
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A new cross-linked enzyme aggregate biocatalyst for NAD+-booster production†
Ana-Belén Martínez-Moñino ab,
Rubén Zapata-Pérezab,
Antonio-Ginés García-Sauraab,
Juana Cabanesab and
Álvaro Sánchez-Ferrer*ab
ab,
Rubén Zapata-Pérezab,
Antonio-Ginés García-Sauraab,
Juana Cabanesab and
Álvaro Sánchez-Ferrer*ab
aDepartment of Biochemistry and Molecular Biology-A, Faculty of Biology, Regional Campus of International Excellence “Campus Mare Nostrum”, University of Murcia, Campus Espinardo, E-30100 MURCIA, Spain. E-mail: alvaro@um.es; Fax: +34 868884147; Tel: +34 868884777
bMurcia Biomedical Research Institute (IMIB), 30120 Murcia, Spain
First published on 3rd March 2017
Abstract
Nicotinamide adenine dinucleotide (NAD+) is not only a redox cofactor/coenzyme but also a nutritional sensor molecule. Compounds that raise NAD+ levels (NAD+-boosters) are used as dietary supplements, since they have shown great potential as calorie restriction mimetics to treat numerous age-related conditions. In this paper, an enzymatic bioprocess using recombinant NMN deamidase immobilized in the form as cross-linked enzyme aggregates (CLEAs) was developed to obtain one of the above compounds, nicotinic acid mononucleotide (NaMN). The immobilization process was optimized using different precipitants and dispersing technologies. CLEAs with full deamidase activity were recovered in short period by co-aggregation with albumin combined with a reciprocating dispersing methodology. These CLEAs showed high activity, storage and operational stability for the production of NaMN, suggesting the first time that CLEA technology could be used to obtain highly valuable NAD+-boosters. This is also the first report on a Ser/Lys catalytic dyad enzyme immobilized as CLEAs.
1. Introduction
Nicotinamide adenine dinucleotide (NAD+) is a not only a key cellular redox factor for the intermediary metabolism, but also a key substrate for signaling enzymes, including protein deacetylases (sirtuins), poly-ADPribosyl-polymerases (PARPs), ADPribosyl-transferases (mARTs), and cADPR synthases (CD38 and CD157).1,2 The biomedical importance of NAD+ was established early with the discovery of pellagra and a dietary factor called niacin (vitamin B3).2 Nicotinic acid and nicotinamide are now recognized as different forms of vitamin B3. Recently, a new form of vitamin B3 has been proposed, nicotinamide riboside (NR).3 This nucleoside has been found in yeast-containing foods and in milk-derived products, such as whey fractions.4Over the last few years, NR has been demonstrated as a molecule capable of raising intracellular NAD+ levels (NAD+ booster) in animal and tissue models involving dietary supplementation.5 Surprisingly, NR provided broad improvements in the prevention of high-fat diet-induced glucose deregulation,6 DNA damage,7 and cardiac injury.8 It also improved cognitive function in a mouse model of Alzheimer disease, and led to stem cell-niche depletion and a modest (∼5%) but significant increase in longevity.9 These findings suggest that NR might serve as a potent agent for the treatment of neurodegenerative diseases and metabolic disorders associated with mitochondrial dysfunction.10 In addition, it has recently been claimed that other NAD+ precursors, such as nicotinic acid adenine dinucleotide (NaAD) or nicotinic acid riboside (NaR), both as yet untested, may prove to be equally or even more efficacious.5 By analogy, nicotinamide mononucleotide (NMN), which was seem to protect 2-year old mice against a high-fat diet and to restore youthful levels of mitochondrial function through SIRT1 activation,11 and its corresponding deamidated nucleotide, nicotinic acid adenine mononucleotide (NaMN), could serve as dietary supplement after proper assessment. In order to carry out such experiments with NaMN on a commercial scale, a cost-effective enzyme technology process for its production is necessary. Among enzymes able to produce NaMN, nicotinamide mononucleotide (NMN) deamidases (EC 3.5.1.42), also known as CinAs, has been a recent focus of interest.12,13 These bacterial Ser/Lys amidohydrolases catalyze the conversion of NMN into NaMN and ammonia, playing a dual role in decreasing the intracellular level of NMN to avoid inhibition of NAD+-dependent DNA ligases, and at the same time, securing a continuous NAD+ supply for the ligases by priming NaMN into the Preiss–Handler pathway to produce new NAD+.14
After selecting the enzyme, an effective method for enzyme immobilization is crucial for its repeated use in the production of NaMN. The use of cross-linked enzyme aggregates (CLEAs) rather than carrier-bound immobilization techniques seems to be advantageous as an immobilization method due to the high volumetric productivity associated, a substantial space time yield, better functional and storage stability (temperature and pH), excellent recyclability/recoverability, amenability for easy scaling-up, and the proven fact that the method does not require a highly pure enzyme.15 CLEAs are mainly prepared by precipitating enzyme solution with specific precipitating agents like water-miscible organic solvents, inorganic salts and non-ionic polymers followed by cross-linking with a bifunctional cross-linking agent, generally glutaraldehyde.16 This CLEA technology has been used to immobilize different enzymes, such as aldolases, catalases, esterases, galactosidases, laccases, lipases, nitrilases and oxynitrilases.17–22 However, only three hydrolases acting on carbon–nitrogen bonds other than peptide bonds have been immobilized as CLEAs,15 penicillin amidase (EC 3.5.1.11), Aspergillus melleus aminoacylase (EC 3.5.1.14) and Rhodococcus erythropolis amidase (EC 3.5.1.4), none of them a Ser/Lys amidohydrolase.
In order to assess the potential of CLEA technology in the biomedical and dietary use of NAD+-boosters, the present work describes an easy method for obtaining highly catalytically active, cheap and stable cross-linked enzyme aggregates of Escherichia coli nicotinamide mononucleotide deamidase (EcCinA). In addition, several parameters, including precipitant agent, additives such as bovine serum albumin (BSA) and sugars, cross-linking concentration and time, were tested in order to maximize activity recovery of this enzyme with a very low number of lysines, five, only one of which is involved in the catalysis. The results showed that co-aggregation with BSA and dispersion with a reciprocating cell disruptor (FastPrep) of the EcCinA–BSA–CLEAs provided a robust biocatalyst for the production of NaMN with high reusability and no by-products. This process is of potential biotechnological interest, given that the commercial value of NaMN is ten times that of NMN. In addition, and to the best of our knowledge, this is the first report on a Ser/Lys catalytic dyad enzyme immobilized as CLEAs.
