DOI:
10.1039/C7RA00710H
(Paper)
RSC Adv., 2017,
7, 14046-14052
Supramolecular catalysis in the methylation of meta-phenylene ethynylene foldamer containing N,N-dimethylaminopyridine†
Received
17th January 2017
, Accepted 24th February 2017
First published on 2nd March 2017
Abstract
Density functional theory calculations were performed to elucidate the mechanism of the methylation reaction of the N,N-dimethylaminopyridine (DMAP)-modified meta-phenylene ethynylene foldamer with eight methyl sulfonate esters with different alkyl groups. The helical structure of the DMAP-modified mPE foldamer results from multiple intramolecular π–π stacking interactions between m-phenylene ethynylene arms, which can be characterized by the helical energy. The noncovalent interactions between the foldamer and the substrate can stabilize the transition state and result in an acceleration of the methylation reaction of the foldamer. Due to the different shapes of the alkyl chains of the methylating agents, the methylation rates of the foldamers with linear and branched substrates show different rules. It is expected that these mechanistic insights into supramolecular catalysis can be used in the design and preparation of supramolecular catalysts and reactors.
Introduction
As a rich research model system, phenylene ethynylene oligomers have been applied extensively in supramolecular chemistry and nanoscale materials.1–6 They include para-, meta-, and ortho-connected types and can be used to construct various architectures. The fully conjugated para-phenylene ethynylene oligomers have been identified as excellent candidates for molecular electronics.1–3 The artificial meta- and ortho-phenylene ethynylene oligomers with unique helical structures, namely foldamers, have also been used to mimic the behaviors of natural biopolymers, such as molecular recognition and catalysis.4–6 Hence, foldamer chemistry is also a fascinating field of supramolecular chemistry and supramolecular catalysis.7,8
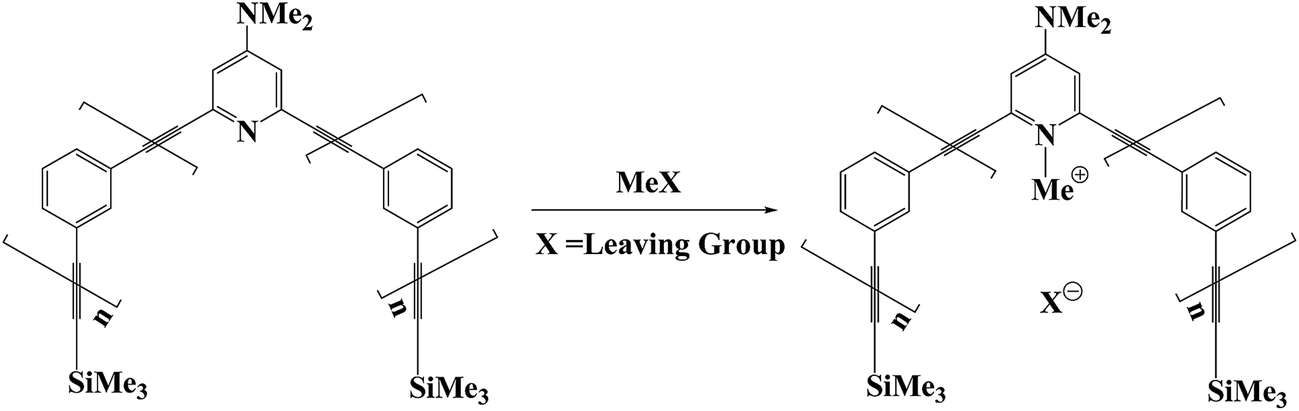
At the same time, methylation is one of the most important reactions in biological processes, such as DNA and RNA methylation.9–11 Recently, the meta-phenylene ethynylene (mPE) foldamer that contains N,N-dimethylaminopyridine (DMAP) was used as a supramolecular reactor to accelerate the methylation of DMAP with alkyl methanesulfonate esters (Fig. 1).12–17 As a reactive sieve, the DMAP-modified mPE foldamer has a well-defined helical structure with a tubular cavity and an active site to recognize and bind guest molecules. The mPE foldamer can enhance the methylation rate of DMAP up to 1600-fold relative to that of the reference reactions.15 Despite these advances in foldamer catalysis experiments, the exact nature of this rate acceleration is unknown. Detailed theoretical studies are required to understand the origin of the observed enhancement.
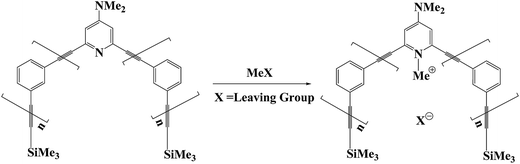 |
| | Fig. 1 Methylation of DMAP-modified mPE foldamer. “n” represents the number of aromatic ring units. | |
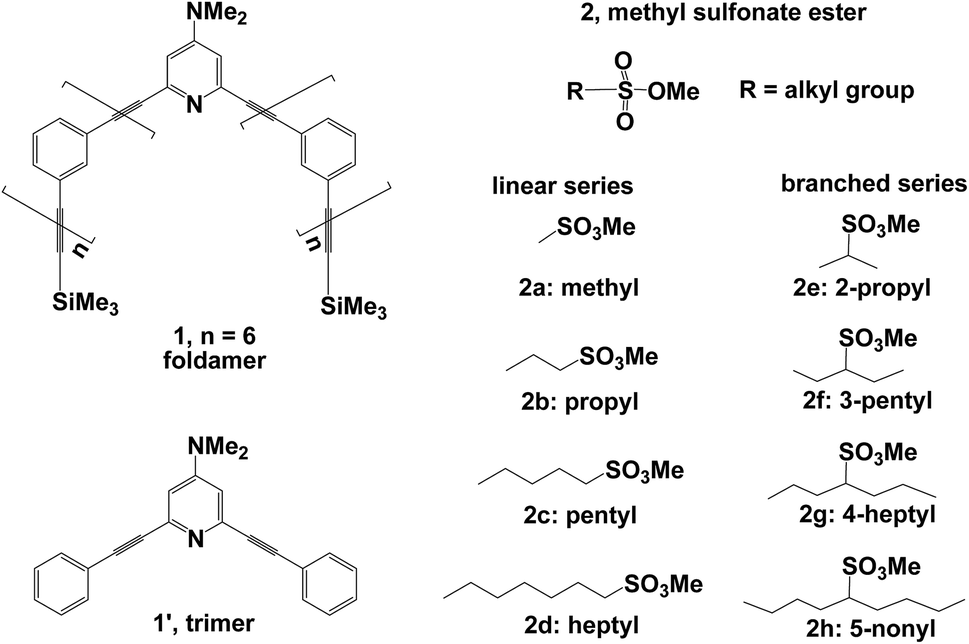
Several theoretical investigations of phenylene ethynylene foldamers exist, which are only focused on helix formation and dynamic behavior.18–25 In this work, we investigate mechanistic details of the methylation reaction of DMAP-modified mPE foldamer with different methyl sulfonate esters (Fig. 2). The details were described in the Computational section. For methylation reactions in mPE foldamer 1, we chose eight methyl sulfonates with linear (2a, 2b, 2c, and 2d) and branched (2e, 2f, 2g, and 2h) alkyl groups as methylating agents. The reaction of the DMAP-modified trimer 1′ with each substrate can be considered to be a reference reaction, because of its unfolded conformation. We focus on the multiple effects of the host–guest interaction on the free energy profile of the methylation reaction of the DMAP-modified mPE foldamer. Calculated results from this study will provide insight into the role of noncovalent interactions between the foldamer and guest molecules in accelerating the methylation reaction.
