
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pyridazinediones deliver potent, stable, targeted and efficacious antibody–drug conjugates (ADCs) with a controlled loading of 4 drugs per antibody†
Eifion Robinson‡
a,
João P. M. Nunes‡a,
Vessela Vassilevab,
Antoine Maruani a,
João C. F. Nogueiraa,
Mark E. B. Smitha,
R. Barbara Pedleyb,
Stephen Caddicka,
James R. Baker*a and
Vijay Chudasama*a
a,
João C. F. Nogueiraa,
Mark E. B. Smitha,
R. Barbara Pedleyb,
Stephen Caddicka,
James R. Baker*a and
Vijay Chudasama*a
aDepartment of Chemistry, University College London, London, WC1H 0AJ, UK. E-mail: j.r.baker@ucl.ac.uk; v.chudasama@ucl.ac.uk; Tel: +44 (0)20 7679 2653 Tel: +44 (0)20 7679 2077
bUCL Cancer Institute, 72 Huntley Street, London, UK
First published on 30th January 2017
Abstract
Herein we report the use of pyridazinediones to functionalise the native solvent accessible interstrand disulfide bonds in trastuzumab with monomethyl auristatin E (MMAE). This method of conjugation delivers serum stable antibody–drug conjugates (ADCs) with a controlled drug loading of 4. Moreover, we demonstrate that the MMAE-bearing ADCs are potent, selective and efficacious against cancer cell lines in both in vitro and in vivo models.
Antibody–drug conjugates (ADCs) are a promising class of biotherapeutics primarily aimed at cancer treatment. ADCs combine the cell-killing potency of a toxic drug with the target selectivity of an antibody towards overexpressed receptors on cancer cell membranes. Their relevance is demonstrated by two FDA-approved ADCs already in the market, brentuximab vedotin (Adcetris™) and trastuzumab emtansine (Kadcyla™), and over 40 ADCs undergoing clinical trials.1,2
The choice of conjugation method to covalently attach a drug to an antibody has a direct effect on the control over drug loading and affects several properties of the resulting ADC, such as potency, selectivity, stability, clearance rate and therapeutic index.3–5 Lysine conjugation is the method employed to produce Kadcyla™ but the high abundance of lysines results in a heterogeneous distribution of antibody conjugates with different drug loadings and therapeutic indexes.6–8 Maleimide conjugation to cysteines of an interstrand reduced IgG1 exploits the lower abundance of solvent accessible disulfides to reduce heterogeneity. This is exploited in the synthesis of Adcetris™ by first reducing the solvent accessible disulfides in the antibody, followed by conjugation to a maleimide-functionalised drug-linker construct.9 Despite the reduction in heterogeneity, the resulting ADC is still composed of multiple species with loss of interstrand disulfide bonds and an overall suboptimal performance in pharmacokinetics and therapeutic index.10–14 The existence of high drug to antibody ratio (DAR) species has been recognised to lead to physical instability, faster clearance and lower therapeutic index, highlighting the need to carefully control drug loading during ADC synthesis.8,10,11,15 In fact, homogeneity of DAR species has already been demonstrated to improve therapeutic index.16–18 The linker joining the drug to the conjugation site must be stable in circulation and enable drug release following internalisation into the target cell, with the linker stability being recognised as a major challenge. This is exemplified following the data on the acyl hydrazone linkers that were employed in Mylotarg™,3,19 and thioether-succinimide conjugates, employing classical maleimides for cysteine conjugation, undergoing retro Michael (leading to deconjugation and transfer onto thiols such as albumin20).18,21