2. Experimental
2.1. Reagents, microorganisms, and plasmids
Genomic DNA was isolated from Escherichia coli K12. The pET28a cloning vector was from Novagen (EMD Millipore, Madrid, Spain). The QIAquick PCR purification kit and QIAprep spin miniprep kit were from Qiagen (Valencia, CA, USA). KAPA HiFi polymerase was from KapaBiosystems (Boston, MA, USA). Nicotinamide mononucleotide and nicotinic acid mononucleotide were from Santa Cruz Biotechnology (Heidelberg, Germany). High purity glutaraldehyde, ammonium sulfate (BioUltra), and other reagents were from Sigma-Aldrich (Madrid, Spain).2.2. Enzyme expression and purification
The cloning and transformation techniques used were essentially those previously described.20 Genomic DNA from Escherichia coli strain K12 was used as the source of cinA gene (UniProt entry: P0A6G3). The coding sequence (498 bp) was amplified by PCR using KAPA HiFi DNA polymerase, and the corresponding forward (5′-GCGGGCTAGCATGACTGACAGTGAACTGATG-3′) and reverse (5′-GCGCCTCGAGTCAAGTGTTTTGTAGAAATTG-3′) primers, which include NheI and XhoI restriction site extensions (underlined). The resulting PCR product was purified and digested with the above restriction enzymes, ligated to the digested pET28a, and transformed into Escherichia coli Rosetta™ 2(DE3) competent cells. A selected clone harboring the correct sequence was denoted as pET28-EcCinA.The above cells harboring the recombinant plasmid pET28-EcCinA were grown in Luria Broth (LB) with 50 μg mL−1 kanamycin and 34 μg mL−1 chloramphenicol at 37 °C with constant shaking until an OD600 of 0.8 was reached. Then, the culture was transferred to 1 L of Terrific Broth (TB) supplemented with the above-described antibiotics. These cultures were grown at 37 °C, allowed to reach an OD600 of 4.0, and induced by addition of isopropyl-β-D-thiogalactoside (IPTG) to a final concentration of 0.8 mM at 25 °C. The resulting culture was harvested by centrifugation, resuspended in 50 mM Tris–HCl pH 8.5 with 150 mM NaCl, disrupted by sonication (450D Sonifier, BRANSON), and centrifuged at 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 40 min at 4 °C. The enzyme was purified by Ni2+-chelating affinity chromatography (HiPrep IMAC 16/10 FF column) (GE Healthcare, Inc.) equilibrated with the above lysis buffer. The protein was eluted with imidazole in lysis buffer and fractions containing the NMN deamidase activity were pooled, desalted, concentrated and stored at −80 °C with 10% glycerol. The recombinant enzyme thus obtained was determined to be electrophoretically pure by SDS-PAGE. Gel filtration (Superdex 200, GE Life Sciences) was used to confirm the homogeneity and the molecular mass of the purified enzyme. The protein concentration was determined using Bradford's reagent (Bio-Rad) and BSA as standard.
000g for 40 min at 4 °C. The enzyme was purified by Ni2+-chelating affinity chromatography (HiPrep IMAC 16/10 FF column) (GE Healthcare, Inc.) equilibrated with the above lysis buffer. The protein was eluted with imidazole in lysis buffer and fractions containing the NMN deamidase activity were pooled, desalted, concentrated and stored at −80 °C with 10% glycerol. The recombinant enzyme thus obtained was determined to be electrophoretically pure by SDS-PAGE. Gel filtration (Superdex 200, GE Life Sciences) was used to confirm the homogeneity and the molecular mass of the purified enzyme. The protein concentration was determined using Bradford's reagent (Bio-Rad) and BSA as standard.
2.3. CLEAs production
Cross-linked aggregates of EcCinA were prepared at 4 °C by dissolving 1 mL of enzyme solution (15 mg mL−1) in 50 mM Tris–HCl pH 8.5 with the required quantity of ammonium sulfate to bring the mixture to the desired degree of saturation. Precipitation with acetone, DMSO, acetonitrile or 2-propanol (final concentration up to 90% v/v) were carried out in the same way except for the addition of the cold solvent instead of salt. Co-aggregation was carried out by addition of 1 mg mL−1 of BSA or 1 mg mL−1 of different sugars to the EcCinA solution, before adding ammonium sulfate. After 1 h at 4 °C with no stirring, the appropriate amount of glutaraldehyde stock solution (25% v/v in water) was slowly added to the final concentration of 1% (v/v). The mixture was kept at 4 °C for cross-linking for 2 h without stirring. The resultant suspensions were centrifuged at 12000g for 10 min at 4 °C. The precipitates and supernatants were collected separately and checked for NMN deamidase activity. The insoluble EcCinA–CLEAs were washed three times with 20 mM Tris–HCl pH 8.5. To study the best disruption procedure for the aggregates, a vortex at medium speed or a FastPrep Cell Disruptor (www.mpbio.com) at 6 m s−1 were used. The final preparation of CLEAs was stored at 4 °C, at which temperature they were stable for at least one month. The experiments were made in triplicate and the error bar represents the percentage error in each set of readings.
