
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective metabolism of the UV-filter 2-ethylhexyl 4-dimethylaminobenzoate and its metabolites in rabbits in vivo and vitro†
Yiran Liang,
Jing Zhan,
Xueke Liu,
Zhiqaing Zhou,
Wentao Zhu,
Donghui Liu and
Peng Wang *
*
Beijing Advanced Innovation Center for Food Nutrition and Human Health, Department of Applied Chemistry, China Agricultural University, Department of Applied Chemistry, China Agricultural University, No. 2 Yuanmingyuan West Road, Beijing 100193, P. R. China. E-mail: wangpeng@cau.edu.cn; Tel: +86-10-62732937
First published on 17th March 2017
Abstract
The stereoselective metabolism of the enantiomers of the UV-filter 2-ethylhexyl 4-dimethylaminobenzoate (EDP) in rabbits was studied. The two major metabolites 4-(N,N-dimethylamino) benzoic acid (DMP) and 4-methylaminobenzoic acid (MMP) were also investigated in vivo and in vitro. Cytotoxicity of EDP and its two metabolites was also investigated in hepatocytes. The results showed that EDP degraded rapidly to its metabolites (DMP and MMP) and could not be detected in blood at 5 min after intravenous administration to rabbit in vivo. In almost all the tissue samples, EDP, DMP and MMP could not be detected at 3 h expect DMP was found in the liver and kidney at about 1 mg kg−1 level. EDP was found to be degraded to DMP rapidly in plasma and liver microsome in vitro with t1/2 less than 20 and 5 min and the whole process was enantioselective with preference of (+)-form. DMP was observed to be further degraded to MMP in liver microsome in the presence of NADPH. The cytotoxic effects of EDP, DMP and MMP were carried out using buffalo rat liver cell line. The results from cell viability assays indicated that the degradation of EDP was a detoxification process.
1. Introduction
Long-term exposure to ultraviolet (UV) radiation can cause immunosuppression, DNA damage and photoaging. It is estimated that about 6000 people die per year from malignant skin cancer because of too much sun exposure.1 Thus, with the growing awareness of the risk of ultraviolet radiation, more and more sunscreen products have been consumed. The main functional composition of UV radiation protection in sunscreen products is UV-filter. In general, based on the composition, UV filters can be classified into two groups, one is organic compounds which can absorb and stabilize UV radiation, and the other is inorganic compounds which reflect and scatter UV radiation. Generally, both kinds of UV filters give good protection against UV radiation. Many commercial sunscreen products contain both organic and inorganic UV filters to provide broad UV spectrum protection.2,3 Organic UV filters have desirable effects without leaving marks onto the skin.Along with the widespread and steadily increasing use of organic UV filters worldwide, the concerns about their toxicity are also increasing. Organic UV filters have been known to cause photoallergic contact dermatitis.4,5 One of the earliest evidence of photoallergic contact dermatitis caused by exogenous agents was reported 60 years ago and the allergen was the organic UV filter para-amino benzoic acid.6 Since then, many studies have reported that organic UV filters are responsible for photoallergic contact dermatitis.7,8 Furthermore, the developmental and reproductive toxicity of organic UV-filters have attracted more and more attention in recent years since an increasing number of in vivo and vitro studies identified their endocrine disrupting effects.9–15 Because of the highly exposure frequency, the risk of organic UV-filters should be fully evaluated.
The UV-filter 2-ethylhexyl 4-dimethylaminobenzoate (Fig. 1), also known as octyldimethyl-EDP or EDP, is one of the most commonly used UV filters for its effective protection from UV radiation.16 EDP is designed for external application on the skin, and a recent study has shown that it could be absorbed through skin.17,18 However, the fate and pharmacokinetic parameters of EDP and its metabolites in animals are still unclear. The cytotoxicity of EDP and its two main metabolites, 4-(N,N-dimethylamino) benzoic acid (DMP) and 4-methylaminobenzoic acid (MMP), have not been studied. In addition, EDP exists in two enantiomeric forms, (+)-EDP and (−)-EDP. Enantiomers may have different biological properties in uptake, metabolism, excretion and also may differ in toxicity.19 Enantioselectivity has been widely researched in medicine, pesticide and environmental pollutants,20–24 but little is known about UV-filter. Despite of the chirality, EDP has been used in racemate form and the stereochemistry has not been considered in pharmacokinetic and toxicology studies.
