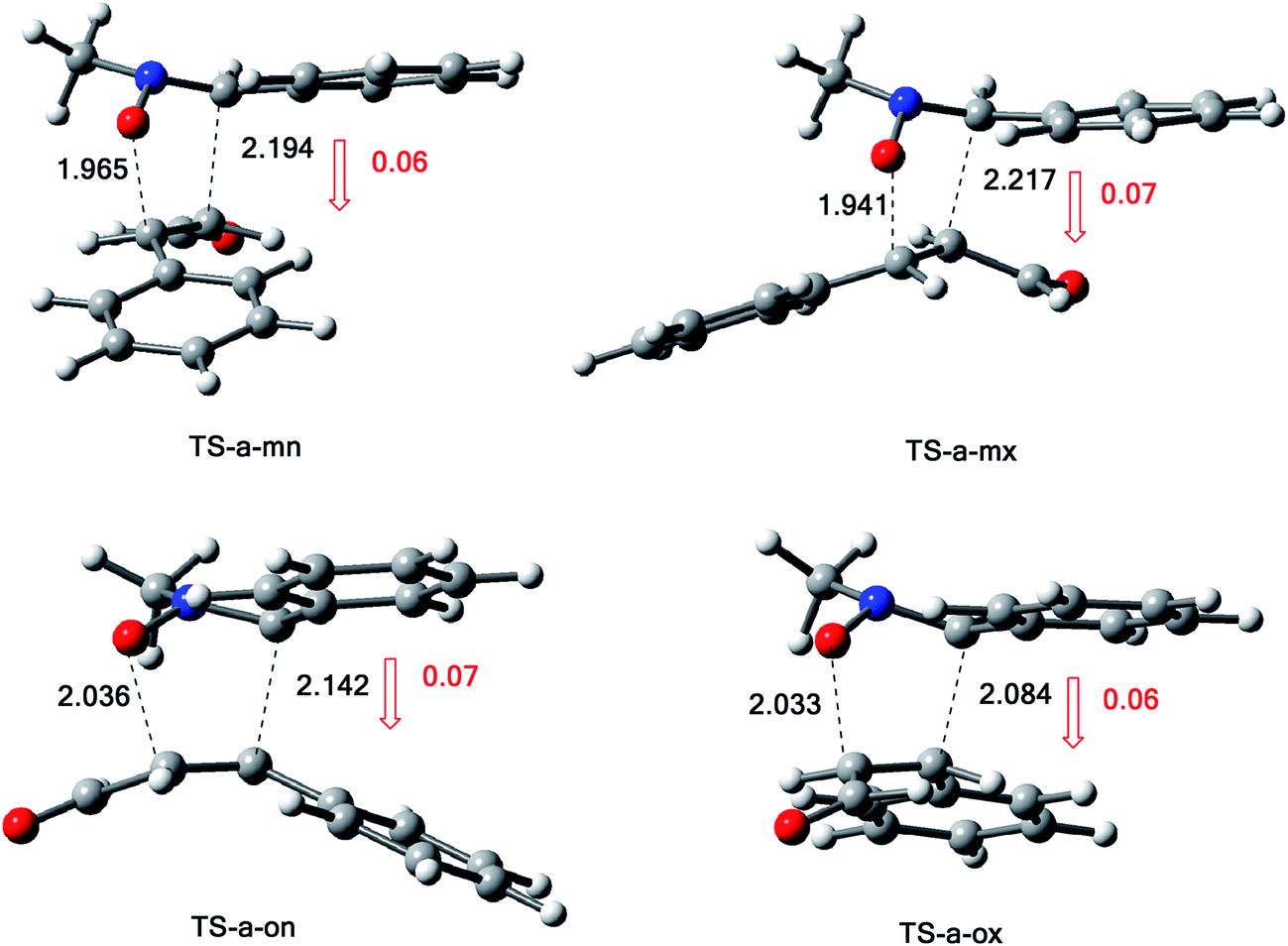 | ||
| Fig. 3 Structure of TSs involved in the 13DC reaction between nitrone 1 and alkene 2a. | ||
The polarity of this 13DC reaction has been analyzed by computing the global electron density transfer (GEDT) at the TSs. Along a polar reaction there is an electron-density transfer from the nucleophile partner toward the electrophile partner. This transfer can be analyzed through the GEDT value, in which, the high value of the GEDT at the TSs indicate for the high polar character of the reaction.
The GEDT along these possible pathways using Natural Population Analysis (NPA).16 The natural atomic charges at the TSs were calculated from the residual charge at the alkene 2a. The negative sign of the GEDT account that the flux of GEDT takes place from nitrone 1 toward alkene 2a. Therefore, nitrone 1 is considered as a nucleophile, whereas, alkene 2a as an eletrophile. The values of GEDT are 0.06e at TS-a-ox, 0.07e at TS-a-on, 0.07e at TS-a-mx and 0.06e at TS-a-mn. These low values accounts for a non-polar character for this 13DC reaction, justifying the obtained high activation energies and the experimental low yield.
 | ||
| Scheme 10 13DC reaction between nitrone 1 and alkene 2d. | ||
 | ||
| Scheme 11 The possible regio- and stereoisomeric pathways for the 13DC reaction of nitrone 1 with alkene 2d. | ||
 | ||
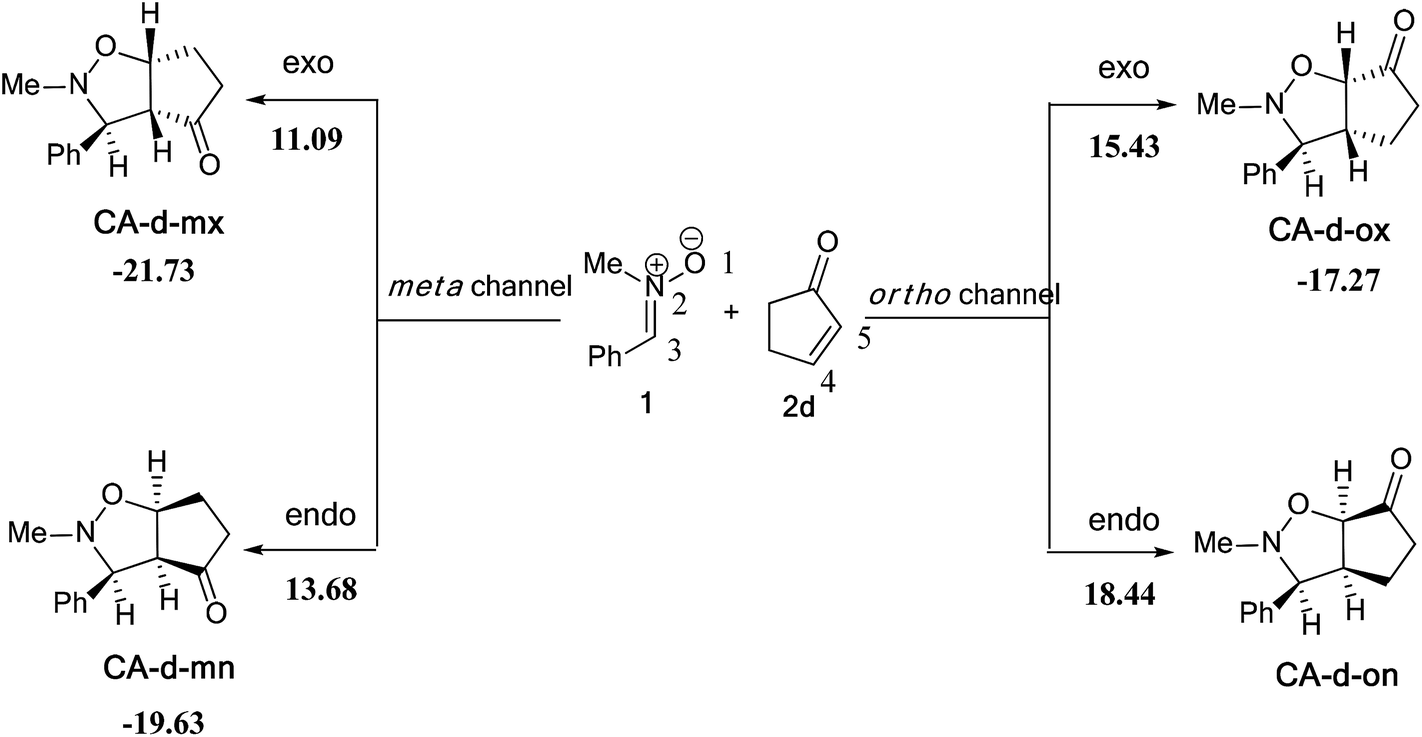
| Fig. 4 Structure of TSs involved in the 13DC reaction between nitrone 1 and alkene 2d. | ||
 | ||
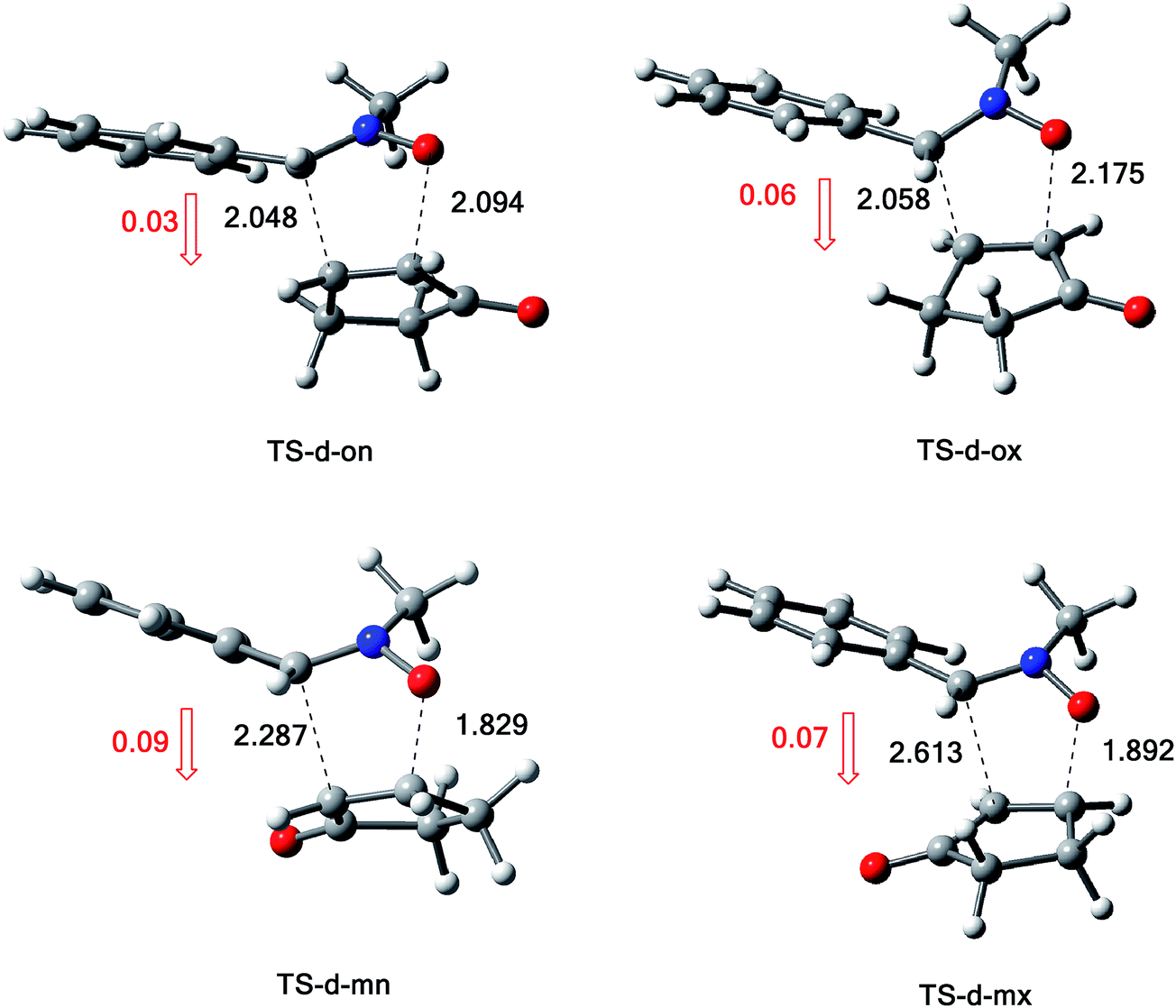
| Fig. 5 Energy profiles for the 13DC reaction between nitrone 1 and alkene 2d. | ||
