DOI:
10.1039/C7RA00779E
(Paper)
RSC Adv., 2017,
7, 16155-16162
How does the interplay between bromine substitution at bay area and bulky substituents at imide position influence the photophysical properties of perylene diimides?†
Received
18th January 2017
, Accepted 5th March 2017
First published on 14th March 2017
Abstract
This article reports a comparative study on the synthesis, self-assembly, and photophysical properties of perylene diimides (PDIs) symmetrically tethered with long alkyl chains or polyhedral oligomeric silsesquioxanes (POSS) at the imide position and/or bromo substitutions at 1,7-positions of the bay area. This series of samples include dodecyl–PDIH–dodecyl (1), dodecyl–PDIBr–dodecyl (2), POSS–PDIH–POSS (3), and POSS–PDIBr–POSS (4). In solution, the PDIs with bromine substitution at bay area (2, 4) exhibit red-shifted absorption maximum compared to those without (1, 3), which is consistent with a twisted perylene chromophore as revealed by molecular simulation. Similar bathochromatic shift was observed on the solid crystal state emission of 2 as compared to 1. However, in crystals, the emission spectrum of 4 exhibits a seemingly hypochromatic shift relative to that of 3, which could be rationalized by their packing in the crystals. The bromo substitution is believed to partially quench the fluorescence and the relatively loose packing of the twisted π-plane of 4 may not be able to confine π-plane in place, leaving multiple pathways for fluorescent quenching rather than red-shifted emission. While both 3 and 4 exhibit a unique dimer packing scheme, the dimers have quite different longitudinal offset and transverse offset of the π-plane. The longitudinal offset in dimers of 4 is so large that the naphthalene moieties in the dimer almost adopt a face-to-face arrangement and their mutual interactions are considered relatively independent. All these contribute to the less red-shifted fluorescent emission and the lower fluorescent yields in crystals of 4 relative to 3 as compared to that in solution. The study shall shed light into the complicated mutual interactions among intrinsic electronic structure, microscopic molecular packing, and the macroscopic optoelectronic properties.
1 Introduction
In recent decades, significant technical advances in microelectronics have pushed up the demand of optoelectronic devices like light-emitting diodes, fluorescence sensors, display devices, and biochemical probes.1–4 These components have traditionally been developed from luminescent materials based on inorganic oxide powders.1 However, the cost of these inorganic luminescence materials is usually high and they often require complicated processing techniques onto rigid substrates. Alternative cost-effective sources of luminescent materials are thus highly demanded, especially for flexible electronics. Various organic π-conjugated molecules, such as pentacence, phthalocyanine, pentacence, prophrin, perylene, have shown desirable optical properties and high versatility in the design and engineering of their properties.2,5 Among them, perylene diimides (PDIs) have been investigated intensively and extensively due to many desirable features such as high fluorescence quantum yields, superb chemical, thermal and photochemical stability, and facile tunability of colors via core substitution.6–12 However, the photoluminescent properties are often significantly compromised in solid state due to quenching as a result of continuous π,π-stacking.13–16 It is necessary to design molecules to tune their emission and to improve the fluorescent properties in the solid state.
In general, strong π–π interactions between PDIs lead to tightly stacked arrays of PDI and result in fluorescence quenching in the solid state. Breaking the stack can be achieved either by reducing the planarity of PDI π-plane via bay area modification or by introducing bulky substitutions at the N-imides that can prohibit the continuous packing of molecules via steric hindrance.14,17,18 The former is very effective in tuning the energy level of PDI cores and the packing of PDIs. It usually leads to a core-twisted π-system. Typical substituents include fluorine, chlorine, bromine, and cyano group.17,19 Among them, PDI with 1,7-bromo substitution is perhaps the most widely utilized due to the ease of preparation under mild conditions and further derivatization by nucleophilic substitution.20,21 The downside is the undesired broadening of the lowest energy absorption band and the loss of vibronic fine structure of the parent PDI due to core distortion arising from steric crowding at the bay area.22 A balance should be achieved for optimal properties. For the latter method, a large variety of functional groups, such as phenyl substituents,17,23 alkyl group, polyglycerol,24 and polyhedral oligomeric silsesquioxanes (POSS),25–29 have been reported. PDI with attached POSS cages displays outstanding properties like strong red fluorescence with quantum yield approaching unity in solution and ∼0.48 in crystals. A unique dimer packing scheme was observed in the crystals of PDI tethered with POSS cages and was proposed to account for the high fluorescence quantum yield of the crystal.28,30,31 Recently, our group has reported mono-POSS-tethered PDI with two reactive groups (anhydride and bromine) with enhanced fluorescence as an intermediate for other materials.31 Further substitution of bromo with pyrrolidines limits the intermolecular π–π interaction, leading to much improved photophysical properties for optoelectronic applications.32 To the best of our knowledge, there is only very few reports on a systematic study of the bay-area substitution and N-substitution on the final material properties, especially with respect to their self-assembled structures both in solution and in the solid states.
In this article, we report a study of PDI derivatives with/without bromo-substitution at the bay area and with two types of bulky groups (namely, dodecyl group and POSS) at the N-imide positions. We strive to understand the fine roles of the type and position of substituents on the chromophore arrangement and the resulting change in properties. Four samples were thus designed and synthesized (Chart 1), as dodecyl–PDIH–dodecyl (1), dodecyl–PDIBr–dodecyl (2), POSS–PDIH–POSS (3), and POSS–PDIBr–POSS (4). The photophysical properties were studied using UV-vis absorption and fluorescence spectrometry and the crystal structure of 4 was determined and compared to that of 3 to illustrate the impact of molecular packing on the final material properties.
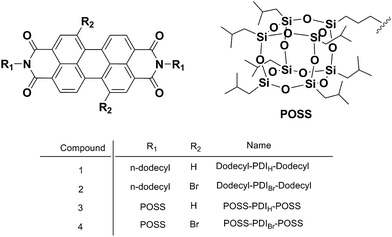 |
| | Chart 1 Chemical structures of the compounds. | |
2 Results and discussion
2.1 Molecular design and chemical synthesis
The synthesize of POSS–PDIH–POSS compounds connected via both flexible and rigid linkages has been previously reported.28 Both of them show strong tendency toward dimerization and adopt a unique dimer packing scheme in crystals. POSS-tethered PDI shows high fluorescence quantum yield due to the steric hindrance of POSS that isolates the chromophore and prohibits the potential structural relaxation process. Functionalization of PDI at bay area with bromine is an important way to expand the scope of PDI materials by allowing incorporation of various substituents on PDI chromophores. Meanwhile, it also leads to a twisted core. It is intriguing how it would affect the self-assembly behaviors and molecular packing in crystal. This is compared to compound 3 with no bromine substitution. The effect of a three-dimensional POSS cage versus a one-dimensional linear chain substitution on their photophysical properties is illustrated by the comparison between compound 3 (4) and compound 1 (2). These samples can be conveniently synthesized using conventional condensation methods according to previous work.33 In particular, the synthesis of compound 4 is discussed below.