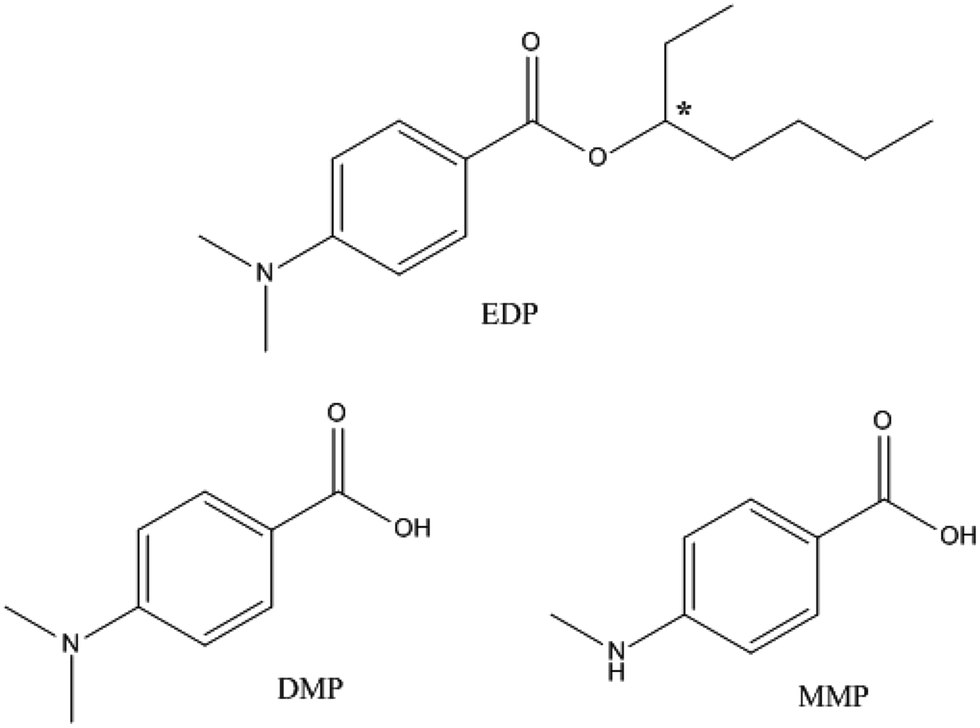 | ||
| Fig. 1 Chemical structures of EDP and its metabolites (DMP and MMP). Chiral center is denoted by an asterisk (*). | ||
In this study, the enantiomeric separation of EDP on Chiralpak IC column by HPLC was investigated, and the individual enantiomers were identified by an optical rotatory dispersion detector. The chiral residue analysis methods for EDP and its two metabolites (DMP and MMP) in animal tissues were set up. In order to investigate the potential impacts, the metabolism of EDP in rabbit was conducted in vivo and vitro on an enantiomeric level, and its two main metabolites were also determined. For a more integrative assessment of the risk of EDP, cytotoxicity of EDP and its main metabolites (DMP and MMP) was assessed via the methyl-thiazol-tetrazolium (MTT) assay.
2. Materials and methods
2.1. Chemicals and reagents
Racemic 2-ethylhexyl 4-dimethylaminobenzoate (rac-EDP, 99%, technical grade), 4-dimethylaminobenzoic acid (98.0%, technical grade) and 4-(methylamino)benzoic acid (98.0%, technical grade) were purchased from J&K Chemical (Beijing, China). Acetonitrile, n-hexane, 2-propanol and trifluoroacetic acid (TFA) were obtained from Fisher Scientific (Fair Lawn, NJ, USA). Tris, β-nicotinamide adenine dinucleotide phosphate (NADPH), Tween-80, rat tail collagen, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and heparin were purchased from Sigma-Aldrich (St Louis, MO, USA). Tris–HCl solution (PH 7.4) was prepared using Milli-Q water (18 MΩ cm, Laikie Instrument Co., Ltd., Shanghai, China). Dimethyl sulfoxide (DMSO), collagenase, fetal calf serum (FCS), insulin, N-(2-hydroxyethylpiperazine)-N′-(2-ethanesulfonic acid) (HEPES), antibiotic and Dulbecco's Modified Eagle's Medium (DMEM) were purchased from GIBCO (Grand Island, NY, USA). All other chemicals and solvents were of analytical grade and purchased from commercial sources.2.2. Chiral separation
The mobile phase was a mixture of n-hexane and isopropanol. The effluent was monitored using UV detection at 240 nm with a flow rate of 0.6 mL min−1. The elution orders of rac-EDP were determined by the optical rotatory dispersion (ORD) detector. The following parameters were measured:| Rs (resolution factor) = 2(t2 − t1)/(w1 + w2) |
| Capacity factor of the first eluted enantiomer k1 = (t1 − t0)/t0 |
| Capacity factor of the second eluted enantiomer k2 = (t2 − t0)/t0 |
Tri-t-butylbenzene was used to determine the t0-value
| Selectivity factor α = k2/k1 |
2.3. Animal experiments
Male Japanese white rabbits weighting 1.8–2.0 kg were purchased from Experimental Animal Research Institute of China Agriculture University and were kept in well-ventilated cages at a constant temperature (20 ± 2 °C) with a 12 h light/dark cycle. All the animals were acclimatized to laboratory conditions for seven days with standard pellet diet and water ad libitum before the experiments. Racemic EDP was dissolved in sterile saline (5% Tween 80, v/v), and then given to rabbits by intravenous injection in the ear vein at a dose of 50 mg kg−1 body weight. Blood samples were taken from heart and collected in heparinized tubes at 5, 15, 30, 45, 60, 90, 120 and 180 min. Plasma was separated by centrifugation at 4000 rpm for 15 min, and then immediately transferred to labeled polypropylene tubes. Brain, liver, kidney, lung, urine and feces were gathered at 3 h. All the samples were stored immediately at −20 °C until analysis. Animal experiments were approved and performed in accordance with the guidelines of Institutional Animal Care and Use Committee of China Agricultural University.2.4. Plasma and microsome incubation
