
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Discovery of phosphorodiamidate mustard-based O2-phosphorylated diazeniumdiolates with potent anticancer activity†
Yu Zoua,
Chang Yana,
Edward E. Knausb,
Huibin Zhang*a,
Yihua Zhang*a and
Zhangjian Huang *a
*a
aState Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, Jiangsu Key Laboratory of Drug Screening, China Pharmaceutical University, Nanjing 210009, PR China. E-mail: zhangjianhuang@cpu.edu.cn; zyhtgd@163.com; zhanghb80@163.com
bFaculty of Pharmacy and Pharmaceutical Sciences, University of Alberta, Edmonton, Alberta T6G 2E1, Canada
First published on 29th March 2017
Abstract
Nitric oxide (NO) has recently joined the clinical arena of cancer therapy because high levels of NO could not only induce cytotoxicity and apoptosis of cancer cells, but also sensitize the cells to chemo- and radio-therapies. Diazeniumdiolates are an important class of NO donors, and the O2-alkylation, arylation and sulfonylation of diazeniumdiolates result in more stable and potent anticancer agents. However, O2-phosphorylation has so far not been reported yet. Herein, we describe the design, synthesis and biological evaluation of a group of phosphorodiamidate mustard-based O2-phosphorylated diazeniumdiolates, 6–9. The most active compound, 7, was comparable, or even more potent, than a known anticancer agent, O2-2,4-dinitrobenzene diazeniumdiolate JS-K, against six cancer cell lines. Furthermore, 7 released larger amounts of NO, caused more significant DNA damage and cancer cell apoptosis than JS-K in the cancer cells. Our findings suggest that this new type of O2-substituted diazeniumdiolate could be potentially applied in the fight against cancer.
Introduction
Nitric oxide (NO) is known to mediate numerous physiological processes, including vasodilation, neurotransmission and immune response.1 Currently, it has become clear that high levels of NO can damage biomacromolecules such as proteins, DNA and lipids, and cause cell death by triggering apoptosis.2 Additionally, NO is capable of interacting with reactive oxygen species (ROS) to form a variety of highly reactive nitrogen oxides (NOx), involving peroxynitrite anion (ONOO−), nitrogen dioxide (NO2), and dinitrogen trioxide (N2O3), etc. which are responsible for cytotoxic nitration and nitrosylation.3–5 Therefore, NO is considered as a potential anticancer agent and has joined the clinical arena of cancer therapy.6–8 Since NO is implicated in a myriad of biological processes, however, NO donor compounds should release large amounts of NO localized at cancer sites in order to exert anticancer activity and avoid side effects.Diazeniumdiolates are an important class of NO donors, which spontaneously release two molecules of NO under physiological conditions (pH 7.4, 37 °C) with a range of half-lives from a few seconds to several hours.9 To enhance their stability and selectivity, a large number of O2-derived diazeniumdiolates have been synthesized (Fig. 1), and these derivatives can be enzymatically cleaved to produce NO. For instance, O2-carbonyloxymethylated (1), O2-glycosylated diazeniumdiolates (2), and O2-arylated (JS-K, 3) could be selectively triggered by esterases,10 glycosidases,11 and glutathione/glutathione S-transferases,12–14 respectively, to generate diazeniumdiolate anion, which spontaneously liberate NO in situ exhibiting potent biological activity. Among them, JS-K displayed significant anticancer activity in a broad spectrum of human cancer cell lines in vitro as well as in several mouse xenograft models in vivo.15
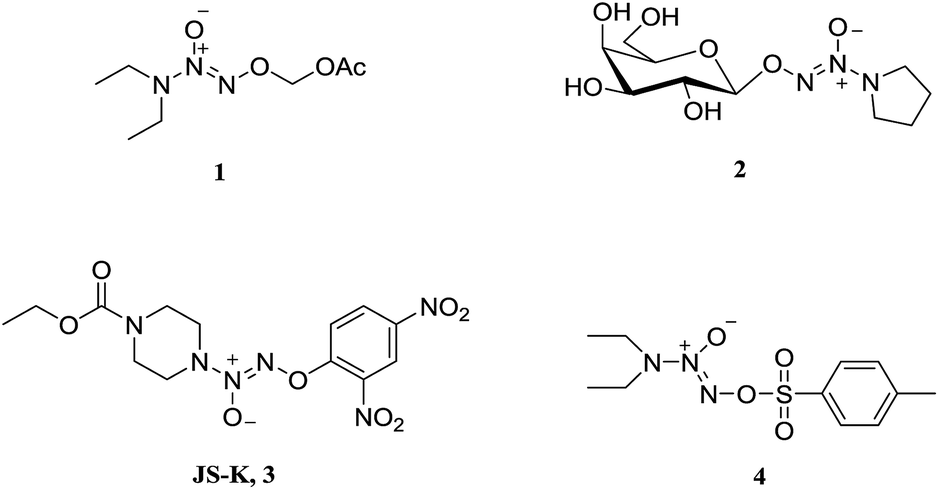 | ||
| Fig. 1 Structures of some important O2-derived diazeniumdiolates. | ||
O2-Derived diazeniumdiolates (Fig. 1) are generally synthesized by O2-alkylation (for 1 and 2), arylation (for 3) or sulfonylation (for 4).16 As far as we know, O2-phosphorylation has not yet been documented. It was therefore of interest to examine whether O2-phosphorylation of diazeniumdiolates could be achieved and exert anticancer activity.
In this article, we would like to report the design, synthesis and biological evaluation of O2-phosphorylated diazeniumdiolates.