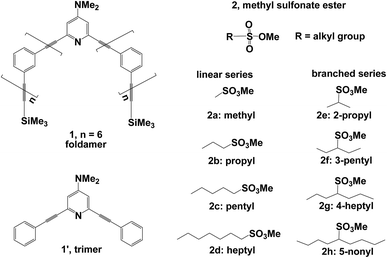 |
| | Fig. 2 DMAP-modified mPE foldamer 1, trimer 1′, and linear (2a, 2b, 2c, and 2d) and branched (2e, 2f, 2g, and 2h) methyl sulfonate esters as methylating agents. | |
Results and discussion
Structure of DMAP-modified mPE oligomer
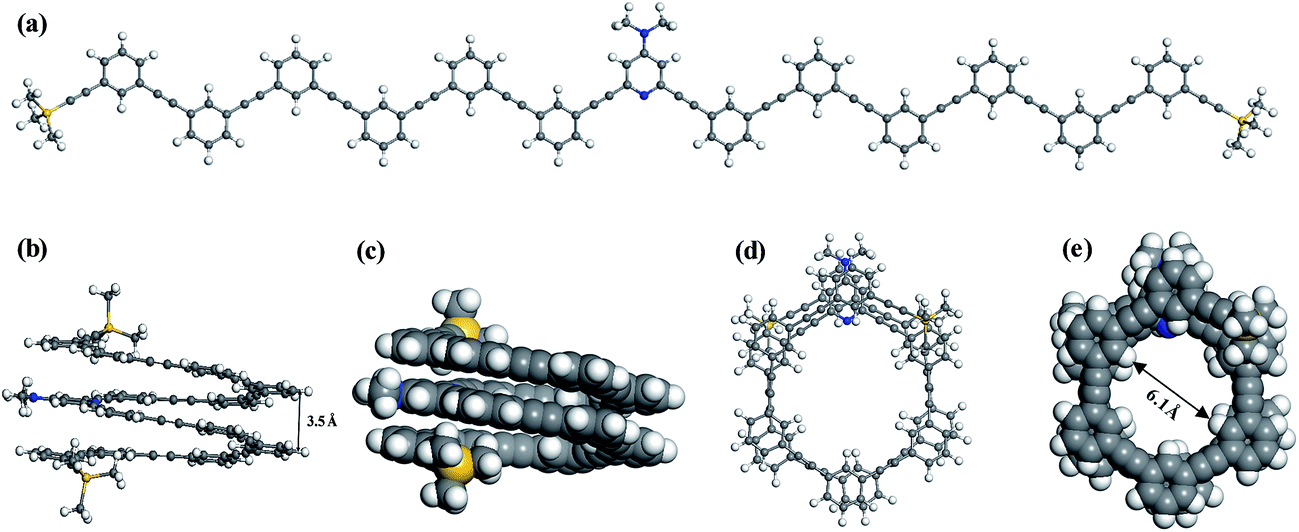
In acetonitrile solvent, the DMAP-modified mPE oligomer with different monomer units will fold into a helical conformation with a cavity by multiple intramolecular π–π stacking interactions between meta-phenylene ethynylene arms.17 To verify the role of π–π interactions, we optimized the folded and unfolded structures of the DMAP-modified mPE oligomer with 13 monomer units using two density functionals, namely, B3LYP and M06-2X, and the same basis set, 6-31G(d,p). The B3LYP results show that the helical conformation of foldamer 1 has a mean center-to-center distance of ∼4.2 Å and an almost equal energy with the unfolded conformation of foldamer 1, indicating that the B3LYP functional neglects π–π stacking interactions and cannot describe this type of weak interaction.26 In contrast, the M06-2X functional can reasonably describe π–π stacking interactions.26–28 The M06-2X results show that the energy of foldamer 1 is lower by 45.5 kcal mol−1 than that of the corresponding unfolded structure (Fig. 3). The helical structure has a center-to-center distance of ∼3.5 Å and a cylinder with the diameter of ∼6.1 Å under considering van der Waals radius of hydrogen atoms of the inner wall of the cylinder.29 All these findings are consistent with experimental structures.30 It should be noted that the calculated potential energy of the foldamer may further decrease by using a larger basis set. To gain a compromise between accuracy and computational cost, all calculation results reported below were obtained at the M06-2X/6-31G(d,p) level.