The blank plasma was separated from drug-free heart blood by centrifugation at 4000 rpm for 15 min at 4 °C and diluted by Tris–HCl buffer (50 mM, pH 7.4 at 37 °C) to obtain a 10% plasma solution (v/v). The plasma solution (990 μL) was transferred to a 15 mL plastic centrifuge tube and preincubated at 37 °C for 10 min before the incubation. The reaction started when 10 μL of alcohol-dissolved EDP was added to a 10 mg kg−1 spiked level. Samples (1 mL) were then taken at 0, 5, 10, 20 40 and 60 min, and 2 mL of ice-cold acetonitrile was immediately added to terminate the enzyme–substrate reaction.Rabbit liver microsome was prepared according to the previously described method.25 In brief, after rabbits were sacrificed, livers were removed and immediately placed on ice and thoroughly washed with ice-cold 1.15% KCl solution. The pooled livers were minced and homogenized in four times of ice-cold EDTA solution (1 mM EDTA and 50 mM Tris–HCl, pH 7.4). The microsomal fraction was prepared using differential centrifugation. The pellet was suspended in 50 mmol L−1 Tris–HCl buffer (pH 7.4) containing 20% glycerol (v/v) solution, then quickly frozen and stored at −80 °C until use. Protein concentration was determined according to the method of Bradford.26
The rabbit liver microsome was diluted by Tris–HCl buffer (50 mM, pH 7.4 at 37 °C) to obtain a microsomal suspension that containing 0.2 mg L−1 protein. After the microsomal suspension (990 μL) was preincubated at 37 °C for 10 min, the reaction was initiated by the addition of 10 μL of alcohol-dissolved EDP (10 mg L−1 of EDP in the reaction system). Samples (1 mL) were taken after incubation at 37 °C for 0, 5, 10, 20, 40 and 60 min, respectively, and 2 mL of ice-cold acetonitrile was added to terminate the reaction.
The metabolism of DMP in rabbit liver microsome was also conducted. Alcohol-dissolved DMP (10 μL) was added to 900 μL of microsomal suspension to achieve a concentration of 5 mg kg−1 DMP in the reaction mixture (1 mL). After preincubation at 37 °C for 10 min, the reaction was then generated with the addition of 90 μL of NADPH at a concentration of 1.0 mM. The time point and the termination of the reaction were the same as the metabolism of EDP mentioned above. The extraction procedure was described hereunder.
2.5. Cytotoxicity study
The buffalo rat liver (BRL-3A) cell line was cultured in high-glucose DMEM containing 10% FCS, 100 U mL−1 penicillin, 10 μg mL−1 insulin and 100 μg mL−1 streptomycin at 37 °C in an atmosphere of 5% CO2. After the cell viability was demonstrated to be greater than 90%, each 100 μL of BRL-3A cell suspension (5 × 104 cells per well) was placed into a 96-well microplate precoated with collagen and incubated in a 5% CO2 incubator at 37 °C for 24 h to obtain the adhesive BRL-3A cell monolayers. Then the BRL-3A cell monolayers were treated with a series of concentrations of EDP (0.5, 0.25, 0.13, 0.06 and 0.03 μg mL−1), DMP (100, 50, 25, 12 and 6 μg mL−1) and MMP (295, 148, 74, 37 and 18 μg mL−1). DMSO was used to dissolve EDP, DMP and MMP. After 72 hours, 15 μL of 5 mg mL−1 MTT solution was added. The plates were then incubated for 4 hours in 5% CO2. After the medium was removed from the wells, 200 μL of DMSO was added into each well. The absorbance was measured at 492 nm with a spectrophotometer (Multiskan MK3, Thermo Scientific, Pittsburgh, PA) after 10 min of shaking at room temperature.272.6. Sample preparation
Plasma (1 mL) and microsome (1 mL) samples were transferred to 15 mL plastic centrifuge tubes with the addition of 2 mL of acetonitrile. To obtain a better extraction, 100 μL of HCl (1 mol L−1) was added to the samples. An aliquot of 0.2 g of sodium chloride was added for better liquid–liquid separation. After vortexed for 5 min, the tubes were centrifuged at 4000 rpm for 5 min to get supernatant acetonitrile phase. The same extraction step was repeated once with another 2 mL of acetonitrile. The combined acetonitrile was evaporated at 35 °C under a gentle nitrogen stream. The analytes were reconstituted in 0.5 mL of 2-propanol, and finally filtered through a 0.22 μm filter prior for HPLC determination.Urine (1 mL) and 0.5 g of liver, kidney, lung, brain, and feces samples were extracted as mentioned above and reconstituted in 0.5 mL of acetonitrile. As matrix interferences existed in these samples, a solid-phase extraction (SPE) method using CNWBOND LC-C18 SPE tube was used to clean up. The SPE tubes were preconditioned with 5 mL of methanol and 400 μL of the extract was then loaded. After the tubes were washed with 5 mL of 0.1% acetic acid, the target compounds were eluted with 5 mL of methanol. The eluent was evaporated at 35 °C under a gentle nitrogen stream and the residue was dissolved in 400 μL of 2-propanol, and finally filtered through a 0.22 μm filter prior for HPLC determination.
