
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot oligoamides syntheses from L-lysine and L-tartaric acid
R. Olivaa,
M. A. Ortenzib,
A. Salvini *a,
A. Papacchinia and
D. Giomia
*a,
A. Papacchinia and
D. Giomia
aDipartimento di Chimica “Ugo Schiff”, Università di Firenze, Via della Lastruccia 3-13, 50019 Sesto Fiorentino (FI), Italy. E-mail: antonella.salvini@unifi.it; Tel: +39 055 457 3455
bCRC Materiali Polimerici (LaMPo), Dipartimento di Chimica, Via Golgi, 19 – 20133 Milano, Italy
First published on 20th February 2017
Abstract
Oligoamides based on natural raw materials, L-lysine and L-tartaric acid, were synthesized using one-pot processes. An L-lysine diketopiperazine structure was obtained with good selectivity without protection/deprotection steps. Reactions were performed using two methods, the first involving the use of solvent and catalyst under a nitrogen atmosphere, the second exploiting the “raw” monomers under vacuum. The reaction products were characterized through FT-IR, NMR and MALDI spectroscopies, acid–base titration, SEC and polarimetric analyses. Thermal properties were evaluated by DSC and TGA analyses.
1. Introduction
In recent years, the interest associated with the development of biopolymers has encouraged the use of products derived from renewable sources as starting materials. Therefore, natural polymers (starch, cellulose, chitin) or natural monomers have been used as such or after chemical transformations1–13 to obtain new polymeric materials.Polyamides are a widely studied polymeric class showing excellent mechanical and physical characteristics and good versatility, that allows their use in various application fields.14 Biobased polyamides were obtained using hydroxylated diacids, selected from natural compounds or their derivatives, as L-tartaric acid, or D-(+)-glucaric acid and α,α-trehaluronic acid with commercial diamines as 1,n-alkylenediamines, aromatic diamines, branched alkylenediamines, diaminoethers and polyamines,15–25 or using amino acids and their derivatives.26–28 These products can be used in a wide application range,29–31 from standard plastic materials to the biomedical field.
Reactions involving amino acids typically require several protection/deprotection steps, which cause long reaction times, by-products formation and consequently make the industrial scale-up hardly feasible.
Gachard et al.31 described the synthesis of polyamides from two natural amino acids, L-lysine and L-aspartic acid; both monomers were protected to avoid secondary reactions. These products could be used in biomedical field as new materials for sutures and drug-delivery systems since these biopolymers should be subject to enzymatic or hydrolytic degradation with formation of metabolizable compounds.
Bou et al. reported several polyamides syntheses based on L/D-tartaric acid and L-lysine. At first, a linear polyamide32 was obtained from the polycondensation reaction between N,N-bis(trimethylsilyl)-L-lysine ethyl ester and bis(pentachlorophenyl)di-O-methyl-L-tartrate in chloroform solution. Subsequently33 the aregic (ar), isoregic (ir) and syndioregic (sr) linear polyamides were obtained starting from L-lysine modified as methyl L-lysinate dihydrochloride or N-benzyloxycarbonyl methyl L-lysinate hydrochloride, and D- and L-tartaric acid, modified as bis(pentachlorophenyl)di-O-methyl tartaric acid or di-O-methyl tartaric anhydride.
On the contrary, cyclic oligomers based on L-lysine 2,5-diketopiperazine and tartaric acid have been synthesized by Bou et al.30 through a process that requires several step. L-Lysine was selectively N-protected (–NH2 in ε position) and esterified with methanol via copper complex, then it was self-condensed and treated with HBr in acetic acid to obtain the diketopiperazine structure. At last, polycondensation of 2,5-diketopiperazine structure with pentachlorophenyl ester of di-O-methyl tartaric acid was carried out in dry DMF, with triethylamine, at room temperature, for 3 days, with 50% yield.
Other 2,5-diketopiperazine derivatives were obtained by the reaction between pairs of amino acids, using protection/deprotection steps,34,35 by activation of carboxylic group as methylester,34,36 since using the monomer as such34,37 lower yields were obtained.
2,5-Diketopiperazines may be used as “monomers” in the syntheses of polymers such as polyamides and polyesters, according to their endings.38,39
Starting from amino acids (aspartic acid, serine, lysine, glycine, and tyrosine) modified as methyl ester salts, resins with properties and cost similar to the common industrial nylons that show great potential as high-performance polymers to be used in films, fibers, and composites, were synthesized.40
Other polymers with good thermal stability and high glass transition temperatures, compared to those of common aliphatic nylon were obtained41 by reaction between diketopiperazine (obtained by L-glutamic acid dimerization) and various diamines.
All the syntheses described require many steps and processes which are not always in agreement with the principles of green chemistry and with the requirement of industrial sustainability. These synthetic methods are sometimes necessary to obtain products with specific properties but are not essential for their use in several applications as an adhesive or a consolidant for conservation purposes. With the goal of obtaining polymers to be used in these application fields, in the present work oligoamides from an aldaric acid derivative (L-tartaric acid dimethyl ester) and an amino acid (L-lysine) have been synthesized through one-pot polycondensation reactions, without any protection/deprotection step, obtaining products completely based on natural raw material.
Two different polymerization methods have been used: one was performed with solvent and catalyst in a Sovirel® tube under nitrogen atmosphere and the other one in a vacuum evaporator without solvent and catalyst, i.e. using bulk polymerization, normally used in the industrial polycondensation.42
Among the derivatives of aldaric acid dimethyl-L-tartrate was chosen for its availability, for the simple structure of the precursor and for the ease of production of the corresponding diester.43
L-Lysine is a polar, essential amino acid, with chiral structure, industrially produced by fermentation technologies,44,45 that can easily forms diketopiperazine structures.9,34–37,39,46
2. Experimental
2.1. Materials
L-Tartaric acid, boric acid, L-lysine and D2O were purchased from Aldrich. Triethylamine was purchased from Carlo Erba and methanol from Normapur.All chemicals were reagent grade and were used without further purification.
Dimethyl L-tartrate was synthesized according to literature methods.15
2.2. Instruments
1H-NMR, 13C-NMR, gCOSY and gHSQC spectra were recorded with a Varian Mercury Plus 400 spectrometer working at 399.921 MHz. The chemical shifts are reported in ppm and referred to TMS as internal standard.Number average molecular weight (Mn) and the degree of polymerization (DP) were estimated using 1H-NMR integral calculations.
Spectra elaboration was performed with the software Mestre-C 4.3.2.0.
FT-IR spectra were recorded with a Shimadzu FT-IR-8400S model and elaborated with the Spectrum v.3.0202. Spectra of solid samples were recorded as KBr pellets.
DSC analyses were conducted using a Mettler Toledo DSC1, on samples weighting from 7 to 10 mg each. For dynamic DSC analyses, samples were first heated from 25 °C to 100 °C at 10 °C min−1 and maintained at 100 °C for 2 min to eliminate residual water, then cooled down to 25 °C at 20 °C min−1 and maintained at 25 °C for 2 min; for Tm and Tg determination a second thermal cycle from 25 °C to 250 °C was then used.