Results and discussion
Rationale for the design of O2-phosphorylated diazeniumdiolates
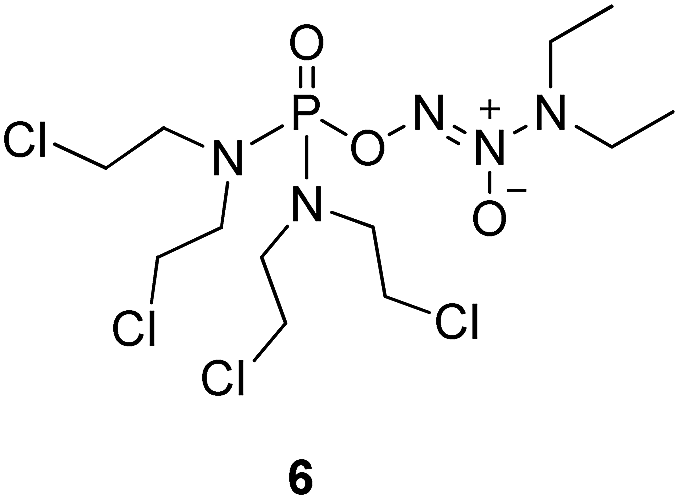
We initially investigated the reaction of 1-(N,N-diethylamino)diazen-1-ium-1,2-diolate sodium with bis(dimethyl-amino)phosphoryl chloride, that could afford O2-phosphorylated diazeniumdiolate 5 (Scheme S1 in ESI†). Based on this success, it was therefore of interest to extend the scope of O2-phosphorylated diazeniumdiolate derivatives using various substituted phosphoryl chlorides.It is known that phosphoramide and phosphorodiamidate mustards are cytotoxins, which can be produced from enzymatic activation of some known anticancer drugs. For example, N,N,N′,N′-tetrakis(2-chloroethyl)phosphorodiamidic acid is derived from the glutathione S-transferase π (GSTπ) activation of canfosfamide (Fig. 2A) via a Tyr 7 triggered deprotonation and β-elimination cleavage.17 In this context, we hypothesized that phosphorodiamidate mustard-based O2-phosphorylated diazeniumdiolates might produce phosphorodiamidate mustard and diazeniumdiolate moieties after in vivo metabolism, exerting significant anticancer activity (Fig. 2B).
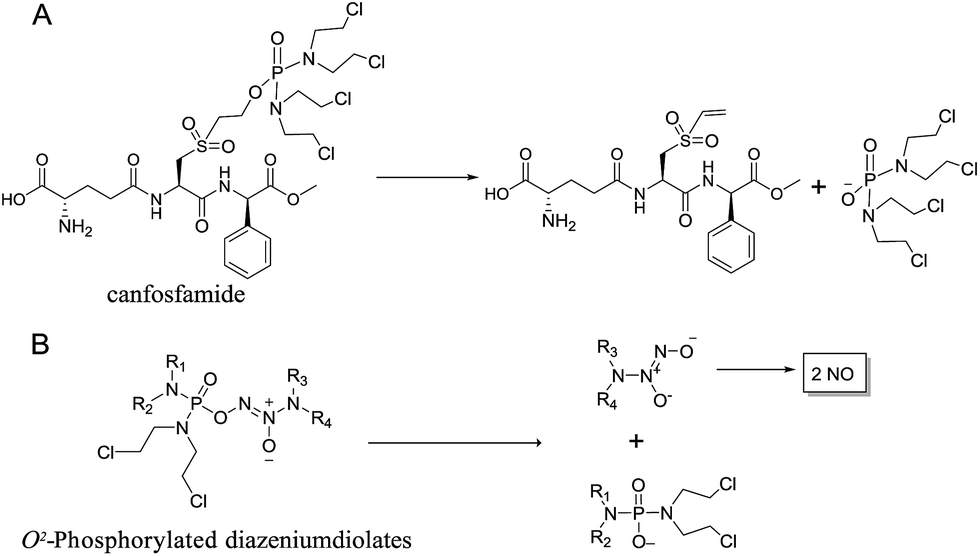 | ||