2.7. Sample analysis
Analysis was performed using an Agilent 1200 series HPLC equipped with a G1322A degasser, G1311A quatemary pump, a G1329A automatic liquid sampler and G1314B variable wavelength UV detector. Column temperature was controlled with an AT-930 column oven (Tianjin Automatic Science Instrument, China). Agilent 1200 Chemstation software was used to process the chromatographic data. The separation of racemic EDP and its metabolites was performed on Chiralpak IC chiral column (cellulose tris-(3,5-dichorophenylcarbamate), 250 × 4.6 mm, 5 μm, Daicel Chemical Industries, Ltd., Osaka, Japan). The polarimeter was provided by Chiralyser-MP Optical Rotation Detector (IBZ Messtechnik, Germany). Normal phase mode was used with the mobile phase of n-hexane/2-propanol/TFA (v/v/v, 90/10/0.1), at a flow rate of 0.6 mL min−1 at 20 °C, and the UV detection wavelength was 240 nm.2.8. Method validation
The linearities for the determination of EDP, DMP and MMP over the range of 0.5–50 mg kg−1 were generated from the working standard solution. Calibration curves were generated by plotting peak area versus concentration. The linear regression analysis was performed using Excel 2007 (Microsoft).3. Results and discussion
3.1. Enantioseparation of EDP enantiomers
The two enantiomers of EDP could be completely separated by Chiralpak IC on HPLC using n-hexane/2-propanol as mobile phase. The elution order of EDP was (−)/(+) based on ORD signal at 426 nm (shown in Fig. S1†). The enantioseparation results using different compositions of mobile phase (n-hexane/2-propanol) were summarized in Table S1.† With 2-propanol decreasing from 20% to 2%, the separation factor (α) increased slightly and the resolution factors (Rs) increased from 1.18 to 2.36. The influence of temperature from 5–30 °C on the chiral separation and retention time was investigated using the mobile phase of n-hexane/2-propanol/(90/10, v/v) (Table S2†). To get a better understanding of the mechanism of the chiral separation, thermodynamic parameters were calculated using Van't Hoff equations as follows.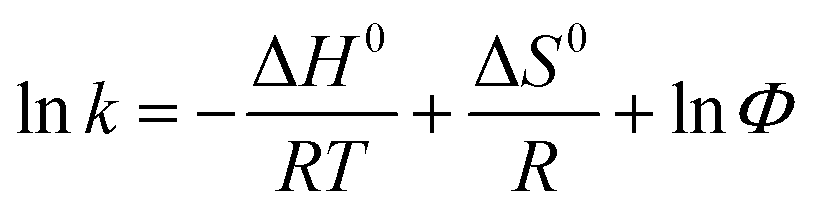
where ΔH0 and ΔS0 represent the changes of standard enthalpy and entropy in the process of analyte from the mobile phase to the stationary phase, ΔΔH0 and ΔΔS0 are the differences of the enantiomers in enthalpy and entropy changes. Φ is the phase ratio, R is the gas constant and T is the absolute temperature. If the plots of ln
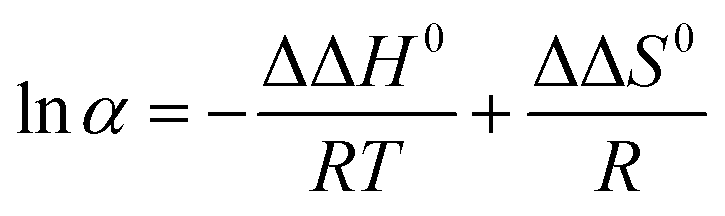
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k versus 1/T could fit straight lines, −ΔH0/R and ΔS0/R + lnΦ are the slope and intercept. For the linear plots of lnα versus 1/T, the slope and intercept are −ΔΔH0/R and ΔΔS0/R, respectively. Therefore, the thermodynamic parameters and equations can be calculated. Negative values of ΔH0 suggest that the transfer of the solutes from the mobile phase to the stationary phase is an enthalpy-driven process. The calculated ΔΔH0 and ΔΔS0 were shown in Table 1, indicating that the separation of EDP enantiomers on Chiralpak IC in n-hexane/2-propanol was enthalpy-driven.