TGA analyses were conducted on a Perkin Elmer TGA4000 under nitrogen atmosphere on samples weighting 6 mg, heating from 25 °C to 600 °C (20 °C min−1).
SEC analyses were performed using a system having Waters 1515 Isocratic HPLC pump and a four styragel columns set (HR3-HR4-HR5-HR2) with a Waters 2487 Dual λ Absorbance Detector set at 244 nm using a flow rate of 1 mL min−1 and 20 μL as injection volume; samples were prepared dissolving 20 mg of product in 1 mL of anhydrous CH2Cl2. Before the analysis, the solution was filtered with 0.45 μm filters. o-Dichlorobenzene was used as internal reference (5 μL mL−1). Molecular weights were determined using a calibration made with monodisperse polystyrene standards. In order to dissolve polyamides in anhydrous CH2Cl2, samples underwent Schultz N-trifluoroacetylation reaction.47
Potentiometric titration was performed with Metrohm Titrino 751. Samples were prepared dissolving 120 mg of product in 40 mL of water.
Amino groups were titrated with HCl (0.01 M); acid groups were back-titrated with NaOH (M 0.01).
MALDI analysis were performed with Bruker Daltonic Ultraflex MALDI TOF/TOF spectrometer. ESI LC-MS analysis were performed with LTQ Thermo Scientific with a Spray voltage of 5 kV (capillary temperature 290 °C).
Polarimetric measurements were performed on a JASCO DIP-370. Samples were prepared dissolving 20 mg of product in 2 mL of water.
2.3. Oligoamide synthesis
Several polycondensation reactions were performed using general procedures as described in method 1 and method 2 and using different reaction conditions as reported in Table 1.| Product | Methoda | T (°C) | t (days) | Molar ratiob | Solvent | Cat. | Yield (%) | DP | MW (g mol−1) | Tg (°C) |
|---|---|---|---|---|---|---|---|---|---|---|
| a 1: in a Sovirel® tube, under nitrogen atmosphere; 2: in a rotary evaporator, under vacuum.b Molar ratio diester: L-lysine.c N.D.: not determined.d N.E.: not evaluable. | ||||||||||
| 1 | 1 | 80 | 1 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
MeOH | NEt3 | 40.5 | 3 | 1366 | — |
| 2 | 1 | 80 | 1 | 1:2 |
MeOH | NEt3 | 53.6 | 3 | 1366 | 121.7 |
| 3 | 1 | 80 | 3 | 1:1 |
MeOH | NEt3 | 46.5 | 4 | 1736 | — |
| 4 | 1 | 80 | 3 | 1:2 |
MeOH | NEt3 | 66.3 | 3 | 1366 | 120.8 |
| 5 | 1 | 80 | 7 | 1:1 |
MeOH | NEt3 | 53.8 | 5 | 2105 | — |
| 6 | 1 | 80 | 7 | 1:2 |
MeOH | NEt3 | 71.8 | 4 | 1736 | 120.2 |
| 7 | 2 | 60 | 9 hours | 1:2 |
— | — | 36.0 | 2 | 996 | N.D.c |
| 8 | 1 | 80 | 7 | 0:1 |
MeOH | NEt3 | 0 | N.E.d | N.E.d | N.E.d |
| 9 | 2 | 60 | 9 hours | 0:1 |
— | — | 0 | N.E.d | N.E.d | N.E.d |
| 10 | 2 | 60 | 12 hours | 0:1 |
— | — | 0 | N.E.d | N.E.d | N.E.d |
Method 1: with a slight overpressure (dimethyl L-tartrate
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :L-lysine = 1:1). Into a Sovirel® tube, triethylamine (58.70 mg, 0.58 mmol) was added, under nitrogen atmosphere and under continuous stirring, to 4.00 mL of a methanol solution of L-lysine (314.30 mg, 2.15 mmol). A methanol solution (4.00 mL) of dimethyl L-tartrate (382.70 mg, 2.15 mmol) was added and the mixture was allowed to react at 80 °C for 24 hours (product 1). After cooling to room temperature, the mixture was filtered on a Büchner funnel and the solid obtained was washed with methanol and dried at room temperature (282.30 mg, 41% yield).
:L-lysine = 1:1). Into a Sovirel® tube, triethylamine (58.70 mg, 0.58 mmol) was added, under nitrogen atmosphere and under continuous stirring, to 4.00 mL of a methanol solution of L-lysine (314.30 mg, 2.15 mmol). A methanol solution (4.00 mL) of dimethyl L-tartrate (382.70 mg, 2.15 mmol) was added and the mixture was allowed to react at 80 °C for 24 hours (product 1). After cooling to room temperature, the mixture was filtered on a Büchner funnel and the solid obtained was washed with methanol and dried at room temperature (282.30 mg, 41% yield).The reaction was repeated increasing the reaction time to 3 days (product 3) and 7 days (product 5), respectively with 47% yield (324.20 mg) and 54% yield (375.00 mg). The solids obtained were analysed by 1H-NMR (Table 2), 13C-NMR (Table 3), FT-IR (Table 4), MALDI, SEC, thermal analyses (DSC and TGA) and acid–base titration.