| Fig. 2 (A) Activation of canfosfamide to form phosphoro-diamidate mustard. (B) Rationale for the design of phosphorodiamidate mustard-based O2-phosphorylated diazeniumdiolates and the proposed mechanism of action. | ||
Chemistry
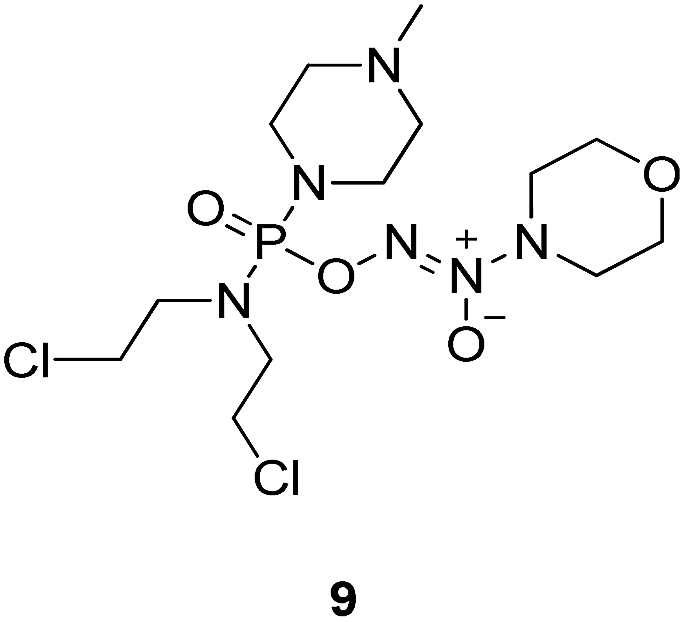
To verify the above hypothesis, we synthesized phosphorodiamidate mustard-based O2-phosphorylated diazeniumdiolates 6–9 as depicted in Scheme 1. Compound 10 was prepared by the reaction of phosphorous oxychloride with nitrogen mustard hydrochloride in toluene in the presence of triethylamine at room temperature, then treatment of 10 with another molecule of nitrogen mustard hydrochloride under the same conditions except that reaction temperature was raised to 110 °C, yielding phosphorodiamidate mustard 11.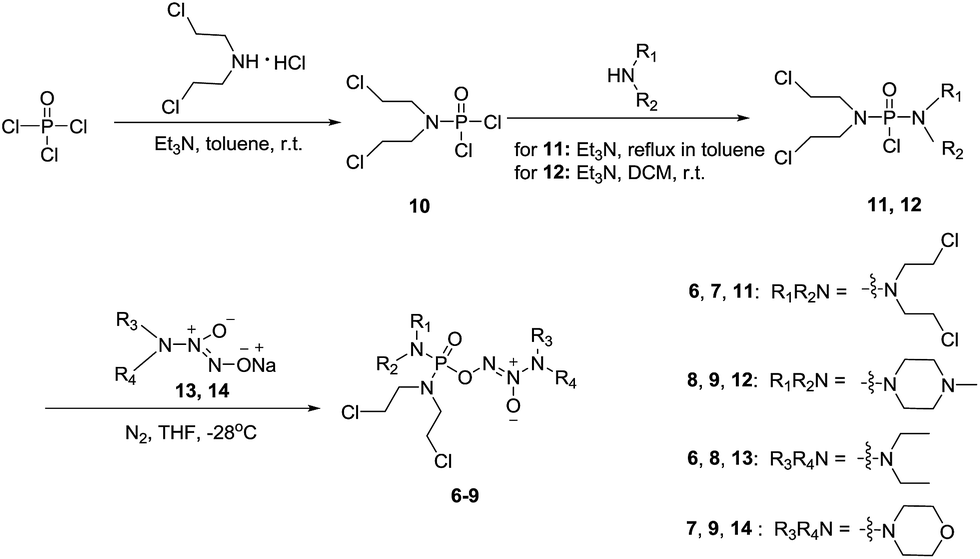 | ||
| Scheme 1 The synthetic route for target compounds 6–9. | ||
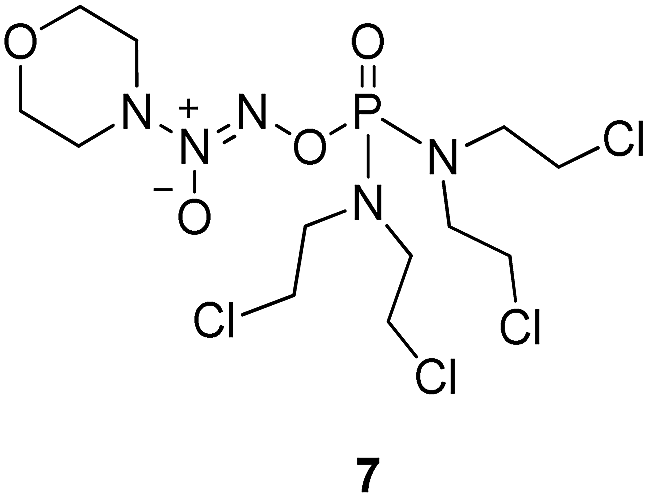
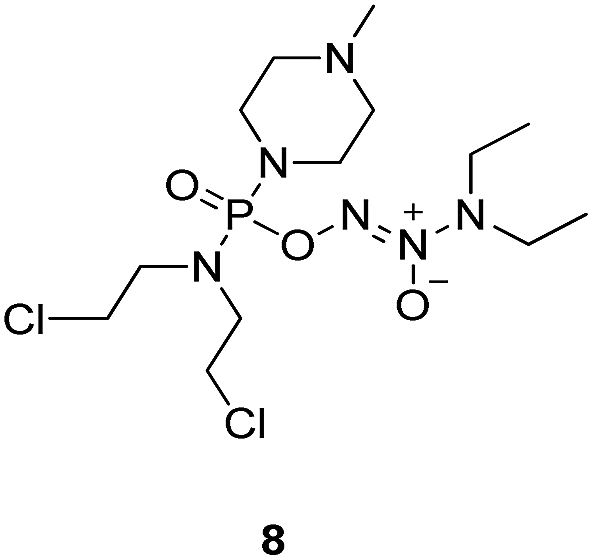
Compound 12 was obtained by reaction of 10 with N-methyl piperazine in dichloromethane in the presence of triethylamine at room temperature. On the other hand, the corresponding secondary amines were treated with NO gas (50 psi) in the presence of NaOMe, forming diazeniumdiolate sodium salts 13 and 14.9 Finally, condensation of 11 and 12 with 13 and 14 in tetrahydrofuran under the nitrogen protection at −28 °C furnished 6–9, respectively.