k versus 1/T could fit straight lines, −ΔH0/R and ΔS0/R + lnΦ are the slope and intercept. For the linear plots of lnα versus 1/T, the slope and intercept are −ΔΔH0/R and ΔΔS0/R, respectively. Therefore, the thermodynamic parameters and equations can be calculated. Negative values of ΔH0 suggest that the transfer of the solutes from the mobile phase to the stationary phase is an enthalpy-driven process. The calculated ΔΔH0 and ΔΔS0 were shown in Table 1, indicating that the separation of EDP enantiomers on Chiralpak IC in n-hexane/2-propanol was enthalpy-driven.
| Enantiomer | lnk = −ΔH0/RT + ΔS0/R |
ΔH0, kJ mol−1 | ΔS0, J mol−1 | lnα = −ΔΔH0/RT + ΔΔS0/R |
ΔΔH0, kJ mol−1 | ΔΔS0, J mol−1 K−1 |
|---|---|---|---|---|---|---|
| + | lnk1 = 1463.1/T − 4.19 |
−12.16 | −4.19 | lnα = 103.37/T − 0.28 |
−0.86 | −2.31 |
| R2 = 0.9981 | ||||||
| − | lnk2 = 1566.4/T − 4.41 |
−13.02 | −4.41 | R2 = 0.9985 | ||
| R2 = 0.9982 |
3.2. Chiral residue analysis method and assay validation
Good linearities were obtained over the range of 0.5–50 mg L−1 for each analyte, with the correlation coefficients (R2) all above 0.998. The recoveries for all the analytes in plasma, liver microsome, brain, kidney, lung, liver, urine and feces at three levels ranged from 81% to 109% with RSD of 2% to 13% (Table S3†). The limit of detection (LOD) and quantitation (LOQ) for all enantiomers were 0.2 and 0.5 mg kg−1 respectively.3.3. In vivo metabolism of EDP in rabbit
After the intravenous injection of rac-EDP to rabbit at 50 mg kg−1 body weight, EDP could not be detected in plasma at 5 min (Fig. 2). The overwhelming amount of the hydrolysis metabolites DMP and a small amount of MMP were detected in rabbit.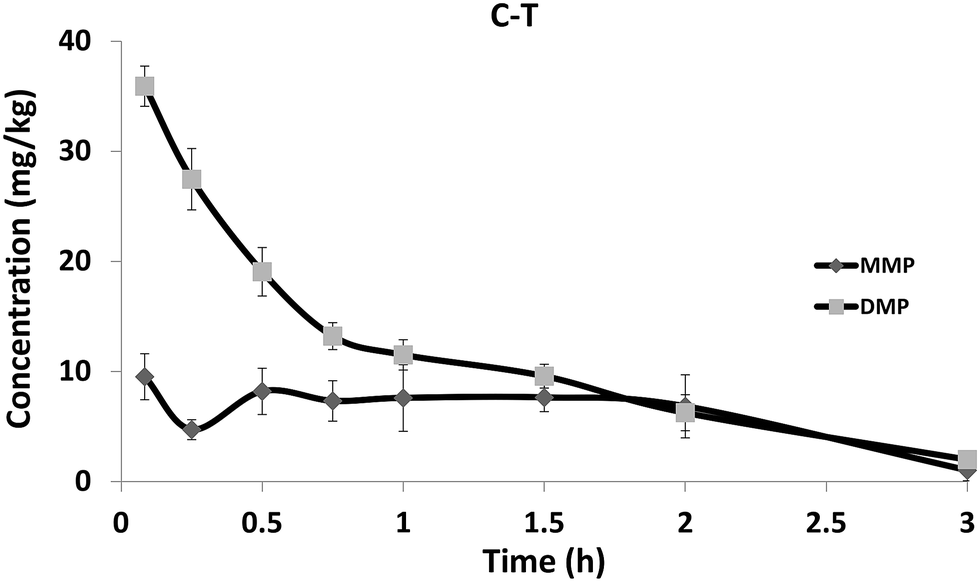 | ||
| Fig. 2 The concentration–time curves of DMP and MMP in blood after intravenous administration. Each point represents the mean ± SD (n = 3). Blood DMP and MMP were not detectable through the whole study. | ||
The concentration of DMP and MMP both decreased by more than 90% within 3 hours in plasma. The data of the residues of the analytes in the tissues such as liver, kidney, lung and brain at 3 h were shown in Table S4.† EDP and MMP were not detectable in all tissues, and DMP could only be detected in liver (1.08 mg kg−1) and kidney (0.72 mg kg−1). Both DMP and MMP were found in urine and feces. Based on a rough calculation (according to the amount of urine or feces, the molarity of EDP, DMP and MMP in urine or feces and the administered dose of EDP), the rabbit excreted approximately 1.44% (in the form of DMP) and 0.35% (in the form of MMP) of the administered dose of EDP (Table S5†).
The quick degradation of EDP and generation of DMP found in rabbit in vivo indicated that EDP could be hydrolyzed rapidly and DMP was the main metabolite of EDP. DMP was then metabolized to MMP by certain enzymes such as cytochrome P450 enzyme through demethylation and this process was relatively slow causing higher level of DMP. The concentration of DMP was higher in liver than in other tissues which might lead to toxic effect on the liver. The relative low amount of DMP and MMP in urine and feces might be attributed to the existence of phase II biotransformation pathways.17 The relative low residue level of DMP and MMP in tissues and plasma at 3 h indicated that single exposure to EDP would not be accumulated.
