
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microwave-assisted synthesis of polypyridyl ruthenium(II) complexes as potential tumor-targeting inhibitors against the migration and invasion of Hela cells through G2/M phase arrest†
Jieqiong Cao‡
a,
Qiong Wu‡ b,
Wenjie Zheng*abd,
Li Lic and
Wenjie Mei*c
b,
Wenjie Zheng*abd,
Li Lic and
Wenjie Mei*c
aCollege of Pharmacy, Jinan University, Guangzhou, China. E-mail: tzhwj@jnu.edu.cn
bIntegrated Chinese and Western Medicine Postdoctoral Research Station, Jinan University, Guangzhou, China
cSchool of Pharmacy, Guangdong Pharmaceutical University, Guangzhou, China. E-mail: wenjiemei@126.com
dDepartment of Chemistry, Jinan University, Guangzhou, China
First published on 17th May 2017
Abstract
A series of polypyridyl ruthenium(II) complexes coordinated by phenanthroimidazole derivatives [Ru(phen)2(R)](ClO4)2 (where 1 R = IP, 2 R = PIP, 3 R = p-HPIP, 4 R = p-OCH3PIP) was synthesized with an average yield of >85% under microwave irradiation at 140 °C for 30 min. The inhibitory effect of these complexes against various tumour cells were evaluated by MTT assay, and the results showed that these polypyridyl ruthenium(II) complexes exhibited acceptable inhibition against different tumour cells, especially 4, with an IC50 of 18.4 μM for Hela cells. The results showed that 4 inhibited the growth of cervical cancer Hela cells by inducing G2/M phase arrest, which was followed by slight apoptosis. Further studies showed that 4 displayed better inhibition against the invasion and metastasis of Hela cells than NAMI-A. Studies on the in vivo distribution and metabolism indicated that 4 was rapidly distributed in the entire body, absorbed by the tumour tissue and had only a small accumulation of toxicity in the body. These results demonstrated that this type of ruthenium(II) complex can block the growth of Hela cells and inhibit their migration and invasion through G2/M phase arrest, which suggests the complex could act as a potential tumour-targeting inhibitor in future clinical applications.
Introduction
Cervical cancer, especially metastatic cervical cancer with a high invasive potential and infinite proliferation,1–4 is the most common primary malignant tumour in women worldwide.5–11 The worldwide incidence rate of cervical cancer has been on the rise in recent years, and its metastasis remains one of the main pathological conditions endangering human life worldwide. The treatment of cervical cancer faces one of the most challenging problems in clinical oncology in that there are few effective treatments to block the metastasis of cervical cancer cells.12–15Ruthenium complexes have always been regarded as some of the most potential antitumour inhibitors owing to their decent antitumour activity16–20 and low toxicity.21–24 A number of Ru complexes have been designed and their anti-metastasis activities against various tumours have been investigated extensively.25–27 For example, NAMI-A28,29 and KP1019![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30 have entered phase II clinical studies as NAMI-A can selectively reduce tumour metastasis and inhibit tumour cell invasion in vitro and KP1019 can inhibit the migration and invasion of MDA-MB-231 breast cancer cells by reducing the release of the extracellular matrix (MMP-2/9). In addition, the Ru complex RM175 also exhibits tumour metastasis inhibition and reduces the invasion and metastasis by promoting cell–cell re-adhesion and by decreasing the release of metalloproteinases (MMPs).31 However, little attention has been paid to the inhibition of the migration and invasion of ruthenium polypyridyl complexes.32
30 have entered phase II clinical studies as NAMI-A can selectively reduce tumour metastasis and inhibit tumour cell invasion in vitro and KP1019 can inhibit the migration and invasion of MDA-MB-231 breast cancer cells by reducing the release of the extracellular matrix (MMP-2/9). In addition, the Ru complex RM175 also exhibits tumour metastasis inhibition and reduces the invasion and metastasis by promoting cell–cell re-adhesion and by decreasing the release of metalloproteinases (MMPs).31 However, little attention has been paid to the inhibition of the migration and invasion of ruthenium polypyridyl complexes.32
In this study, a series of polypyridyl ruthenium(II) complexes, [Ru(phen)2(R)](ClO4)2 (where 1 R = IP, 2 R = PIP, 3 R = p-HPIP, 4 R = p-OCH3PIP) were synthesized and demonstrated to inhibit the invasion and metastasis of Hela cells. The molecular mechanisms through which the ruthenium(II) complexes caused cancer cell death were also elucidated. This study demonstrated that polypyridyl ruthenium(II) complexes display great antitumour activity against cervical cancer cells and effectively inhibit the migration and invasion of Hela cells in vitro. Moreover, further study indicated that the polypyridyl ruthenium(II) complexes induced growth inhibition was mainly caused by cell cycle arrest at the G2/M phase.
Results and discussion
Synthesis of polypyridyl ruthenium(II) complexes
Compared with the conventional heating method, microwave-assisted synthesis heating technology33–36 can significantly improve the yield of the compound and effectively reduce the reaction time. It was observed that the reaction system temperature could instantly reach 130 °C in less than 2 min under microwave irradiation, and the reaction condition stayed almost unchanged during the whole process (Fig. S1†). The average yields of 1, 2, 3 and 4 under microwave irradiation were greater than 80%, which was far more higher than typically obtained under the conventional heating method (Table S1†). Through orthogonal tests, we obtained the optimized reaction conditions, including reaction temperature (130 °C), reaction time (30 min) and the weight ratio of materials (1:1.5), with a high average yield of 92.38%. These results indicated that microwave irradiation could decrease the time consumption and increase the yield.