Assessment of in vitro anti-proliferative activity
Compounds 6–9 were tested for their anti-proliferative activity against six human cancer cell lines (A549, A2780, HepG2, K562, MCF-7 and HCT-116) by MTT assay using JS-K as a positive control (Fig. 3).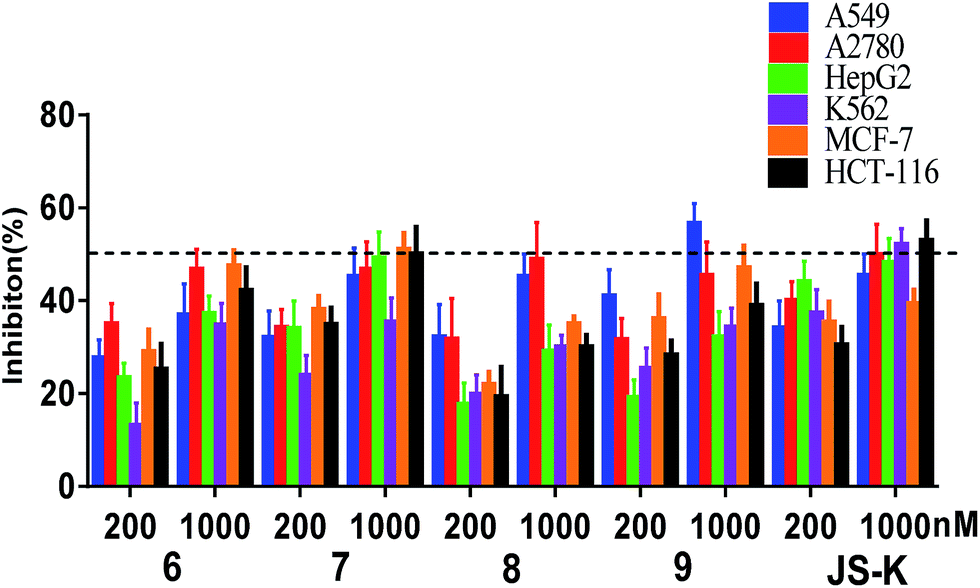 | ||
| Fig. 3 Inhibitory activity of 6–9 at 200 nM and 1000 nM against cancer A549, A2780, HepG2, K562, MCF-7 and HCT-116 cells after incubation for 72 h. Cell proliferation was determined by MTT assay. Data are means ± SD of the inhibition (%) from three independent experiments. | ||
As shown in Fig. 3, the most active compound 7 exhibited significant cytotoxicity on six human cancer cell lines, comparable to JS-K. Thus, we further determined the IC50 values of 7 as shown in Table 1. Evidently, 7 inhibited the proliferation of A549, A2780, HepG2, K562, and HT-29 cell lines as potent as JS-K and more potent against MCF-7 cells than JS-K. Interestingly, 7 exhibited much less antiproliferative activity against normal breast epithelial MCF10A cells (IC50 = 86.5 μM) than breast cancer MCF-7 cells (IC50 = 0.65 μM), suggesting the selective inhibition of compound 7 against cancer cells. Furthermore, the IC50s of 7 against MCF-7 during 24, 48 or 72 h incubation periods were determined as 1.78, 1.26 or 0.65 μM (Table 1), respectively, suggesting that 7 exerted activity in a time-dependent manner.
| 7 | JS-K | |
|---|---|---|
| a The IC50s of 7 and JS-K against several cancer cells and normal breast epithelial MCF10A cells were determined by MTT assay. Data were expressed as the means ± SD of each group of cells from five individual experiments. Without other mentioned, the incubation time is 72 h.b The IC50s of 7 against MCF-7 during 24 or 48 h incubation periods are 1.78 ± 0.13 μM or 1.26 ± 0.11 μM, respectively. | ||
| HCT-116 | 0.847 ± 0.09 | 0.698 ± 0.08 |
| A549 | 1.56 ± 0.21 | 1.53 ± 0.18 |
| A2780 | 1.27 ± 0.10 | 1.59 ± 0.16 |
| HepG2 | 1.29 ± 0.09 | 1.00 ± 0.12 |
| MCF-7 | 0.651 ± 0.07b | 1.60 ± 0.19 |
| MCF10A | 86.5 ± 15.5 | 23.5 ± 4.52 |
Since compound 7 exhibited remarkable inhibitory activity against different cancer cells, especially for MCF-7 cell lines. MCF-7 cells were thus selected for further assays, including NO release, comet assay and apoptosis induction assay.
Assay of intracellular NO release
NO release behavior of 7 and JS-K in MCF-7 cells was examined using both NO-sensitive fluorophore, 4-amino-5-(methylamino)-2′,7′-difluorofluorescein diacetate (DAF-FM DA),18 and Griess reagent.19 It was observed that 7 showed more significant fluorescence (Fig. 4A) and produced greater amounts of nitrite (Fig. 4B), an oxidant product of NO, in MCF-7 cells than JS-K, indicating 7 released a larger amounts of NO than JS-K in MCF-7 cells.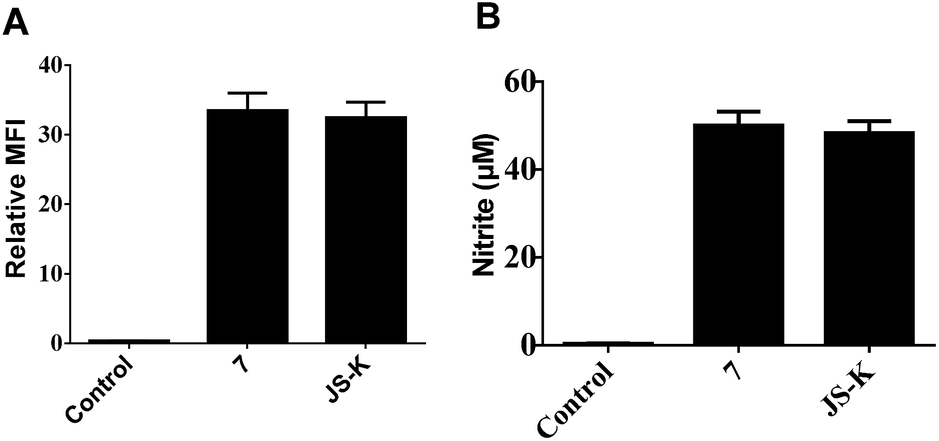 | ||