The activation energies associated with this 13DC reaction are 13.68 (TS-d-mn), 11.09 (TS-d-mx), 18.44 (TS-d-on), and 15.43 kcal mol−1 (TS-d-ox). We can notice that TS-d-mx is more favored by 2.59 kcal mol−1 than the second favoured approach (TS-d-mn) and by 4.34 kcal mol−1 than the more favoured ortho approach (TS-d-ox). These values accounts for a high meta regioselectivity and an exo stereoselectivity. Therefore, the present 13DC reaction favors the formation of a single stereoisomer which generated from meta–exo approach (CA-d-mx), in great agreement with our experimental observation.
The structures of the TSs implanted in the 13DC reaction of nitrone 1 with alkene 2d are given in Fig. 4. For the ortho regioisomeric pathways, the lengths of the O–C and C–C new forming bonds are 2.09 and 2.05 Å at TS-d-on, 2.18 and 2.06 Å at TS-d-ox. For the favoured meta pathways, the lengths of the O–C and C–C newly formed bonds are 1.83 and 1.29 Å at TS-d-mn and 1.98 and 2.61 Å at TS-d-mx. Therefore, for the ortho pathways the formation of O–C and C–C were performed synchronously. However, for the meta pathways, The formation of O–C and C–C were performed asynchronously; where, the new O–C σ bond was more advanced than the C–C one.
As the previous 13DC reaction, here also, we have evaluated the GEDT along these possible pathways using Natural Population Analysis (NPA).16 The natural atomic charges at the TSs were calculated from the residual charge at the alkene 2d. The negative sign of the GEDT indicate that the fluxes of GEDT occur from nitrone 1 toward alkene 2d. Therefore, nitrone 1 will react as a nucleophile, whereas, alkene 2d as an eletrophile. The values of GEDT are 0.06e at TS-d-ox, 0.03e at TS-d-on, 0.07e at TS-d-mx and 0.09e at TS-d-mn. These low values accounts for a non-polar character for this 13DC reaction.