| Product | γ | δ | δterminal | β | εterminal | ε | αD | αL | αterminal | CH–OH | OCH3 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| L-Lysine | 1.36 | — | 1.61 | 2.90 | — | — | — | 3.30 | — | — | |
| 1 and 2 | 1.38 | 1.56 | 1.68 | 1.83 | 2.98 | 3.20–3.40 | 3.68 | 4.22 | — | 4.50 | — |
| 3 and 4 | 1.41 | 1.59 | 1.71 | 1.88 | 3.01 | 3.18–3.38 | 3.72 | 4.21 | — | 4.52 | — |
| 5 and 6 | 1.41 | 1.59 | 1.72 | 1.88 | 3.02 | 3.20–3.40 | 3.73 | 4.22 | — | 4.52 | — |
| 7 | 1.41 | 1.59 | 1.71 | 1.87 | 3.01 | 3.18–3.38 | 3.71 | 4.23 | — | 4.52 | 3.80 |
| 8, 9, 10 | 1.39 | — | 1.65 | 2.95 | — | — | — | 3.35 | — | — |
| Product | γ | δterm. | δ | β | ε | εterm. | α | αterm. | CH–OH | C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
–OCH3 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Spectra recorded at 50 MHz.b N.D.: not determined. | |||||||||||
| L-Lysine | 21.8 | 27.4 | — | 33.2 | — | 39.3 | — | 55.5 | — | 181.8 | |
| 1 and 2 | 21.6 | 26.4 | 28.0 | 30.2 | 38.6 | 39.0 | 54.7 | — | 72.2 | 173.6–175.2 | — |
| 3 and 4a | 20.8 | 25.6 | 27.3 | 29.3 | 37.8 | 38.2 | 53.8 | — | 71.4 | 172.7–174.1 | — |
| 5 and 6a | 20.8 | 25.6 | 27.3 | 29.3 | 37.9 | N.D.b | 53.8 | — | 71.4 | 172.7–173.9 | — |
| 7 | 21.4 | 26.3 | 28.0 | 30.0 | 38.6 | 39.0 | 54.4 | — | 72.4 | 173.5–175.6 | — |
| 8, 9, 10 | 21.8 | 27.1 | — | 32.9 | — | 39.2 | — | 55.3 | — | 181.1 | — |
| Product | N–H stretching | O–H stretching | C–H stretching | –NH3+ aminoacids stretching | –COO− stretching | CO stretching (amide I) |
N–H bending (amide II) | C–O stretching |
|---|---|---|---|---|---|---|---|---|
| L-Lysine | 3350 (m) | — | 2939 (vs) | 2632 (w) | 1582 (vs) | — | 1518 (vs) | — |
| 2862 (vs) | 2135 (w) | |||||||
| 1 and 2 | 3323 (vs) | 3323 (vs) | 2937 (w) | 2632 (w) | (1581) | 1654 (vs) | 1581, 1520 (vs) | 1130 (m) |
| 2864 (w) | 2130 (w) | 1074 (m) | ||||||
| 3 and 4 | 3323 (vs) | 3323 (vs) | 2939 (w) | 2630 (w) | (1583) | 1650 (vs) | 1583, 1520 (vs) | 1132 (m) |
| 2865 (w) | 2135 (w) | 1074 (m) | ||||||
| 5 and 6 | 3323 (vs) | 3323 (vs) | 2939 (w) | 2630 (w) | (1582) | 1645 (vs) | 1582, 1516 (vs) | 1131 (m) |
| 2864 (w) | 2131 (w) | 1075 (m) | ||||||
| 7 | 3321 (vs) | 3321 (vs) | 2939 (w) | 2624 (w) | (1583) | 1650 (vs) | 1583, 1520 (vs) | 1130 (m) |
| 2864 (w) | 2132 (w) | 1068 (m) | ||||||
| 8, 9, 10 | 3354 (m) | — | 2937 (w) | 2620 (w) | 1582 (vs) | — | 1516 (vs) | — |
| 2862 (w) | 2135 (w) |
Method 1: with a slight overpressure (dimethyl L-tartrate
:L-lysine = 1:2). Into a Sovirel® tube, triethylamine (58.70 mg, 0.58 mmol) was added, under nitrogen atmosphere and under continuous stirring to 4.00 mL of a methanol solution of L-lysine (314.30 mg, 2.15 mmol). A methanol solution (2.00 mL) of dimethyl L-tartrate (191.30 mg, 1.07 mmol) was added and the mixture was allowed to react at 80 °C for 24 hours (product 2). After cooling to room temperature, the mixture was filtered on a Büchner funnel and the solid obtained was washed with methanol and dried at room temperature (271.0 mg, 54% yield).The reaction was repeated increasing the reaction time to 3 days (product 4) and 7 days (product 6), respectively with 66% (335.20 mg) and 72% (368.00 mg) yields. The solids obtained were analysed by 1H-NMR (Table 2), 13C-NMR (Table 3), FT-IR (Table 4), SEC, thermal analyses (DSC and TGA) and acid–base titration.
Method 2: under vacuum (dimethyl L-tartrate
:L-lysine = 1:2). Into a 10 mL single-neck flask, L-lysine (146.20 mg, 1.00 mmol) was added to dimethyl L-tartrate (89.07 mg, 0.50 mmol). The mixture was allowed to react at 60 °C for 9 hours under vacuum in a rotary evaporator (product 7). At the end of the reaction, the formation of a yellow solid was observed: the solid was washed with methanol, filtered on a Büchner funnel and dried under vacuum at room temperature (85.20 mg, 36% yield). The solid obtained was analysed by 1H-NMR (Table 2), 13C-NMR (Table 3), FT-IR (Table 4), thermal analysis (DSC) and acid–base titration.
2.4. L-Lysine reactivity
The reaction was repeated increasing the reaction times to 12 hours (product 10). The products obtained were analysed by 1H-NMR (Table 2), 13C-NMR (Table 3), FT-IR (Table 4).
3. Results and discussion
The oligoamides were synthesized by polycondensation between L-lysine and dimethyl L-tartrate using molar ratios dimethyl L-tartrate/L-lysine 1:1 or 1:2. Two different synthetic methods were evaluated: products 1–6 were obtained using method 1, under nitrogen atmosphere, while product 7 was obtained using method 2, under vacuum. The conversion, DP, molecular weight and Tg for each product obtained with different reaction conditions are described in Table 1. The reaction scheme is reported in Fig. 1. The reactivity of L-lysine alone in the reaction conditions used in method 1 (product 8) and method 2 (products 9–10) was also studied as described in Table 1.
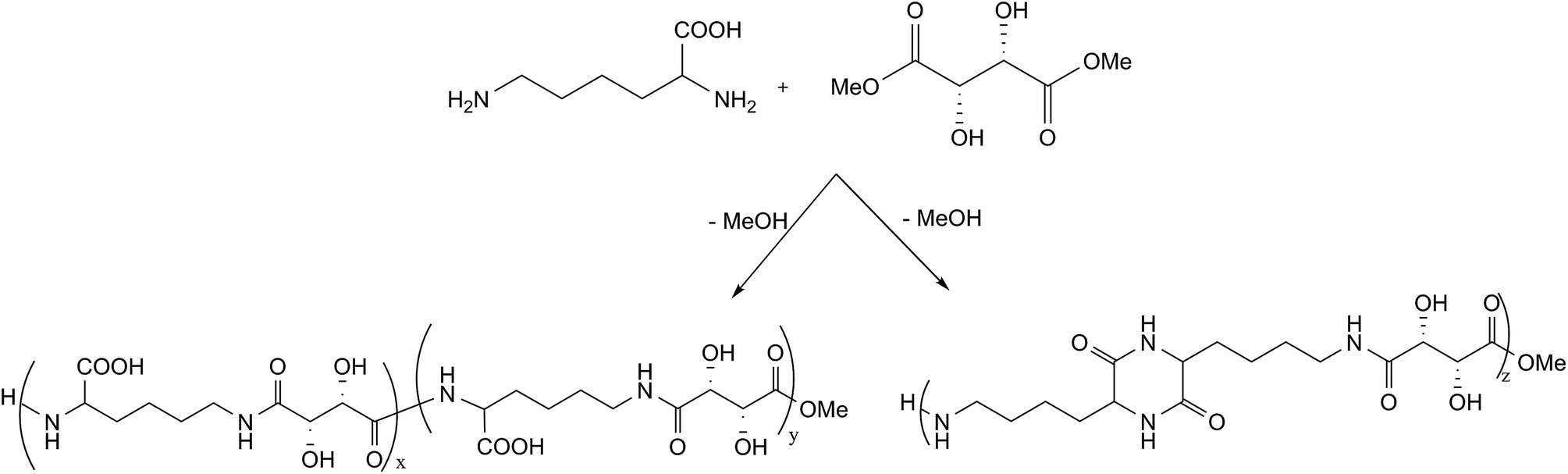 | ||
| Fig. 1 One-pot synthesis of polyamides from L-lysine and L-tartaric acid. | ||
L-Tartaric acid was activated as dimethyl ester and the polycondensation reaction was carried out in mild conditions in order to avoid protection/deprotection steps. In fact, the acid conversion into dimethyl ester promotes the subsequent condensation reactions with the diamino compounds.