2.4. Biochemical characterization of the free and immobilized EcCinA
For the standard assay of the EcCinA activity, the free enzyme (14 nM) and the CLEAs (2.5 μg mL−1) were incubated at 37 °C with the substrate (0.5 mM NMN) in 50 mM Tris–HCl pH 8.5. The reaction was stopped by addition of TFA to a final pH of 3.0 in the case of the free enzyme or by centrifuging out the CLEAs (12000g for 10 min). A sample from the supernatant was injected into an HPLC with a C18 column (Phenomenex Gemini C18, 4.6 × 250 mm) and mobile phase (20 mM ammonium acetate pH 6.9) running at 0.8 mL min−1. Under these conditions, the retention time (RT) for NMN and NaMN were 4.3 and 3.6 min, respectively. One unit of activity was defined as the amount of enzyme required to cleave 1 μmol of NMN releasing 1 μmol of NaMN per minute. Kinetic parameters were obtained after three repeated experiments. The recovered activities in the CLEAs were calculated according to the relation given in eqn (1):
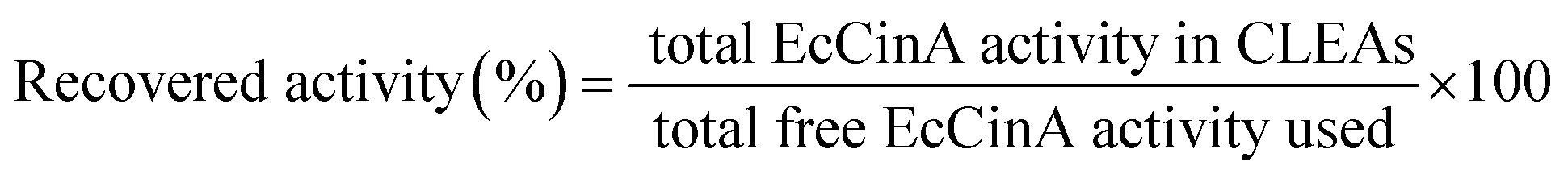 | (1) |
The pH profile and pH-stability were determined by measuring the residual activity of the enzyme after incubation at 37 °C at different pHs (pH 6.0–10.0). The temperature profile and heat-stability assay were carried out by incubating the enzyme in 50 mM Tris–HCl pH 8.5 at different temperatures (20–90 °C). Aliquots were taken at different times, cooled on ice, and the activity was measured as above by HPLC in the standard reaction medium.
The reusability of EcCinA–CLEAs was assessed in fifteen consecutive cycles under the standard reaction conditions described above. After each cycle, CLEAs were recovered by centrifugation at 12000g for 10 min, and washed twice with 50 mM Tris–HCl pH 8.5. Then, they were subjected to a new reaction. The residual enzymatic activity was compared with that of initial CLEAs, which was defined as 100%.
2.5. In silico analysis
Lysines of EcCinA and BSA were calculated using the ProtParam tool from Expasy Proteomic server (http://www.expasy.org/tools/protparam.html).3. Results and discussion
3.1. Optimization of CLEAs production
The cinA gene from E. coli was cloned into pET28a vector and transformed into E. coli Rosetta™ 2(DE3). The soluble recombinant protein (EcCinA) obtained after induction with IPTG was purified in one simple step by affinity chromatography, as described in Experimental section. A highly homogeneous single band migrating according to a mass of 20 kDa was obtained by SDS-PAGE (Fig. S1,† lane 1). This estimated mass agrees with the mass of the protein deduced from the gene sequence (17.5 kDa). This datum together with that obtained by gel filtration (about 40 kDa) (data not shown), confirmed the dimeric nature of EcCinA. This recombinant EcCinA was active towards NMN, rendering NaMN as the sole product in HPLC. In fact, when NMN (retention time 4.3 min) was incubated in the presence of purified EcCinA, a new peak appeared at 3.6 min until complete conversion (Fig. S2,† red line). This latter peak agreed with the retention time of commercial NaMN (data not shown).The first step in CLEAs production involves the physical aggregation of the enzyme by a precipitant. However, precipitation agents and precipitation conditions to obtain an active conformation of the enzyme cannot be generalized for all enzymes, since one precipitating agent can produce different results, depending on the enzyme used.23–26 In this study, several precipitants were tested for EcCinA in pilot conditions (Fig. 1A), either to change the hydration state of the enzyme molecules (90% saturation ammonium sulfate) or to alter the electrostatic constant of the solution with water-miscible organic solvents (acetone, 2-propanol, DMSO, methanol, and acetonitrile at 90% v/v) (Fig. 1A). After 1 hour at 4 °C, the appropriate amount of glutaraldehyde was added to reach a final concentration of 1%, and the activity remaining in the supernatant after 2 h crosslinking was measured. Of the precipitants, ammonium sulfate was the best for preserving