The values of relative enthalpies, entropies and free energies associated to this 13DC reaction are collected in Table 3, while the values of total ones are gathered in Table S4 in ESI.†
| ΔH | ΔS | ΔG | |
|---|---|---|---|
| TS-d-on | 20.39 | −46.97 | 38.40 |
| TS-d-ox | 16.96 | −45.61 | 34.44 |
| TS-d-mn | 15.92 | −45.56 | 33.38 |
| TS-d-mx | 13.07 | −44.86 | 30.27 |
| CA-d-on | −11.01 | −48.64 | 7.64 |
| CA-d-ox | −12.36 | −47.74 | 5.94 |
| CA-d-mn | −14.75 | −48.46 | 3.83 |
| CA-d-mx | −16.73 | −47.27 | 1.39 |
A comparison between the activation enthalpy values indicated that the approach meta–exo is the more favoured reactive pathway, in agreement with the predicted activation energy. In addition, we notice that this 13DC reaction is entropically unfavourable due to bimolecular process. Thus, this 13DC reaction has activation entropy in the range from −46.97 to −44.86 cal mol−1 K−1, thereby, the addition of thermal corrections and entropic contribution to the electronic energies increases the activation free energies by around 18 kcal mol−1, which raises the free activation energy to 38.40 to 34.44, 33.38, 30.27 kcal mol−1 for TS-d-on, TS-d-ox, TS-d-mn and TS-d-mx, respectively. Therefore, these high values do not change the meta–exo selectivity, but it making this 13DC reaction slightly unfavourable. In addition, the obtained cycloadducts are more stables in comparison with these obtained in the 13DC reaction of nitrone 1 with alkene 2a, which explain the experimental good yield.
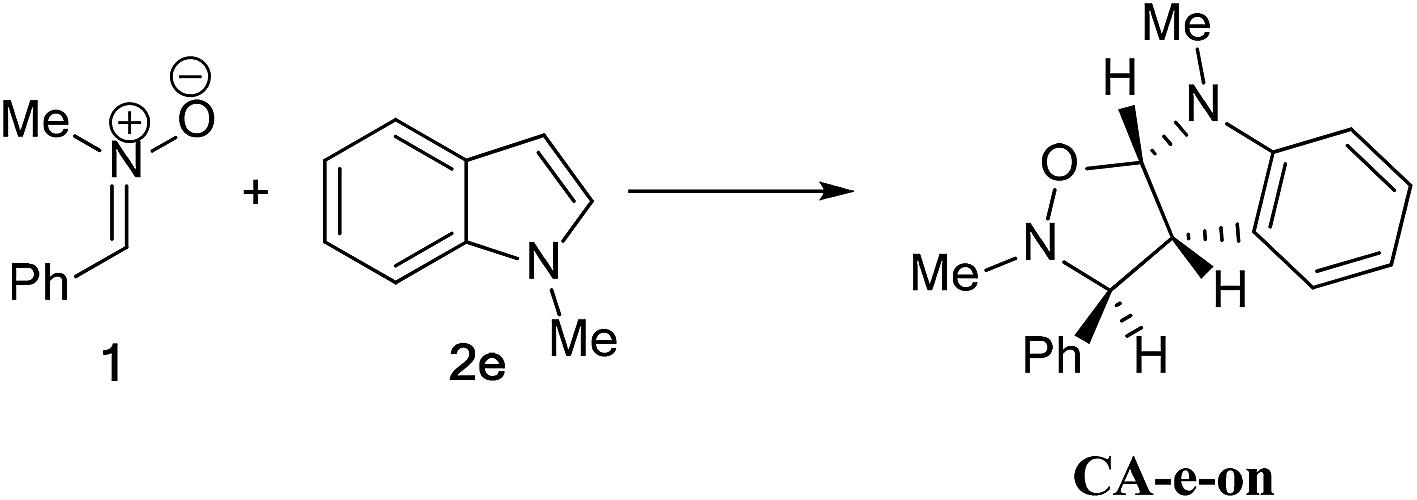 | ||
| Scheme 12 13DC reaction between nitrone 1 and alkene 2e. | ||
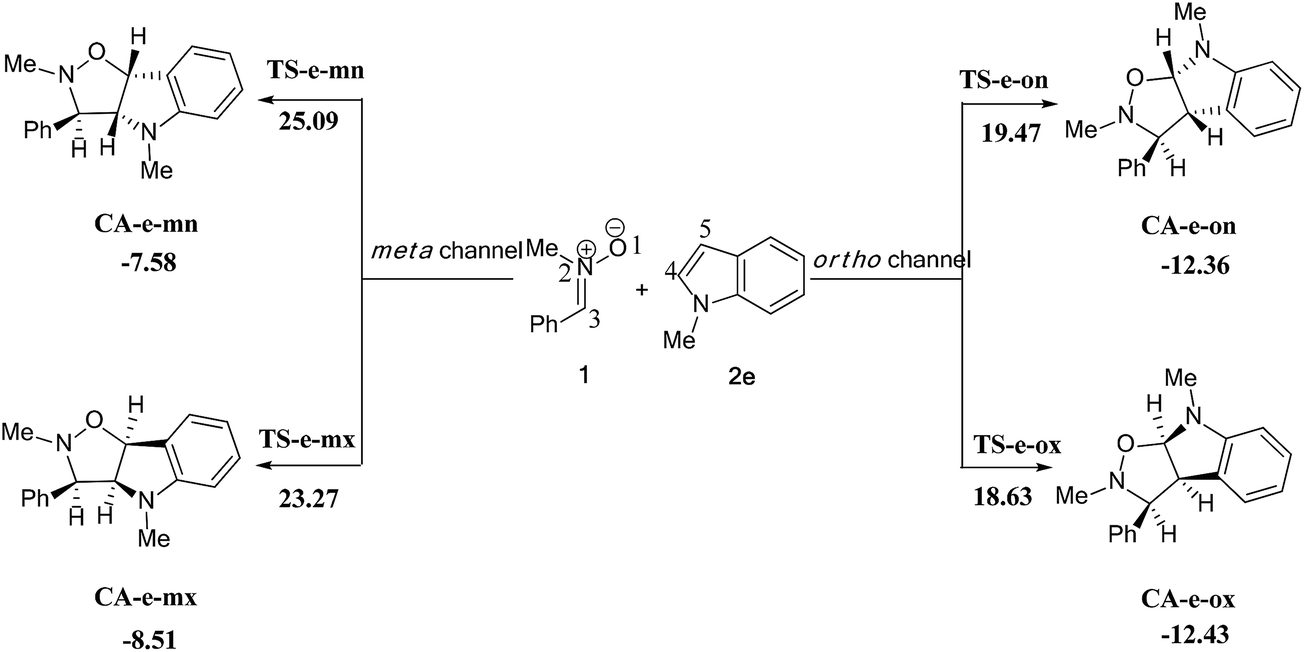 | ||
| Scheme 13 The possible regio- and stereoisomeric pathways of the 13DC reaction between nitrone 1 and alkene 2e. | ||
We can notice from Scheme 13 that the gas phase activation energies associated with the 13DC reaction between nitrone 1 and alkene 2e are 25.09 (TS-e-mn), 23.27 (TS-e-mx), 19.47 (TS-e-on), and 18.63 kcal mol−1 (TS-e-ox), these values accounts for a complete ortho regioselectivity and an exo stereoselectivity (Fig. 6). On the other hand, the comparison between the cycloadducts energies shows that these products are less stables. This fact accounts for the irreversibility of this 13DC reaction. Thereby, this cycloaddition is under both kinetic and thermodynamic controls. Therefore, the present 13DC reaction favoring kinetically and thermodynamically the formation of a single stereoisomer CA-e-ox, in great agreement with experimental data.