It was reported by Würthner et al. that dibromination of PTCDA is not regio-selective and subsequent imidization yields regio-isomeric mixture of 1,7- and 1,6-dibromoperylene diimides as evidenced by 1H NMR spectrometry.21,34 Through repetitive recrystallization, they successfully prepared regio-isomerically pure 1,7-dibromoperylene diimide. Following the literature procedure, the bromination was successfully achieved after three times of recrystallization. Compound 4 was prepared by condensation of PTCDABr with two equivalents of POSS–NH2 in imidazole and ODCB solution. The success of the synthesis was confirmed by molecular characterizations including 1H NMR, 13C NMR, FT-IR, and mass spectrometry (see the ESI†).
2.2 Photophysical properties in solution
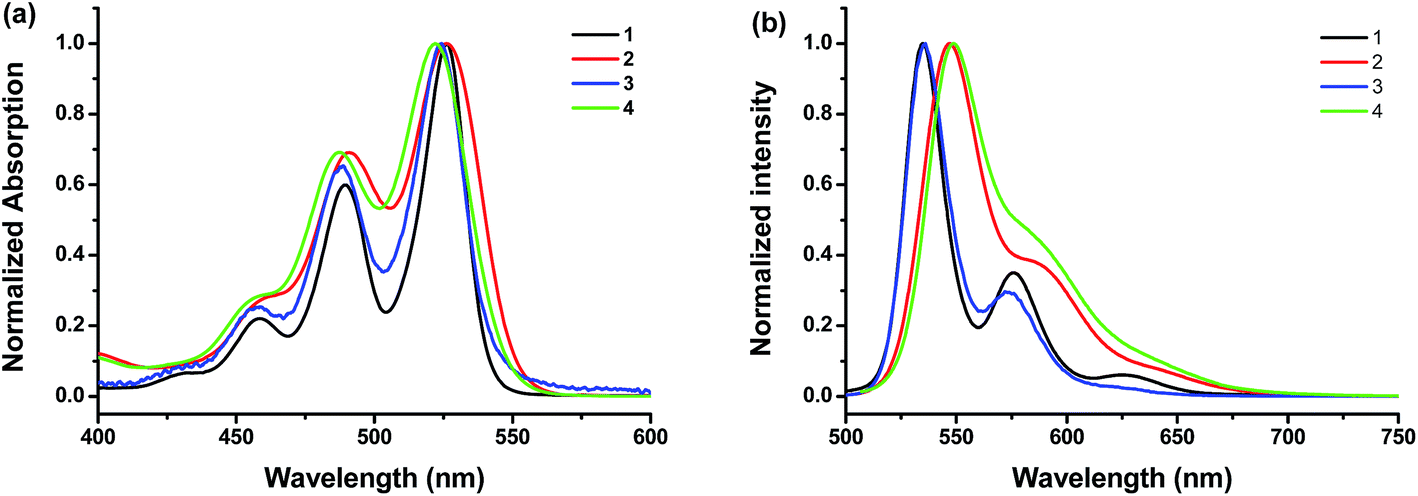
The UV-vis absorption and photoluminescence profiles of these compounds in solution are shown in Fig. 1. All samples clearly shown three characteristic monomeric PDI absorption bands. There was very little difference in absorption spectra between these samples except the peak broadening, which is reported to be due to the loss of planarity upon bromo substitution.35 It also seems that brominated PDI (2 and 4) losses some of the fine features of the electronic structure (Fig. 1a), which is consistent with previous reports.31 In the fluorescence emission spectra (Fig. 1b), all of these samples exhibit resembling mirror image with regard to the corresponding bands in UV-vis absorption spectrum. But it should be noted that the fluorescence spectra of brominated PDI (2 and 4) show considerable bathochromatic shifts relative to that of the unsubstituted PDI (1 and 3). It suggests that the intermolecular π–π interactions are weaker in 2 and 4 due to the presence of the bromine substituents. To rationalize the emission red shift, quantum calculations were performed based on molecular structures by density functional theory (DFT) using Gaussian 09 package. The geometries (Fig. S5†) of the compounds 2 and 4 were optimized by adopting B3LYP method with 6-31G** basic set36,37 via energy minimization. The twist angles between the two naphthalenoid moieties of 2 and 4 are 22.11° and 21.83° respectively, which are close to the reported value in literature.14,38 The twisted perylene plane, caused by the introduction of bromine, leads to different photophysical behaviors between brominating PDIs and unmodified PDI, as demonstrated in the above discussion. In addition, the stoke shift of 4 (2) was larger than that of 3 (1), which indicate that the fluorescence is more sensitive to modification at the imide position with bulky substitution than with linear substituents. This is probably because the former is more effective in weakening the π–π interaction of the PDI core. Fluorescence quantum yields (Φf) in solution were also tested (see Table 1). They all have high Φf values close to unity, demonstrating their potential as highly fluorescent material for wide-ranging applications.