| Fig. 4 Intracellular NO release behaviour of indicated compounds. (A) Quantitative measurement of intracellular NO production. MCF-7 cells were treated with indicated compounds (30 μM) for 6 h, and NO were measured as DAF-NO derivative fluorescence intensities (% of control). Data are expressed as means ± SD from three separate experiments. (B) MCF-7 cells were incubated with the indicated compounds (30 μM) for 24 h and the levels of NO (presented as nitrite) were determined using a colorimetric Griess reaction assay. Data are expressed as means ± SD from three separate experiments. | ||
Comet assay
Subsequently, DNA interstrand crosslink (ICL) activity of 7 and JS-K in the MCF-7 cells was tested using a well-established alkaline comet assay to examine cellular DNA damage. As visually shown in Fig. 5, 7 exhibited significant DNA ICL activity in a dose-dependent manner, and an obvious cellular DNA damage was observed as comet tails at 0.01 μM. In comparison, JS-K showed much lower DNA damage activity than 7, and no obvious cellular DNA damage was found under the same conditions. These results suggest that diazeniumdiolate together with the phosphoramide mustard moiety may display potent effects, making 7 more active in inducing DNA damage than JS-K.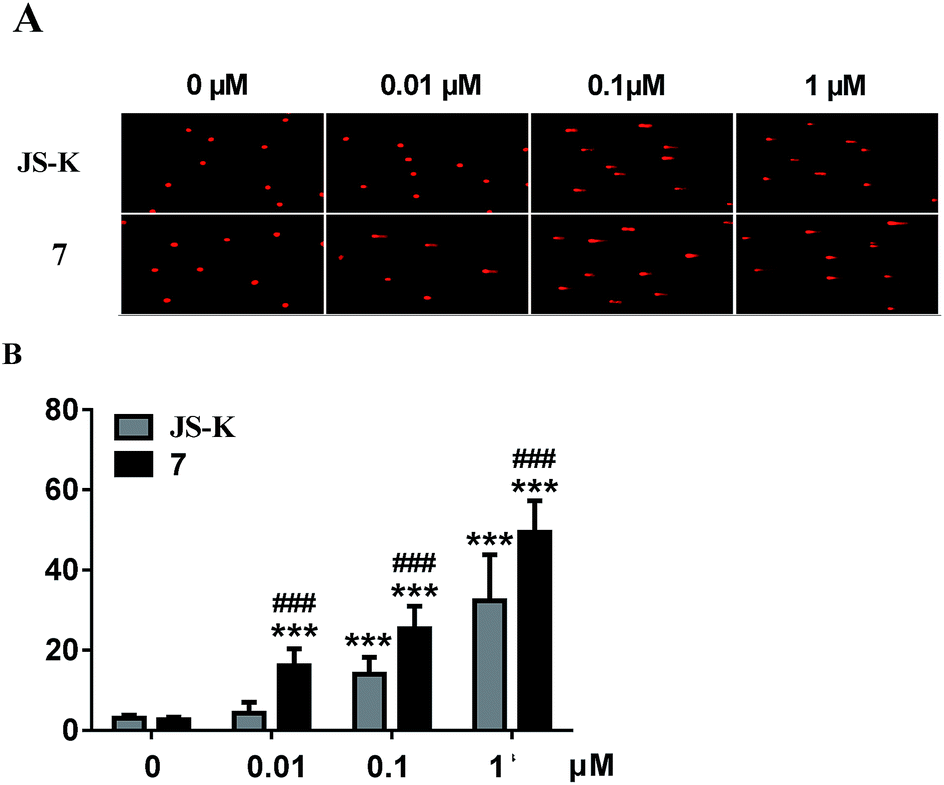 | ||
| Fig. 5 Effect of compounds 7 and JS-K on MCF-7 cell DNA damage. MCF-7 cells were treated for 2 h in the presence or absence of 7 and JS-K (0.01, 0.1, 1 μM). (A) The cells were collected and alkaline comet electrophoresis were performed. (B) The quantitative analysis was performed with the comet analysis software CASP, and the olive tail moment was employed to evaluate DNA damage in 7 and JS-K treated MCF-7 cells. Assays were repeated three times and data were expressed as means ± SD. ***p < 0.001, vs. control, ###p < 0.001, vs. the same concentration of JS-K. | ||
Induction of MCF-7 cell apoptosis
To examine whether the inhibitory effects of 7 on breast cancer cellular proliferation are accompanied by enhanced cancer cell apoptosis, Annexin V–FITC and propidium iodide (PI) staining were carried out and the percentages of apoptotic cells were determined using flow cytometry assay. MCF-7 cells were incubated with different concentrations of vehicle, 7, or JS-K for 24 h. It was observed that treatment with 7 significantly induced apoptosis in MCF-7 cells, more potently than treatment with JS-K (Fig. 6).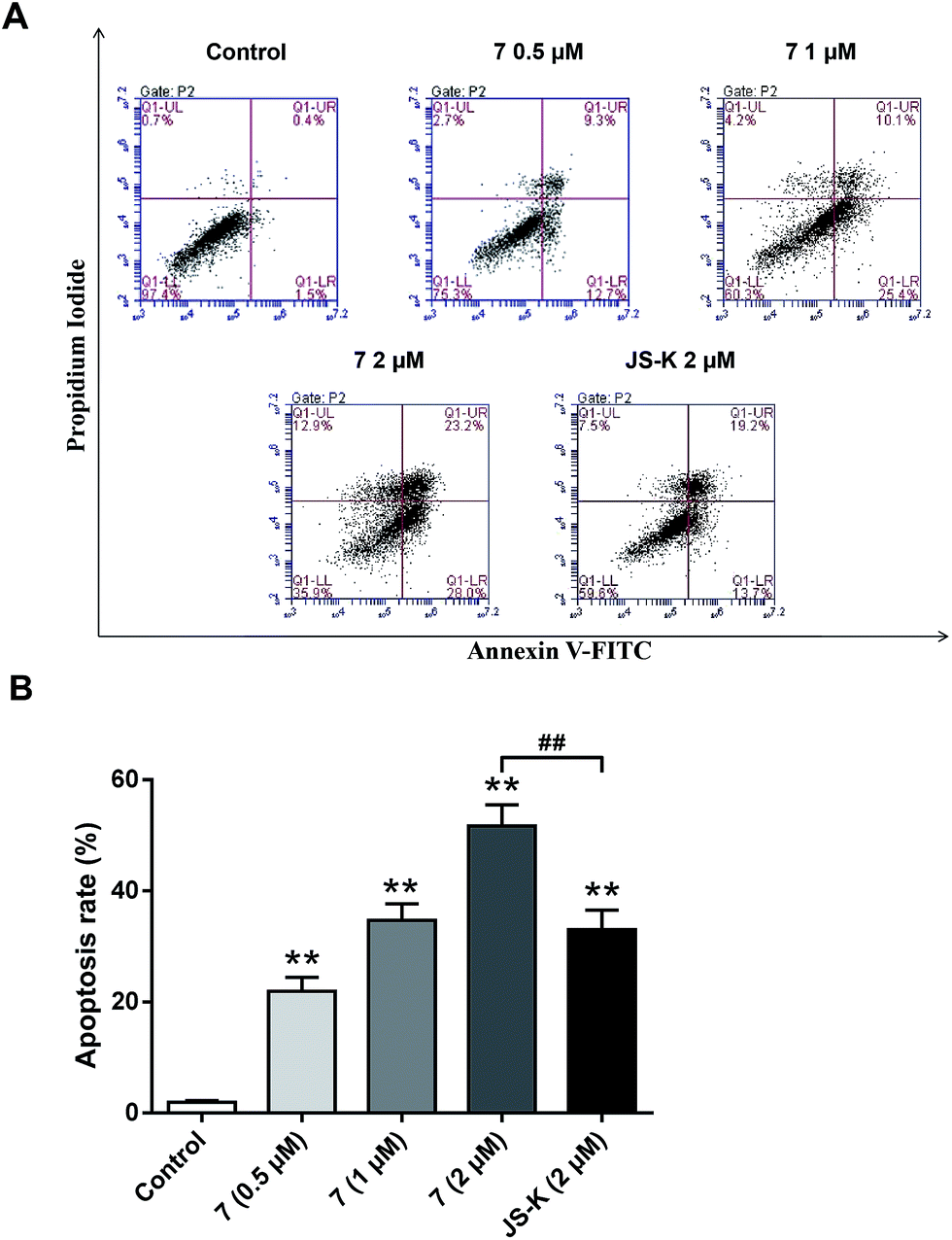 | ||
| Fig. 6 Apoptosis of MCF-7 cells treated with the indicated concentrations of 7 for 24 hours. (A) Annexin V–FITC and propidium iodide (PI) staining were carried out. (B) The apoptosis rate was measured using flow cytometry. The percentage of cells in each population are shown as the means ± SD (n = 3). **P < 0.01, with control group; ##P < 0.01. The data are shown representative of three independent experiments. | ||
Conclusion