activity (55%), whereas water-miscible organic solvents were ineffectual precipitants, less than 21% activity being recovered in CLEA form (Fig. 1A). This last result indicates a possible denaturing effect of these organic solvents on EcCinA, probably caused by removal of the water molecules bound to the protein during the precipitation steps, diminishing its flexibility.26 These results were similar to those recently reported for α-amylase, where, of the seven types of precipitant used, only acetone and saturated ammonium sulfate precipitated-CLEAs showed high activity, whereas those aggregated with the other water-miscible solvents (2-propanol, acetonitrile, ethanol, methanol, and n-butanol) led to low CLEAs activity.25
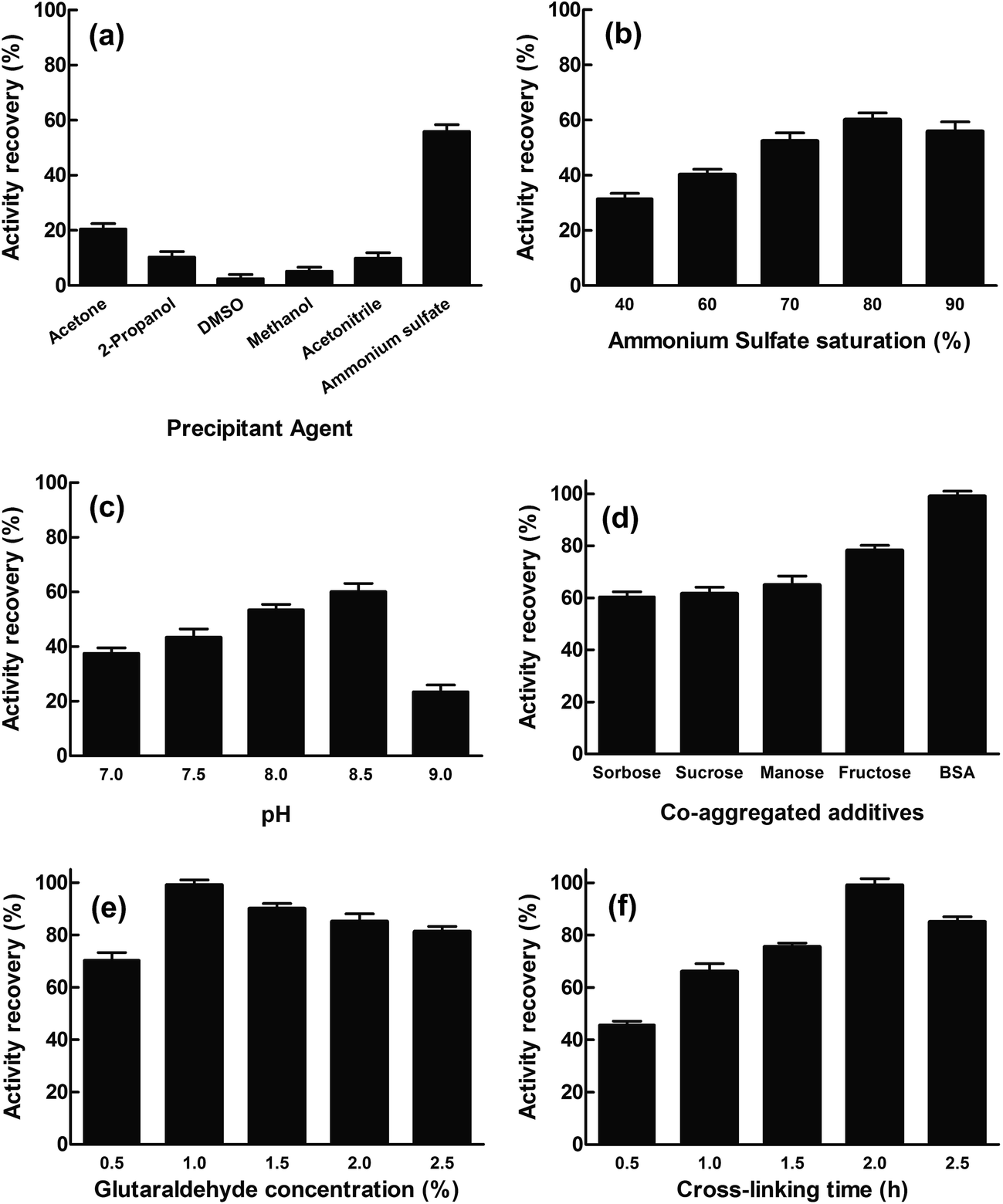 | ||
| Fig. 1 Optimization of EcCinA–CLEA production. (a) Effect of precipitant type. The final concentration of organic solvents was 90% v/v except for ammonium sulfate, which was 90% saturation. (b) Effect of ammonium sulfate saturation. (c) Effect of pH. The buffers (50 mM) used were sodium phosphate pH 7.0–7.5, Tris–HCl pH 8.0–8.5, and glycine–NaOH pH 9.0. (d) Effect of additives. Additives were used at 1 mg mL−1 final concentration. (e) Effect of glutaraldehyde. (f) Effect of cross-linking time. The experiments were made in triplicate and the error bar represents the percentage error in each set of readings. | ||
The ammonium sulfate concentration was also studied since this affects the aggregation process.27 The amount of enzyme trapped in the EcCinA–CLEAs increased as the saturation of ammonium sulfate was raised to 80% salt, but decreased at higher saturation. The activity recovery of CLEAs particles ranged from 31% to 60% at 40% and 80% ammonium sulfate saturation, respectively (Fig. 1B). Thus, 80% ammonium sulfate saturation was selected for further optimization. In addition, the activity recovery was pH-dependent, increasing as the pH increased from pH 7.0 to pH 8.5 (Fig. 1C). However, at pH 9.0, the recovery activity decreased drastically. Surprisingly, this effect was not dependent on the buffer used, since the activity recovery was the same in 50 mM HEPES pH 7.5 and 50 mM sodium phosphate (pH 7.0–7.5), about 45% (data not shown).
In an attempt to increase activity recovery, several additives were used. Among them, sugars are well known as stabilizers of proteins in natural dehydrated environments and during the lyophilization process since they preserve the essential water layer at the protein surface and thus protect the protein structure.28 In this work, three monosaccharides (glucose, fructose, and sorbose) and one disaccharide (sucrose) were tested to prepare sugar-assisted EcCinA–CLEAs with 80% ammonium sulfate in 50 mM Tris–HCl pH 8.5. Compared with conventional CLEAs (Fig. 1B), only fructose-assisted aggregates were able to increase the activity by about 20% (Fig. 1D). This unexpected result is of interest, since only sugar-assisted CLEAs have previously been described as minimizing enzyme activity loss during the precipitation step using water-miscible organic solvents for penicillin G acylase, phenyalanine ammonia lyase, and bovine pancreatic lyase,21,27 but they were never tested during ammonium sulfate precipitation. This positive effect found for fructose could be explained by the “water substitute” mechanism invoked for sugar protection during the protein freeze drying process.29 By replacing water molecules with other hydrogen bond forming compounds, such as sugars, there is a direct interaction between sugars and the surface of the proteins, and the native structure of proteins can be stabilized during the precipitation process.30 Thus, compared with sugar-free precipitation, more native structures of EcCinA were preserved in the presence of the fructose in the form of active enzyme, resulting in a higher activity yield. However, further research is needed using other enzymes in order to clearly understand the difference between fructose and the other tested sugars.