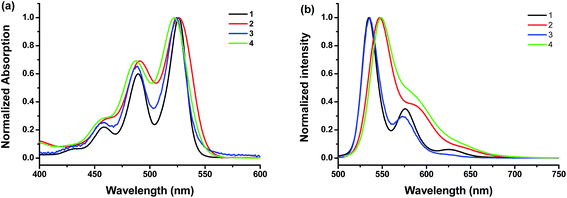 |
| | Fig. 1 (a) Normalized UV-vis absorption spectra and (b) fluorescence spectra of the compounds 1, 2, 3 and 4 at room temperature in CHCl3 at concentrations of 1.6 × 10−6, 1.3 × 10−6, 1.0 × 10−6, and 3.0 × 10−6 M, respectively. The excitation wavelength is 470 nm, 470 nm, 480 nm and 480 nm for 1, 2, 3, and 4, respectively. | |
Table 1 Solution and solid state emission and fluorescence quantum yield (Φf) of the PDI samples
| |
λem,max (nm) |
Φf (%) (λex/nm) |
| Solution |
Crystal |
Solution |
Crystal |
| The data are from ref. 33. |
| 1a |
542 |
647 |
98 (490) |
0.02 (420) |
| 2a |
549 |
661 |
95 (490) |
2.83 (420) |
| 3 |
538 |
624 |
100 (490) |
48 (415) |
| 4 |
552 |
603 |
96 (450) |
8.70 (380) |
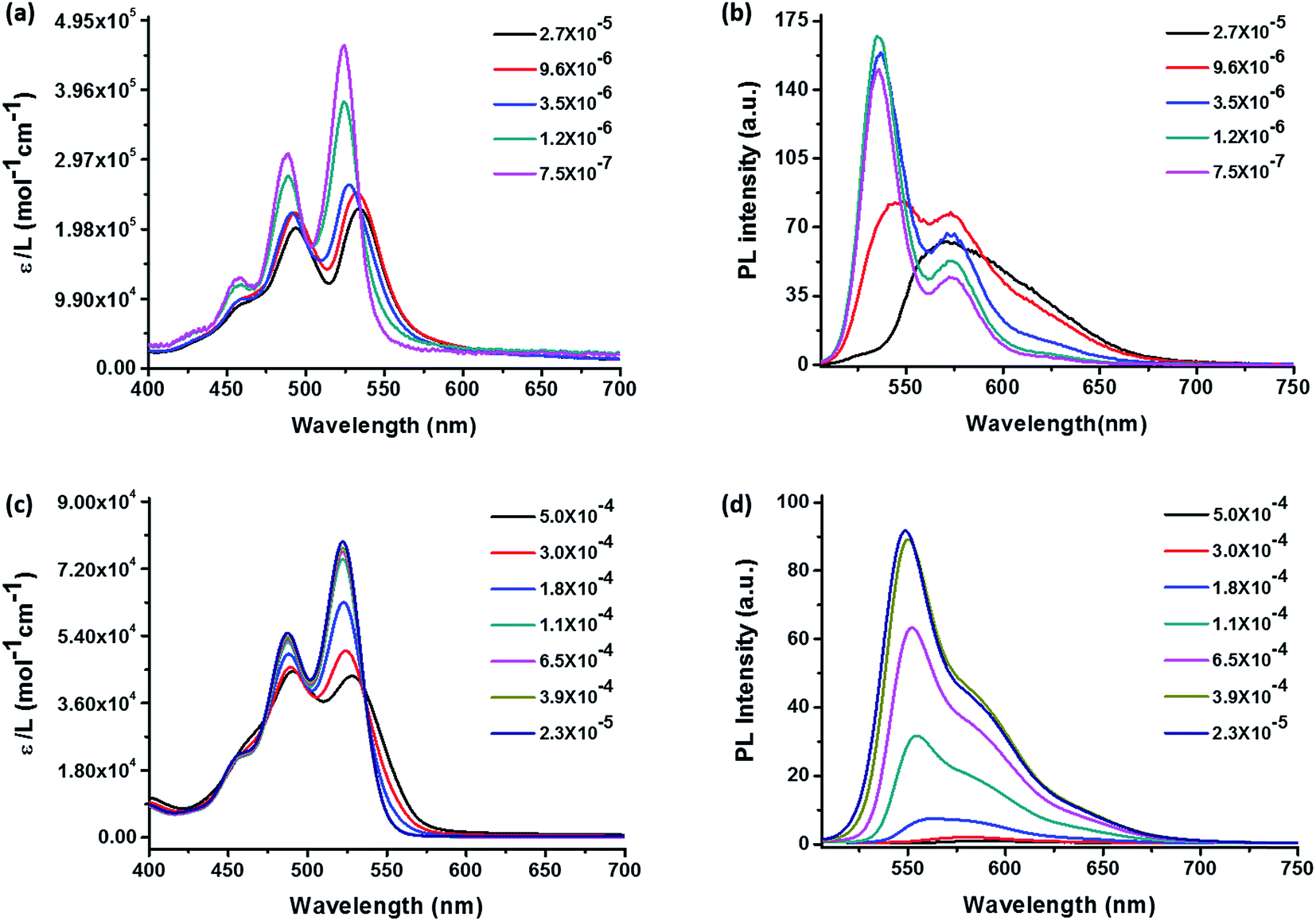
Self-assembly behaviors in solution are affected by concentration, solvent, temperature, etc. It is important to choose a solvent that has appropriate solubility to ensure that aggregation can be observed at low concentrations without precipitation. Concentration-dependent UV-vis absorption and fluorescence spectra were tested in hexanol to determine the aggregation of 3 and 4 with increasing concentration. The results are presented in Fig. 2. The absorption spectra (Fig. 2 left) display at low concentrations are typical monomeric absorption characteristics of PDIs, including three bands at 456 nm, 487 nm, 522 nm, which correspond to the 0–2, 0–1, 0–0 electronic transitions, respectively.39 Sample 4 in hexanol at low concentration also exhibits high extinction efficient since the molecules are monomeric and don't interfere with each other. It was observed that from 2.3 × 10−5 to 1.1 × 10−4 M, the absorption spectra of 4 remain characteristic of the monomeric state (Fig. 2c). While there was a gradual and uniform reduction of peak intensity with increase in concentration, the ratio of peak intensity remains constant. However, as the concentration increases further to 1.8 × 10−4 M, the peak intensity reduction becomes evident and the ratio of absorption maximum at peak to that of the shoulder begins to reduce. This trend persisted at higher concentrations and the absorption maximum reversed at 5.0 × 10−4 M. The shoulder peak is now the maximum peak. It is also noted that the reduction of peak intensity ratio was accompanied by some red shift of both maximums, which may be attributed to the coexistence of dimers and monomers. The isosbestic points were found to be at 535 and 496 nm, respectively. Reduction of absorption peak intensity ratio, the appearance of an isosbestic point, and the maximum peaks reversal are signs for aggregation in concentration-dependent absorption spectra.40 Therefore, upon increasing the concentration, the molecules begin to form aggregates, as evidenced by an apparent decrease in absorption efficient and the ε0–0/ε0–1 become smaller. Besides, the 0–0 and 0–1 band peaks reverse indicating that the molecules form dimers instead of oligomeric aggregates according to previous works and others.39,41 In addition, the ε0–0/ε0–1 represents indirectly the ratio between monomeric molecules and dimers in solution since the strengths of 0–0 and 0–1 electronic transitions are inversely proportional to that of π–π stacking.42 Thus the dimers in solution become more and more significant with increasing concentration. The corresponding concentration-dependent fluorescence spectra of 4 (Fig. 2d) also agree well with the above discussion. The emission spectrum of 4 exhibits a mirror image to absorption spectrum with corresponded 0–0 and 0–1 transitions. The only change was the reduction of peak intensity with increasing the concentration. Significant fluorescent quenching occurred at concentrations higher than 1.8 × 10−4 M without formation of any significant new red-shifted peak, which suggest that the electronic interaction between the chromophores in the aggregates mainly leads to fluorescent quenching.