The potential use of NO-donating drugs in anticancer field has drawn dramatic attention, and a large number of cancer cell lines have been proved to be sensitive to NO-induced cytotoxic effects.20 It is of great importance to develop new O2-substituted diazeniumdiolates and to extend the NO-based anticancer therapy.21 Given that O2-phosphorylation of diazeniumdiolates have not yet been reported, we first designed, synthesized and biologically evaluated a new class of phosphorodiamidate mustard-based O2-phosphorylated diazeniumdiolates 6–9. These compounds showed potent anticancer activity. Among them, 7 was the most potent in inhibiting proliferation of six cancer cell lines, especially against MCF-7, superior to JS-K. Interestingly, 7 exhibited selective antiproliferative activity against MCF-7 cells relative to the normal breast epithelial MCF10A cells. Furthermore, 7 released a greater amount of NO and caused more DNA damage and apoptosis than JS-K in MCF-7 cells. Our findings suggest that 7 might be a potential anticancer agent, and this O2-phosphorylated diazeniumdiolate may provide a better insight into the design of anticancer drug pertaining to diazeniumdiolate-based NO donors.Experimental
General information
1H NMR, 13C NMR and 31P NMR spectra were recorded with a Bruker Avance 300 MHz spectrometer at 300 K, using TMS as an internal standard. MS spectra were recorded on a Mariner mass spectrometer (ESI) and high resolution mass spectrometry (HRMS) spectra on an Agilent Technologies LC/MSD TOF instrument. Analytical and preparative TLC was performed on silica gel (200–300 mesh) GF/UV 254 plates, and the chromatograms were visualized under UV light at 254 and 365 nm. All solvents were reagent grade and, when necessary, were purified and dried by standard methods. The purity of all compounds tested was characterized by high resolution mass spectrometry (Agilent Technologies LC/MSD TOF). Individual compounds with a purity of >95% were used for biological experiments. Compounds 10–12 were prepared according to ref. 22–24 (see the experimental procedures for 10, 11 and 12 in ESI†).General procedure for the synthesis of 6 and 7
A solution of 3 mmol of intermediate 11 in 20 mL of dry THF was cooled to −28 °C under nitrogen. 3 mmol sodium diazeniumdiolate (13 or 14) was added and the reaction mixture was stirred at this temperature for 6–8 h until the starting material was totally consumed as indicated by TLC. The solvent was evaporated and then the crude product was purified by column chromatography [4% (v/v) MeOH–CH2Cl2] to give 6 (331 mg, 24% yield, TLC Rf 0.64: 6% (v/v) MeOH–CH2Cl2) or 7 (401 mg, 29% yield, TLC Rf 0.59: 6% (v/v) MeOH–CH2Cl2).The title compound was obtained in 24% yield as a yellow oil. 1H NMR (300 MHz, CD3OD), δ (ppm): 1.21 (t, J = 9.0 Hz, 6H, 2 × CH3), 3.43–3.71 (m, 12H, 3 × CH2NCH2), 3.78–3.83 (m, 8H, 4 ×
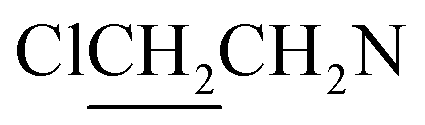 ). 13C NMR (75 MHz, CD3OD), δ (ppm): 49.0, 46.5, 41.3, 10.4. 31P NMR (121.5 MHz, CD3OD), δ (ppm): 17.7. ESI-MS 482.0 [M + Na]+; HRMS calculated for C12H26Cl4N5O3PNa [M + Na]+ 482.0425, found 482.0414 (PPM error of −2.3).