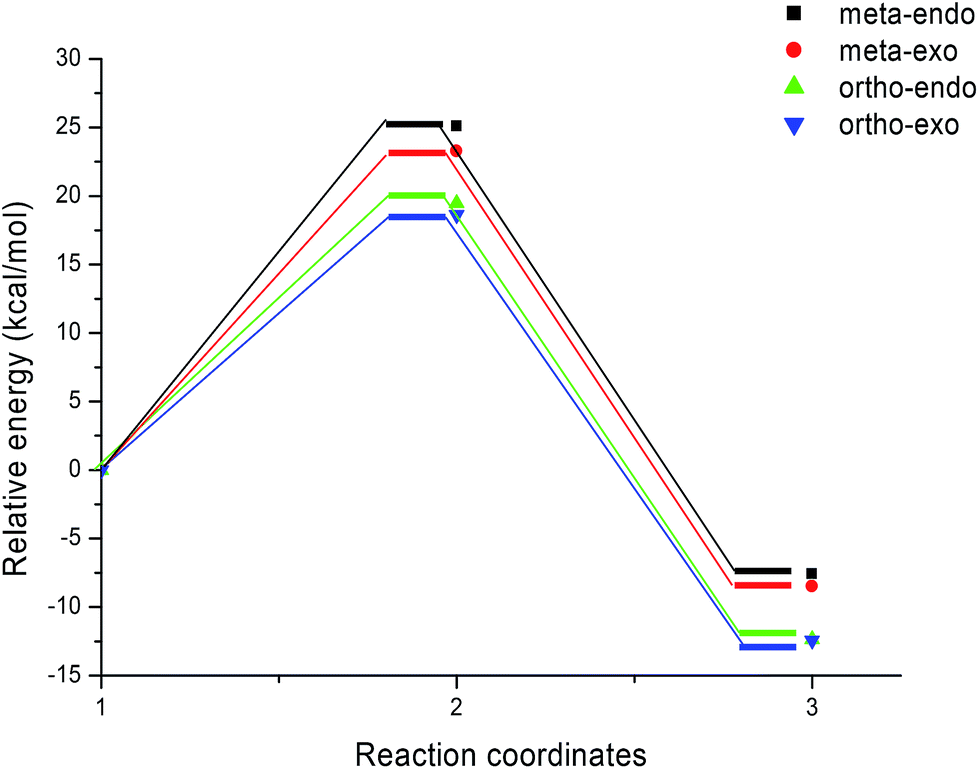 | ||
| Fig. 6 Energy profiles for the 13DC reaction between nitrone 1 and alkene 2e. | ||
The values of the relative enthalpies, entropies and Gibbs free energies of the TSs and cycloadducts involved in the reaction of nitrone 1 with alkene 2e are collected in Table 4, while the total ones are given in Table S6 in ESI.†
| System | ΔH | ΔS | ΔG |
|---|---|---|---|
| TS-e-mn | 27.54 | −45.48 | 44.97 |
| TS-e-mx | 25.02 | −45.13 | 42.31 |
| TS-e-on | 22.31 | −46.54 | 40.15 |
| TS-e-ox | 21.04 | −44.56 | 38.11 |
| CA-e-mn | −2.79 | −47.26 | 15.31 |
| CA-e-mx | −3.73 | −47.50 | 14.47 |
| CA-e-on | −7.35 | −48.12 | 11.09 |
| CA-e-ox | −7.30 | −47.88 | 11.04 |
From Table 2, a comparison between the relative activation enthalpies associated with the four reactive pathways of the 13DC reaction of nitrone 1 with alkene 2e indicates that the most favourable approach mode is always that associated with TS-e-ox (ΔH = 21.04 kcal mol−1). Addition of the entropic contribution to the enthalpy increases the activation Gibbs free energy of this reactive channel to 38.11 kcal mol−1, as a consequence of the unfavourable negative activation entropy, ΔS = −53.9 cal mol−1 K−1. Moreover, this 13DC reaction shows slightly endothermic character, which account for its reversibility, where CA-e-ox is favoured thermodynamically because it is the more stable cycloadduct. Consequently, it is expected to obtain a single isomer (CA-e-ox) from this 13DC reaction, as observed experimentally.
The structures of the TSs implanted in the 13DC reaction of nitrone 1 with alkene 2e are illustrated in Fig. 7. For the favoured ortho regioisomeric pathways, the lengths of the O–C and C–C newly formed bonds are 2.05 and 2.09 Å at TS-e-ox, 2.00 and 2.12 Å at TS-e-on. For the meta pathways, the lengths of the O–C and C–C new forming bonds are 2.16 and 2.02 Å at TS-e-mx and 2.08 and 2.04 Å at TS-e-mn. Therefore, for both the ortho and meta pathways the formation of O–C and C–C were performed synchronously.
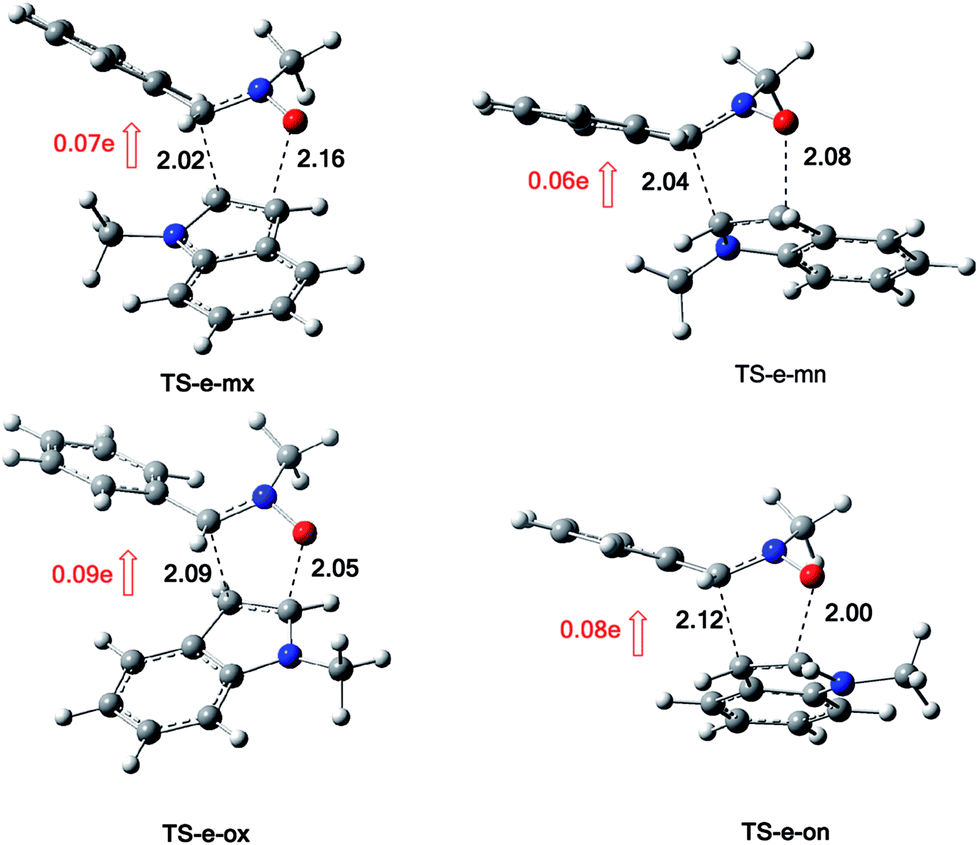 | ||
| Fig. 7 Structure of TSs involved in the 13DC reaction between nitrone 1 and alkene 2e. | ||
The evaluation of the GEDT along these 13DC paths is performed using NPA analysis.16 The natural atomic charges at the TSs were shared between nitrone 1 and alkene 2e. These values are calculated from the sum of the partial residual charge of the nitrone 1 atoms. The negative sign of GEDT indicates that the fluxes of the GEDT occur from alkene 2e toward nitrone 1. The values of GEDT are 0.09e at TS-e-ox, 0.08e at TS-e-on, 0.07e at TS-e-mx and 0.06e at TS-e-mn. These values indicate that this 13DC reaction has a low polar character, which account for a non-polar 13DC reaction and justifying the high activation energy.17
| HOMO | LUMO | μ | η | ω | N | |
|---|---|---|---|---|---|---|
| Nitrone 1 | −5.49 | −1.32 | −3.41 | 4.17 | 1.39 | 4.00 |
| Alkene 2a | −6.58 | −2.09 | −4.33 | 4.49 | 2.09 | 2.91 |
| Alkene 2d | −6.38 | −2.01 | −4.19 | 4.37 | 2.01 | 3.11 |
| Alkene 2e | −5.13 | −0.11 | −2.62 | 5.03 | 0.68 | 4.36 |
The electronic chemical potential of nitrone 1, μ = −3.41 eV is higher than that of alkene 2a, μ = −4.33 eV and alkene 2d, μ = −4.19 eV. Thereby, during these 13DCs, the GEDT will takes place from nitrone 1 toward alkenes 2a and 2b. On the other hand, this value is lower of that of alkene 2e, μ = −2.62 eV, indicating that the GEDT will occur from alkene 2e towards nitrone 1, in great agreement with the GEDT calculated at the TSs.