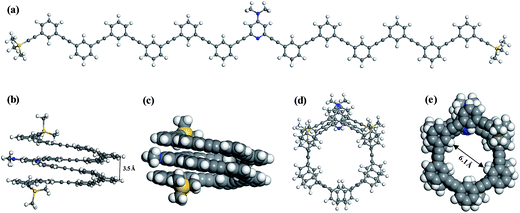 |
| | Fig. 3 The unfolded (a) and folded (b–e) structures of DMAP-modified mPE oligomer with 13 monomers, which were optimized at the M06-2X/6-31(d,p) level. (b–e) represent ball-stick (b and d) and CPK (c and e) models of side and top views of foldamer 1, respectively. | |
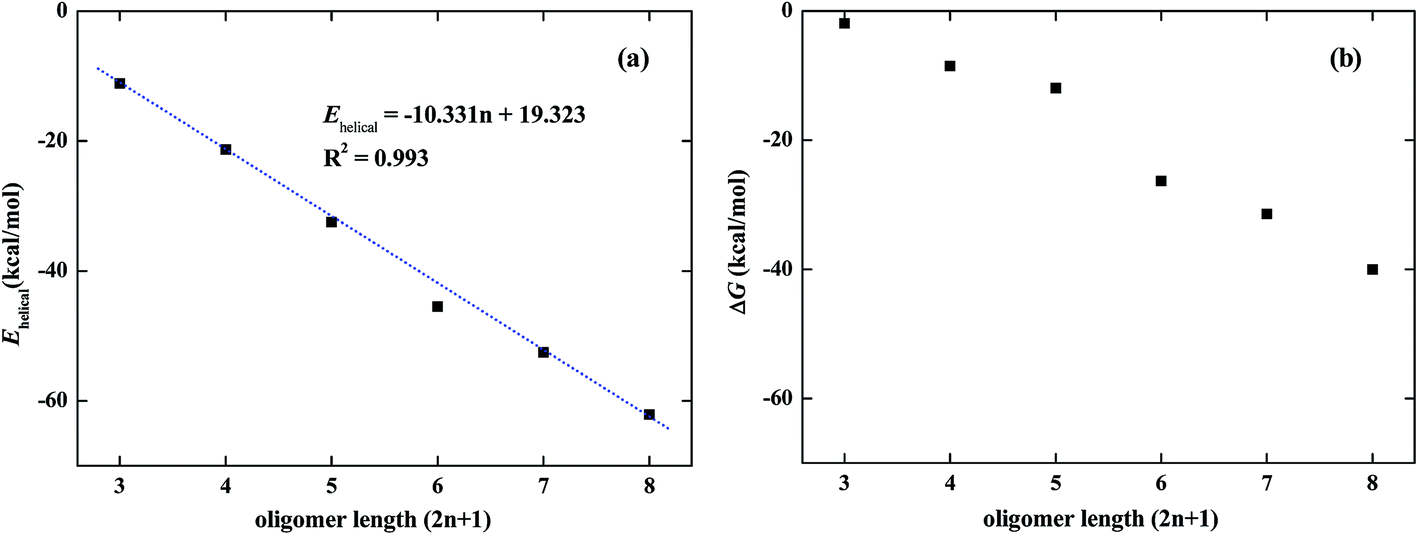
The π–π stacking interaction between meta-phenylene ethynylene arms can be characterized by the energy difference between the folded and unfolded conformations of the DMAP-modified mPE oligomer. We can term this energy difference the helical energy (Ehelical), which can be calculated according to the following formula:
| | |
Ehelical = Efolded − Eunfolded
| (1) |
where
Efolded and
Eunfolded represent the energies of the folded and unfolded conformations of the DMAP-modified
mPE oligomer, respectively. As shown in
Fig. 4, the helical energy of the DMAP-modified
mPE oligomer decreases with the monomer units (2
n + 1,
n = 3, 4, 5, 6, 7, and 8). The contribution of π–π interactions between two
meta-phenylene ethynylene arms is approximately 10.3 kcal mol
−1. Simultaneously, the helical process of the DMAP-modified
mPE oligomer is a process of entropy reduction. When the length (2
n + 1) of the
mPE oligomer is short, the free energy change of the folded process is too small and the corresponding foldamer may be unstable. The optimized structures of foldamers with 7–17 monomers are shown in Fig. S1.
† It can also be seen that even if the oligomer with fewer monomers can be folded, the cavity has very shallow depth and cannot accelerate the reaction. When the monomer unit is up to 13, the foldamer is stable due to the large free energy change of −26.3 kcal mol
−1 and can form a unique cavity. Because of the strong alkalinity of DMAP, the unique cavity is able to recognize and bind linear and branched methyl sulfonates, which can be termed a reactive sieve.
15,16
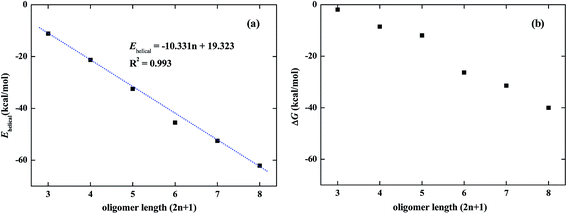 |
| | Fig. 4 Helical energy (a) of DMAP-modified mPE oligomer with different monomer units (2n + 1, n = 3, 4, 5, 6, 7, and 8) and the free energy change (b) between unfolded and folded conformations. | |
Methylation of DMAP-modified mPE foldamer with different linear substrates
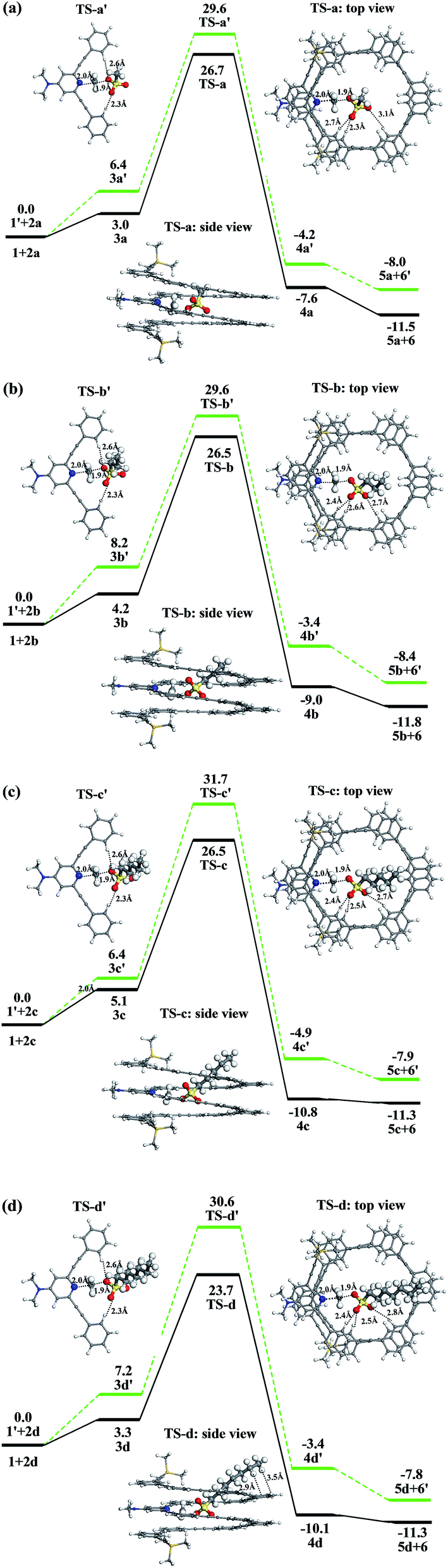
We firstly explored the reaction mechanism for the methylation of mPE foldamer 1 with four linear methylating agents (2a–2d). For comparison, we also studied the corresponding reference reactions, the uncatalyzed methylation reactions of the DMAP-modified trimer 1′, to illustrate the role of the host–guest interaction in the rate enhancement. The free energy profiles of the methylation reactions are displayed in Fig. 5.