As previously reported,15 L-tartaric acid dimethyl ester was obtained from L-tartaric acid through a reaction with H3BO3, which catalyzes the selective esterification of α-hydroxycarboxylic acids.
The condensation reactions were carried out with molar ratios between diester and amino acid 1:1 or 1:2 and with two different synthetic methods (see Table 1). Method 1 proceeds through a reaction in Sovirel® tube, under nitrogen atmosphere, with a slight overpressure. This method, previously used for the synthesis of various oligoamides,15,48 requires the presence of triethylamine as catalyst and methanol as solvent. An advantage of this method relies on the presence of the solvent, which guarantees a good temperature control preventing the formation of products with intense colours and leading to a good reproducibility of the results. Method 2 is a process without solvent and catalyst, more similar to industrial polycondensation processes. The reaction was performed using the vacuum evaporation technique to remove the by-products (i.e. methanol) formed and to let the polycondensation proceed. Advantages of this method rely on the absence of solvent and catalyst, on lower reaction times and on milder reaction temperatures (60 °C).
1H-NMR spectra of all products obtained with different synthetic procedures (method 1 or 2, molar ratio diester:amino acid equal to 1:1 or 1:2) were in agreement with the presence of a similar mixture of polycondensation products with a prevailing ratio L-lysine:L-tartrate 2:1 (Fig. 2); therefore, on the basis of the integrals of the characteristic signals, the presence as prevalent compound of an oligotartaramide containing L-lysine-diketopiperazine monomers could be hypothesized. The presence of an amide moiety was confirmed by the signals between 3.20 and 3.40 ppm (CH2NHCO, ε in the Fig. 2), and at 3.68 ppm (CHNHCO, α in the Fig. 2); signals related to Hγ, Hδ and Hβ at about 1.38, 1.56–1.68 and 1.83 ppm and to CHOH at 4.50 ppm were also present. At last, CH2NH2 group of L-lysine at 2.98 (Hε terminal) was present as chain-end: this signal can be taken as a reference for integrals calculations. Signals related to a different chain-end as an ester (at about 3.75 ppm) or an α amino group (at about 3.35 ppm) were not present; the lack of this last signal, which could actually be shifted and overlapped with other signals, was definitely confirmed by gCOSY analysis.
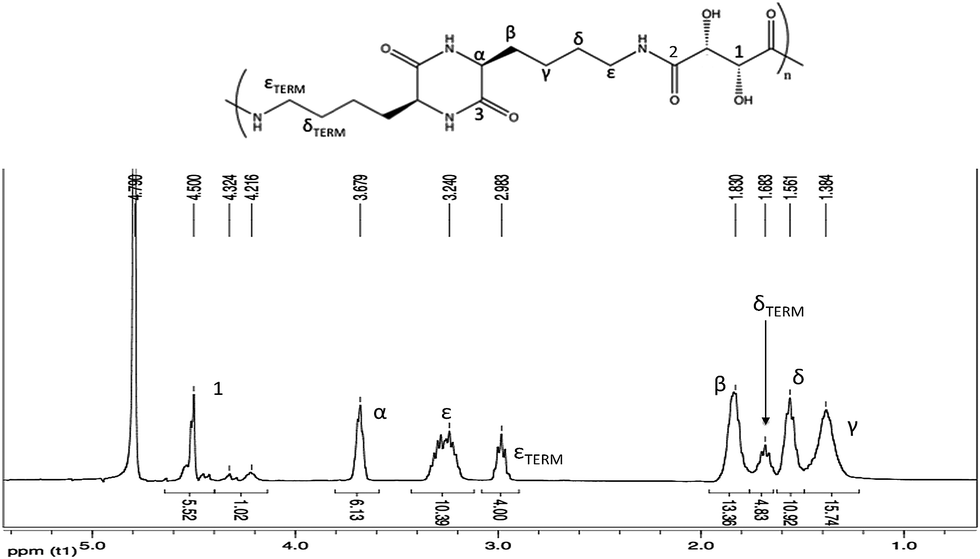 | ||
| Fig. 2 1H-NMR spectrum in D2O of product 1 (400 MHz). | ||
gCOSY spectrum (Fig. 3) confirms the attribution made through 1H-NMR analysis for the presence of coupling between the CH2NH2 group (ε terminal) with terminal Hδ and between CH2NHCO group (ε internal) with internal Hδ. Furthermore, it is possible to observe the correlation between the Hβ with the CHNHCO group (α amide diketopiperazine structure defined as “αD”) at 3.68 ppm and with another signal at 4.22 ppm. This last value could be attributed at the CHNHCO group of the linear oligoamide (“αL”) (Fig. 4), as reported in the literature for a similar structure.32
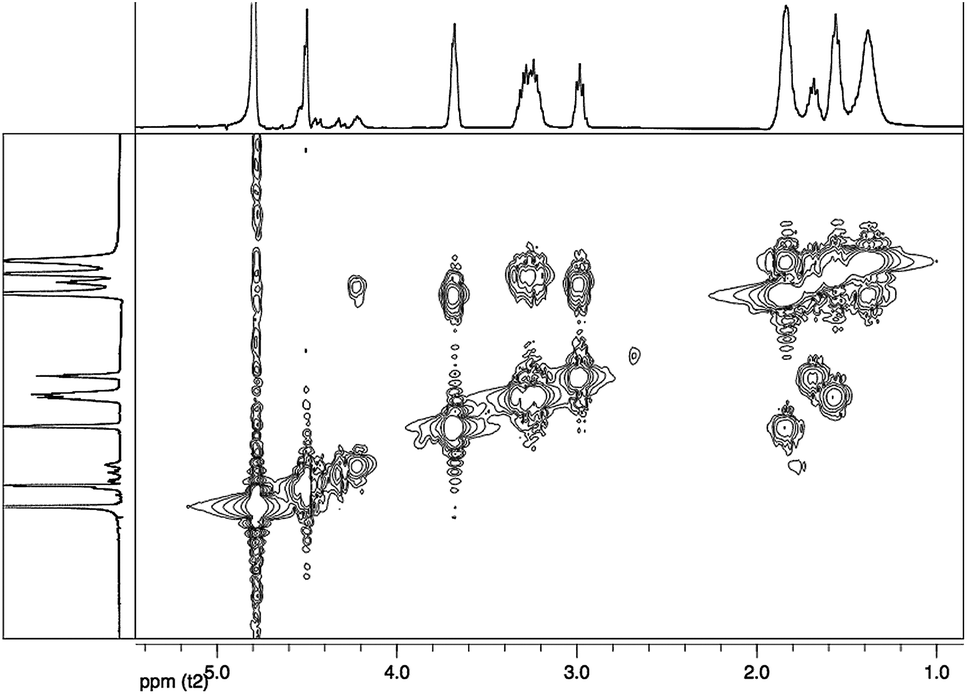 | ||
| Fig. 3 gCOSY spectrum in D2O of the product 1. | ||
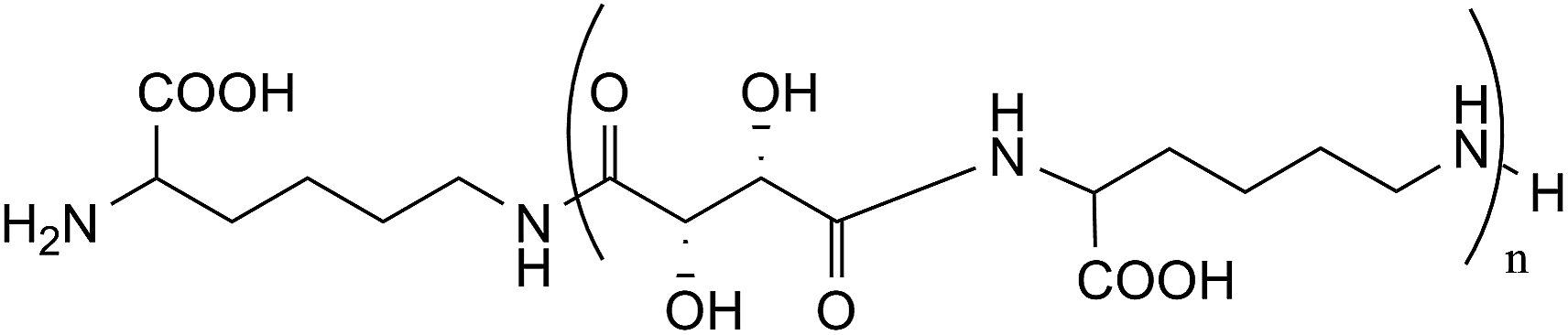 | ||
| Fig. 4 Linear oligotartaramide (ratio L-lysine/L-tartrate 1:1). | ||