New conjugation strategies have been developed to address these challenges, such as engineered cysteines through site-directed mutagenesis8,22 and non-natural amino acids for bioorthogonal conjugation.13,23,24 These strategies rely on protein engineering and thus represent a move away from the simple concept of a chemical strategy for post-translational modification of a native antibody. We feel that chemical methods that are employed on native antibody scaffolds which reduce heterogeneity and afford complete drug loading control are highly desirable for ADC production. Site-selective disulfide bridging with small molecules fits these requirements. We have developed two classes of molecules used for functional disulfide re-bridging, namely next-generation maleimides (NGMs)25–32 and pyridazinediones (PDs).33–37 Godwin and co-workers have also pioneered bis-sulfones as reagents for disulfide bridging and ADC synthesis.17,38
Recently, we have shown the use of NGMs for ADC synthesis,30 and have shown the ADCs to have good selectivity towards creating DAR 4 conjugates and to have stability in blood serum, as well as in vitro potency.26 NGMs require stabilisation against thiol scavenging conditions by hydrolysing to maleamic acid conjugates under basic pH.25,26,28 PD-conjugates on the other hand are stable to hydrolysis, less reactive in thiol scavenging conditions and also stable against retro Michael. Thus, PDs do not require any additional modification beyond conjugation to deliver stable ADCs; although (prior to this communication) they were only shown to be stable in blood plasma mimicking conditions.37 In addition, PDs have been shown to be amenable to forming 2-in-1 reagents for both disulfide reduction and stapling to reduce ADC heterogeneity as well as to be amenable to making controlled DAR 2 ADCs.34 The PD scaffold has also been shown to incorporate dual click handles, which enables controlled modification of each PD bridged disulfide with two distinct agents.37 However, to date, there has been no in vivo data on PD conjugates and no serum stability or hydrophobic interaction chromatography (HIC) analysis.
Herein, we demonstrate the synthesis and evaluation of PD-based ADCs with an industry relevant drug monomethyl auristatin E (MMAE). We show that these ADCs are synthesised almost exclusively as DAR 4 species (by HIC and mass spec), have stability in blood serum and are selective and potent against HER-2 expressing tumour cells. Moreover, we demonstrate for the first time that both NGM- and PD-based ADC potency and selectivity translate well into therapeutic efficacy in an in vivo mouse model. Both ADC formats showed excellent tumour regression.
Both NGM and PD based ADCs are prepared by a common and simple method (Scheme 1). The four solvent accessible disulfide bonds in an IgG1 are reduced using tris-2-carboxyethylphosphine (TCEP), followed by functional re-bridging with either a NGM or a PD molecule. Conjugation with NGM is followed by hydrolysis to stabilise the conjugate, whereas PD conjugates themselves are stable.
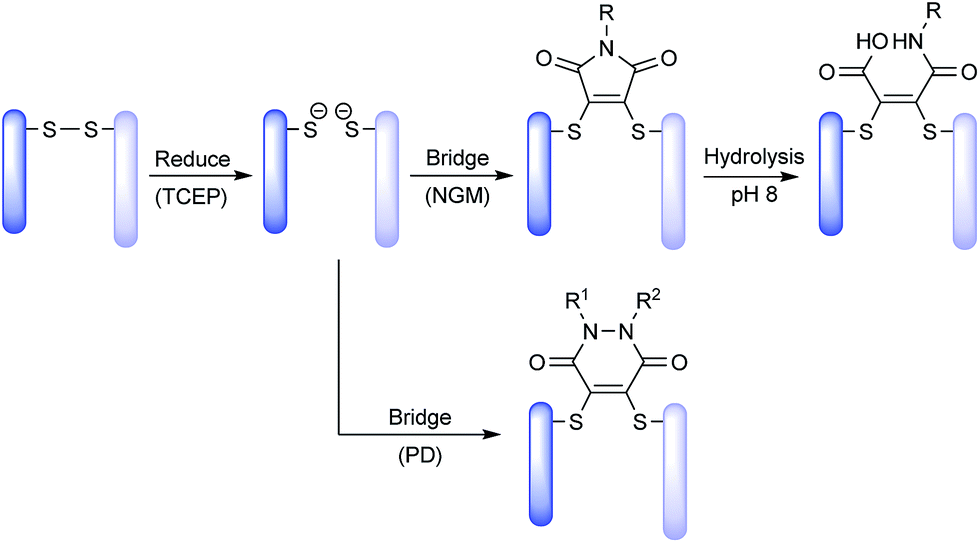 | ||
| Scheme 1 Exemplification of NGM and PD technology platforms for insertion into a disulfide bond. | ||
We chose trastuzumab as our antibody platform to demonstrate disulfide modification with PDs. Trastuzumab (Herceptin™) is a monoclonal IgG1 antibody targeting HER2 and is used for breast cancer therapy both as antibody alone39 and as the antibody component of ADC Kadcyla™.40 We chose anti-cancer drug MMAE as a suitable payload for ADC design.9,38,41 Lastly, we chose to compare cleavable and non-cleavable linker designs. The cleavable linker design is the widely used cathepsin B labile valine–citruline linker.8,9,38,42,43
It has been suggested that MMAE requires release as a free drug from the antibody linker conjugate to reach anywhere near its full potency inside the target cell.44 However, our previously reported NGM-MMAE ADC 1 showed high potency in vitro despite using a non-cleavable linker.26 Therefore, we chose to prepare both non-cleavable and cleavable linker versions of PD-MMAE ADCs (ADCs 3 and 4, Fig. 1), and re-synthesise NGM-MMAE ADC 1 for further evaluation. We also prepared an analytical amount of classical maleimide-based ADC 2 (a common conjugation strategy employed to make ADCs) to compare its HIC profile against our technology.