). 13C NMR (75 MHz, CD3OD), δ (ppm): 49.0, 46.5, 41.3, 10.4. 31P NMR (121.5 MHz, CD3OD), δ (ppm): 17.7. ESI-MS 482.0 [M + Na]+; HRMS calculated for C12H26Cl4N5O3PNa [M + Na]+ 482.0425, found 482.0414 (PPM error of −2.3).The title compound was obtained in 29% yield as a colorless oil. 1H NMR (300 MHz, CD3OD), δ (ppm): 3.47–3.58 (m, 12H, 3 × CH2NCH2), 3.72–3.76 (m, 8H, 4 ×
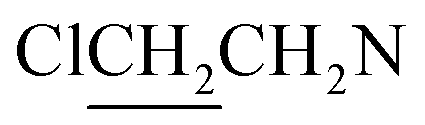 ), 3.84 (t, J = 11.8 Hz, 4H, CH2OCH2). 13C NMR (75 MHz, CD3OD), δ (ppm): 66.5, 52.3, 50.4, 42.8. 31P NMR (121.5 MHz, CD3OD), δ (ppm): 18.1. ESI-MS 496.0 [M + Na]+; HRMS calculated for C12H24Cl4N5O4PNa [M + Na]+ 496.0218, found 496.0226 (PPM error of 1.6).
), 3.84 (t, J = 11.8 Hz, 4H, CH2OCH2). 13C NMR (75 MHz, CD3OD), δ (ppm): 66.5, 52.3, 50.4, 42.8. 31P NMR (121.5 MHz, CD3OD), δ (ppm): 18.1. ESI-MS 496.0 [M + Na]+; HRMS calculated for C12H24Cl4N5O4PNa [M + Na]+ 496.0218, found 496.0226 (PPM error of 1.6).General procedure for the synthesis of 8 and 9
A solution of 3 mmol of intermediate 12 in 20 mL of dry THF was cooled to −28 °C under nitrogen. 3 mmol of diazeniumdiolate (13 or 14) was added and the reaction mixture was stirred at this temperature for 6–8 h until the starting material was totally consumed as indicated by TLC. The solvent was evaporated and then the crude product was purified by column chromatography [4% (v/v) MeOH–CH2Cl2] to give 8 (264 mg, 21% yield, TLC Rf 0.52: 6% (v/v) MeOH–CH2Cl2) or 9 (351 mg, 27% yield, TLC Rf 0.47: 6% (v/v) MeOH–CH2Cl2).The title compound was obtained in 21% yield as a yellow oil. 1H NMR (300 MHz, CD3OD), δ (ppm): 1.17 (t, J = 5.2 Hz, 6H, 2 × CH3), 2.33 (s, 3H, CH3), 2.49 (t, J = 3.0 Hz, 4H, 2 × CH2), 3.28–3.35 (m, 4H, CH2NCH2), 3.42–3.54 (m, 8H, 2 × NCH2), 3.69–3.74 (m, 4H, 2 × CH2Cl). 13C NMR (75 MHz, CD3OD), δ (ppm): 54.4, 49.0, 46.7, 44.9, 44.0, 41.4, 10.6. 31P NMR (121.5 MHz, CD3OD), δ (ppm): 16.6. ESI-MS 419.2 [M + H]+; HRMS calculated for C13H30Cl2N6O3P [M + H]+ 419.1494, found 419.1501 (PPM error of 1.7).
The title compound was obtained in 27% yield as a colorless oil. 1H NMR (500 MHz, CD3OD), δ (ppm): 2.37 (s, 3H, N–CH3), 2.53–2.59 (m, 4H, 2 × –NCH2), 3.31–3.34 (m, 4H, 2 × -PNCH2), 3.44–3.50 (m, 4H, 2 × CH2, chloroethyl), 3.56 (t, J = 4.5 Hz, 4H, CH2NCH2, morpholine), 3.71–3.73 (m, 4H, 2 × CH2Cl, chloroethyl), 3.84 (t, J = 4.8 Hz, 4H, CH2OCH2, morpholine). 13C NMR (75 MHz, CD3OD), δ (ppm): 67.0, 56.3, 52.8, 50.3, 46.6, 45.8, 43.3. 31P NMR (121.5 MHz, CD3OD), δ (ppm): 16.9. ESI-MS 455.1 [M + Na]+; HRMS calculated for C13H27Cl2N6O4PNa [M + Na]+ 455.1106, found 455.1098 (PPM error of −1.8).
MTT assay
The inhibitory effects on cell proliferation of tested compounds were investigated by the MTT method. All cells were obtained from BIORN Life Science Co. Ltd. Several final density of 1.0 × 104 cells per well were placed in 96-well cell plates overnight and treated with or without different concentrations of tested compounds for various periods of time. During the last 4 h culture, the cells were exposed to MTT (5 mg mL−1), and the resulting formazan crystals were dissolved in 150 μL of DMSO and measured using a spectrophotometer (Tecan) at a test wavelength of 570 nm. Experiments were conducted in triplicate. Inhibition rate (%) = [(Acontrol − Atreated)/Acontrol] × 100%.NO release measurement in MCF-7 cells
Comet assay
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 21372261 and No. 81673305) and Jiangsu Province Funds for Distinguished Young Scientists (BK20160033). Part of the work was supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), Program for New Century Excellent Talents in University (NCET-13-1033), College Students Innovation Project for the R & D of Novel Drugs (J1030830), and Jiangsu Shuang Chuang team.Notes and references
- E. Culotta and D. E. Koshland Jr, Science, 1992, 258, 1862–1865 CAS.