Biological activity
The in vitro inhibitory activities of the polypyridyl ruthenium(II) complex against cervical cancer cells (Hela, Siha, Caski) were evaluated by MTT assay. As shown in Table 1, it was found that all of the four ruthenium(II) complexes exhibited moderate anti-proliferative activity against several cancer cells, and that this effect was dosage-dependent. It was seen that the inhibitory activity of these complexes against the growth of Hela cells followed the sequence 1 < 2 < 3 < 4, indicating that the planarity of the main ligand and electron-donating groups in the main ligand play a key role in enhancing the inhibitory activity of these complexes. Compared with 1 and 2, with the introduction of a benzene ring at the end of the imidazole ring, 2 exhibited better antitumour activity. Moreover, the most active complex was 4 with a methoxy group at the para-position in the end benzene ring, which displayed a broad spectrum growth inhibition against several cancer cells, especially for Hela cells, with an IC50 value of 18.4 μM, compared to the biological activity with the other ruthenium(II) complexes, such as [(Ru(phen)2)2(tpphz)]4+, which had an IC50 value to Hela cells of >200 μM,37 and [(C6H6)Ru(p-NMe2PIP)Cl]Cl·2H2O, which had an IC50 value to Hela cells of 88.5 μM.38 These results were reasonable given that the introduction of a benzene ring and the presence of donor substituents can improve the antitumour activity.| Comp. | IC50/μM | logP |
||
|---|---|---|---|---|
| Hela | Siha | Caski | ||
| 1 | 84.8 | >100 | 73.8 | −0.35 |
| 2 | 48.8 | >100 | 53.1 | 0.05 |
| 3 | 24.5 | 83.2 | 17.4 | −0.23 |
| 4 | 18.4 | 49.4 | 21.9 | −0.12 |
| NAMI-A | >100 | >100 | >100 | −2.57 |
Lipophilicity often exerts an effect on a compound's bioavailability and effectiveness. Thus, we investigated the octanol/water partition coefficients of these complexes by ultraviolet detection. The lipophilicity partition coefficients for 1, 2, 3, 4 and NAMI-A were approximately −0.35, 0.05, −0.23, −0.12 and −2.57, respectively. Therefore, we concluded that the molecular polarity changes with the size of the substituents as well as with their electron-donating power.25
Inhibition of migration and invasion against Hela cells