Bovine serum albumin (BSA) was also tested as a proteic feeder, since it is known to facilitate CLEA preparation when the enzyme activity is vulnerable to high concentrations of glutaraldehyde,31,32 or when a severe drop in activity is results from the modification of catalytic amino acid (lysine) by glutaraldehyde,17,20 or when the enzyme has a low content of lysine residues.33 These two latter conditions concur in EcCinA, which has only five lysines in its sequence and a Ser/Lys catalytic dyad.13 In fact, activity recovery of EcCinA–CLEAs increased up to 99% with the addition of 1 mg mL−1 BSA (Fig. 1D). This increase in activity recovery was similar to that described for other enzymes, such as aminoacylases, lipases, lyases, nitrilases, penicillin acylase and aldolases, when BSA, which contains 66 lysines in its sequence, was chosen to increase the content of lysine residues, and the consequent cross-linking efficiency.18,20,33 This benefit of BSA contrasts with the decrease in activity recovery described for the white rot fungus Coriolopsis polyzona laccase in the presence of BSA, which was explained by mass transfer limitations.34
Cross-linker concentration and cross-linking time were also studied for EcCinA–BSA–CLEAs (Fig. 1E and F). The activity recovery increased as glutaraldehyde concentration increased, with a maximum at 1% (v/v) glutaraldehyde (Fig. 1E). However, higher concentrations seemed to be harmful for activity recovery (Fig. 1E). Previous reports showed that excessive glutaraldehyde could produce an excess of enzyme cross-linking, resulting in a loss of flexibility.16,24,33 In addition, cross-linking time is an important factor in activity recovery (Fig. 1E). For example, activity recovery in EcCinA–BSA–CLEAs increased as cross-linking time increased up to 2 h, above which it decreased (Fig. 1E), demonstrating that excessive cross-linking time could reduce active site availability. This decrease in the activity recovery caused by excessive cross-linking periods has also been described for galactosidases, trehalose synthase, and lipases.16,24,35,36
The final step in CLEAs optimization was the dispersion time and device used to recover maximal activity, since after cross-linking reactions, washing periods, centrifugations and cleaning steps, the size of CLEAs could result in internal mass-transfer limitations that limit the diffusion of the substrate.36 Our group previously demonstrated that the activity recovery of CLEAs prepared with reciprocating cell disruptors (FastPrep) based on a precession movement was higher than that of CLEAs prepared with vortexing.19,20,37 This was also the case with EcCinA–BSA–CLEAs. One hour was needed to recover 90% activity with a vortex, whereas only 50 s where required using a FastPrep system to recover 99% activity (Table S1†). The number of repeated pulses (10 s) in FastPrep at 6 m s−1 needed to recover maximal activity was five (Table S1†). The number of pulses needed is related with the optimal particle size. Thus if the CLEAs size is too big, the inner enzymes of the CLEAs are unable to form complexes with the substrates, whereas if the particle size of the CLEAs is too small, it is difficult to isolate and recover CLEAs from the reaction medium.
3.2. Biochemical characterization of EcCinA–BSA–CLEAs
The influence of pH on the activity of free and immobilized EcCinA was assayed from pH 6.0 to pH 10.0. The same optimum pH (pH 8.5) was found for both free enzyme and EcCinA–BSA–CLEAs (Fig. 2A). Cross-linked EcCinA exhibited an improved enzyme activity over a broader pH range, especially at pH 6.0–8.0 (Fig. 2A, circles), with a slight decrease in activity at alkaline pH values compared to the free enzyme (Fig. 2A, squares). This displacement of optimum pH towards neutral pHs in CLEAs is of relevance for the long term stability of the product (NaMN). This broad range of optimum pH when EcCinA was immobilized as CLEAs was similar to that previously reported for trehalose synthase, bovine pancreatic lipase and β-galactosidase.24,35,38 As regards pH-stability, both free and EcCinA–BSA–CLEAs appeared to be very stable at neutral-basic pH values (from pH 7.5 to 8.5), when more than 80% of their activity was maintained for 72 hours (Fig. 2B), with EcCinA–BSA–CLEAs in 50 mM Tris–HCl pH 8.5 having slightly higher stability (Fig. 2B, open circles).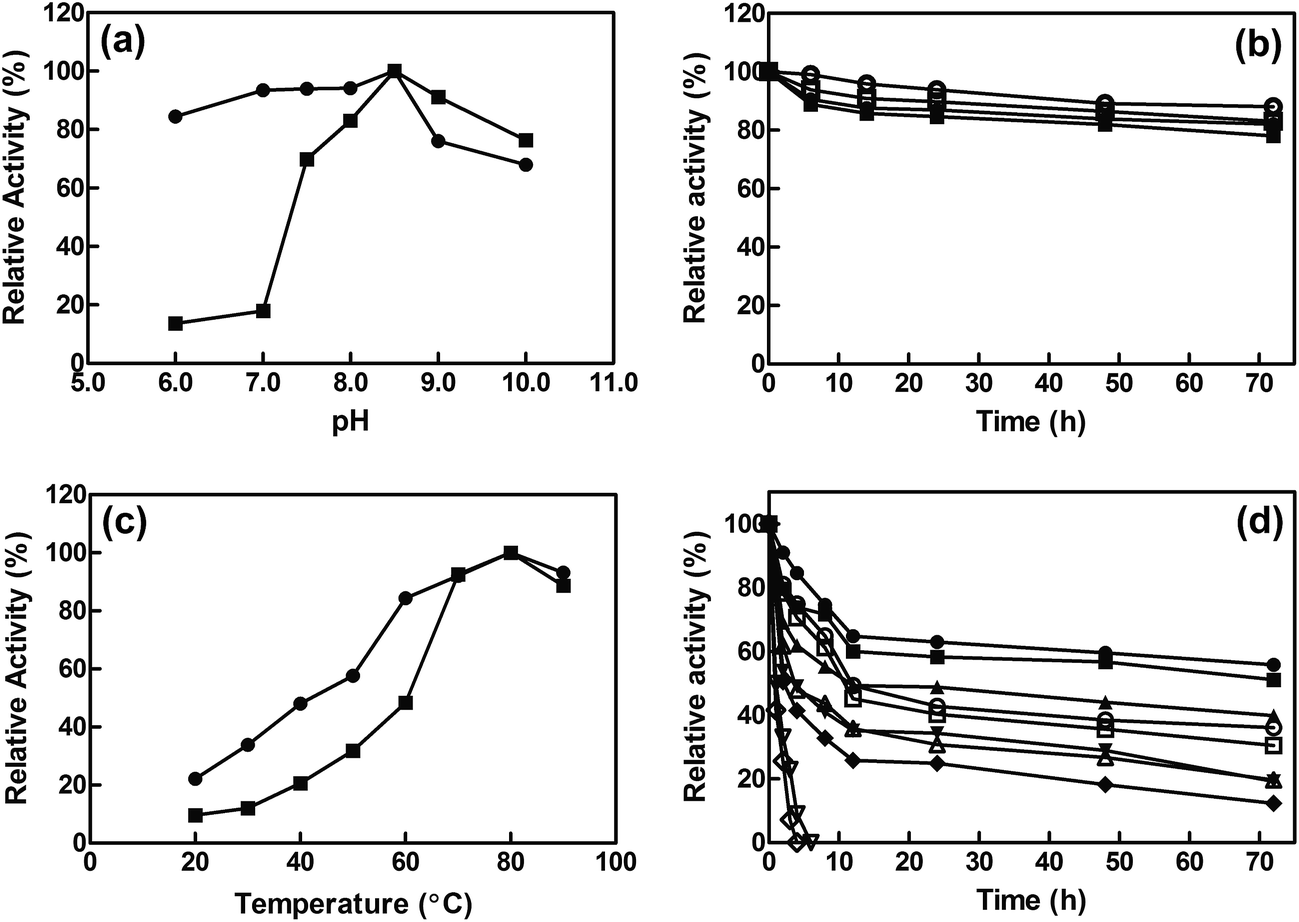 | ||