13C-NMR (Fig. 5) and gHSQC (Fig. 6) spectra confirm oligoamide formation, thanks to the presence of the signals at 38.6 and 54.7 ppm, related to CH2NHCO (ε in Fig. 5) and CHNHCO (α in Fig. 5), respectively. Moreover, in the gHSQC spectrum, correlations between the signal at 54.7 ppm with two signals at 3.68 and 4.22 ppm confirm the presence of small amounts of linear oligotartaramide together with the diketopiperazine oligotartaramide. It is also possible to assign the signal at 21.6 ppm to Cγ, at 26.4 ppm to terminal Cδ, at 28.0 ppm to internal Cδ and at 30.2 ppm to Cβ. In the carbonyl amide range there are two signals, one at 173.6, the other one at 175.2 ppm, with the same intensity. The 13C-NMR spectrum confirms the absence of the ester termination due to the absence of signals at about 171.5 ppm and of the amino termination (CH(COOH)NH2) given the absence of a signal at about 55.5 ppm. Signals related to the L-lysine carboxyl group at about 181 ppm (COOH) are not detectable. Therefore, NMR data are in agreement with the conversion of L-lysine in two oligoamides, containing Hα with two very different chemical environments.
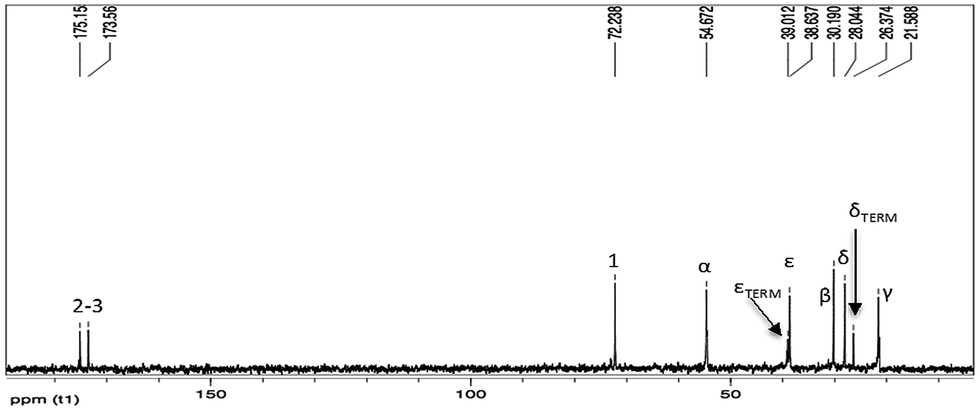 | ||
| Fig. 5 13C-NMR spectrum in D2O of the product 1. | ||
 | ||
| Fig. 6 gHSQC spectrum in D2O of the product 1: magnification of area between 2.50–5.50 ppm. | ||
In fact, in the oligoamide with diketopiperazine structure, present in higher concentration, HαD has a classic “amide-type” neighbourhood, while in the linear oligomer diester/L-lysine, present in lower concentration (Fig. 4), HαL has a higher de-shielding effect due to the presence of a free carboxylic group. The relative amounts of the two structures can be evaluated by the ratio between the integrals of HαD and HαL in CHNHCO signals related to the presence of 2H and 1H, respectively.
The determination of the size (i.e. average numeral molecular weight, Mn) of the synthesized molecules was performed via calculation of the integrals of the signals in the 1H-NMR spectrum. In fact, the signals for the different end groups (–CO2CH3, –CH2NH2, –CHNH2) can be used as reference values to determine the average molecular weight, comparing them with the integrals of the signals related to protons inside the polymer chain.
In the 1H and 13C-NMR analyses of the products, only the signal related to the terminal amino group (CH2NH2) is present. Based on this consideration, it is possible to calculate the number (n) of repeating units, according to the following calculations for the oligoamide with the diketopiperazine structure:
| Integral of Hγ signal = 4(n + 1) |
| Integral of Hβ signal = 4(n + 1) |
| Integral of Hδ internal signal = 4n |
| Integral of Hδ terminal signal = 4 |
| Integral of Hε in CH2NH2 signal = 4 |
| Integral of Hε in CH2NHCO signal = 4n |
| Integral of HαD in CHNHCO signal (αD) = 2(n + 1) |
| Integral of CHOH signal = 2n |
Similarly, it is possible to calculate the number (n) of repeating units, according to the following calculations, for the oligotartaramide with linear oligomer and ratio L-lysine/L-tartrate 1:1, structure present in low concentration:
| Integral of Hγ signal = 2(n + 1) |
| Integral of Hβ signal = 2(n + 1) |
| Integral of Hδ internal signal = 2(n − 1) |
| Integral of Hδ terminal signal = 4 |
| Integral of Hε in CH2NH2 signal = 4 |
| Integral of Hε in CH2NHCO signal = 2(n − 1) |
| Integral of HαL in CHNHCO (αL) = (n + 1) |
| Integral of CHOH signal = 2n. |
Considering the overlapping of almost all the signals of the two structures, the calculation of the molecular weight must be made considering the percentage contribution of the two structures for each signals and evaluating the correct value for integral of HαD and HαL in CHNHCO signal with respect to the corresponding Hε terminal contribute. Therefore, integrals elaboration by 1H-NMR was performed with the correction due to the presence of two different structures. However, due to the lower concentration of the linear structure, DP was calculated via 1H-NMR only for the diketopiperazine derivative.