As is well known, metastasis is the major cause of death from cervical cancer, and is a process that includes migration and invasion. As shown in Fig. 1A, the inhibitory effect of 4 against the migration of Hela cells was evaluated by wound healing assays.39 It was found that Hela cells treated without 4 showed a decreasing distance of wound closure at 72 h; however, an increasing distance of wound closure was observed with the further addition of 4. For Hela cells treated with 4 ([Ru] = 2 μM), the healing rate of scratches was about 47.9%, which was far lower than the healing rate for Hela cells treated without 4 (60%). These results can be compared to Hela cells treated with NAMI-A ([Ru] = 10 μM) with a healing rate of 41.5%. When the concentration of 4 was increased up to 8 μM, the scratches healing degree was significantly reduced to 27.2% (Fig. 1B). These data suggest that 4 can inhibit the migration of Hela cells effectively.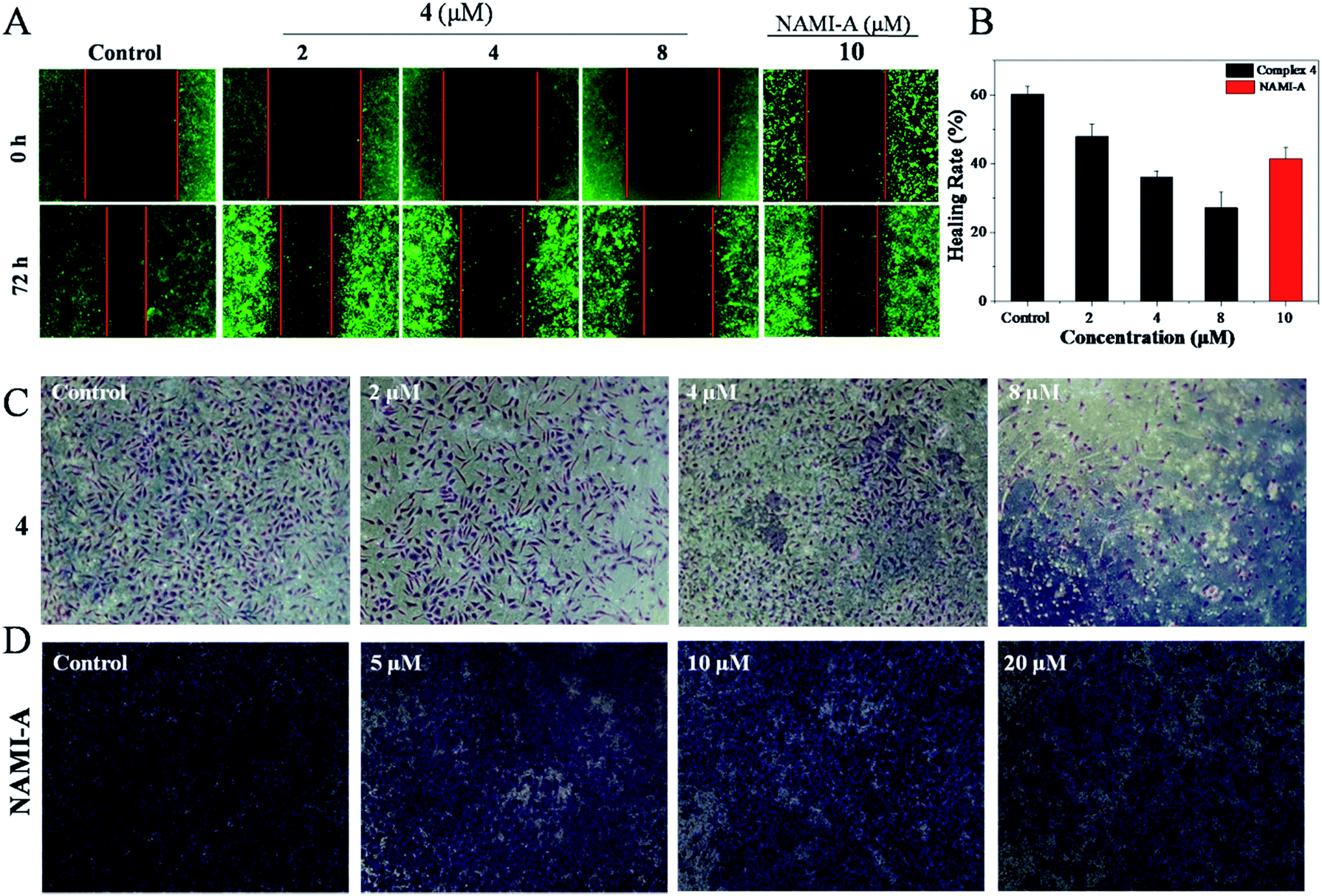 | ||
| Fig. 1 (A) The wound healing assay of Hela cells after treatment with 4 (0, 2, 4 and 8 μM) and [NAMI-A] = 10 μM. (B) The healing rate of Hela cells treated with 4 and NAMI-A. (C) The transwell assay of Hela cells after treatment with 4 (0, 2, 4 and 8 μM) and (D) [NAMI-A] = (0, 5, 10 and 20 μM). | ||
The transwell invasion assay is usually applied to observe the number of invasive tumour cells. In general, cells with high invasion rates can form invadopodia to release MMPs, which degrade the matrigel and migrate to the other side of the membrane. The more cells migrate to other membranes, the stronger the invasion of the cells. Compared with the control, the invasion of Hela cells was blocked with the increasing concentration of 4. After being treated with 2 μM of 4, the number of Hela cells that degraded the matrigel decreased markedly. With the increase in 4, the invasion ability of Hela cells was clearly blocked as could be observed from the fact that few cells migrated to another membrane (Fig. 1C). Treatment with 4 even exhibited better inhibition of invasion at 8 μM with the healing rate of 27.2% than NAMI-A with a healing rate of 41.5%, (5 μM) which is an anti-metastasis agent in vivo (Fig. 1D). These data revealed that the invasion abilities of Hela cells could be blocked by 4 significantly.