3.4. In vitro metabolism of EDP in plasma and liver microsome
The concentration–time curves for EDP and its metabolite DMP in 10% rabbit plasma were shown in Fig. 3A. In the plasma, EDP was rapidly metabolized and the degradation of (+)-EDP (t1/2 of 15.3 min) was faster than (−)-EDP (t1/2 of 25.4 min). Due to the quick degradation of EDP, the concentration of DMP was almost equally increased. The concentration–time curves for EDP and its metabolite in rabbit liver microsomal suspension were shown in Fig. 3B. Similarly, the concentration of EDP decreased fast accompanied by the generation of DMP with the t1/2 of 3.0 min and 5.8 min for (+) and (−)-form showing (+)-EDP was preferentially metabolized. To further study the reason for the occurrence of MMP, the metabolism of DMP in liver microsome was studied. It was proved MMP came from the metabolism of DMP in liver microsome (Fig. 3C).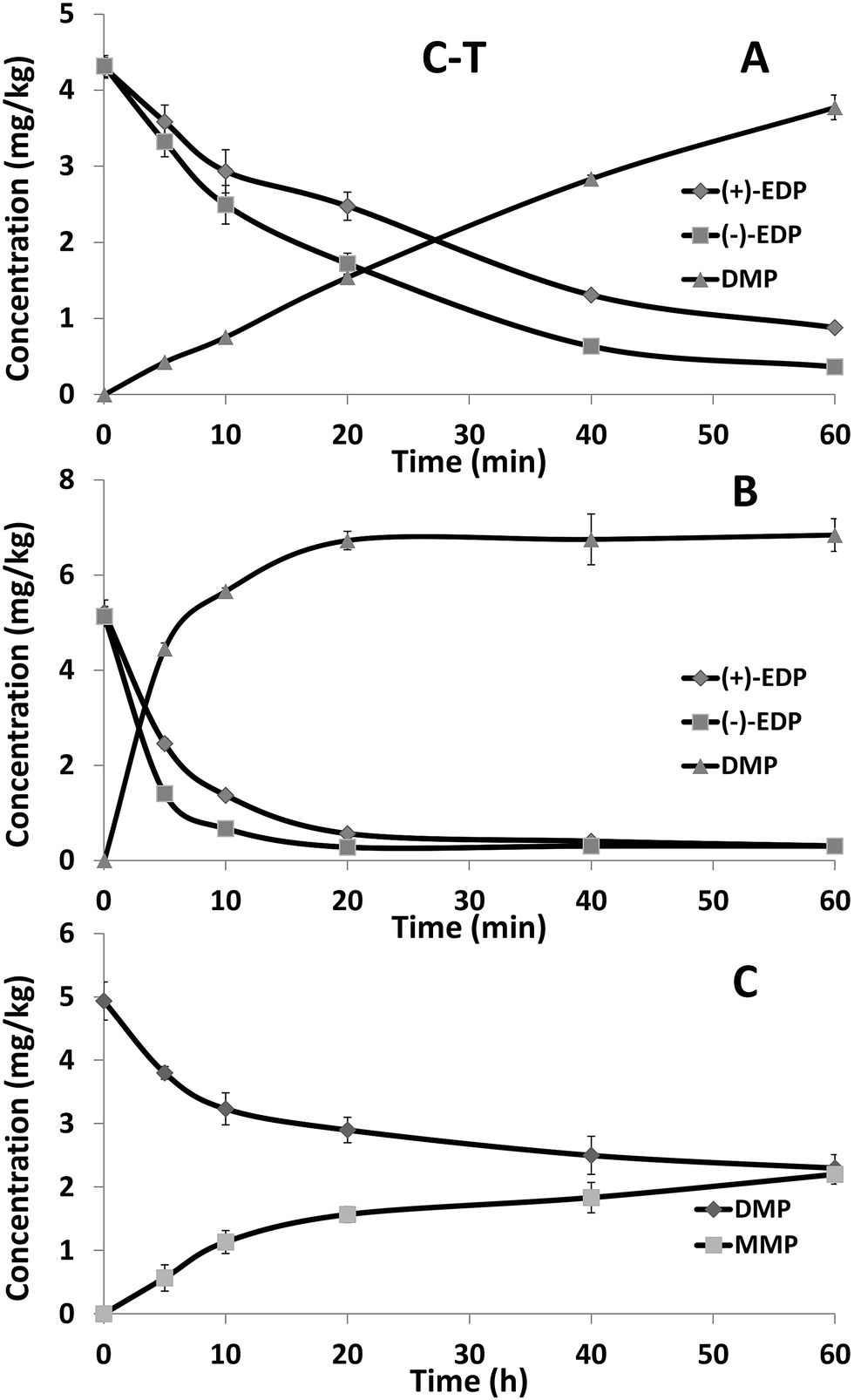 | ||
| Fig. 3 The concentration–time curves of EDP enantiomers in rabbit plasma (A) and liver microsome (B) and DMP in liver microsome (C). Each point represents the mean ± SD (n = 3). | ||
Esterases are widely distributed in animals which can hydrolyze a number of aliphatic and aromatic esters including drugs and toxicants.28,29 Among them, the esterases found in plasma and liver show an especially high level of stereoselectivity and activity.30 Thus, plasma and liver microsome are usually chosen to explore the enantioselective degradation of chiral ester drugs. In this study, EDP was degraded rapidly in plasma and liver microsome with the preference of (+)-EDP. The rapid degradation of EDP should be esterases involved hydrolysis process in plasma and liver. It was found that DMP could be further degraded to MMP only with NADPH in liver microsome. Thus, cytochrome P450 enzyme should be the reason for the demethylation of DMP and the generation of MMP. The results of both in vivo and vitro studies suggested that DMP and MMP were the main formation of EDP. Therefore, not only EDP but also DMP and MMP should be taken into account to make a full risk assessment of EDP contained cosmetics.