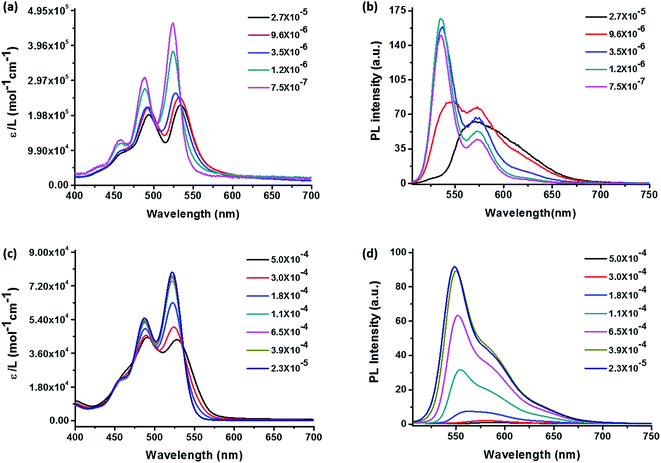 |
| | Fig. 2 Concentration-dependent UV-vis absorption spectra (left) and fluorescence emission spectra (right) in hexanol of POSS–PDIH–POSS (a, b) and POSS–PDIBr–POSS (c, d). The excitation wavelength is 460 nm and 487 nm for (b) and (d), respectively. The label ε stands for extinction coefficient in the absorption spectra and PL stands for photoluminescence. | |
Comparing the results of 3 (Fig. 2a and b) and 4 (Fig. 2c and d), several distinct features can be identified. First, the aggregation of 4 occurs at much higher concentration (1.8 × 10−4 M) than 3 (1.2 × 10−6 M), as evidenced by the onset of peak intensity ratio change. In fact, the reduction of the peak intensity ratio in 3 was so quick that even at 3.5 × 10−6 M the bands already start to show signs of maximum peak reversal. Second, new absorption bands and fluorescent emission bands occur in 3 whereas complete fluorescent quenching was observed in 4. The results suggest that due to the presence of bromo group at the bay area, 4 has much higher solubility and is less prone to aggregate than 3. The aggregation of 4 at higher concentrations leads to broadening of the fluorescent emission and shifting of the maximum to ∼575 nm with profound quenching effect. By contrast, the new emission band with red-shifted emission maximum and significant tailing to longer wavelengths could be observed at a concentration of 2.7 × 10−5 M (Fig. 2b black line), which could be attributed to the aggregation of compound 3.28 It suggests that while the presence of POSS substituents imposes considerable steric hindrance to disfavor continuous π–π stacking, so the twisted PDI core in 4 may lead to a completely different aggregate structure than 3. We then proceed to study their molecular packing and photophysical properties in condensed states, especially, in crystals.
2.3 Photophysical properties in aggregated and condensed states
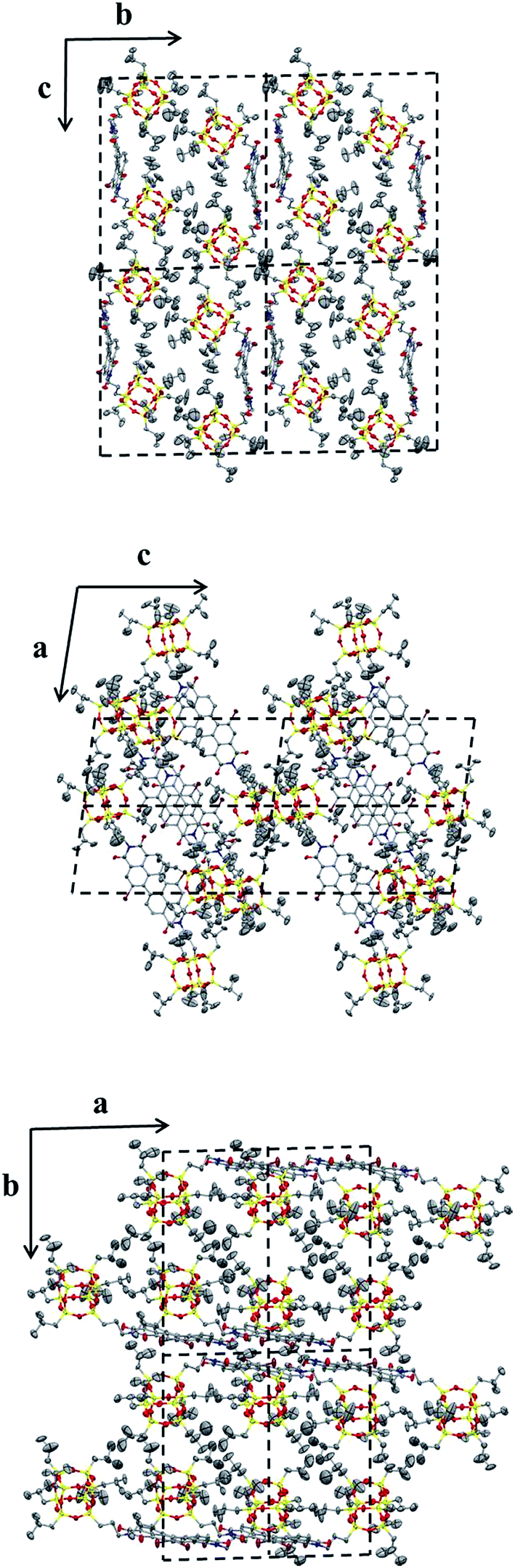
2.3.1 Morphology and crystal structure of POSS–PDIBr–POSS. A slow-diffusion-induced crystallization between two solvent phases was used to grow single crystals of 4 in MeOH/chloroform, as outlined in the ESI.† The SEM images clearly (Fig. 3) reveal a needle-like crystal morphology for 4. The crystal is also strongly birefringent, indicating a high degree of molecular anisotropy within the crystal and that the optical axis is along the direction of π–π stacking.43,44 In addition, the crystals exhibit strong red fluorescence, which indicates that the π–π stacking does not dramatically quench the fluorescence and that the molecules may be well-organized in the solid state.45,46 Using single crystal X-ray diffraction experiments, the crystal structure and molecular packing was determined to be a triclinic unit cell with a = 1.11 nm, b = 2.16 nm, c = 2.43 nm and α = 92.6°, β = 99.0°, γ = 90.7°. This is close but not identical to that of similar POSS–PDI–POSS derivatives reported previously,28 which is reasonable considering the very similar structures of the two compounds. Fig. 4 shows the molecular packing scheme of 4 along different axes in its crystal lattice using Mercury 3.0 software. The structural motif consists of two molecules. Because of steric hindrance of POSS cages and the relatively flexible linkages, two molecules of 4 pack together via π–π interaction. The POSS cages must bend away from PDI onto two sides, preventing it from continuous packing beyond dimers. Therefore, the dimer is a sandwiched structure with two POSS layers and a middle PDI layer. This is very much like the crystal structure of 3. However, the existence of bromo atoms at 1,7 positions leads to fine differences in the precise structure of the dimer, which may lead to differences in the photophysical properties between the crystals of 3 and 4.