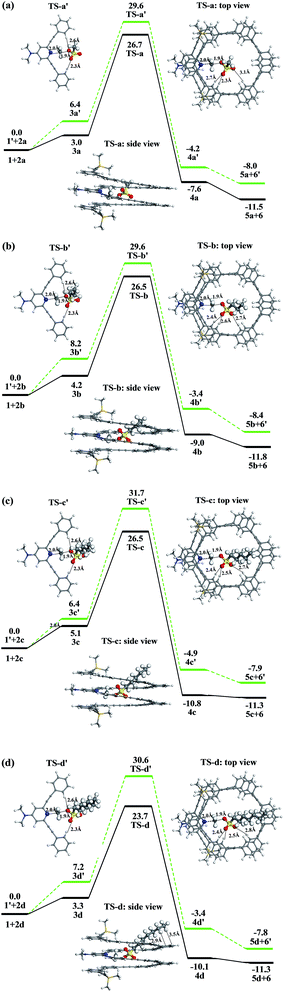 |
| | Fig. 5 Free energy profiles (ΔG in kcal mol−1, (a–d)) of methylation reactions of DMAP-modified mPE foldamer 1 and trimer 1′ with different linear methyl sulfonate esters (2a–2d). The black and green lines represent the methylation reactions of foldamer 1 and trimer 1′, respectively. | |
We use methyl heptylsulfonate 2d as methylating agent to rationalize the methylation reaction of the foldamer. When methyl heptylsulfonate 2d is added to the DMAP-modified mPE foldamer 1, a host–guest complex 3d is formed, which is less stable by 3.3 kcal mol−1 than the reactants. In the intermediate 3d, the methyl heptylsulfonate is close to the DMAP unit with a C⋯N distance of 2.9 Å, which indicates that the methyl agent is preorganized for subsequent methylation. The methyl group of 2d is transferred to the DMAP unit of the foldamer via the transition state TS-d with a free energy barrier of 23.7 kcal mol−1. This is a bimolecular nucleophilic substitution (SN2) process, in which the lone pair of the pyridine nitrogen atom attacks the methyl of sulfonate, and at the same time, sulfonate leaves. In the transition state TS-d, the C⋯N distance is 2.0 Å, which indicates that C–N bond formation is in process. As shown in Fig. 5d, the linear methyl heptylsulfonate 2d reclines in the cavity and does not erect in the cavity of the foldamer. Three C–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S hydrogen bonds exist between the substrate and the foldamer with distances of 2.4, 2.5, and 2.8 Å. The alkyl chain of the substrate is bent to achieve C–H⋯π interaction with the meta-phenylene ethynylene arm of foldamer 1. The distance between the hydrogen atom of the alkyl chain and the center of the benzene ring is 2.9–3.5 Å. Lastly, an ionic compound 4d that is composed of the sulfonate anion 5d and the foldamer cation 6 is formed. The overall reaction is exergonic by −11.3 kcal mol−1 and has an activation free energy barrier of 23.7 kcal mol−1 with respect to the initial reactants.
S hydrogen bonds exist between the substrate and the foldamer with distances of 2.4, 2.5, and 2.8 Å. The alkyl chain of the substrate is bent to achieve C–H⋯π interaction with the meta-phenylene ethynylene arm of foldamer 1. The distance between the hydrogen atom of the alkyl chain and the center of the benzene ring is 2.9–3.5 Å. Lastly, an ionic compound 4d that is composed of the sulfonate anion 5d and the foldamer cation 6 is formed. The overall reaction is exergonic by −11.3 kcal mol−1 and has an activation free energy barrier of 23.7 kcal mol−1 with respect to the initial reactants.
To understand which factors accelerate methylation of the foldamer 1, we have investigated the free energy profile of the uncatalyzed methylation reaction of DMAP-modified trimer 1′ with methyl heptylsulfonate 2d, shown in Fig. 5d. First, these two reactants would also form an encounter complex 3d′, in which the C⋯N distance is 2.9 Å. The ionic compound 4d′ is formed via the transition state TS-d′. In the transition state TS-d′, the C⋯N distance is the same as that in the TS-d. But only two C–H⋯OS hydrogen bonds exist between the substrate and the trimer 1′ with distances of 2.3 and 2.6 Å. The activation barrier for the methylation reaction is 30.6 kcal mol−1. This barrier is higher than that of the methylation reaction of the foldamer 1 by 6.9 kcal mol−1. The barrier of the reference reaction is contributed to by an enthalpic term of 17.1 kcal mol−1 and an entropic term of 13.5 kcal mol−1. In contrast, the corresponding enthalpic and entropic terms in the methylation reaction of the foldamer are 7.7 and 16.0 kcal mol−1, respectively. As a result, the enthalpic contribution (7.7 minus 17.1) offsets the disadvantageous entropic contribution (16.0 minus 13.5) and plays a major role in stabilizing the transition state in the foldamer. A comparison of the optimized geometries of TS-d and TS-d′ shows that enthalpic stabilization may result because of three hydrogen bonds and C–H⋯π interactions between substrate and foldamer in TS-d, but only pure hydrogen bonds in TS-d′.