3.5. Cytotoxicity of DMP and MMP
MTT assay is a typical method for assessing cytotoxicity activity based on the conversion of MTT (yellow) to formazan (purple) in live cells. In this study, MTT assay was used to assess the cytotoxicity of EDP, DMP and MMP (Table 2). The survival rate of BRL-3A cells decreased along with the increasing concentration of EDP, DMP and MMP, which manifested that these compounds had obvious cytotoxicity. BRL-3A cells had a significantly lower viability when exposed to EDP, indicating that EDP was more toxic than DMP and MMP. Several previous studies have reported the toxicity effects of EDP,31–33 but the toxicology of the metabolites still has not been well known. In terms of the higher cytotoxicity of EDP than its two metabolites found in this work, the rapid metabolism of EDP to DMP and MMP was a detoxification process and led to less liver cell damage. However, as cosmetics are frequently applied to large areas of skin, the penetration of EDP may cause long-time and low-level exposure of DMP and MMP, the chronic toxicology of DMP and MMP still needs further consideration.| Concentration (mg kg−1) | EDP | Concentration (mg L−1) | DMP | Concentration (mg L−1) | MMP | |||
|---|---|---|---|---|---|---|---|---|
| Survival rate (%) | RSD (%) | Survival rate (%) | RSD (%) | Survival rate (%) | RSD (%) | |||
| 0.50 | 12.23 | 0.41 | 100.00 | 65.05 | 0.75 | 295.00 | 56.15 | 0.41 |
| 0.25 | 12.78 | 0.34 | 50.00 | 69.39 | 1.08 | 147.50 | 60.22 | 0.34 |
| 0.13 | 12.84 | 0.28 | 25.00 | 75.18 | 0.27 | 73.75 | 71.52 | 0.28 |
| 0.06 | 13.26 | 0.22 | 12.50 | 77.85 | 0.17 | 36.88 | 72.96 | 0.22 |
| 0.03 | 13.58 | 1.15 | 6.25 | 80.34 | 0.64 | 18.44 | 79.56 | 1.15 |
4. Conclusion
The two enantiomers of the UV-filter EDP were separated on HPLC with a Chiralpak IC chiral column and the chromatographic conditions were optimized. Based on the chiral analysis method developed, the stereoselective metabolism of EDP in rabbit in vivo and vitro was studied and the two metabolites DMP and MMP were also monitored. It is found that EDP could be rapidly metabolized both in vivo and vitro via the hydrolysis by esterases, and DMP was the main metabolite of EDP. The generation of MMP might result from the demethylation effect of cytochrome P450 enzyme. The low residue of EDP and its metabolites were found in the tissues of rabbit might be attributed to the existence of phase II biotransformation pathways. In order to investigate the potential toxic effects of EPD and its metabolites, the cytotoxicity using BRL-3A cell line as the model was determined and the results showed that the degradation of EDP was a detoxification process. The data of this research would offer some information for a more comprehensive assessment of the toxicology and health risks of EDP contained cosmetics. Investigation of the metabolism behaviors of the personal care products on enantiomeric level as well as the toxicity evaluation of the metabolites should be paid more attention.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Contract Grants 21277171, 21337005), supported by New-Star of Science and Technology and by Beijing Nova program YETP0323.References
- F. Lohezic-Le Devehat, B. Legouin, C. Couteau, J. Boustie and L. Coiffard, J. Photochem. Photobiol., B, 2013, 120, 17–28 CrossRef CAS PubMed.
- J. R. Villalobos-Hernandez and C. C. Mueller-Goymann, Int. J. Pharm., 2006, 322, 161–170 CrossRef CAS PubMed.
- S. A. Wissing and R. H. Muller, Pharmazie, 2001, 56, 783–786 CAS.
- T. Wong and D. Orton, Clin. Dermatol., 2011, 29, 306–310 CrossRef PubMed.