In vivo distribution and metabolism
Furthermore, tests on the in vivo distribution and metabolism of 4 were conducted on tumour-bearing nude mice. As shown in Fig. 2A, with the injection of 4 into the tail vein, the drug was quickly distributed in the whole body, even passing the blood–brain barrier in the head, with the maximum concentration occurring at 6 h. However, as time goes on, the complex is progressively metabolized out, with increasingly weaker fluorescence in the body, indicating that 4 can be metabolized normally and dropped out of the mouse body. Then at 48 h, little fluorescence of the complex was observed, indicating that the complex was almost totally eliminated from the mice. Moreover, further studies showed that at 6 h, (Fig. 2B), 4 was mainly accumulated in the liver, kidney and tumour tissue together with a small distribution in the heart, lung and spleen. After 48 h, the complexes still had a large amount of accumulation of tumour tissue in vivo, suggesting that 4 could enrich in tumour tissue, but also can be accumulated in the liver (Fig. 2C). The above results indicated that 4 was easily distributed in nude mice and enriched in the tumour tissue, and could then be majorly metabolized by the liver and excreted by the kidney.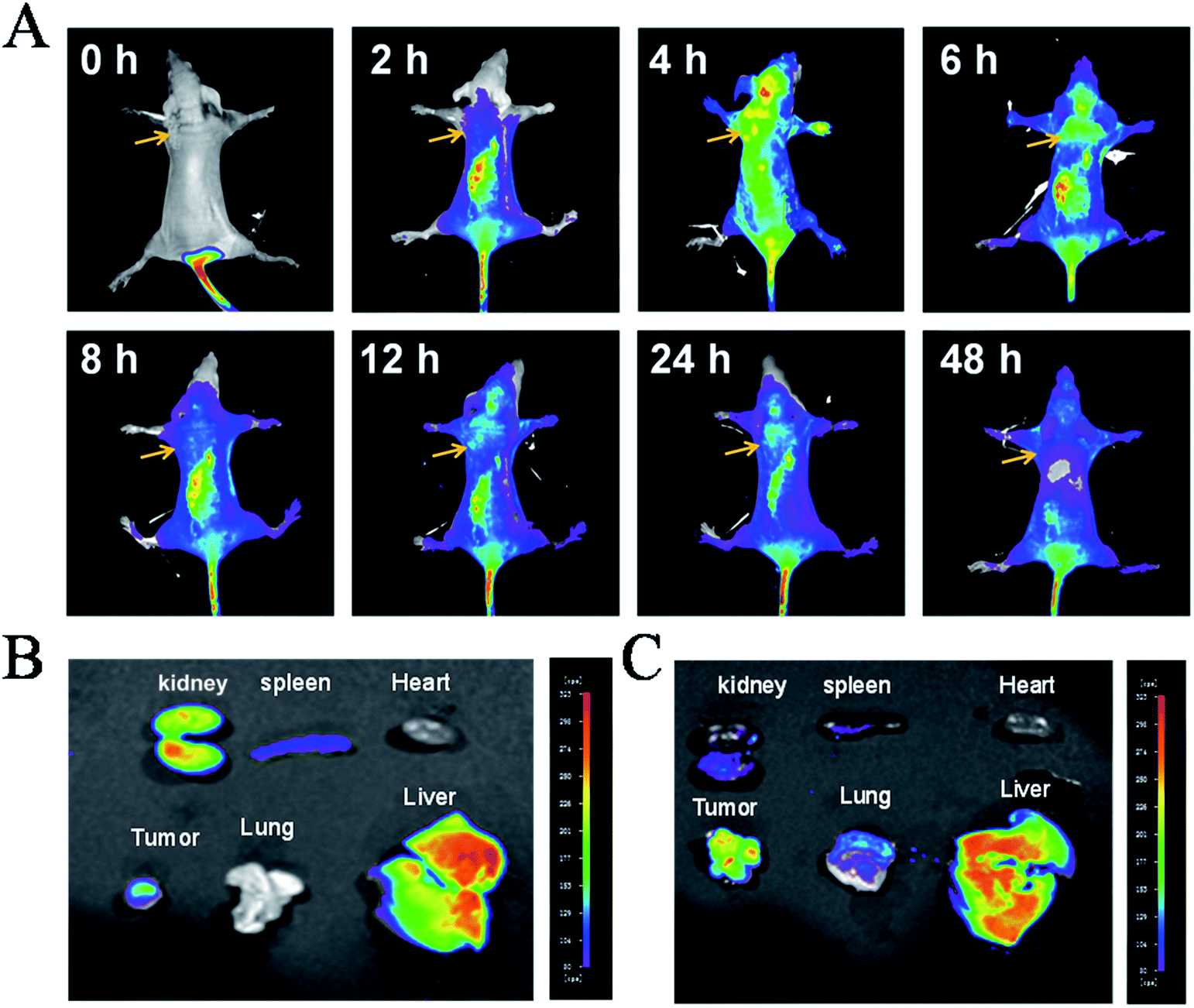 | ||
| Fig. 2 In vivo distribution and metabolism of polypyridyl Ru(II) 4. (A) The distribution and localization of 4 (5 mg kg−1) in vivo. Tissue distribution of 4 for 6 h (B) and 48 h (C). (Excited by 475 nm, emission at 600 nm). | ||
Cellular localization
To clarify the underlying mechanism of 4 against the migration and invasion of Hela cells, the cellular localization was first studied. After the incubation of 4 for 6 h, it was observed that 4 (red) was distributed in the whole cell, and enriched in the surface of the cell membrane, and there was a certain number of 4 also distributed in the nucleus (blue) and the mitochondria (green) (Fig. 3A). Further study showed that with increasing the concentration of 4, there was little influence on the mitochondrial membrane potential as observed by JC-1 tests (Fig. 3B). These results suggested that 4 was mainly distributed in the surface of the cell membrane, which induced the death of tumour cells through a non-mitochondria-mediated pathway.40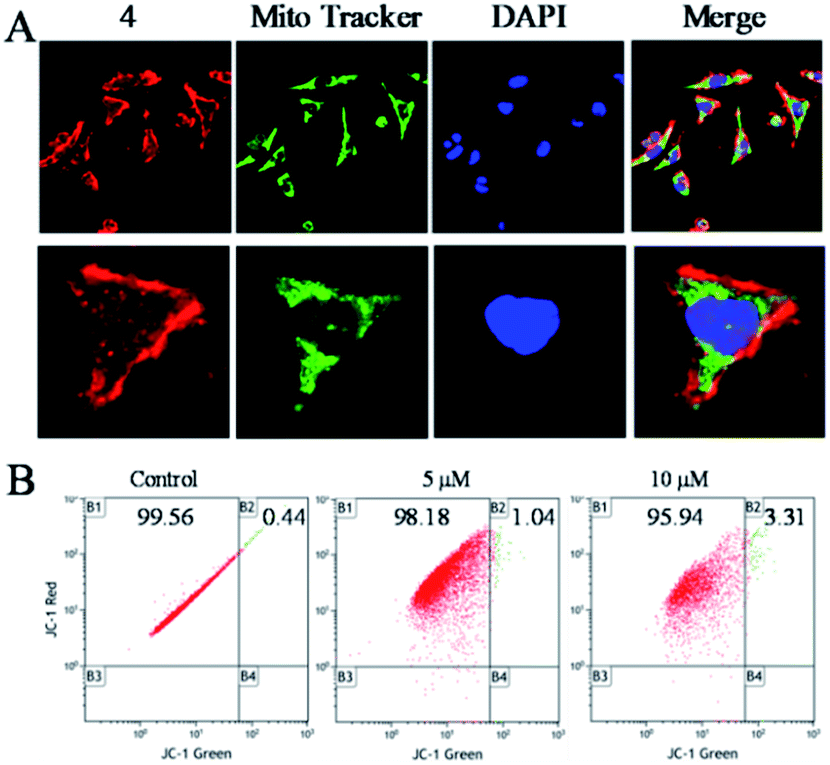 | ||