To reduce the error resulting from the superposition of signals of two different structures and from the approximations made in the calculation, the evaluation of the DP was made considering the average of the values obtained for all the signals present in the spectrum (Table 5). The final value for DP and Mn of the synthesized products are reported in Table 1.
| C.P.a | DP CH–OH | DP CH2 (ε) | DP β, γ, δ | DP CH (αD) | DP average | Dik.b (%) |
|---|---|---|---|---|---|---|
| a Code product.b Diketopiperazine oligoamide (%). | ||||||
| 1 | 3.2 | 3.4 | 3.6 | 3.2 | 3.4 | 83.0 |
| 2 | 2.8 | 2.9 | 3.1 | 2.8 | 2.9 | 81.3 |
| 3 | 4.0 | 3.8 | 4.0 | 4.1 | 4.0 | 75.5 |
| 4 | 2.9 | 2.9 | 3.3 | 2.7 | 2.9 | 84.8 |
| 5 | 4.6 | 5.0 | 5.2 | 4.6 | 4.8 | 82.9 |
| 6 | 3.7 | 3.9 | 4.0 | 3.5 | 3.8 | 89.9 |
| 7 | 2.2 | 2.2 | 2.4 | 2.2 | 2.3 | 94.3 |
MALDI analysis (Table 6) and ESI LC-MS (Fig. 7) were performed on the sample 5 and allowed to identify the presence of oligomers with ratio diester: L-lysine 1:1. In particular, the data obtained using MALDI allow to quantify the molecular weight of the linear oligoamide, that was detected with more difficulty in the NMR spectra, probably because it is present in low quantities. However, peaks at m/z 407 (406 + 1) and 667 (666 + 1) are partially overlapped to the matrix and then not easily visible in the MALDI spectrum while the presence of these peaks was detectable in the ESI LC-MS spectrum. On the contrary, the oligomers based on tartaric-lysine diketopiperazine structure were not detectable by MALDI or ESI LC-MS probably due to a low stability of the diketopiperazine system in the analytical conditions.
| Molecular mass (m/z) | Assignment (repeating units) | MW (g mol−1) |
|---|---|---|
| a Peaks also detectable in the ESI LC-MS spectrum. | ||
| 407a | 1 | (260) + 145 + 1 = 406 |
| 667a | 2 | (260 × 2) + 145 + 1 = 666 |
| 928 | 3 | (260 × 3) + 145 + 1 = 926 |
| 1188 | 4 | (260 × 4) + 145 + 1 = 1186 |
| 1448 | 5 | (260 × 5) + 145 + 1 = 1446 |
| 1708 | 6 | (260 × 6) + 145 + 1 = 1706 |
| 1969 | 7 | (260 × 7) + 145 + 1 = 1966 |
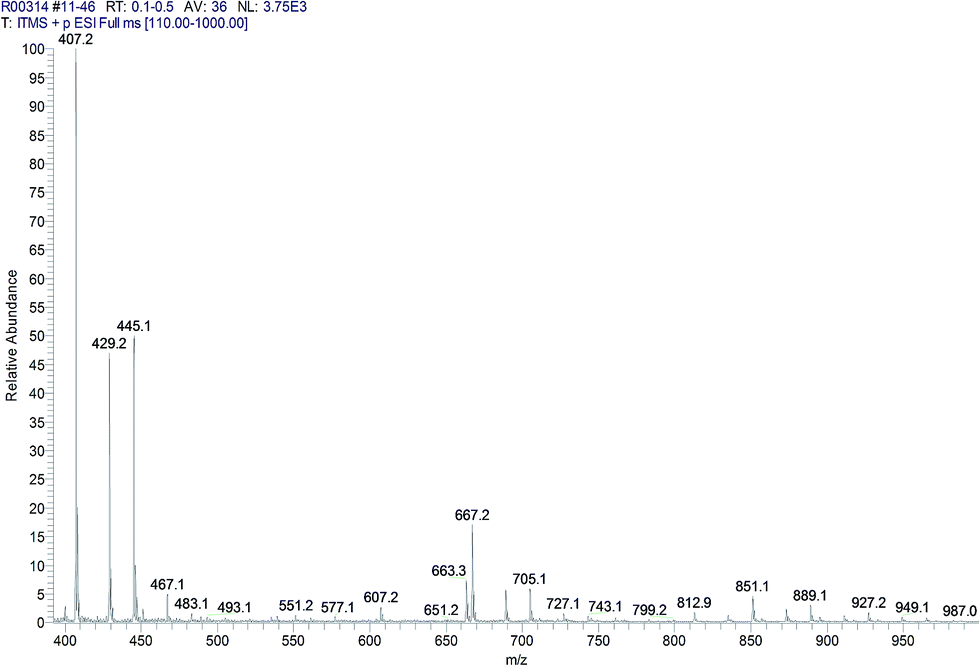 | ||
| Fig. 7 ESI LC-MS spectrum of sample 5. | ||
Potentiometric titration confirms the presence of two types of amino groups and one carboxyl group in all products. However, the presence of only one terminal amino group was detected using dilute solution (<2.5 mg mL−1) whereas performing the analysis with higher amounts of sample (over 3 mg mL−1) two types of amino groups and a carboxyl group were detected.
Also this result can be correlated with the presence of very low amounts of linear oligoamide structure, detectable only at relatively high concentrations.
SEC analyses were performed on the samples 5 and 6 (Fig. 8): the shape and hydrodynamic volume of the peaks confirm that the products obtained have low molecular weights. Nevertheless, the analyses show the presence of more peaks, indicating that more than one product is present. Peaks for product 5 (molar ratio between reagents 1:1) and product 6 (molar ratio between reagents 1:2) have the same retention time and slightly different intensity that is correlated to their concentration in the final product. Since molecular weights obtained via SEC are not absolute values, molecular weights evaluation was performed only by NMR or MALDI, as explained before.