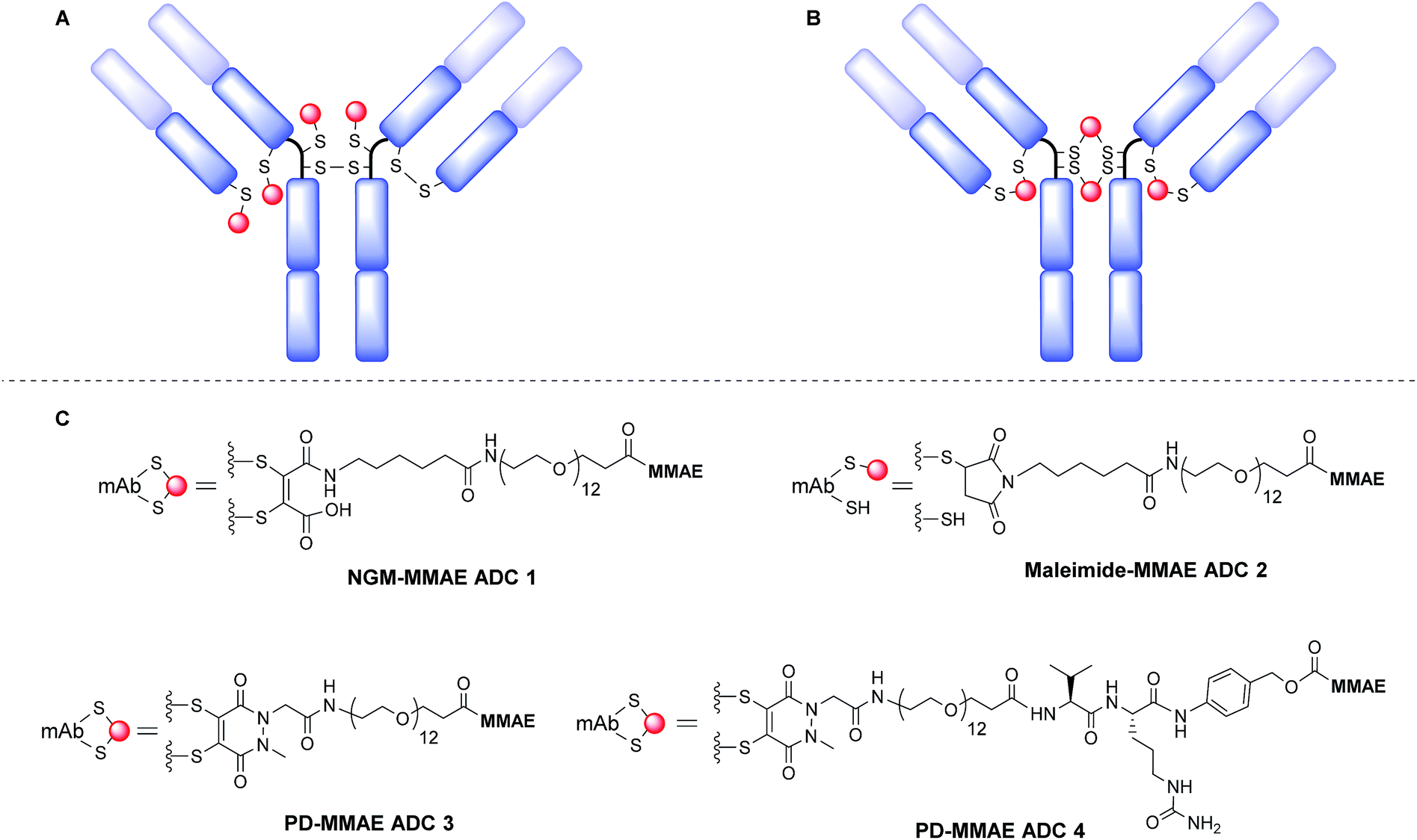 | ||
| Fig. 1 Modification of reduced disulfide bonds in a IgG1 antibody with: (A) classical maleimides leading to a heterogeneous mixture and loss of structural bonds; (B) NGMs and PDs leading to more homogeneous and retention of structural bonds. (C) NGM-MMAE and PD-MMAE ADC conjugates prepared in this work. | ||
The synthesis and characterisation of NGM-MMAE ADC 1 was previously described.26 Synthesis of classical maleimide-MMAE ADC 2 was adapted from a reported protocol (Fig. 1, see ESI for details†).9 PD based bridging reagents, with cleavable or non-cleavable linkers, were synthesised from a previously reported PD scaffold36 in reasonable yields (see ESI for details†). ADCs 3 and 4 were obtained by treatment of TCEP-reduced trastuzumab with the relevant bridging reagent as illustrated in Scheme 1. To our delight, in line with the best industry standard, mass spectrometry analysis of the conjugates confirmed ADCs 3 and 4 to each have a DAR of 4 as the most predominant species by far (Fig. 2A and B, respectively).
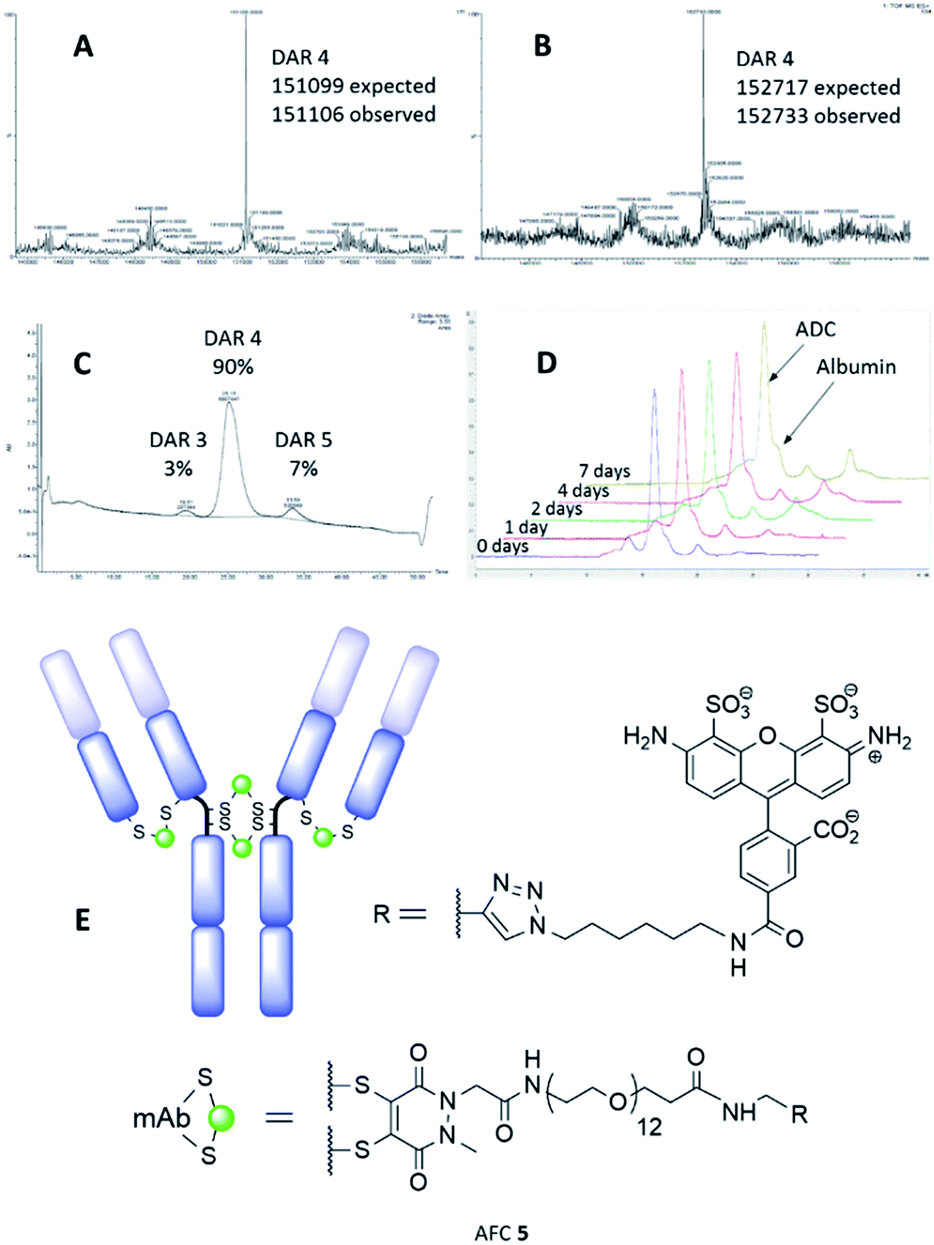 | ||