With methyl methylsulfonate 2a, methyl propylsulfonate 2b, methyl pentylsulfonate 2c, and methyl heptylsulfonate 2d as methylating agents, the methylation rate within foldamer 1 is also accelerated. For 2a, the activation barrier of the methylation reaction decreases from 29.6 kcal mol−1 with trimer 1′ to 26.7 kcal mol−1 in foldamer 1. For 2b, the barrier of the methylation reaction decreases from 29.6 to 26.5 kcal mol−1. For 2c, the methylation barrier also decreases from 31.7 to 26.5 kcal mol−1. As listed in Table 1, the activation energies (Ga) of the methylation reactions of trimer 1′ and foldamer 1 decrease by 2.9, 3.1, 5.2, and 6.9 kcal mol−1 for 2a–2d, respectively. This increased rule of linear methylating agents can provide a reasonable explanation of the methylation rate enhancement of the foldamer. Meanwhile, the activation energies for foldamer 1 with substrates 2a, 2b, and 2c are very similar, yet when the size is increased to heptyl in 2d, the activation energy is ∼3 kcal mol−1 lower. The reason is that the methyl sulfonate with long alkyl chain has stronger C–H⋯π interactions with the foldamer, which also can be seen from side-view structures of transition states in Fig. 5.
Table 1 Activation free energies (Ga in kcal mol−1) and their energy differences (ΔGa in kcal mol−1) for the methylation reactions of DMAP-modified mPE foldamer 1 and trimer 1′ with different linear methyl sulfonate esters (2a–2d)
| |
Ga (1′) |
Ga (1) |
ΔGa |
| 2a |
29.6 |
26.7 |
−2.9 |
| 2b |
29.6 |
26.5 |
−3.1 |
| 2c |
31.7 |
26.5 |
−5.2 |
| 2d |
30.6 |
23.7 |
−6.9 |
Methylation of DMAP-modified mPE foldamer with different branched substrates
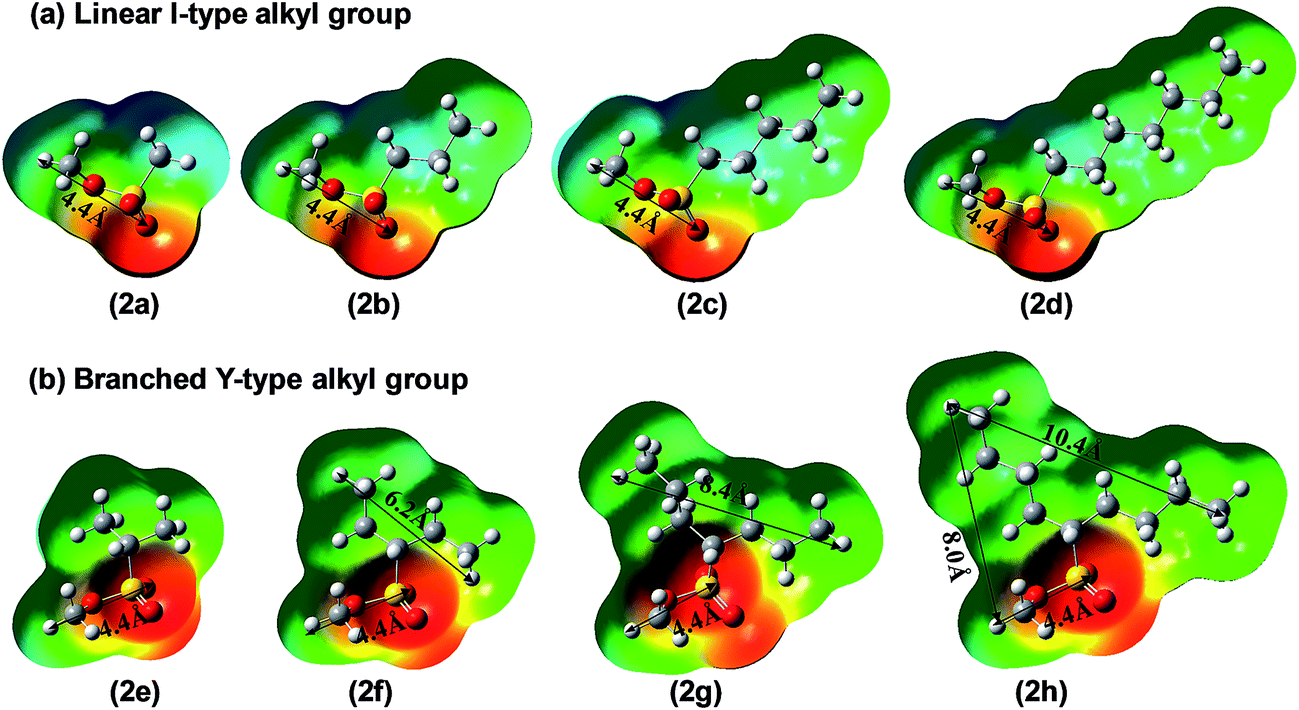
Similarly, the pyridine ring of foldamer 1 can also recognize the branched methylating agents 2e–2h through their methylsulfonate groups and should show the same methylation behavior with linear methyl sulfonate esters. Previous opinion thought that long alkyl groups of branched methylating agents, such as 2h, can first go through the cavity and then the sulfonate groups enter the cavity of the foldamer.15 However, Fig. 6 shows that long alkyl groups of branched methyl sulfonates cannot enter and go through the cavity (∼6.1 Å) of the foldamer due to the limitations of the shape and size of methyl sulfonate esters. Another factor may be that the foldamer lacks the function of recognizing the alkyl groups, although alkyl groups can be artificially stuffed into the cavity of the foldamer. In contrast, the sulfonate groups of branched methyl sulfonates can enter the cavity of the foldamer because the size (4.4 Å) of the sulfonate group can meet the requirement of the cavity, similar to the linear methyl sulfonate esters. In fact, the real reactive site of the foldamer is the nitrogen atom of the pyridine ring, which can only recognize the sulfonate group of the methylating agents, not the alkyl group. All these can also be seen from the electrostatic potential maps of linear and branched methylating agents. As shown in Fig. 6, the sulfonate groups have negative charges and the alkyl groups have positive charges. Thus, the cavity of the foldamer can recognize sulfonate groups prior to the alkyl groups, whether linear or branched methyl sulfonate esters.