 |
| | Fig. 3 SEM images of crystals of 4 at (a) ×1000 magnification and (b) ×10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 magnification. 000 magnification. | |
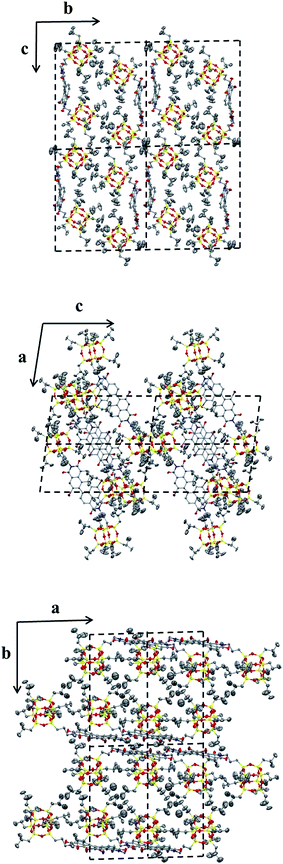 |
| | Fig. 4 Molecular packing of compound 4 in the crystal lattice. Top: projections from the bc-plane; middle: projections from the ac-plane; bottom: projections from the ab-plane. Each projection contains four unit cells. | |
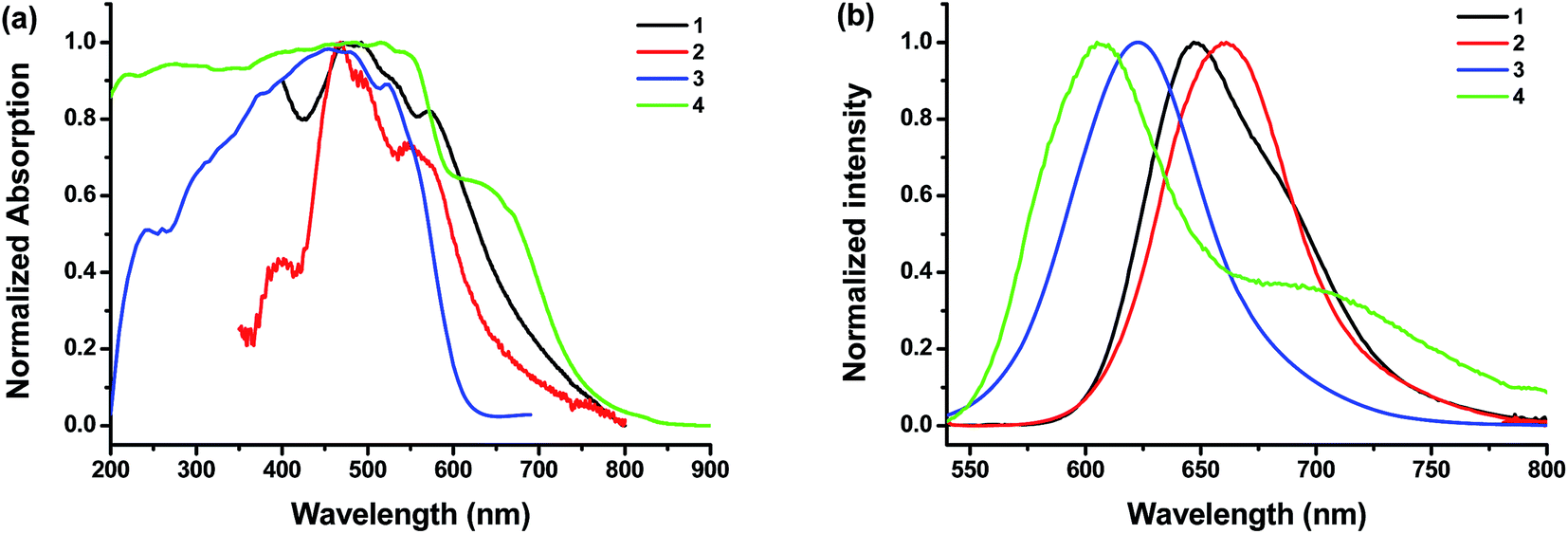
2.3.2 Characterization of photophysical properties. Solid state UV-vis absorption and fluorescence spectra of 1–4 were tested and compared. Their solid state (crystals) fluorescence quantum yield (Φf) were also measured (Fig. 5). The results are summarized in Table 1. All samples have broad but similar UV-vis absorption profiles. However, their fluorescent emission profiles are quite different. The emission maximums of 1 and 2 appear at longer wavelengths of 647 nm and 661 nm with low Φf of only 0.02 and 2.83, respectively. In the solid state, the emission maxima of 2 is red-shifted as compared to 1. The trend is the same in solution where the emission maxima is 525 nm for 1 and 550 nm for 2 (Fig. 1). For both compounds 1 and 2, the π,π-stacking between PDI planes are so strong that the fluorescences are already significantly quenched in the solid state. This effect is much more profound than that from the heavy bromo atom. However, the steric hindrance from bay area bromo substitution of 2 leads to twisted PDI plane, which could not pack as closely as that in 1. Therefore, it alleviates the influence of π,π-stacking and leads to a higher fluorescence quantum yield in 2. By contrast, the emission maximums of 3 and 4 are at 624 nm and 603 nm, respectively. The Φf values are generally higher for POSS-bound PDIs (3 and 4) than linear alkyl bound PDIs (1 and 2). There are two surprising observations: (1) the emission maximum of 4 appears at shorter wavelengths than that of 3; (2) the Φf value of 4 (8.7%) is much lower than that of 3 (48%). The order is opposite to that in dilute solutions (Fig. 1). Nevertheless, it could be understood from the concentration-dependent aggregation experiments. On the one hand, the lower Φf value of 4 in the solid state is consistent with the significant fluorescent quenching of 4 in solution with increasing concentration. On the other hand, the emission red-shift of 4 with increasing concentration in solution is not as profound as that of 3. The fluorescent emission of 3 has a long tailing into longer wavelengths, which is consistent with the solid state emission maximum at about 600 nm. To rationalize this, we speculate that the introduction of bromine atoms at bay positions are not only expected to twist the PDI plane and reduce the π–π stacking, but also play the role as a typical heavy atom perturbation to quench the fluorescence to certain extent.47 To shed light into this unique change in fluorescence spectra, we further turn to the molecular packing in the crystals.28
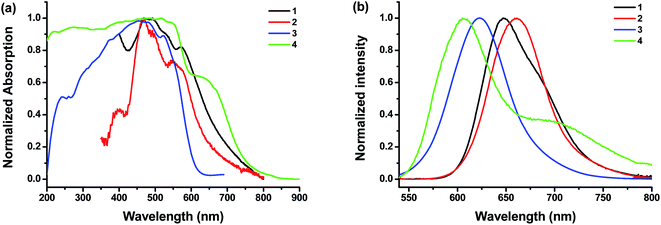 |
| | Fig. 5 Solid state UV-vis absorption (a) and fluorescence spectra (b) of the compounds (λex = 480 nm). | |