| Fig. 2 Effect of pH and temperature on the free and immobilized EcCinA activities. (a) Effect of pH. Activity was assayed at 37 °C in the standard reaction medium. Free (■) and immobilized (●) EcCinA. (b) Effect of pH on enzyme stability at 37 °C. Enzyme was incubated at pH 7.5 (free enzyme ■, CLEAs ●), and at pH 8.5 (free enzyme □, CLEAs ○) at different time intervals. (c) Effect of temperature. The reactions were carried out at pH 8.5 for free (■) and immobilized (●) EcCinA, respectively. (d) Effect of temperature on enzyme stability. CLEAs are represented as filled symbols at 4 °C (●), 20 °C (■), 37 °C (▲), 50 °C (▼), and 60 °C (♦). Free enzyme is represented by the corresponding open symbols. Data points are the average of triplicate measurements, and error bars have been omitted for clarity. | ||
The effect of temperature on the relative activities of free and immobilized enzyme was monitored by measuring enzymatic activities as a function of temperature in the range of 20 °C to 90 °C (Fig. 2C). Both free and CLEAs enzymes, showed a high optimum temperature (80 °C) and a similar temperature profile. These results were related with enzyme activity, since no autohydrolysis of NMN occurred in HPLC when it was incubated at 80 °C for 3 h (data not shown). However, at temperatures below 60 °C, the relative activity of EcCinA–BSA–CLEAs was about double than that found for free EcCinA (Fig. 2C), and almost 3-fold higher at 30 °C. A similar pH profile between the free and CLEAs was also described for β-galactosidases and lipases.24
The thermostability of both free and immobilized EcCinA was further investigated by incubating the enzymes without substrate at different temperatures (4–60 °C). Fig. 2D shows that the EcCinA–BSA–CLEAs were more thermostable than the free EcCinA at all the temperatures tested, but especially at high temperatures. Thus, at 60 °C, free enzyme completely lost its activity after 4 h, whereas CLEAs maintained 41% at the same time (Fig. 2D). At 4 °C, the stability was also higher in CLEAs compared with free enzyme (55% vs. 36% at 72 h, respectively) (Fig. 2D). This 55% activity in EcCinA–BSA–CLEAs was maintained for at least one month at 4 °C (data not shown). These results indicate that efficient cross-linking provided a more stable conformation of EcCinA, resulting in better biocatalyst stabilization.24,27,32
The activity of both free and immobilized EcCinA showed a Michaelis–Menten kinetic when substrate concentration was increased (Fig. S3†). The Km obtained for NMN was 23.2 ± 1.3 μM with a kcat of 5.31 ± 0.2 s−1 and a kcat/Km of 222.2 mM−1 s−1 at pH 8.5 for free EcCinA (Table 1). These kinetic data for the free enzyme were similar to those previously described for this enzyme.13 However, when EcCinA was immobilized in the form of CLEAs, this Km value increased about 15-fold (Table 1), with a concomitant decrease in catalytic efficiency, since kcat remained the same. This increase in Km is common feature in CLEA technology, and has previously been observed in several enzymes.19,27,31,33 This high Km in CLEAs (Table 1) suggests that molecular hindrances could be involved in the binding of NMN to the enzyme active site after cross-linking with glutaraldehyde. Surprisingly, the kcat found in EcCinA–BSA–CLEAs was unchanged, clearly indicating that the attack on the catalytic lysine of EcCinA was prevented by the use of BSA as protein feeder. The overall decrease in catalytic efficiency usually described in CLEAs of different enzymes does not affect their commercial use,19,31,33 and contrasts with the kcat/Km increase described for some laccases.39
| Km (μM) | kcat (s−1) | kcat/Km (mM−1 s−1) | |
|---|---|---|---|
| Free EcCinA | 23.2 ± 1.3 | 5.3 ± 0.2 | 222.2 |
| EcCinA–BSA–CLEAs | 355.3 ± 6.1 | 5.4 ± 0.3 | 15.1 |
3.3. Production of NaMN