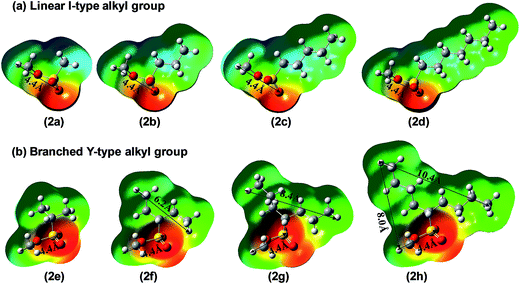 |
| | Fig. 6 Optimized structures and electrostatic potential maps of methyl sulfonate esters with linear l-type ((a), 2a–2d) and branched Y-type ((b), 2e–2h) alkyl groups. The numbers represent the distances between two atoms. | |
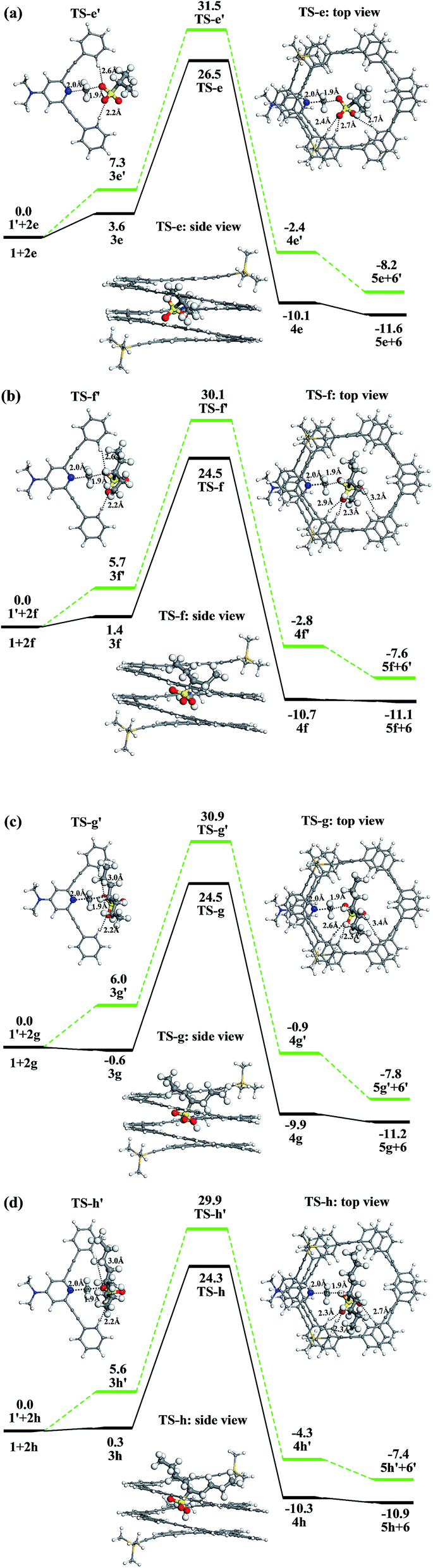
Fig. 7 shows the Gibbs free energy profiles of the methylation reactions of foldamer 1 and trimer 1′ with 2e–2h. These results indicate that the methylation of foldamer 1 with branched methyl sulfonates can be catalyzed by its cavity. In contrast to the linear methyl sulfonates, the reaction barrier of the methylation of foldamer 1, relative to trimer 1′, firstly decreases and then increases with branched methyl sulfonates. As listed in Table 2, from the trimer to the foldamer, the decreased barriers of methylation are 5.0, 5.6, 6.4, and 5.6 kcal mol−1 for 2e–2h, respectively. For example, branched methyl sulfonate 2g shows a maximal decrease of activation energy with foldamer 1, which should show a maximum accelerated rate of methylation. The difference essentially results from the fact that the shape of the alkyl group of branched methylating agents is Y-type and that of linear methyl sulfonates is l-type (Fig. 6). This can also be seen from the structures of the transition states (TS) of the methylation reactions. As shown in Fig. 7a, the shortest alkyl group of the branched methyl sulfonate, 2e, can tilt in the cavity, not vertically insert into the cavity. When the alkyl length increases, such as for 2f, 2g, and 2h, the alkyl group cannot go through the cavity and should stay outside the cavity of foldamer. Similarly, the sulfonate group of the branched methyl sulfonate can easily enter into the cavity of the foldamer. The branched methyl sulfonates can interact with foldamer 1 through C–H⋯OS hydrogen bonds and C–H⋯π interaction. Due to the Y-type structure, the branched methyl sulfonates need to adjust the position and direction of the methylsulfonate and alkyl groups to accommodate the cavity of foldamer 1. Meanwhile, as listed in Table 2, the activation energies for foldamer 1 with substrates 2f, 2g, and 2h are very similar, further indicating that the sulfonate group can enter into the cavity of the foldamer and the Y-type branched alkyl groups can interact with the foldamer. All these interactions can stabilize the transition state and lower the overall barrier of methylation.