The electrophilicity and nucleophilicity indices of nitrone 1 are ω = 1.39 and N = 4.00 eV, allow its classification as a moderate electrophile and a strong nucleophile based on the electrophilicity20 and nucleophilicity21 scales. In addition, the electrophilicity and nucleophilicity indices of alkene 2a are ω = 2.09 and N = 2.91 eV, and for alkene 2b are ω = 2.01 and N = 3.11 eV, being classified as a moderate electrophiles and as a moderate nucleophiles. Therefore, nitrone 1 will react as a strong nucleophile and alkenes 2a and 2d as a moderate electrophiles. Consequently, these 13DC reactions will presents relatively moderate activation energy and proceeds via a slightly polar mechanism.
On the other hand, the electrophilicity and nucleophilicity indices of alkene 2e are ω = 0.68 and N = 4.36 eV, being classified as a marginal electrophile and as a strong nucleophile. Therefore, in the 13DC reaction between nitrone 1 and alkene 2e, the dipole (nitrone 1) will react as an electrophile and the dipolarophile (alkene 2e) as a nucleophile. Tacking account that both nitrone 1 and alkene 2e are a strong nucleophiles, this 13DC reaction will demand a strong experimental conditions, and proceeds via a non-polar mechanism and high activation energy, in agreement with experimental data and GEDT analysis, which allowing to explain the obtained low yield.
In order to predict the regioselectivity in cycloaddition reactions, recent studies indicates that the most favorable reactive channel is that implied the initial two-center interaction between the most electrophilic and nucleophilic centers of both reagents.22 Recently, Domingo et al.23 proposed the electrophilic P+k and nucleophilic P−k Parr functions derived from the changes of spin electron-density occurred via the GEDT process from the nucleophile to the electrophile as powerful tools in the study of the local reactivity. Therefore, the electrophilic P+k Parr functions for alkene 2a, alkene 2d, and nitrone 1 and the nucleophilic P−k Parr functions for alkene 2e and nitrone 1 are analysed in order to predict the most favourable electrophile/nucleophile two-center interaction in these 13DC reactions and, accordingly to explain the regioselectivity observed experimentally.
The tridimensional (3D) maps of the atomic spin density (ASD) of both systems; the radical cation of nitrone 1 and alkene 2e and the radical anion of alkene 2a, alkene 2d and nitrone 1, including the values of nucleophilic Parr functions of nitrone 1 and alkene 2e and the electrophilic Parr functions of alkene 2a, alkene 2d and nitrone 1 are given in Fig. 8.
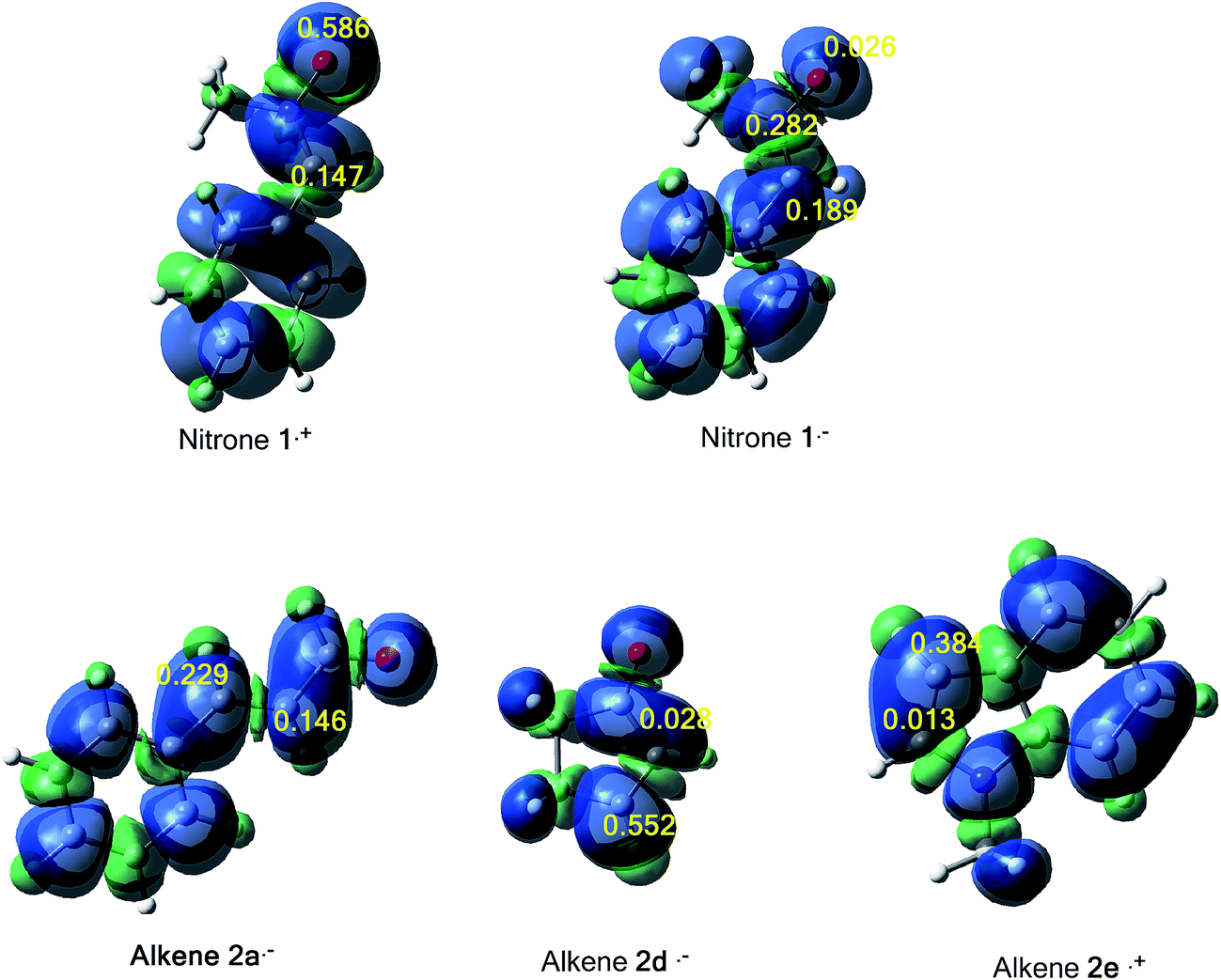 | ||
| Fig. 8 3D Maps of ASD for cation radicals nitrone 1˙+, alkene 2e˙+ and anion radical nitrone 1˙− and alkene 2d˙− and alkene 2a˙+ with the nucleophilic P−k of nitrone 1˙+ and alkene 2e˙+ and electrophilic P+k Parr functions for nitrone 1˙−, alkene 2a˙+ and alkene 2d˙−. | ||
From Fig. 8, we can notice that along these 13DC reactions, analysis of electrophilic Parr functions of alkenes 2a and 2b indicates that the C4 carbon atom (see Schemes 9 and 11 for atom numbering) is the most electrophilic one of these molecules, P = 0.229 for alkene 2a and P = 0.552 for alkene 2b. On the other hand, the nucleophilic Parr functions of nitrone 1 are mainly concentrated at the O1 oxygen atom (P = 0.586), account for a meta regioselectivity of these 13DC reactions. For the 13DC reaction between nitrone 1 and alkene 2e, analysis of electrophilic Parr functions of nitrone 1 indicates that the C3 carbon atom is the most electrophilic center (P = 0.189). In addition, for the alkene 2e, the most nucleophilic center is located at the C5 atom (see Scheme 13 for atom numbering). Thereby, the most favourable nucleophilic/electrophilic interaction will occur between C3 carbon atom of nitrone 1 and C5 atom of alkene 2e leading to the formation of the ortho regioisomers, in good agreement with the experimental outcome.