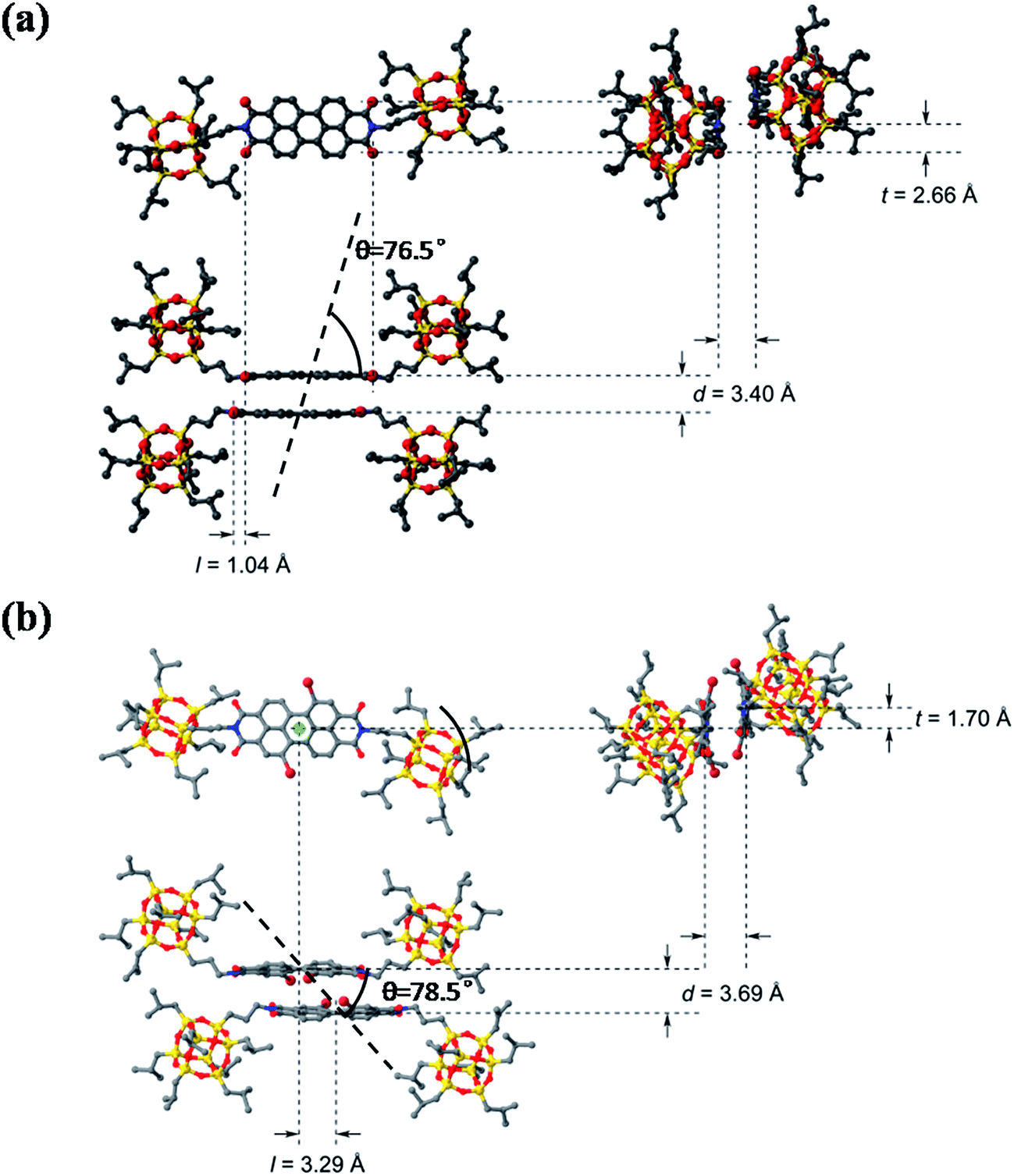
A comparison of the dimer motifs of 3 and 4 in crystal is shown in Fig. 6. The parameters, including longitudinal offset (l), transverse offset (t), interplanar spacing (d) and the rotating angle (α), are listed in Table 2. It can be found that POSS–PDIBr–POSS has a larger π-stacking distance than POSS–PDIH–POSS and other reported PDI chromophores owing to the twisted π-plane with bulky bromo substitution at the bay area. Compound 4 also shows much larger longitudinal offset but slightly smaller transverse offset than 3 (Fig. 6b). The θ value, defined as the angle of the transition dipole moments with respect to their geometrical arrangement, is 51.7°.48–50 Since both 3 and 4 form dimers rather than continuously packed oligomers, it may not be accurate to describe them with reference to the conventional H- and J-aggregates. By examining the dimer structure of 4 even more closely (Fig. S6†), one can find that the longitudinal offset is so large that the two neighboring naphthalene units in the dimer are almost face-to-face. If we consider only the naphthalene planes, the θ value is 78.5°. This unique geometry leads to relatively independent interaction between the naphthalene units, which may account for less red-shifted emission in crystal as compared to that in solution. The similarity between the fluorescent emission spectra of 4 and that of certain reported naphthalene derivatives also offers some indirect evidences to this hypothesis.51,52
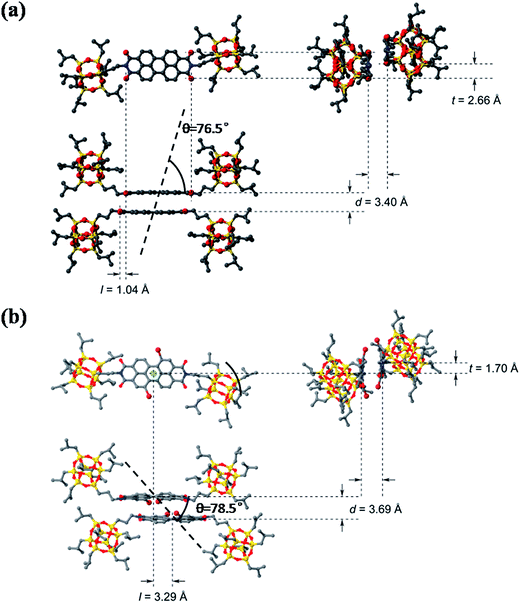 |
| | Fig. 6 The molecular packing structure of dimer of 3 (a) and 4 (b) in the unit cell of crystal with longitudinal offset, l, and transverse offset, t, interplanar spacing, d and rotation angle (θ). | |
Table 2 The packing parameters of the dimer motifs of compounds 3 and 4 in the crystal
| Compound |
d/Å |
l/Å |
t/Å |
θ/° |
| 3 |
3.40 |
1.04 |
2.66 |
76.5 |
| 4 |
3.69 |
3.29 |
1.70 |
78.5 |
3 Conclusion
In this paper, we report the synthesis of perylene diimides with bromine substitutions at 1,7 positions and POSS's symmetrically tethered at the imide positions and compared their self-assembly and photophysical properties with a variety of control samples. The results show that the bulky POSS particles can constrain π–π stacking due to steric hindrance. Compounds 3 and 4 exhibit very similar packing schemes in the crystal state, however, 4 exhibits apparently hypochromatic shifts relative to 3, whereas dodecyl-substituted 2 exhibits bathochromatic shifts relative to 1 in fluorescence emission spectra. It should be noted that in dilute solution, both 2 and 4 exhibit bathochromatic shifts in fluorescence emission maxima relative to 1 and 3, respectively. Therefore, the apparent hypochromatic shift of emission maximum of 4 relative to 3 is again understood from the unique dimer packing scheme of the crystals. It would lead to two consequences: (1) the bromine atoms in the dimer act as a quencher to partially quench the fluorescence, which is responsible for the lower fluorescence quantum yield; (2) the