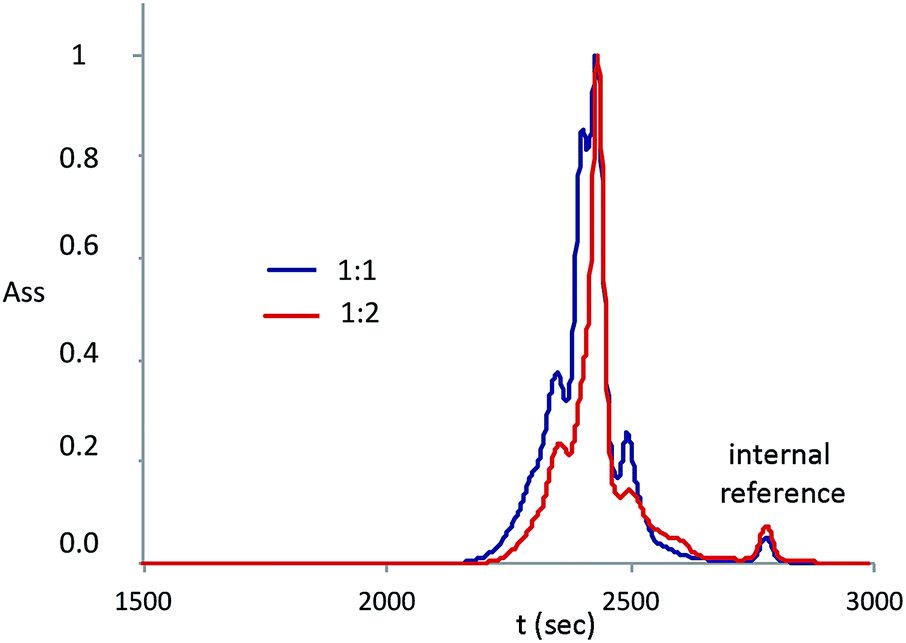 | ||
| Fig. 8 GPC of the samples 5 and 6 (peaks normalized around the main peak). | ||
Products were characterized also by polarimetric analysis, to evaluate their optical activity, and all of them showed values ranging between +54° and +56°.
Thermal properties of the oligomers synthesized were evaluated via DSC, measuring glass transition (Tg) and melting temperature (Tm). Tg, having values slightly higher than 120 °C, become visible when higher MWs are obtained (products 2, 3 and 5), since when the oligomer chain is very short (DP = 2) no glass transitions exist (product 7). Tm is not visible before 290 °C; over this temperature, melting and degradation probably occur at the same time. Thermal degradation of product 1 was studied by TGA (Fig. 9) and two weight losses were observed: the first between 175–280 °C and the second between 300–450 °C.
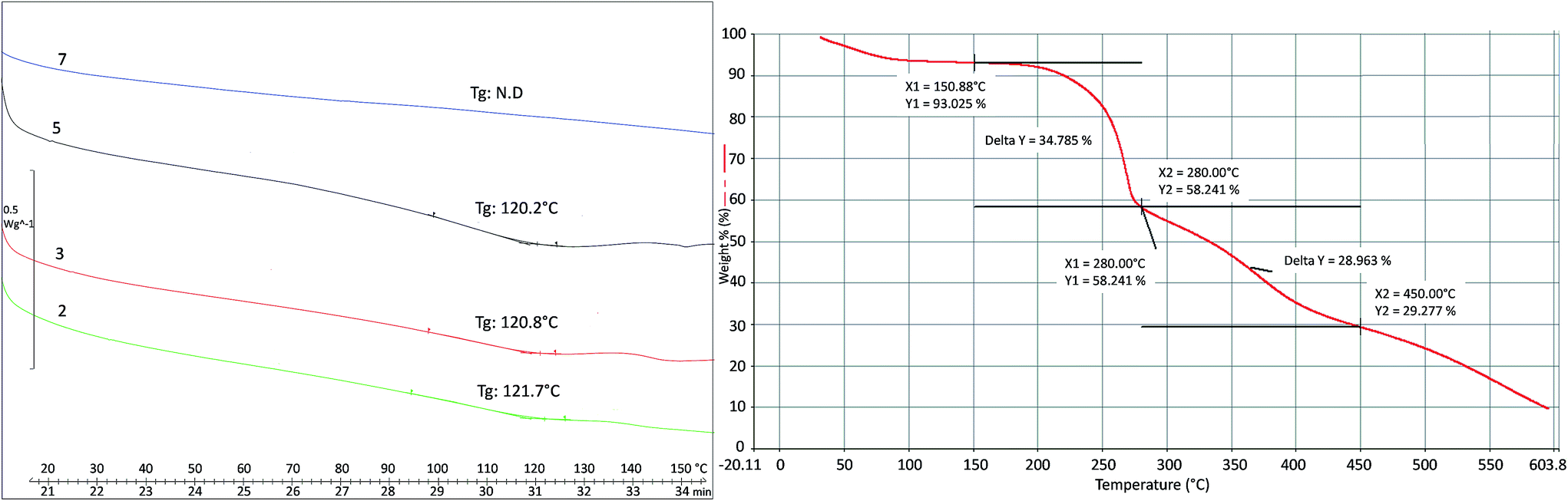 | ||
| Fig. 9 Glass transition temperature (Tg) of products 2, 3, 5, 7 (left) and thermal degradation studied by TGA of product 1 (right). | ||
Finally, the behaviour of L-lysine when heated alone in the same reaction conditions reported in method 1 and method 2 was also studied; in both cases oligoamides were not formed, and unreacted L-lysine was recovered. This confirms that the presence of dimethyl L-tartrate promotes the formation of oligoamides and in particular of diketopiperazine structures.
4. Conclusions
Oligoamides deriving from L-lysine and L-tartaric acid have been synthesized in a new one-pot process. Two synthetic methods have been reported, leading to the same type of polycondensation product, that is water soluble, optically active and has high glass transition temperature; the oligotartaramides were mainly composed by L-lysine diketopiperazine monomer units (about 80–90%) with low amount (about 10–20%) of the linear oligoamide obtained by polycondensation from dimethyl L-tartrate and L-lysine in ratio 1:1. Method 1, used for all previously oligoamide syntheses, guarantees a good temperature control, preventing formation of intense colours of the products and maintaining a good reproducibility of the results. Method 2, using bulk polymerization, relies on the absence of the solvent and catalyst, the reduction of reaction times and the use of lower temperatures. The presence of dimethyl L-tartrate is required to enable the formation of oligoamides in mild reaction conditions. The prevailing formation of diketopiperazine structures could be in agreement with an initial activation of the amino group in ε and a consequent reaction between the amino and carboxylic groups in α position, of two different L-lysine molecules, to obtain a diketopiperazine structure.
These results appear significant because they allow to obtain new oligoamides applying the green chemistry principles. Further studies are in progress in our laboratory to their use within the scope of the preservation of cellulosic material and to optimize the reaction conditions with the aim to obtain higher molecular weights as well as higher yields for the title compounds.
Acknowledgements
The authors thank the Ente Cassa di Risparmio di Firenze for financial support.Notes and references
- G. Cipriani, A. Salvini, P. Baglioni and E. Bucciarelli, J. Appl. Polym. Sci., 2010, 118, 2939 CrossRef CAS.