| Fig. 3 (A) The distribution of 4 in Hela cells for 6 h, blue: DAPI (10 nM), green: Mito-Tracker (10 nM), red: 4 = 5 μM. (B) Change in mitochondrial membrane potential of Hela cells incubated with 4 (0, 5 and 10 μM) for 24 h as tested by flow cytometric analysis. | ||
Cell phase arrest induced by the Ru(II) complex
The growth inhibition or death of cells is the result of apoptosis, cell cycle arrest or a combined action of both.41 Therefore, flow cytometry analysis was carried out to determine the action of 4. According to the results, it was revealed that 4 induced growth inhibition was mainly caused by cell cycle arrest at the G2/M phase. After Hela cells were exposed to different concentrations of 4, a significant increase in the cell proportion in the G2/M phase of Hela cells was observed (Fig. 4A). For instance, when the concentration of 4 reached 4 μM, the G2/M phase cells peak increased from 14.79% to 29.67%, while there was a significant decrease in cell proportion in the G0/G1 phase, while the S-phase proportion of cells showed no obvious change with different concentrations of 4.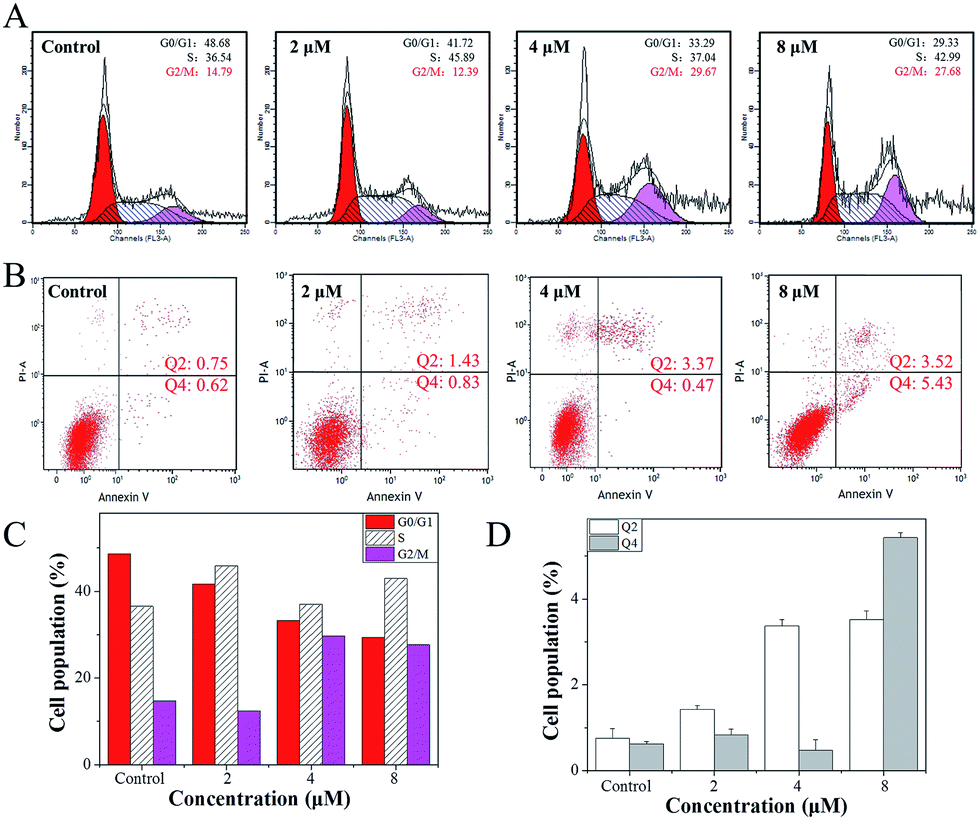 | ||
| Fig. 4 (A) Cell cycle distribution of Hela cells incubated with 4 (0, 2, 4 and 8 μM) for 24 h tested by flow cytometric analysis. (B) Apoptosis of Hela cells induced by 4 (0, 2, 4 and 8 μM); (C) change in cell cycle distribution of Hela cells induced by 4 (0, 2, 4 and 8 μM). (D) Cells population of apoptosis of Hela cells induced by 4 (0, 2, 4 and 8 μM). | ||
Furthermore, a large number of studies in the literature42–45 have reported that small molecule drugs that induce G2/M phase arrest could eventually induce tumour cell apoptosis. However, after Hela cells were treated with 4, little apoptosis was observed (Fig. 4B). With the addition of 4, when the concentration reached 2 μM, the Hela cells early apoptosis rate was just 0.83, while the late apoptosis rate was 1.43, and even when the concentration of 4 was increased to 8 μM, the Hela cells early apoptosis rate was only 5.43, while the late apoptosis rate was 3.52. These results indicate that 4 mainly inhibits the growth of Hela cells through G2/M phase arrest.