- W. Xu, L. Z. Liu, M. Loizidou, M. Ahmed and I. G. Charles, Cell Res., 2002, 12, 311–320 CrossRef PubMed.
- B. Oronsky, G. R. Fanger, N. Oronsky, S. Knox and J. Scicinski, Clin. Transl. Oncol., 2014, 7, 167–173 CrossRef PubMed.
- R. A. Serafim, M. C. Primi, G. H. Trossini and E. I. Ferreira, Curr. Med. Chem., 2012, 19, 386–405 CrossRef CAS PubMed.
- A. B. Seabra, R. de Lima and M. Calderon, Curr. Top. Med. Chem., 2015, 15, 298–308 CrossRef CAS PubMed.
- D. Hirst and T. Robson, J. Pharm. Pharmacol., 2007, 59, 3–13 CrossRef CAS PubMed.
- S. Huerta, S. Chilka and B. Bonavida, Internet J. Oncol., 2008, 33, 909–927 CAS.
- R. Scatena, P. Bottoni, G. E. Martorana and B. Giardina, Expert Opin. Invest. Drugs, 2005, 14, 835–846 CrossRef CAS PubMed.
- J. E. Saavedra, T. M. Dunams, J. L. Flippenanderson and L. K. Keefer, J. Org. Chem., 1992, 57, 6134–6138 CrossRef CAS.
- J. E. Saavedra, P. J. Shami, L. Y. Wang, K. M. Davies, M. N. Booth, M. L. Citro and L. K. Keefer, J. Med. Chem., 2000, 43, 261–269 CrossRef CAS PubMed.
- R. S. Nandurdikar, A. E. Maciag, S. Y. Hong, H. Chakrapani, M. L. Citro, L. K. Keefer and J. E. Saavedra, Org. Lett., 2010, 12, 56–59 CrossRef CAS PubMed.
- P. J. Shami, J. E. Saavedra, L. Y. Wang, C. L. Bonifant, B. A. Diwan, S. V. Singh, Y. Gu, S. D. Fox, G. S. Buzard, M. L. Citro, D. J. Waterhouse, K. M. Davies, X. Ji and L. K. Keefer, Mol. Cancer Ther., 2003, 2, 409–417 CAS.
- A. E. Maciag, H. Chakrapani, J. E. Saavedra, N. L. Morris, R. J. Holland, K. M. Kosak, P. J. Shami, L. M. Anderson and L. K. Keefer, J. Pharmacol. Exp. Ther., 2011, 336, 313–320 CrossRef CAS PubMed.
- T. Kiziltepe, T. Hideshima, K. Ishitsuka, E. M. Ocio, N. Raje, L. Catley, C. Q. Li, L. J. Trudel, H. Yasui, S. Vallet, J. L. Kutok, D. Chauhan, C. S. Mitsiades, J. E. Saavedra, G. N. Wogan, L. K. Keefer, P. J. Shami and K. C. Anderson, Blood, 2007, 110, 709–718 CrossRef CAS PubMed.
- P. J. Shami, J. E. Saavedra, C. L. Bonifant, J. Chu, V. Udupi, S. Malaviya, B. I. Carr, S. Kar, M. Wang, L. Jia, X. Ji and L. K. Keefer, J. Med. Chem., 2006, 49, 4356–4366 CrossRef CAS PubMed.
- R. A. D'Sa, Y. Wang, P. H. Ruane, B. M. Showalter, J. E. Saavedra, K. M. Davies, M. L. Citro, M. N. Booth, L. K. Keefer and J. P. Toscano, J. Org. Chem., 2003, 68, 656–657 CrossRef PubMed.
- D. F. Dourado, P. A. Fernandes, M. J. Ramos and B. Mannervik, Biochemistry, 2013, 52, 8069–8078 CrossRef CAS PubMed.
- H. Kojima, Y. Urano, K. Kikuchi, T. Higuchi, Y. Hirata and T. Nagano, Angew. Chem., Int. Ed. Engl., 1999, 38, 3209–3212 CrossRef CAS PubMed.
- D. S. Bredt and S. H. Snyder, Annu. Rev. Biochem., 1994, 63, 175–195 CrossRef CAS PubMed.
- P. J. Shami, D. L. Sauls and J. B. Weinberg, Leukemia, 1998, 12, 1461–1466 CAS.
- L. K. Keefer, ACS Chem. Biol., 2011, 6, 1147–1155 CrossRef CAS PubMed.
- M. H. Lyttle, A. Satyam, M. D. Hocker, K. E. Bauer, C. G. Caldwell, H. C. Hui, A. S. Morgan, A. Mergia and L. M. Kauvar, J. Med. Chem., 1994, 37, 1501–1507 CrossRef CAS PubMed.
- A. Satyam, M. D. Hocker, K. A. Kane-Maguire, A. S. Morgan, H. O. Villar and M. H. Lyttle, J. Med. Chem., 1996, 39, 1736–1747 CrossRef CAS PubMed.
- X. L. Chen, J. W. Yuan, L. B. Qu, Z. B. Qu, S. H. Xu, F. J. Wang and Y. F. Zhao, Phosphorus, Sulfur Silicon Relat. Elem., 2012, 187, 245–254 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The synthetic route for compound 5; experimental procedures for 5, 10, 11 and 12; 1H NMR, 13C NMR, 31P NMR and HRMS spectra of 6–9. See DOI: 10.1039/c7ra00401j |
| This journal is © The Royal Society of Chemistry 2017 |