- L. Yu, K. Dean and L. Li, Prog. Polym. Sci., 2006, 31, 576 CrossRef CAS.
- Q. Wang, J. S. Dordick and R. J. Linhardt, Chem. Mater., 2002, 14, 3232 CrossRef CAS.
- H. Gruber and S. Knaus, Macromol. Symp., 2000, 152, 95 CrossRef CAS.
- U. R. Vaidya and M. Bhattacharya, Biodegradable compositions of synthetic and natural polymers, PCT Pat. Int. Appl., WO9323456 A1, 1993.
- J. Helin, J. Natunen, R. Lehtilae, M. Roslund and R. Leino, Novel carbohydrate compositions and a process of preparing same, PCT Pat. Int. Appl., WO2004031244 A1, 2004.
- A. R. Alborzi, S. Zahmatkesh, K. Zare and J. Sadeghi, Amino Acids, 2011, 41, 485 CrossRef CAS PubMed.
- J. Puiggalì and J. A. Subirana, J. Pept. Sci., 2005, 11, 247 CrossRef PubMed.
- T. Kunisaki, K. Kawai, K. Hirohata, K. Minami and K. Kondo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 927 CrossRef CAS.
- K. Terada, E. B. Berda, K. B. Wagener, F. Sanda and T. Masuda, Macromolecules, 2008, 41, 6041 CrossRef CAS.
- F. Rafiemanzelat, A. Fathollahi Zonouz and G. Emtiazi, Polym. Degrad. Stab., 2012, 97, 72 CrossRef CAS.
- S. Dhamaniya and J. Jacob, Polym. Bull., 2012, 68, 1287 CrossRef CAS.
- S. Muñoz-Guerra, High Perform. Polym., 2012, 24, 9 CrossRef.
- E. C. Schule, in Encyclopedia of polymer science and technology, 1969, vol. 10, p. 468 Search PubMed.
- G. Cipriani, A. Salvini, M. Fioravanti, G. Di Giulio and M. Malavolti, J. Appl. Polym. Sci., 2013, 127, 420 CrossRef CAS.
- J. J. Bou, I. Iribarren and S. Muñoz-Guerra, Macromolecules, 1994, 27, 5263 CrossRef CAS.
- J. J. Bou, A. Rodriguez-Galan and S. Muñoz-Guerra, Macromolecules, 1993, 26, 5664 CrossRef CAS.
- M. S. Marques, C. Regano, J. Nyugen, L. Aidanpa and S. Muñoz-Guerra, Polymer, 2000, 41, 2765 CrossRef CAS.
- K. Terada, F. Sanda and T. Masuda, J. Macromol. Sci., Part A: Pure Appl. Chem., 2007, 44, 789 CrossRef CAS.
- D. W. Morton and D. E. Kiely, J. Appl. Polym. Sci., 2000, 77, 3085 CrossRef CAS.
- D. W. Morton and D. E. Kiely, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 604 CrossRef CAS.
- C. Regaño, A. Martínez De Ilarduya, J. I. Iribarren and S. Muñoz-Guerra, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2687 CrossRef.
- A. Alla, A. Rodriguez-Galan, A. Martinez de Ilarduya and S. Muñoz-Guerra, Polymer, 1997, 38, 4935 CrossRef CAS.
- A. Alla, J. Oxelbark, A. Rodríguez-Galán and S. Muñoz-Guerra, Polymer, 2005, 46, 2854 CrossRef CAS.
- D. Esquivel, J. J. Bou and S. Muñoz-Guerra, Polymer, 2003, 44, 6169 CrossRef CAS.
- Z. Bohak and E. Katchalski, Biochemistry, 1963, 2, 228 CrossRef CAS PubMed.
- S. Jin, P. M. Mungara and K. E. Gonsalves, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 499 CrossRef CAS.
- A. El-Faham, H. H. Hassan and S. N. Khattab, Chem. Cent. J., 2012, 6, 128 CAS.
- S. G. Roy and P. De, J. Appl. Polym. Sci., 2014, 131, 41084 CrossRef.
- M. A. Majó, J. J. Bou, C. Herranz and S. Muñoz-Guerra, Macromol. Chem. Phys., 2006, 207, 615 CrossRef.
- I. Gachard, B. Coutin and H. Sekiguchi, Polym. Bull., 1997, 38, 643 CrossRef CAS.
- J. J. Bou and S. Muñoz-Guerra, Polymer, 1995, 36, 181 CrossRef CAS.
- M. A. Majó, A. Alla, J. J. Bou, C. Herranz and S. Muñoz-Guerra, Eur. Polym. J., 2004, 40, 2699 CrossRef.
- A. D. Borthwick, Chem. Rev., 2012, 112, 3641 CrossRef CAS PubMed.
- E. Katchalski, I. Grossfeld and M. Frankel, J. Am. Chem. Soc., 1946, 68, 879 CrossRef CAS PubMed.
- J. Akerlund, S. Harmeier, J. Pumphrey, D. C. Timm and J. I. Brand, J. Appl. Polym. Sci., 2000, 78, 2213 CrossRef CAS.
- D. A. Parrish, PhD thesis, University of Southern Mississippi, 2002.
- V. Crescenzi, V. Giancotti and F. Quadrifoglio, Makromol. Chem., 1968, 120, 220 CrossRef CAS.
- A. Ciana, V. Crescenzi, V. Giancotti, E. Russo and L. Salvestrini, US Pat., US3763091, 1973.
- J. Akerlund, S. Harmeier, J. Pumphrey, D. C. Timm and J. I. Brand, J. Appl. Polym. Sci., 2000, 78, 2213 CrossRef CAS.
- D. A. Parrish and L. J. Mathias, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2000, 41, 1311 CAS.
- M. Jiang, G. Chen, P. Lu and J. Dong, J. Appl. Polym. Sci., 2014, 131, 39807 Search PubMed.
- T. A. Houston, B. L. Wilkinson and J. T. Blanchfield, Org. Lett., 2004, 6, 679 CrossRef CAS PubMed.
- J. Xu, M. Han, J. Zhang, Y. Guo, H. Qian and W. Zhang, J. Chem. Technol. Biotechnol., 2014, 89, 1924 CrossRef CAS.
- F. Käβ, I. Hariskos, A. Michel, H. J. Brandt, R. Spann, S. Junne, W. Wiechert, P. Neubauer and M. Oldiges, Bioprocess Biosyst. Eng., 2014, 37, 1151 CrossRef PubMed.
- T. Badel, S. Jeol and F. Touraud, Supramolecular polymers and compositions containing said polymers, US Pat.,US9012567 B2, 2011.
- H. Schuttemberg and R. C. Schultz, Angew. Chem., 1976, 88, 848 CrossRef.
- R. Oliva, F. Albanese, G. Cipriani, F. Ridi, D. Giomi, M. Malavolti, L. Bernini and A. Salvini, J. Polym. Res., 2014, 21, 496, DOI:10.1007/s10965-014-0496-2.
| This journal is © The Royal Society of Chemistry 2017 |