3. Conclusions
In this work, we have investigated experimentally and theoretically the regio- ans stereoselectivities of the 1,3-dipolar cycloaddition of C-phenyl-N-methylnitrone with different substituted alkenes.For the synthesis of isoxazolidines, the pure products were purified using chromatography technique and the structures were determined using spectroscopic methods. The principal conclusions are the followings:
(a) The 13DC reactions of nitrone 1 with alkenes 2a, 2b and 2d are completely regioselective and stereoselective leading to the formation of the cycloadduct generated from meta–endo and meta–exo approaches, respectively.
(b) The 13DC reaction between nitrone 1 and alkene 2c is completely regioselective but it has a poor stereoselectivity leading to the formation of a mixture of both meta–endo and meta–exo cycloadducts in a ratio of 80/20.
(c) The 13DC reactions of nitrone 1 with alkene 2e are completely regioselective and stereoselective leading to the formation of the cycloadduct generated from ortho–exo approach.
For the theoretical investigation, we have performed a computational study of the mechanism and the regio- and stereoselectivities of the 13DC reactions of nitrone 1 with alkenes 2a, 2d and 2e using DFT methods at the B3LYP/6-31G(d) theoretical level. The main conclusions which can be extracted from our results are the following:
(i) The 13DC reaction of nitrone 1 with alkene 2e is completely ortho-regioselective and an exo stereoselective leading kinetically and thermodynamically to the formation of a single diastereoisomer CA-e-ox, in a low yield.
(ii) The 13DC reactions of nitrone 1 with alkenes 2a and 2d is completely meta regioselective and an endo stereoselective for the alkene 2a and exo stereoselective for the alkene 2d, leading kinetically and thermodynamically to the formation of a single diastereoisomer CA-a-mn and CA-d-mx, respectively, in a great agreement with experimental observations.
(iii) An analysis of the relative free energies of the TSs involved in these 13DCs reveals that the inclusion of the thermal corrections and entropic contribution to the electronic energies does not modify the selectivity found with the gas-phase electronic energies. The main change is the increase of the activation energies, as a consequence of the negative values of entropies associated with the bimolecular character of these 13DC reactions.
(iv) The endothermic character account for the reversibility of these 13DC reactions, which reveals that they also under thermodynamic control, in which the kinetic product is also favoured thermodynamically.
(v) Analysis of GEDT and geometries of TSs indicates that these 13DC reactions proceed via a non-polar synchronous mechanism for the 13DC reaction between nitrone 1 and alkene 2e, as a consequence of the nucleophilic character of both reagents. For the 13DC reaction between nitrone 1 and alkenes 2a and 2d, the favorable meta paths proceeds with an asynchronous non-polar mechanism and a relatively low activation energies, as a consequence for the electrophilic character of both alkene 2a and alkene 2d.
(vi) Analysis of the DFT reactivity indices at the ground state of the reagents involved in these 13DC reactions indicates that both nitrone 1 and alkene 2e are a strong nucleophile and alkenes 2a and 2d are a good electrophiles which explain the obtained activation energies and the polar character of these 13DC reactions.
(vii) The analysis of local reactivity indices based on Parr functions method predicts correctly the regioselectivity observed experimentally.
(viii) The present study suggests that the electronic and steric effects have an important role in the determination of the regioselectivity and the stereoselectivity, as well as the reactivity of the regents.
4. Experimental section
4.1. General
All reactions were carried out in dried reaction glassware under argon, using a parallel reactor that will allow us to run multiple reactions simultaneously and under the same conditions; same temperature and same stirring speed. The temperatures of the reaction are reported as the temperature of the hot plate used. The commercial products were obtained from Sigma Aldrich, Alfa Aesar and was typically used without further purification except the reagent trans-cinnamaldehyde 2a and toluene solvent which were used freshly distilled. The spectra of 1H NMR, 13C NMR, cosy (2D: 1H–1H) and HMQC (2D: 1H–13C) were recorded on a Bruker Ultra shield 400 MHz. Chemical shifts (δ) are expressed on parts per million (ppm) relative to external reference TMS. Coupling constants (J) are given in hertz. The NMR spectra were performed in CDCl3 and referenced to the residual peak of CHCl3 at δH = 7.26 ppm and δC = 77.00 ppm for 1H and 13C, respectively. Infrared spectra (IR) were recorded with a spectrometer Perkin Elem spectrum 1000. Mass spectra and high-resolution mass spectra were obtained from the mass spectrometer operated by the Laboratory of Physics Measurement of the University Montpellier 2. All the reactions were monitored by thin-layer chromatography (TLC) on silica gel 60 F254 TLC plates; the revelation was attended up with UV light, and a colored solution of potassium permanganate or a ninhydrin solution followed by simple heating. Chromatography was carried out with silica gel 60 A (35–70 μm). Petroleum ether and ethyl acetate were used as eluent for purification with column chromatography.4.2. Procedure for the synthesis of nitrone 1
In a ball bicol of 250 ml under magnetic stirring, the benzaldehyde (0.02 mol) was dissolved in an anhydrous toluene, then the N-methylhydroxylamine hydrochloride (CH3NHOH, HCl) (0.02 mol), was added. When the refluxing began, a solution of triethylamine (NEt3) (0.02 mol) dissolved in dry toluene was added dropwise using a dropping funnel. The reaction mixture is brought to refluxed toluene for 2 hours. We use a Dean Stark for removing the water molecules, which generated during the reaction. Once the reaction is completed, the mixture is immersed in an ice bath, the precipitate removed by vacuum filtration, the solvent was evaporated under reduced pressure. The crude product was purified by re-crystallization in diethyl ether. The nitrone 1 was separated by filtration as crystals. The characterization data and spectroscopic detail for the nitrones 1 are given in ESI.†4.3. General procedure for the synthesis of isoxazolidines heterocycles 3a–e