Conclusions
In conclusion, a series of ruthenium(II) complexes [Ru(phen)2(R)](ClO4)2 (where 1 R = IP, 2 R = PIP, 3 R = p-HPIP, 4 R = p-OCH3PIP) were synthesized with high efficiency and high yield under microwave irradiation. It was demonstrated that all of these complexes, especially 4, exhibited promising inhibitory effect against Hela cells. Further studies showed that 4 could also inhibit the migration and invasion of Hela cells effectively. Moreover, in vivo studies showed that 4 could be enriched in tumour tissue. It was also confirmed that 4 could inhibit the growth of Hela cells through G2/M phase arrest. Therefore, these complexes may be developed as potential targeting inhibitors against the migration and invasion of Hela cells through inducing G2/M phase arrest.Experimental
Reagents and materials
All the reagents were purchased from commercial suppliers without further purification. Solvents were dried and purified by conventional methods prior to use. Ruthenium chloride hydrate was obtained from Mitsuwa Chemicals. 1,10-Phenanthroline, formaldehyde, benzaldehyde, 4-methoxy benzaldehyde and 4-hydroxybenzaldehyde were purchased from Aladdin. Ammonium acetate, ammonia, sodium perchlorate, sulfuric acid, nitric acid, potassium bromide, anhydrous ethanol and anhydrous methanol were purchased from Guangzhou Chemical Reagent factory.Instruments
The compound was synthesized using an Anton Paar Monowave 300 microwave reactor. The 1H NMR and 13C NMR spectra were recorded in d6-DMSO on a Bruker DRX2500 spectrometer operating at room temperature. Cell apoptosis was measured using a flow cytometer (Beckman Coulter, Miami, FL, USA).Synthesis and characterization
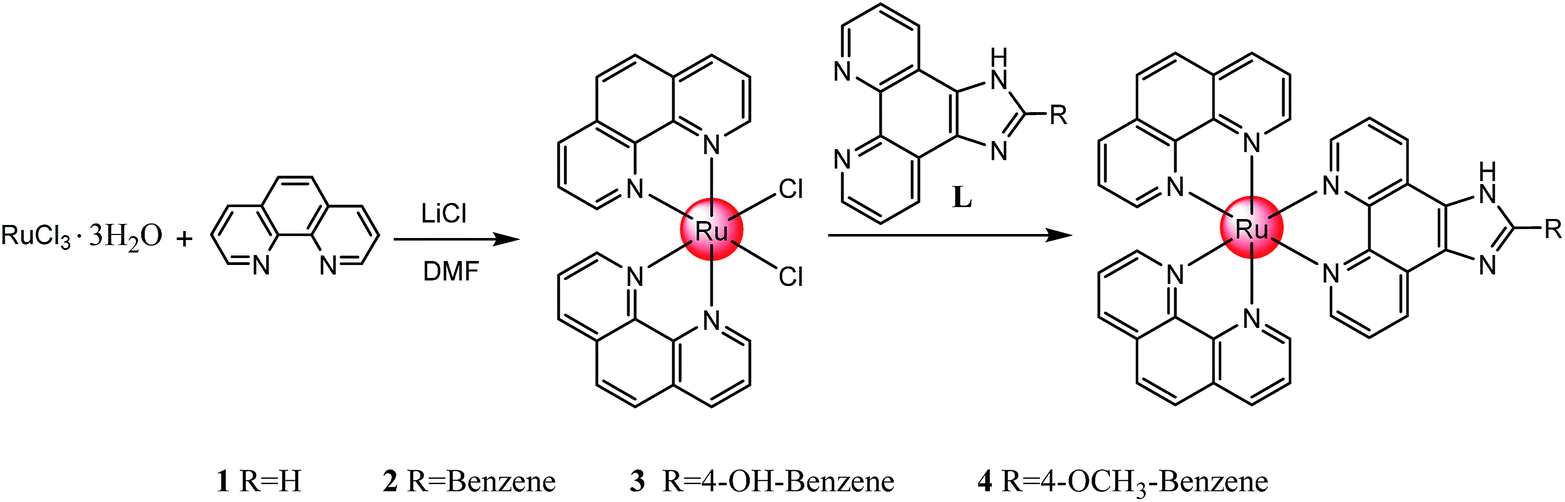 | ||
| Scheme 1 Synthesis route of the polypyridyl ruthenium(II) complexes. | ||
MTT assay
All the complexes were dissolved in DMSO with stock solution at 1 mM, and the cell viability was determined by measuring the ability of the cells to transform MTT into a purple formazan dye. Cells were seeded in 96-well tissue culture plates for 24 h, and then the cells were incubated with different concentrations of the complex for 72 h. After incubation, 20 mL per well of MTT solution (5 mg mL−1 phosphate buffered saline) was added and incubated for 5 h. The colour intensity of the formazan solution, which reflects the cell growth conditions, was measured at 570 nm using a microplate spectrophotometer (SpectroAmaxt 250).Amphiphilicity measurements
Octanol and distilled water (volume ratio = 1:1) were thoroughly mixed in an oscillator for 24 h, then separated into a two-phase solution. The test compound was added dropwise to the aqueous and octanol phases to give a 20 mM solution with mixing. After separation, the final concentration of the water phase was denoted as Cw and the concentration of the octanol phase was denoted as Co. Both Co and Cw were tested by ultraviolet-visible (UV-vis) spectrophotometry, and the partition coefficient (Po/w) for the complex was calculated according to the equation: Po/w = Ao/Aw.
Wound healing assay
Cells were seeded in a 6-well tissue culture plate marked on the back (1 × 105 cells per well) until the monolayer cells spread to more than 80% of the bottom of the culture plate. A line was scratched orthogonally using a tip (200 mL) to mark the plate. The cells were then incubated with the tested compounds at different concentrations (0, 2, 4, 8 μM) for 48 h. Migrating cells were imaged in the same visual field every 12 h for three days. The healing rate were calculated according to the formula 1 − [D48]/[D0], [D48] = wound distance at 48th hour, [D0] = wound distance at 0th hour.Transwell assay
Up to 50 mL of Matrigel was added into the top chamber of the 24-well transwell on ice, and the plate was then incubated at 37 °C and with 5% CO2 for 30 min. Afterward, the cells were placed in the top chamber with serum-free media having different concentrations (0, 2, 4, 8 μM), while the bottom chamber was added with DMEM containing 10% FBS and incubated for 24 h. Cell invasion was imaged using a fluorescence microscope.In vivo biodistribution and target studies
BALB/c nude mice were provided by Guangdong Medical Laboratory Animal Center. The injection was prepared (50 mL) using physiological saline, DMF and twain (88:10:2), with 2.5 mg of the drug dissolved in 5 mL injection. Each rat was injected 200 μL, and every 2 h (2–48 h) an in vivo imaging system (Bruker) was used to detect drug absorption and distribution in the nude mice tissues and organs. When the drug was observed with a large distribution in the tumour tissue site, the heart, liver, spleen, lung and kidney were taken out and photographed. The relevant ethical protocols used for the in vivo study for the BALB/c nude rat were followed according to the relevant laws. The animals were maintained in accordance with the Guide for the Care and Use of Laboratory Animals issued by the National Institutes of Health, and approved by the Laboratory Animal Ethics Committee of Jinan University.