To investigate the potential of EcCinA–BSA–CLEAs for the production of NaMN in aqueous medium, the deamidation reaction of NMN into NaMN was studied at 37 °C and at high NMN concentration (10 mM). EcCinA–BSA–CLEAs completely transformed this NMN into the corresponding NaMN in 1 h (Fig. 3). This short period of time contrasts with the 48 h needed using free CinA from Propionibacterium shermanii40 at 37 °C. Also the large amount of protein necessary for this last reaction (18 mg mL−1)40 compares unfavourably with the small amount of EcCinA–BSA–CLEAs (2.5 μg mL−1) used in our reaction, underlining the advantages of using CLEAs. Having demonstrated NaMN production with EcCinA–BSA–CLEAs, their stability during multiple reuses was also investigated under the same conditions, since reusability of the immobilized enzymes is a key factor in commercial applications, because simplification of downstream processing results in a reduction of enzyme costs.15 Typically, reactions (1 mL) were completed within 1 h, and at least 15 cycles were possible without any significant loss of activity (Fig. 3, inset). This strongly supports the value of using CLEAs in combination with the FastPrep system for dispersion, to yield EcCinA–BSA–CLEAs with excellent resistance to mechanical stress, allowing high recoveries of catalyst after many cycles of use. Ten cycles without loss of activity in CLEAs has also been considered as technologically relevant for lipase, β-galactosidase, and N-acetyl-D-neuraminic acid aldolase,20,24 and contrasts with the low number of reuses described for α-amylase, T. versicolor and S. putrefaciens laccases and Roystonea regia peroxidase.25,39,41 Together with its high storage stability, these results demonstrate that EcCinA–BSA–CLEAs are a robust biocatalyst for NaMN production.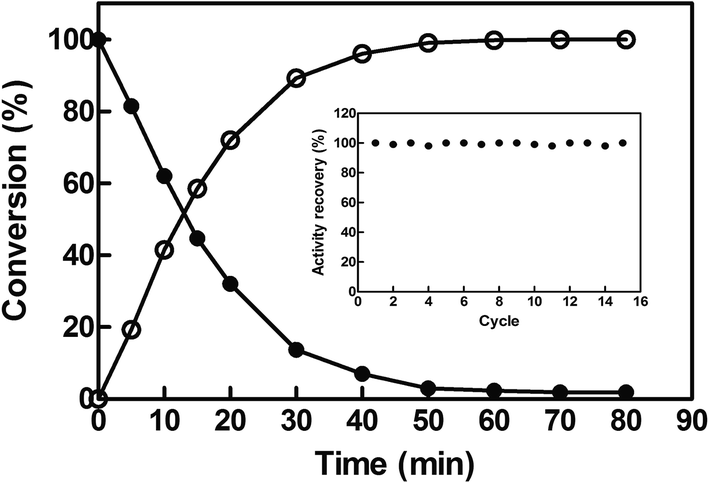 | ||
| Fig. 3 Time-course of NMN conversion into NaMN catalyzed by EcCinA–BSA–CLEAs. The reaction medium at 37 °C contained 10 mM of NMN and 2.5 μg mL−1 EcCinA–BSA–CLEAs in 50 mM Tris–HCl pH 8.5. (●) NMN and (○) NaMN. (Inset) reusability of EcCinA–BSA–CLEAs. The activity was measured under the standard reaction conditions at 37 °C. After 1 h, CLEAs were centrifuged, and the pellet was resuspended in a new reaction solution with 10 mM NMN and the activity measured. | ||
4. Conclusions
In a straightforward procedure, full NMN deamidase activity CLEAs was recovered by using BSA as a proteic feeder combined with an efficient dispersing methodology (FastPrep) in a short period of time (50 s). These EcCinA–BSA–CLEAs showed high activity, storage and operational stability for the production of NaMN, confirming for the first time that CLEA technology could be used for obtaining NAD+-boosters, such as NaMN. These finding open up new opportunities for producing this and other NAD+-boosters enzymatically for using as dietary supplements in metabolic and neurodegenerative diseases.Acknowledgements
This study was partially supported by Spanish grants from MINECO-FEDER (BIO2013-45336-R) and from the Ayudas a los Grupos y Unidades de Excelencia Científica de la Región de Murcia, Fundación Séneca-Agencia de Ciencia y Tecnología de la Región de Murcia (19893/GERM/15, Programa de Apoyo a la Investigación 2014). A.-B. M.-M. has a pre-doctoral research contract associated with the above MINECO-FEDER (BIO2013-45336-R) grant. R. Z.-P. and A. G.-G.-S. are supported by the corresponding predoctoral contracts (FPU-UMU) from the University of Murcia.References
- C. Canto, K. J. Menzies and J. Auwerx, Cell Metab., 2015, 22, 31–53 CrossRef CAS PubMed.
- Y. Yang and A. A. Sauve, Biochim. Biophys. Acta, 2016, 1864, 1787–1800 CrossRef CAS PubMed.
- D. B. Conze, J. Crespo-Barreto and C. L. Kruger, Hum. Exp. Toxicol., 2016, 35, 1–12 Search PubMed.
- Y. Chi and A. A. Sauve, Curr. Opin. Clin. Nutr. Metab. Care, 2013, 16, 657–661 CrossRef CAS PubMed.
- M. S. Bonkowski and D. A. Sinclair, Nat. Rev. Mol. Cell Biol., 2016, 17, 679–690 CrossRef CAS PubMed.
- C. Canto, R. H. Houtkooper, E. Pirinen, D. Y. Youn, M. H. Oosterveer, Y. Cen, P. J. Fernandez-Marcos, H. Yamamoto, P. A. Andreux, P. Cettour-Rose, K. Gademann, C. Rinsch, K. Schoonjans, A. A. Sauve and J. Auwerx, Cell Metab., 2012, 15, 838–847 CrossRef CAS PubMed.
- K. S. Tummala, A. L. Gomes, M. Yilmaz, O. Grana, L. Bakiri, I. Ruppen, P. Ximenez-Embun, V. Sheshappanavar, M. Rodriguez-Justo, D. G. Pisano, E. F. Wagner and N. Djouder, Cancer Cell, 2014, 26, 826–839 CrossRef CAS PubMed.
- W. Xu, T. Barrientos, L. Mao, H. A. Rockman, A. A. Sauve and N. C. Andrews, Cell Rep., 2015, 13, 533–545 CrossRef CAS PubMed.
- H. Zhang, D. Ryu, Y. Wu, K. Gariani, X. Wang, P. Luan, D. D'Amico, E. R. Ropelle, M. P. Lutolf, R. Aebersold, K. Schoonjans, K. J. Menzies and J. Auwerx, Science, 2016, 352, 1436–1443 CrossRef CAS PubMed.
- S. Srivastava, Clin. Transl. Med., 2016, 5, 25 CrossRef PubMed.