An equimolecular amount (1 mmol) of the alkene and the nitrone 1 was introduced on tube of a parallel reactor and dissolved in anhydrous toluene (5 ml) under argon atmosphere. The mixture was refluxed of the solvent with stirring. The reaction evolution was monitored by TLC. After completion of the reaction, the reaction mixture was then cooled to room temperature, evaporated of solvent under reduced pressure. The crude product was purified by column chromatography (silica gel) which afforded the corresponding isoxazolidines. The reported yields were determined based on the isolated pure products and in the case of the mixture, the relative proportions were determined from 1H NMR spectrum.4.4. Characterization data and spectroscopic detail for the obtained compounds
:ethyl acetate 80:20 eluent) afforded 3a as brick red oil in 10% yield as one diastereoisomer (CA-a-mn). Rf = 0.29 (petroleum ether:ethyl acetate: 80:20 eluent). 1H NMR (CDCl3, 400 MHz): δ 2.70 (s, 3H, N–CH3), δ 2.56 (m, 1H, C4–H), δ 4.32 (d, J = 3.68 Hz, 1H, C3–H), δ 5.37 (d, J = 5.8 Hz, 1H, C5–H), δ 7.27–7.35 (m, 10H, Harom), δ 9.85 (s, 1H, CHO). 13C NMR (CDCl3, 400 MHz): δ 42.70 (N–CH3), 44.52 (C3), 61.24 (C4), 62.5 (C5), 125.73 (Carom), 127.73 (Carom), 127.81 (Carom), 128.31 (Carom), 128.77 (Carom), 128.85 (Carom), 128.92 (Carom), 129.04 (Carom), 135 (CHO). IR (thin film): 2832, 1827, 1660, 1340, 1040 cm−1. HRMS (ESI+): m/z calcd for [C17H17NO2 + H]+: 268.1338; found 268.1337.:20) given 3b as red oil in 12% yield as a single regio- and stereoisomer (CA-b-mn). Rf = 0.17 (petroleum ether:ethyl acetate: 80:20 eluent). 1H NMR (CDCl3, 400 MHz): δ 1.25 (s, 3H, CH3), δ 3.18 (q, 1H, C4–H), δ 3.80 (s, 3H, O–CH3), δ 4.68 (d, J = 5.6 Hz, 1H, C3–H), δ 6.05 (d, J = 14.4 Hz, 1H, C5–H), δ 6.84 (m, 2H, CHarom), δ 7.03–7.36 (m, 7H, CHarom), δ 8.42 (s, 1H, CHO). 13C NMR (CDCl3, 100 MHz): δ 38.67 (N–CH3), 55.35 (O–CH3), 70.53 (C4), 72.98 (C5), 80.94 (C3), 110.86 (Carom), 114.22 (Carom), 114.29 (Carom), 126.63 (Carom), 127.02 (Carom), 150 (Carom), 190.36 (CHO) IR (thin film): 2830, 1720, 1550, 1135, 1018 cm−1. HRMS (ESI+): m/z calcd for [C18H19NO3 + H]+: 298.1443; found 298.14.:20 of ratio, respectively, but only one diastereoisomer was isolated as an yellow oil. Rf = 0.44 (PE/AcOEt eluent: 80:20). 1H NMR (CDCl3, 400 MHz): δ 2.73 (s, 3H, N–CH3), δ 3.38 (m, 1H, C4–H), δ 3.84 (d, J = 8.4 Hz, 1H, C3–H), δ 5.90 (d, J = 12 Hz, 1H, C5–H) for CA-c-mn, δ 5.95 (d, J = 4 Hz, 1H, C5–H) for CA-c-mx, δ 7.28 (m, 5H, Harom), δ 7.47 (m, 1H, Harom), δ 7.75 (m, 1H, Harom), δ 8.14 (d, J = 1.2 Hz, 1H, Harom), δ 8.33 (d, J = 8 Hz, 1H, Harom), δ 10.02 (d, J = 2.4 Hz, 1H, CHO), 13C NMR (CDCl3, 400 MHz): δ 42.3 (N–CH3), 72.20 (C5), 74.65 (C5), 76.15 (C3), 124.91 (Carom), 127.84 (Carom), 128.40 (Carom), 128.53 (Carom), 128.93 (Carom), 129.04 (Carom), 134.34 (Carom), 136.35 (Carom), 139.45 (Carom), 198.14 (CHO). IR (thin film): 2850, 1726, 1521, 1345, 1302 cm−1. HRMS (ESI+): m/z calcd for [C17H16N2O4 + H]+: 313.1188; found 313.1192.:15) to give 3d in 90% yield as a single regio- and diastereoisomer (CA-d-mx). Rf = 0.26 (PE:AcOEt: 85:15 eluent) as yellow oil. 1H NMR (CDCl3, 400 MHz): δ 1.97 (m, 2H, CH2), δ 2.01 (m, 2H, CH2), δ 2.54 (s, 3H, N–CH3), δ 2.90 (t, 1H, C4–H), δ 3.42 (d, J = 5.6 Hz, 1H, C3–H), δ 4.86 (q, 1H, C5–H), δ 7.17 (m, 5H, Harom). 13C NMR (CDCl3, 400 MHz): δ 24.14 (CH2), 29.66 (CH2), 35.24 (N–CH3), 42.89 (C4), 64.94 (C5), 77.56 (C3), 127.66 (Carom), 127.74 (Carom), 128.03 (Carom), 128.42 (Carom), 138.84 (Carom), 217.83 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). IR (thin film): 2848.83, 1736, 1154.64 cm−1. HRMS (ESI+): m/z calcd for [C13H15NO2 + H]+: 218.1181; found 218.1186.:15) to give 3e in 23% yield as one diastereoisomer. Rf = 0.36 (PE/AcOEt: 85/15) as pink oil. 1H NMR (CDCl3, 400 MHz): δ 3.68 (s, 3H, N–CH3), δ 3.71 (s, 3H, N–CH3), δ 3.86 (d, J = 10.8 Hz, 1H, C4–H), δ 4.21 (d, J = 0 Hz, 1H, C3–H), δ 5.82 (d, J = 0 Hz, 1H, Harom), δ 6.52 (s, 1H, Harom), δ 6.80 (t, 1H, Harom), δ 7.19 (m, 2H, Harom), δ 7.29 (m, 5H, Harom). 13C NMR (CDCl3, 400 MHz): δ 29.71 (N–CH3indole), 32.57 (N–CH3isoxa), 51.43 (C4), 80.00 (C3), 109.03 (C5), 118.62 (Carom), 120.03 (Carom), 121.40 (Carom), 126.00 (Carom), 128.25 (Carom), 135.12 (Carom), 136.31 (Carom), 148.64 (Carom). IR (thin film): 3110, 3015, 1660, 1545 cm−1. HRMS (ESI+): m/z calcd for [C19H17N2O + H]+: 289.1341; found 289.1340.
O). IR (thin film): 2848.83, 1736, 1154.64 cm−1. HRMS (ESI+): m/z calcd for [C13H15NO2 + H]+: 218.1181; found 218.1186.:15) to give 3e in 23% yield as one diastereoisomer. Rf = 0.36 (PE/AcOEt: 85/15) as pink oil. 1H NMR (CDCl3, 400 MHz): δ 3.68 (s, 3H, N–CH3), δ 3.71 (s, 3H, N–CH3), δ 3.86 (d, J = 10.8 Hz, 1H, C4–H), δ 4.21 (d, J = 0 Hz, 1H, C3–H), δ 5.82 (d, J = 0 Hz, 1H, Harom), δ 6.52 (s, 1H, Harom), δ 6.80 (t, 1H, Harom), δ 7.19 (m, 2H, Harom), δ 7.29 (m, 5H, Harom). 13C NMR (CDCl3, 400 MHz): δ 29.71 (N–CH3indole), 32.57 (N–CH3isoxa), 51.43 (C4), 80.00 (C3), 109.03 (C5), 118.62 (Carom), 120.03 (Carom), 121.40 (Carom), 126.00 (Carom), 128.25 (Carom), 135.12 (Carom), 136.31 (Carom), 148.64 (Carom). IR (thin film): 3110, 3015, 1660, 1545 cm−1. HRMS (ESI+): m/z calcd for [C19H17N2O + H]+: 289.1341; found 289.1340.5. Computational methods
The structures of all systems involved in these 13DC reactions were optimized using the B3LYP24 functional together with the 6-31G(d) basis set.25 The nature of the stationary points were confirmed by frequency calculations in order to distinguished between the minimums and transition states, which have zero and one imaginary frequency, respectively. The electronic structures of the TSs were analyzed by the natural bond orbital (NBO) method.26 Values of enthalpies, entropies, and Gibbs free energies in chloroform were calculated with standard statistical thermodynamics at 383 K and 1 atmosphere over the optimized gas-phase structures27 All computations were carried out with the Gaussian 09 suite of programs.28The global electrophilicity index,29 ω, is given by the following expression, ω = (μ2/2η), in terms of the electronic chemical potential μ and the chemical hardness η. Both quantities may be approached in terms of the one-electron energies of the Frontier molecular orbitals HOMO and LUMO, εH and εL, as μ ≈ (εH + εL)/2 and η = (εL − εH), respectively.30 The empirical (relative) nucleophilicity index,31 N, based on the HOMO energies obtained within the Kohn–Sham scheme,32 is defined as N = εHOMO(Nu) − εHOMO(TCE), where tetracyanoethylene (TCE) is the reference because it presents the lowest HOMO energy in a long series of molecules already investigated in the context of polar organic reactions. The electrophilic P+k and nucleophilic P−k Parr functions22 were obtained through the analysis of the Mulliken atomic spin densities (ASD) of the corresponding radical anion and radical cation by single-point energy calculations over the optimized neutral geometries.