Cellular localization
Cells in incomplete growth medium at 2 × 105 cells per mL were incubated with the tested complex at different concentrations (0, 2, 4, 8 μM) for 6 h, then, the cells were stained with DAPI and Mitotracker green FM for another 10 min. Next, they were removed from the medium and washed with PBS 3 times, and finally luminescence imaging was carried out by confocal microscopy.Flow cytometry analysis of the mitochondrial membrane potential
Treated tumour cells were trypsinized, with 10 ug mL−1 of 0.5 g mL−1 JC-1 mL PBS buffer solution (0.5 mL) suspension and then incubated at 37 °C for 10 min, immediately followed by centrifuging to remove the suspended solids. Then, the cell suspension in PBS buffer solution was subjected to flow cytometry analysis. Green fluorescence intensity produced by the JC-1 monomer showed cell membrane potential losses ΔΨ.Flow cytometry analysis of cycle arrest
The cell cycle distribution was analyzed by flow cytometry as previously reported.46 Treated or untreated cells were trypsinized, washed with PBS, and fixed with 70% ethanol overnight at 4 °C. Fixed cells were washed with PBS and stained with propidium iodide (PI) (1.21 mg mL−1 Tris, 700 U mL−1 RNase, 50.1 mg mL−1 PI, pH = 8.0) for 15 min in the dark. After staining the cells, they were analyzed using the Epics XL-MCL flow cytometry instrument (Beckman Coulter, Miami, FL, USA). Cell cycle distribution was analyzed using the MultiCycle software (Phoenix Flow Systems, San Diego, CA, USA). Apoptotic cells with hypodiploid DNA content were measured by quantifying the sub-G1 peak in the cell cycle pattern.Acknowledgements
This work was supported by the National Nature Science Foundation of China (81572926), the Provincial Major Scientific Research Projects in Universities of Guangdong Province (2014KZDXM053), the Science and Technology Project of Guangdong Province (2014A020212312), Natural Science Fund of Guangdong (408182136045), Guangdong Medical Scientific Research Fund (A2015550), the Guangzhou City Science and Technology Plan (2013J4100072), the Science and Technology Projects of Yuexiu District (2014-WS-039) and the Joint Natural Sciences Fund of the Department of Science and Technology and the First Affiliated Hospital of Guangdong Pharmaceutical University (GYFYLH201309).Notes and references
- G. Ronco, J. Dillner, K. M. Elfström, S. Tunesi, P. J. Snijders, M. Arbyn, H. Kitchener, N. Segnan, C. Gilham and P. Giorgi-Rossi, Lancet, 2014, 383, 524–532 CrossRef.
- J. Ferlay, H. R. Shin, F. Bray, D. Forman, C. Mathers and D. M. Parkin, Int. J. Cancer, 2010, 127, 2893–2917 CrossRef CAS PubMed.
- A. Serranoolvera, L. Cetina, J. Coronel and A. Dueñasgonzález, Expert Opin. Emerging Drugs, 2015, 20, 1–18 CrossRef.
- D. G. Allen, R. S. Planner, P. T. Tang, J. P. Scurry and T. Weerasiri, Best Pract. Res. Clin. Obstet. Gynaecol., 2016, 33, 228 Search PubMed.
- S. E. Waggoner, Lancet, 2003, 361, 2217–2225 CrossRef.
- F. X. Bosch, J. Natl. Cancer Inst., 1995, 87, 796–802 CrossRef CAS PubMed.
- R. Das, K. Bhattacharya, S. K. Samanta, B. C. Pal and C. Mandal, Cancer Lett., 2014, 351, 81–90 CrossRef CAS PubMed.
- D. M. Parkin, F. Bray, J. Ferlay and P. Pisani, Ca-Cancer J. Clin., 2005, 55, 74–108 CrossRef PubMed.
- B. Li, Ca-Cancer J. Clin., 2011, 61, 33–64 Search PubMed.
- A. Jemal, F. Bray, M. M. Center, J. Ferlay, E. Ward and D. Forman, Ca-Cancer J. Clin., 2011, 61, 69–90 CrossRef PubMed.
- R. Sankaranarayanan, A. M. Budukh and R. Rajkumar, Bull. World Health Organ., 2015, 79, 954–962 Search PubMed.
- K. S. Tewari, M. W. Sill, H. J. L. Iii, R. T. Penson, H. Huang, L. M. Ramondetta, L. M. Landrum, A. Oaknin, T. J. Reid and M. M. Leitao, N. Engl. J. Med., 2014, 370, 734–743 CrossRef CAS PubMed.
- Y. Dang, X. Yuan, R. Tian, L. I. Dongguang and W. Liu, Exp. Ther. Med., 2015, 9, 1470–1476 CAS.
- R. N. Eskander and K. S. Tewari, Clin. Ther., 2015, 37, 20–38 CrossRef CAS PubMed.
- J. Manzomerino, A. Contrerasparedes, E. Vázquezulloa, L. Rochazavaleta, A. M. Fuentesgonzalez and M. Lizano, Arch. Med. Res., 2014, 45, 525–539 CrossRef CAS PubMed.
- L. Zeng, Y. Chen, J. Liu, H. Huang, R. Guan, L. Ji and C. Hui, Sci. Rep., 2016, 6, 19449 CrossRef CAS PubMed.
- Y. Chen, R. Guan, C. Zhang, J. Huang, L. Ji and H. Chao, Coord. Chem. Rev., 2016, 310, 16–40 CrossRef CAS.
- J. Liu, Y. Chen, G. Li, P. Zhang, C. Jin, L. Zeng, L. Ji and H. Chao, Biomaterials, 2015, 56, 140–153 CrossRef CAS PubMed.