twisted π-plane of 4 does not pack as tightly as that of 3 and POSS may not be sufficient to confine the π-plane in place, leaving multiple pathways for fluorescent quenching rather than red-shifted emission. Notably, the longitudinal offset in 4 is so large that the naphthalene moieties in the dimer almost adopt a face-to-face arrangement and their mutual interactions are relatively independent, which may also partially contribute to the less red-shifted fluorescent emission in crystal as compared to that in solution.
Acknowledgements
This work was supported by the Program for Changjiang Scholars and Innovative Research Team in University (T2011079, IRT1221), Science and Technology Commission of Shanghai Municipality (16JC1400700).
Notes and references
- C. Feldmann, T. Jüstel, C. R. Ronda and P. J. Schmidt, Adv. Funct. Mater., 2003, 13, 511–516 CrossRef CAS.
- A. L. Kanibolotsky, I. F. Perepichka and P. J. Skabara, Chem. Soc. Rev., 2010, 39, 2695–2728 RSC.
- W. J. Mulder, G. J. Strijkers, G. A. Van Tilborg, D. P. Cormode, Z. A. Fayad and K. Nicolay, Acc. Chem. Res., 2009, 42, 904–914 CrossRef CAS PubMed.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547–1562 CrossRef CAS PubMed.
- Y. Zhong, M. T. Trinh, R. Chen, W. Wang, P. P. Khlyabich, B. Kumar, Q. Xu, C.-Y. Nam, M. Y. Sfeir, C. Black, M. L. Steigerwald, Y.-L. Loo, S. Xiao, F. Ng, X. Y. Zhu and C. Nuckolls, J. Am. Chem. Soc., 2014, 136, 15215–15221 CrossRef CAS PubMed.
- G. Horowitz, F. Kouki, P. Spearman, D. Fichou, C. Nogues, X. Pan and F. Garnier, Adv. Mater., 1996, 8, 242–245 CrossRef CAS.
- C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386–2407 CrossRef CAS PubMed.
- H. Langhals, Chem. Phys. Lett., 1988, 150, 321–324 CrossRef CAS.
- H. Langhals, Heterocycles, 1995, 40, 477–500 CrossRef CAS.
- D. Schlettwein, A. Back, B. Schilling, T. Fritz and N. Armstrong, Chem. Mater., 1998, 10, 601–612 CrossRef CAS.
- F. Würthner, Chem. Commun., 2004, 1564–1579, 10.1039/b401630k.
- X.-D. Zhuang, Y. Chen, B. Zhang, Y. Li, B. Yu and W. Qiao, New J. Chem., 2010, 34, 1120–1124 RSC.
- Z. Chen, U. Baumeister, C. Tschierske and F. Würthner, Chem.–Eur. J., 2007, 13, 450–465 CrossRef CAS PubMed.
- Z. Chen, V. Stepanenko, V. Dehm, P. Prins, L. D. Siebbeles, J. Seibt, P. Marquetand, V. Engel and F. Würthner, Chem.–Eur. J., 2007, 13, 436–449 CrossRef CAS PubMed.
- T. E. Kaiser, H. Wang, V. Stepanenko and F. Würthner, Angew. Chem., Int. Ed., 2007, 46, 5541–5544 CrossRef CAS PubMed.
- F. Wuerthner, C. Bauer, V. Stepanenko and S. Yagai, Adv. Mater., 2008, 20, 1695–1698 CrossRef CAS.
- V. Dehm, Z. Chen, U. Baumeister, P. Prins, L. D. Siebbeles and F. Würthner, Org. Lett., 2007, 9, 1085–1088 CrossRef CAS PubMed.
- F. Würthner, Z. Chen, V. Dehm and V. Stepanenko, Chem. Commun., 2006, 1188–1190, 10.1039/b517020f.
- R. Schmidt, J. H. Oh, Y.-S. Sun, M. Deppisch, A.-M. Krause, K. Radacki, H. Braunschweig, M. Koenemann, P. Erk, Z. Bao and F. Wuerthner, J. Am. Chem. Soc., 2009, 131, 6215–6228 CrossRef CAS PubMed.
- Á. J. Jiménez, M.-J. Lin, C. Burschka, J. Becker, V. Settels, B. Engels and F. Würthner, Chem. Sci., 2014, 5, 608–619 RSC.