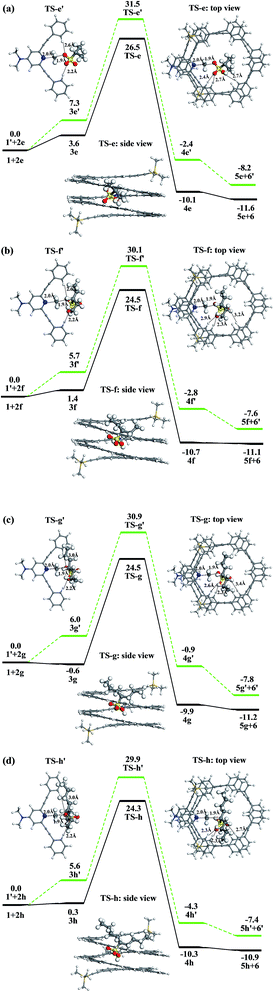 |
| | Fig. 7 Free energy profiles (ΔG in kcal mol−1, (a–d)) of methylation reactions of DMAP-modified mPE foldamer 1 and trimer 1′ with different branched methyl sulfonate esters (2e–2h). The black and green lines represent the methylation reactions of foldamer 1 and trimer 1′, respectively. | |
Table 2 Activation free energies (Ga in kcal mol−1) and their energy differences (ΔGa in kcal mol−1) for methylation reactions of DMAP-modified mPE foldamer 1 and trimer 1′ with different branched methyl sulfonate esters (2e–2h)
| |
Ga (1′) |
Ga (1) |
ΔGa |
| 2e |
31.5 |
26.5 |
−5.0 |
| 2f |
30.1 |
24.5 |
−5.6 |
| 2g |
30.9 |
24.5 |
−6.4 |
| 2h |
29.9 |
24.3 |
−5.6 |
Methylation of DMAP-modified mPE foldamer with different monomer units
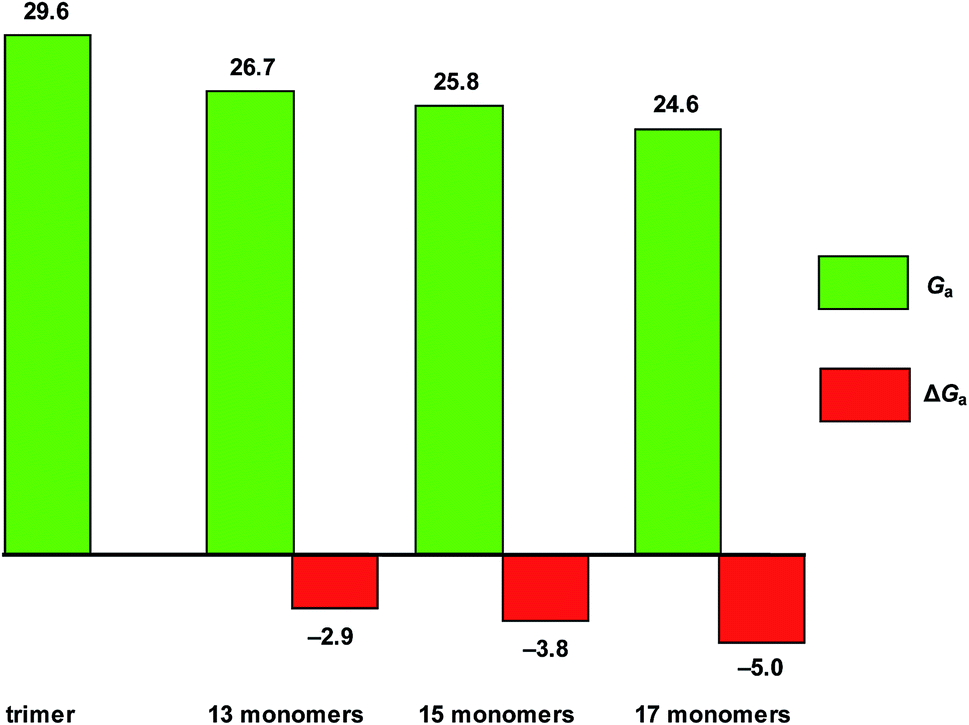
In order to investigate the effect of the depth of the foldamer cavity on the methylation rate, we further studied the methylation reactions of the foldamers consisting of 15 and 17 monomer units. Fig. 8 shows the free-energy barriers of methylation reactions of DMAP-modified mPE foldamers (13, 15, and 17 monomers) with methyl methylsulfonate, respectively. The results show that the barrier of the methylations of DMAP-modified mPE foldamers decrease with the monomer units, indicating that the deeper foldamer cavity can further strengthen the interaction between the foldamer and the substrate and minimize the energy difference between the reactant and the transition state.
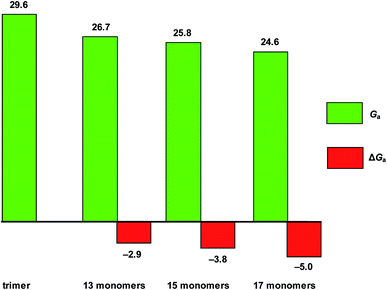 |
| | Fig. 8 Free-energy barriers (Ga in kcal mol−1) of the methylations of DMAP-modified mPE foldamers (13, 15, and 17 monomers) with methyl methylsulfonate (2a) and their barrier differences (ΔGa in kcal mol−1) with the trimer. | |
Experimentally, the methylation rates of foldamer 1 with 2a–2h increase from 40-fold to 1600-fold,15 the corresponding energy barrier should decrease by 2.0–4.4 kcal mol−1 with very small difference. It should be pointed out that these minute difference are based on different reference reactions (1′ with 2a–2h) and result in the difficulty of quantitative comparison with experimental results. Qualitatively, whether linear or branched methyl sulfonates are used, the computational results are in agreement with experimental results. For linear methyl sulfonates, the barriers of the methylation of foldamer decrease with increasing substrate alkyl chain length. As the alkyl chain of the branched substrate, the barriers of the methylation of foldamer first decrease and then increase, relative to that of the small molecule analogue. With the increase of monomer units and the deepening of the cavity, the foldamer has a lower barrier of the methylation.