- L. Xu, X. Chen, J. Wu, J. Wang, L. Ji and H. Chao, Chem.–Eur. J., 2015, 21, 4008–4020 CrossRef CAS PubMed.
- H. Huang, P. Zhang, H. Chen, L. Ji and H. Chao, Chemistry, 2015, 21, 715–725 CrossRef CAS PubMed.
- R. Pettinari, C. Pettinari, F. Marchetti, B. W. Skelton, A. H. White, L. Bonfili, M. Cuccioloni, M. Mozzicafreddo, V. Cecarini and M. Angeletti, J. Med. Chem., 2014, 57, 4532–4542 CrossRef CAS PubMed.
- C. Zheng, Y. Liu, L. Ying, X. Qin, Y. Zhou and L. Jie, J. Inorg. Biochem., 2016, 156, 122–132 CrossRef CAS PubMed.
- G. Sava, S. Zorzet, L. Perissin, G. Mestroni, G. Zassinovich and A. Bontempi, Inorg. Chim. Acta, 1987, 137, 69–71 CrossRef CAS.
- A. Weiss, R. H. Berndsen, M. Dubois, C. Müller, R. Schibli, A. W. Griffioen, P. J. Dyson and P. Nowak-Sliwinska, Chem. Sci., 2014, 5, 4742–4748 RSC.
- Q. Wu, K. Zheng, S. Liao, Y. Ding, Y. Li and W. Mei, Organometallics, 2016, 35, 317–326 CrossRef CAS.
- Q. Wu, J. He, W. Mei, Z. Zhang, X. Wu and F. Sun, Metallomics, 2014, 6, 2204–2212 RSC.
- C. Fan, Q. Wu, T. Chen, Y. Zhang, W. Zheng, Q. Wang and W. Mei, Med. Chem. Commun., 2014, 5, 597–602 RSC.
- A. Bergamo, B. Gava, E. Alessio, G. Mestroni, B. Serli, M. Cocchietto, S. Zorzet and G. Sava, Int. J. Oncol., 2002, 21, 1331–1338 CAS.
- M. Liu, Z. J. Lim, Y. Y. Gwee, A. L. Dr and P. A. L. Prof, Angew. Chem., 2010, 122, 1705–1708 CrossRef.
- A. Bergamo, A. Masi, M. A. Jakupec, B. K. Keppler and G. Sava, Met.-Based Drugs, 2009, 2009, 681270 CAS.
- A. Bergamo, A. Masi, A. Peacock, A. Habtemariam, P. Sadler and G. Sava, J. Inorg. Biochem., 2010, 104, 79–86 CrossRef CAS PubMed.
- A. Bergamo, A. Masi, A. F. Peacock, A. Habtemariam, P. J. Sadler and G. Sava, J. Inorg. Biochem., 2010, 104, 79–86 CrossRef CAS PubMed.
- F. A. Beckford, S. Michael, G. Leblanc, J. Thessing, L. C. Lewisalleyne, A. A. Holder, L. Li and N. P. Seeram, Dalton Trans., 2009, 48, 10757–10764 RSC.
- H. Zhu, X. Wang, Y. Li, Z. Wang, F. Yang and X. Yang, Chem. Commun., 2009, 103, 887–896 Search PubMed.
- Q. Wu, J. Wu, W. J. Mei, Q. Wang, Z. Zhang, X. H. Wu, F. Y. Sun, W. L. Wu, Y. H. Chen and X. Y. Hu, Aust. J. Chem., 2013, 66, 1422–1427 CrossRef CAS.
- S. H. Jhung, J. H. Lee, J. W. Yoon, C. Seree, G. Ferey and J. S. Chang, Adv. Mater., 2007, 19, 121–124 CrossRef CAS.
- M. R. Gill, D. Cecchin and M. G. Walker, et al., Chem. Sci., 2013, 12, 4512–4519 RSC.
- Q. Wu, C. Fan, T. Chen, C. Liu, W. Mei, S. Chen, B. Wang, Y. Chen and W. Zheng, Eur. J. Med. Chem., 2013, 63, 57–63 CrossRef CAS PubMed.
- D. E. F. Van, L. A. Amos and J. Löwe, Nature, 2001, 413, 39–44 CrossRef PubMed.
- F. Marques, L. Cortereal, A. P. D. M. Alves, I. Alho, T. S. Morais, A. I. Tomaz, M. H. Garcia and M. P. Bicho, Microsc. Microanal., 2013, 19, 9–10 CrossRef CAS.
- H. Ling, M. Fabbri and G. A. Calin, Nat. Rev. Drug Discovery, 2013, 12, 847–865 CrossRef CAS PubMed.
- M. Taron, C. Plasencia, A. Abad and C. Martin, Invest. New Drugs, 2000, 18, 139–148 CrossRef CAS PubMed.
- E. Shtivelman, J. Sussman and D. Stokoe, Curr. Biol., 2002, 12, 919–924 CrossRef CAS PubMed.
- Y. Niu, Z. Wang, H. Huang, S. Zhong, W. Cai, Y. Xie and G. Shi, Cancer Lett., 2014, 347, 88–97 CrossRef CAS PubMed.
- N. Y. Tang, Y. T. Huang, C. S. Yu, Y. C. Ko, S. H. Wu, B. C. Ji, J. S. Yang, J. L. Yang, T. C. Hsia and Y. Y. Chen, Anticancer Res., 2011, 31, 1691–1702 CAS.
- X. Wei, D. A. Sipkins, C. M. Pitsillides, J. Novak, I. Georgakoudi and C. P. Lin, Mol. Imaging, 2015, 4, 415–416 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00658f |
| ‡ Equally contribution. |
| This journal is © The Royal Society of Chemistry 2017 |