Acknowledgements
This work was supported by the Ministry of Higher Education and Scientific Research of the Algerian Government [project CNEPRU Code: E01620140051].References
- R. Huisgen, in 1,3-Dipolar Cycloaddition Chemistry, ed. A. Padwa, John Wiley & Sons, New York, 1984, vol. 1, pp. 42–47 Search PubMed.
- J. N. Martin and R. C. Jons, Nitrones, in Synthetic Applications of 1,3-Dipolar Cycloaddition. Chemistry Towards Heterocycles and Natural Products, ed. A. Padwa and W. H. Pearson, John Wiley & Sons, Hoboken, NJ, 2003, ch. 1 Search PubMed.
- (a) M. Frederickson, Tetrahedron, 1997, 53, 403–425 CrossRef CAS; (b) K. V. Gothelf and K. A. Jorgensen, Chem. Rev., 1998, 98, 863–909 CrossRef CAS PubMed.
- J. P. G. Seerden, M. M. M. Boeren and H. W. Scheeren, Tetrahedron, 1997, 53, 11843 CrossRef CAS.
- (a) W. Patterson, P. S. Cheung and M. J. Ernest, J. Med. Chem., 1992, 35, 507 CrossRef PubMed; (b) E. Wagner, L. Becan and E. Nowakowska, Bioorg. Med. Chem., 2004, 12, 265 CrossRef CAS PubMed.
- (a) U. Chiacchio, F. Casuscelli, A. Cocsaro, A. Rescifina, G. Romeo and N. Uccella, Tetrahedron, 1994, 50, 6671–6680 CrossRef CAS; (b) M. Bakavoli, F. Moeinpour, A. Davoodnia and A. Morsali, J. Mol. Struct., 2010, 969, 139–144 CrossRef CAS; (c) K. V. Gothelf and K. A. Jørgensen, Chem. Commun., 2000, 1449–1458 RSC; (d) F. Chafaa, D. Hellel, A. K. Nacereddine and A. Djerourou, Tetrahedron Lett., 2016, 57, 67–70 CrossRef CAS.
- T. Q. Tran, V. V. Diev and A. P. Molchanov, Tetrahedron, 2011, 67, 2391 CrossRef CAS.
- A. K. Nacereddine, C. Sobhi, A. Djerourou, M. Ríos-Gutiérrez and L. R. Domingo, RSC Adv., 2015, 5, 99299 RSC.
- E. Falkowska, M. Y. Laurent, V. Tognetti, L. Joubert, P. Jubault, J. Bouillon and X. Pannecoucke, Tetrahedron, 2015, 71, 8067–8076 CrossRef CAS.
- K. N. Houk, Acc. Chem. Res., 1975, 8, 361 CrossRef CAS.
- (a) R. G. Parr and W. Yang, J. Am. Chem. Soc., 1984, 106, 4049 CrossRef CAS; (b) L. R. Domingo, M. J. Aurell, P. Pérez and R. Contreras, J. Phys. Chem. A, 2002, 106, 6871–6875 CrossRef CAS.
- H. Eyring, J. Chem. Phys., 1935, 3, 107 CrossRef CAS.
- P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793 CrossRef CAS PubMed; D. H. Ess, G. O. Jones and K. N. Houk, Adv. Synth. Catal., 2006, 348, 2337 CrossRef.
- R. C. F. Jones and J. N. Martin, in Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, A. Padwa and W. H. Pearson, John Wiley & Sons, New York, NY, 2002, pp. 1–81 Search PubMed.
- (a) M. P. S. Ishar, G. Singh, K. Kumar and R. Singh, Tetrahedron, 2000, 56, 7817 CrossRef CAS; (b) G. Singh, M. P. S. Ishar, N. K. Girdhar and L. Singh, J. Heterocycl. Chem., 2005, 42, 1047 CrossRef CAS; (c) R. Huisgen, H. Hauck, H. Seidl and M. Burger, Chem. Ber., 1969, 102, 1117 CrossRef CAS; (d) R. Sustmann, R. Huisgen and H. Huber, Chem. Ber., 1967, 100, 1802 CrossRef CAS.
- (a) A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 CrossRef CAS; (b) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- L. R. Domingo, M. J. Aurell and P. Pérez, Tetrahedron, 2014, 70, 4519 CrossRef CAS.
- (a) P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793 CrossRef CAS PubMed; (b) M. Ríos-Gutiérrez, F. Chafaa, A. K. Nacereddine, A. Djerourou and L. R. Domingo, J. Mol. Graphics Modell., 2016, 70, 296–304 CrossRef PubMed.
- L. R. Domingo and J. A. Sáez, Org. Biomol. Chem., 2009, 7, 3576 CAS.
- L. R. Domingo, M. J. Aurell, P. Pérez and R. Contreras, Tetrahedron, 2002, 58, 4417 CrossRef CAS.
- P. Jaramillo, L. R. Domingo, E. Chamorro and P. Pérez, J. Mol. Struct., 2008, 865, 68 CrossRef CAS.
- (a) A. Ghomri and S. M. Mekelleche, Mol. Phys., 2014, 112, 566 CrossRef CAS; (b) L. R. Domingo and P. Pérez, Org. Biomol. Chem., 2013, 11, 4350 RSC; (c) L. R. Domingo and S. R. Emamian, Tetrahedron, 2014, 70, 1267 CrossRef CASS. Bouacha, A. K. Nacereddine and A. Djerourou, Tetrahedron Lett., 2013, 54, 4030 CrossRef CAS; R. Jasinski, Tetrahedron, 2013, 69, 927 CrossRef; (d) F. Chafaa, D. Hellel, A. K. Nacereddine and A. Djerourou, Mol. Phys., 2016, 114, 663 Search PubMed.
- L. R. Domingo, P. Pérez and J. A. Saez, RSC Adv., 2013, 3, 1486 RSC.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- W. J. Hehre, L. Radom, P. V. R. Schleyer, and J. A. Pople, ab initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- (a) A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 CrossRef CAS; (b) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta Jr, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. Fox, J. Gaussian 09, Gaussian, Wallingford, CT, 2009 Search PubMed.
- R. G. Parr, L. V. Szentpaly and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922 CrossRef CAS.
- (a) R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512 CrossRef CAS; (b) R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
- (a) L. R. Domingo, E. Chamorro and P. Perez, J. Org. Chem., 2008, 73, 4615 CrossRef CAS PubMed; (b) L. R. Domingo and P. Pérez, Org. Biomol. Chem., 2011, 9, 7168 RSC.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, 1133 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00258k |
| This journal is © The Royal Society of Chemistry 2017 |