- F. Würthner, V. Stepanenko, Z. Chen, C. R. Saha-Möller, N. Kocher and D. Stalke, J. Org. Chem., 2004, 69, 7933–7939 CrossRef PubMed.
- E. Fron, G. Schweitzer, P. Osswald, F. Würthner, P. Marsal, D. Beljonne, K. Müllen, F. C. De Schryver and M. Van der Auweraer, Photochem. Photobiol. Sci., 2008, 7, 1509–1521 CAS.
- J. Lv, Y. Zhao, G. Li, Y. Li, H. Liu, Y. Li, D. Zhu and S. Wang, Langmuir, 2009, 25, 11351–11357 CrossRef CAS PubMed.
- T. Heek, J. Nikolaus, R. Schwarzer, C. Fasting, P. Welker, K. Licha, A. Herrmann and R. Haag, Bioconjugate Chem., 2013, 24, 153–158 CrossRef CAS PubMed.
- D. Clarke, S. Mathew, J. Matisons, G. Simon and B. W. Skelton, Dyes Pigm., 2012, 92, 659–667 CrossRef CAS.
- F. Du, J. Tian, H. Wang, B. Liu, B. Jin and R. Bai, Macromolecules, 2012, 45, 3086–3093 CrossRef CAS.
- E. Lucenti, C. Botta, E. Cariati, S. Righetto, M. Scarpellini, E. Tordin and R. Ugo, Dyes Pigm., 2013, 96, 748–755 CrossRef CAS.
- X. Ren, B. Sun, C.-C. Tsai, Y. Tu, S. Leng, K. Li, Z. Kang, R. M. V. Horn, X. Li and M. Zhu, J. Phys. Chem. B, 2010, 114, 4802–4810 CrossRef CAS.
- Y. Zhang, L. Zhang, H. Liu, D. Sun and X. Li, CrystEngComm, 2015, 17, 1453–1463 RSC.
- Y. Shao, G.-Z. Yin, X. Ren, X. Zhang, J. Wang, K. Guo, X. Li, C. Wesdemiotis, W.-B. Zhang, S. Yang, M. Zhu and B. Sun, RSC Adv., 2017, 7, 6530–6537 RSC.
- L. M. Wangatia, B. Sun, T. Zeng and M. F. Zhu, Mater. Sci.-Pol., 2015, 33, 113–121 CAS.
- L. M. Wangatia, T. Zeng, B. Sun and M. F. Zhu, Adv. Mater. Res., 2013, 668, 701–705 CrossRef.
- T. Zeng, P. Wu, X. Zhang, L. M. Wangatia, B. Sun and M. Zhu, J. Donghua Univ., 2015, 3, 372–378 Search PubMed.
- R. K. Dubey, A. Efimov and H. Lemmetyinen, Chem. Mater., 2010, 23, 778–788 CrossRef.
- Y. Huang, L. Fu, W. Zou, F. Zhang and Z. Wei, J. Phys. Chem. C, 2011, 115, 10399–10404 CAS.
- M. C. R. Delgado, E.-G. Kim, D. A. da Silva Filho and J.-L. Bredas, J. Am. Chem. Soc., 2010, 132, 3375–3387 CrossRef PubMed.
- V. Sivalmurugan, K. Kazlauskas, S. Jursenas, A. Gruodis, J. Simokaitiene, J. V. Grazulevicius and S. Valiyaveettil, J. Phys. Chem. B, 2010, 114, 1782–1789 CrossRef PubMed.
- Q. Zhao, S. Zhang, Y. Liu, J. Mei, S. Chen, P. Lu, A. Qin, Y. Ma, J. Z. Sun and B. Z. Tang, J. Mater. Chem., 2012, 22, 7387–7394 RSC.
- R. Gvishi, R. Reisfeld and Z. Burshtein, Chem. Phys. Lett., 1993, 213, 338–344 CrossRef CAS.
- M. R. Islam, E. Dahan, S. Saimani and P. R. Sundararajan, Eur. Polym. J., 2012, 48, 1538–1554 CrossRef CAS.
- D. Liu, S. De Feyter, M. Cotlet, A. Stefan, U.-M. Wiesler, A. Herrmann, D. Grebel-Koehler, J. Qu, K. Müllen and F. C. De Schryver, Macromolecules, 2003, 36, 5918–5925 CrossRef CAS.
- A. E. Clark, C. Qin and A. D. Q. Li, J. Am. Chem. Soc., 2007, 129, 7586–7595 CrossRef CAS PubMed.
- K. Balakrishnan, A. Datar, R. Oitker, H. Chen, J. Zuo and L. Zang, J. Am. Chem. Soc., 2005, 127, 10496–10497 CrossRef CAS PubMed.
- S. Yagai, Y. Monma, N. Kawauchi, T. Karatsu and A. Kitamura, Org. Lett., 2007, 9, 1137–1140 CrossRef CAS PubMed.
- A. Datar, K. Balakrishnan, X. Yang, X. Zuo, J. Huang, R. Oitker, M. Yen, J. Zhao, D. M. Tiede and L. Zang, J. Phys. Chem. B, 2006, 110, 12327–12332 CrossRef CAS PubMed.
- P. Yan, A. Chowdhury, M. W. Holman and D. M. Adams, J. Phys. Chem. B, 2005, 109, 724–730 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, 2006 Search PubMed.
- E. Emerson, M. Conlin, A. E. Rosenoff, K. Norland, H. Rodriguez, D. Chin and G. R. Bird, J. Phys. Chem., 1967, 71, 2396–2403 CrossRef CAS.
- A. Mishra, R. K. Behera, P. K. Behera, B. K. Mishra and G. B. Behera, Chem. Rev., 2000, 100, 1973–2012 CrossRef CAS PubMed.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
- X. Chen, J. Wang, G. Zhang, Z. Liu, W. Xu and D. Zhang, New J. Chem., 2013, 37, 1720–1727 RSC.
- E. Sefer and F. B. Koyuncu, Electrochim. Acta, 2014, 143, 106–113 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, detailed characterization of 4, energy-minimized molecular models of 2 and 4, and the X-ray crystallographic data for the structure of 4. CCDC 1527259. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra00779e |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Xinlin Zhang‡a,
Kai Lianga,
Jing Wangb,
Yuejian Linc,
Shuguang Yanga,
Wen-Bin Zhang
a,
Xinlin Zhang‡a,
Kai Lianga,
Jing Wangb,
Yuejian Linc,
Shuguang Yanga,
Wen-Bin Zhang