Conclusions
Through density functional theory calculations, the structure and possible methylation mechanism of DMAP-modified mPE foldamer have been investigated. For comparison, uncatalyzed methylation reactions of the reference trimer in an unfolded conformation have also been studied. The helical structure of DMAP-modified mPE foldamer results from multiple intramolecular π–π stacking interactions between meta-phenylene ethynylene arms, which can be characterized by the helical energy. The unique cavity of the foldamer and the pyridine ring with strong alkalinity can recognize and bind the methyl sulfonates. Noncovalent interactions between the foldamer and the substrate, such as hydrogen bonds and van der Waals interactions, stabilize the transition state and accelerate the confined reaction. As the alkyl chain of the linear substrate increases, the host–guest interaction becomes stronger, which enhances the methylation rates. On the contrary, as the alkyl chain of the branched substrate, the methylation rates of foldamer first increase and then decrease. The difference mainly results from the different shapes, l-type and Y-type, of the alkyl chains of linear and branched substrates. With the increase of monomer units and the deepening of the cavity, noncovalent interactions also become stronger and further accelerate the methylation of the foldamer. We expect that these mechanistic insights into supramolecular catalysis can be used in the design and preparation of supramolecular catalysts and reactors.
Computational section
All stationary points for the methylation reactions of DMAP-modified mPE foldamer 1 and the reference reactions of DMAP-modified mPE trimer 1′ were fully optimized using Gaussian 09 program with M06-2X functional and 6-31G(d,p) basis sets.31 All geometry optimizations were carried out in acetonitrile solvent within the framework of polarizable continuum model (PCM) with the integral equation formalism variant (IEFPCM).32–35 For each stationary point, vibrational frequency calculations were carried out to verify whether it is a minimum or a transition state and to obtain thermodynamic data at 298.15 K and 1 atm. All transition states were verified by intrinsic reaction coordinate calculations.36
Acknowledgements
This work was supported by the Zhejiang Provincial Natural Science Foundation of China (LQ15B030001 and LY17B030003) and the National Natural Science Foundation of China (21571144). We thank the National Supercomputer Center in Guangzhou for providing the computing resources.
Notes and references
- C. Joachim, J. K. Gimzewski and A. Aviram, Nature, 2000, 408, 541–548 CrossRef CAS PubMed.
- D. P. Long, J. L. Lazorcik, B. A. Mantooth, M. H. Moore, M. A. Ratner, A. Troisi, Y. Yao, J. W. Ciszek, J. M. Tour and R. Shashidhar, Nat. Mater., 2006, 5, 901–908 CrossRef CAS PubMed.
- Y. Fu, S. Chen, A. Kuzume, A. Rudnev, C. Huang, V. Kaliginedi, M. Baghernejad, W. Hong, T. Wandlowski, S. Decurtins and S.-X. Liu, Nat. Commun., 2015, 6, 6403 CrossRef CAS PubMed.
- J. C. Nelson, J. G. Saven, J. S. Moore and P. G. Wolynes, Science, 1997, 277, 1793–1796 CrossRef CAS PubMed.
- K. Oh, K.-S. Jeong and J. S. Moore, Nature, 2001, 414, 889–893 CrossRef CAS PubMed.
- E. Yashima, Nat. Chem., 2011, 3, 12–14 CrossRef CAS PubMed.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons, 2nd edn, 2009 Search PubMed.
- P. W. N. M. van Leeuwen, Supramolecular Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2008 Search PubMed.
- D. Gokhman, E. Lavi, K. Prüfer, M. F. Fraga, J. A. Riancho, J. Kelso, S. Pääbo, E. Meshorer and L. Carmel, Science, 2014, 344, 523–527 CrossRef CAS PubMed.
- T. W. Nilsen, Science, 2014, 343, 1207–1208 CrossRef CAS PubMed.
- D. Schübeler, Nature, 2015, 517, 321–326 CrossRef PubMed.
- J. M. Heemstra and J. S. Moore, J. Am. Chem. Soc., 2004, 126, 1648–1649 CrossRef CAS PubMed.
- J. M. Heemstra and J. S. Moore, J. Org. Chem., 2004, 69, 9234–9237 CrossRef CAS PubMed.
- J. M. Heemstra and J. S. Moore, Org. Lett., 2004, 6, 659–662 CrossRef CAS PubMed.
- R. A. Smaldonea and J. S. Moore, J. Am. Chem. Soc., 2007, 129, 5444–5450 CrossRef PubMed.
- R. A. Smaldone and J. S. Moore, Chem.–Eur. J., 2008, 14, 2650–2657 CrossRef CAS PubMed.
- R. A. Smaldone and J. S. Moore, Chem. Commun., 2008, 44, 1011–1013 RSC.
- M. Pickholz and S. Stafström, Synth. Met., 2001, 121, 1271–1272 CrossRef CAS.
- M. Pickholz and S. Stafström, Chem. Phys., 2001, 270, 245–251 CrossRef CAS.
- R. A. Blatchly and G. N. Tew, J. Org. Chem., 2003, 68, 8780–8785 CrossRef CAS PubMed.
- O.-S. Lee and J. G. Saven, J. Phys. Chem. B, 2004, 108, 11988–11994 CrossRef CAS.
- B. Adisa and D. A. Bruce, J. Phys. Chem. B, 2005, 109, 7548–7556 CrossRef CAS PubMed.
- H. H. Nguyen, J. H. McAliley, W. R. Batson III and D. A. Bruce, Macromolecules, 2010, 43, 5932–5942 CrossRef CAS.
- H. H. Nguyen, J. H. McAliley and D. A. Bruce, Macromolecules, 2011, 44, 60–67 CrossRef CAS.
- M. Shravani, S. Balaiah, K. Srinivas, K. Bhanuprakash and I. Huc, ChemPhysChem, 2012, 13, 3526–3534 CrossRef CAS PubMed.
- S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 211–228 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241 CrossRef.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- O. Y. Mindyuk, M. R. Stetzer, P. A. Heiney, J. C. Nelson and J. S. Moore, Adv. Mater., 1998, 10, 1363–1366 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151–5158 CrossRef CAS.
- M. Cossi, G. Scalmani, N. Rega and V. Barone, J. Chem. Phys., 2002, 117, 43–54 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110–114115 CrossRef PubMed.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Optimized structures of foldamers, absolute free energies and atom coordinates of all stationary points. See DOI: 10.1039/c7ra00710h |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Guoyong Fang
*a,
Guoyong Fang