- E. J. H. Collaris and J. Frank, Int. J. Dermatol., 2008, 47, 35–37 CrossRef PubMed.
- E. M. Satulsky, AMA Arch. Dermatol. Syphilol., 1950, 62, 711–713 CrossRef CAS.
- A. Bryden, H. Moseley, S. Ibbotson, M. Chowdhury, M. Beck, J. Bourke, J. English, P. Farr, I. Foulds and D. Gawkrodger, Br. J. Dermatol., 2006, 155, 737–747 CrossRef CAS PubMed.
- F. C. Victor, D. E. Cohen and N. A. Soter, J. Am. Acad. Dermatol., 2010, 62, 605–610 CrossRef CAS PubMed.
- F. Oliver, S. Margret, R. Sasha, H. Manuel, M. Kirsten, D. Stefan and L. Walter, Neurotoxicology, 2009, 30, 249–260 CrossRef PubMed.
- E. Gomez, A. Pillon, H. Fenet, D. Rosain, M. J. Duchesne, J. C. Nicolas, P. Balaguer and C. Casellas, J. Toxicol. Environ. Health, Part A, 2005, 68, 239–251 CrossRef CAS PubMed.
- M. Heneweer, M. Muusse, M. V. D. Berg and J. T. Sanderson, Toxicol. Appl. Pharmacol., 2005, 208, 170–177 CrossRef CAS PubMed.
- L. Hofkamp and B. Timms, Environ. Health Perspect., 2008, 116, 867–872 CrossRef CAS PubMed.
- M. Inui, T. Adachi, S. Takenaka, H. Inui, M. Nakazawa, M. Ueda, H. Watanabe, C. Mori, T. Iguchi and K. Miyatake, Toxicology, 2003, 194, 43–50 CrossRef CAS PubMed.
- H. Klammer, C. Schlecht, W. Wuttke and H. Jarry, Toxicology, 2005, 215, 90–96 CrossRef CAS PubMed.
- H. Holbech, U. Norum, B. Korsgaard and P. Bjerregaard, Pharmacol. Toxicol., 2002, 91, 204–208 CAS.
- V. E. Reeve, M. Bosnic, C. Boehm-Wilcox and R. D. Ley, J. Invest. Dermatol., 1991, 97, 624–628 CrossRef CAS PubMed.
- Z. Leon-Gonzalez, C. Ferreiro-Vera, F. Priego-Capote and M. Dolores Luque de Castro, J. Chromatogr. A, 2011, 1218, 3013–3021 CrossRef CAS PubMed.
- Z. Leon, J. de Vlieger, A. Chisvert, A. Salvador, H. Lingeman, H. Irth and M. Giera, Chromatographia, 2010, 71, 55–63 CAS.
- L. A. Nguyen, H. He and C. Pham-Huy, J. Biomed. Sci. Eng., 2006, 2, 85 CAS.
- J. Ye, M. R. Zhao, L. L. Niu and W. P. Liu, Chem. Res. Toxicol., 2015, 28, 325–338 CrossRef CAS PubMed.
- I. Ali, H. Y. Aboul-Enein and A. Ghanem, Curr. Pharm. Anal., 2005, 1, 109–125 CrossRef CAS.
- I. Kania-Korwel and H. J. Lehmler, Environ. Sci. Pollut. Res., 2016, 23, 2058–2080 CrossRef CAS PubMed.
- H. Huhnerfuss and M. R. Shah, J. Chromatogr. A, 2009, 1216, 481–502 CrossRef PubMed.
- I. Ali, V. K. Gupta, H. Y. Aboul-Enein, P. Singh and B. Sharma, Chirality, 2007, 19, 453–463 CrossRef CAS PubMed.
- W. Zhu, Z. Dang, J. Qiu, Y. Liu, C. Lv, J. Diao and Z. Zhou, Xenobiotica, 2009, 39, 649–655 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- M. M. Yallapu, S. F. Othman, E. T. Curtis, B. K. Gupta, M. Jaggi and S. C. Chauhan, Biomaterials, 2011, 32, 1890–1905 CrossRef CAS PubMed.
- M. A. Sogorb and E. Vilanova, Toxicol. Lett., 2002, 128, 215–228 CrossRef CAS PubMed.
- C. Nebbia, Vet. J., 2001, 161, 238–252 CrossRef CAS PubMed.
- M. Hosokawa, T. Maki and T. Satoh, Mol. Pharmacol., 1987, 31, 579–584 CAS.
- P. Y. Kunz and K. Fent, Aquat. Toxicol., 2006, 79, 305–324 CrossRef CAS PubMed.
- R. Ma, B. W. Cotton and M. Schlumpf, Toxicol. Sci., 2003, 74, 43–50 CrossRef CAS PubMed.
- R. Schreurs, E. Sonneveld, J. H. J. Jansen, W. Seinen and B. van der Burg, Toxicol. Sci., 2005, 83, 264–272 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00431a |
| This journal is © The Royal Society of Chemistry 2017 |