- J. Yoshino, K. F. Mills, M. J. Yoon and S. Imai, Cell Metab., 2011, 14, 528–536 CrossRef CAS PubMed.
- G. Sanchez-Carron, A. B. Martinez-Monino, A. Sola-Carvajal, H. Takami, F. Garcia-Carmona and A. Sanchez-Ferrer, PLoS One, 2013, 8, e82705 Search PubMed.
- L. Sorci, L. Brunetti, L. Cialabrini, F. Mazzola, M. D. Kazanov, S. D'Auria, S. Ruggieri and N. Raffaelli, FEBS Lett., 2014, 588, 1016–1023 CrossRef CAS PubMed.
- L. Galeazzi, P. Bocci, A. Amici, L. Brunetti, S. Ruggieri, M. Romine, S. Reed, A. L. Osterman, D. A. Rodionov, L. Sorci and N. Raffaelli, J. Biol. Chem., 2011, 286, 40365–40375 CrossRef CAS PubMed.
- R. A. Sheldon, Appl. Microbiol. Biotechnol., 2011, 92, 467–477 CrossRef CAS PubMed.
- O. Barbosa, C. Ortiz, A. Berenguer-Murcia, R. Torres, R. C. Rodrigues and R. Fernandez-Lafuente, RSC Adv., 2014, 4, 1583–1600 RSC.
- C. Mateo, J. M. Palomo, L. M. van Langen, F. van Rantwijk and R. A. Sheldon, Biotechnol. Bioeng., 2004, 86, 273–276 CrossRef CAS PubMed.
- R. A. Sheldon, Biochem. Soc. Trans., 2007, 35, 1583–1587 CrossRef CAS PubMed.
- S. Montoro-Garcia, F. Gil-Ortiz, J. Navarro-Fernandez, V. Rubio, F. Garcia-Carmona and A. Sanchez-Ferrer, Bioresour. Technol., 2010, 101, 331–336 CrossRef CAS PubMed.
- M. I. Garcia-Garcia, A. Sola-Carvajal, G. Sanchez-Carron, F. Garcia-Carmona and A. Sanchez-Ferrer, Bioresour. Technol., 2011, 102, 6186–6191 CrossRef CAS PubMed.
- L. Cao, L. M. van Langen, F. van Rantwijk and R. A. Sheldon, J. Mol. Catal. B: Enzym., 2001, 11, 665–670 CrossRef CAS.
- L. Wilson, G. Fernández-Lorente, R. Fernández-Lafuente, A. Illanes, J. M. Guisán and J. M. Palomo, Enzyme Microb. Technol., 2006, 39, 750–755 CrossRef CAS.
- S. Talekar, A. Pandharbale, M. Ladole, S. Nadar, M. Mulla, K. Japhalekar, K. Pattankude and D. Arage, Bioresour. Technol., 2013, 147, 269–275 CrossRef CAS PubMed.
- L. Li, G. Li, L. C. Cao, G. H. Ren, W. Kong, S. D. Wang, G. S. Guo and Y. H. Liu, J. Agric. Food Chem., 2015, 63, 894–901 CrossRef CAS PubMed.
- S. S. Nadar, A. B. Muley, M. R. Ladole and P. U. Joshi, Int. J. Biol. Macromol., 2016, 84, 69–78 CrossRef CAS PubMed.
- S. Velasco-Lozano, F. López-Gallego, C. Mateos-Díaz Juan and E. Favela-Torres, Biocatalysis, 2016, 1, 166 Search PubMed.
- J. D. Cui, R. L. Liu and L. B. Li, Korean J. Chem. Eng., 2016, 33, 610–615 CrossRef CAS.
- W. Q. Sun and P. Davidson, Biochim. Biophys. Acta, 1998, 1425, 235–244 CrossRef CAS.
- L. Chang, D. Shepherd, J. Sun, D. Ouellette, K. L. Grant, X. C. Tang and M. J. Pikal, J. Pharm. Sci., 2005, 94, 1427–1444 CrossRef CAS PubMed.
- B. Wang, S. Tchessalov, M. T. Cicerone, N. W. Warne and M. J. Pikal, J. Pharm. Sci., 2009, 98, 3145–3166 CrossRef CAS PubMed.
- S. Shah, A. Sharma and M. N. Gupta, Anal. Biochem., 2006, 351, 207–213 CrossRef CAS PubMed.
- B. S. Aytar and U. Bakir, Process Biochem., 2008, 43, 125–131 CrossRef CAS.
- T. Dong, L. Zhao, Y. Huang and X. Tan, Bioresour. Technol., 2010, 101, 6569–6571 CrossRef CAS PubMed.
- H. Cabana, J. P. Jones and S. N. Agathos, J. Biotechnol., 2007, 132, 23–31 CrossRef CAS PubMed.
- J. Zheng, Y. Chen, L. Yang, M. Li and J. Zhang, Appl. Biochem. Biotechnol., 2014, 174, 2067–2078 CrossRef CAS PubMed.
- J. D. Cui and S. R. Jia, Crit. Rev. Biotechnol., 2015, 35, 15–28 CrossRef CAS PubMed.
- M. I. García García, A. Sola Carvajal, F. García Carmona and Á. Sánchez Ferrer, Process Biochem., 2014, 49, 90–94 CrossRef.
- Y. Chen, C.-p. Xiao, X.-y. Chen, L.-w. Yang, X. Qi, J.-f. Zheng, M.-c. Li and J. Zhang, J. Mol. Catal. B: Enzym., 2014, 100, 84–90 CrossRef CAS.
- Z. A. Sinirlioglu, D. Sinirlioglu and F. Akbas, Bioresour. Technol., 2013, 146, 807–811 CrossRef CAS PubMed.
- H. C. Friedmann, Methods Enzymol., 1971, 18B, 192–197 Search PubMed.
- A. Morales, O. Barbosa, N. Rueda, Z. Fonseca, R. Torres, R. C. Rodrigues, C. Ortiz and R. Fernandez-Lafuente, RSC Adv., 2015, 5, 53047–53053 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00505a |
| This journal is © The Royal Society of Chemistry 2017 |