| Fig. 2 MS data for: (A) PD-MMAE ADC 3, and; (B) PD-MMAE ADC 4. (C) HIC analysis of PD-MMAE ADC 3. (D) SEC-HPLC analysis data from stability study of AFC 5. (E) AFC 5 conjugate. | ||
Further to this, key HIC analysis of PD-MMAE ADC 3 showed this conjugate to be synthesised almost exclusively as a DAR 4 species (Fig. 2C). This stands in stark contrast with the typical mixture of different DAR species obtained for classical maleimide-MMAE conjugate 2 (see ESI for full HIC data†). HIC analysis of PD-MMAE ADC 4 displays a more complex pattern to ADC 3; this conjugate appears as a wider peak with a shoulder. Previous HIC analysis of disulfide bridged MMAE ADCs bearing the valine–citrulline linker in the literature also show a similar pattern and those results were interpreted by Badescu et al. as DAR 4 species.38 Furthermore, HIC analysis of NGM-MMAF ADCs in the literature interpreted these as isomorphs of DAR 4 species.16 Furthermore, we have shown that LCMS analysis of PD-MMAE 4 to reveal the DAR 4 species almost solely (Fig. 2B); this represents the best control of loading for such reagents in the field, to the best of our knowledge.
Following this, we examined the stability of antibody-PD conjugates in blood serum. To do this, an alkyne bearing PD reagent was synthesised (PD-propargylamide, see ESI for details†), which would simulate the linker properties of the reagent used for PD-MMAE ADC 3. PD-propargylamide was conjugated to trastuzumab, and then click functionalised with AlexaFluor® 488-azide to give antibody-fluorophore conjugate (AFC) 5 (Fig. 2E). The conjugate was incubated in blood serum over 7 days and compared side-by-side with a classical maleimide counterpart conjugate as control. Whereas the classical maleimide control displayed significant transfer of fluorescence from trastuzumab to serum albumin (see ESI for details†), the PD conjugate showed minimal transfer (Fig. 2D), and is certainly at the very least comparable to the best reagents in the field.
The in vitro potency of all our ADCs was next evaluated against HER2-positive human breast cancer cell line HCC1954. The HER2-negative cell line MCF-7 was used as a negative control. Both cell lines are sensitive to MMAE with IC50 values of 0.2 nM and 0.9 nM for HCC1954 and MCF-7, respectively. Gratifyingly, the ADCs proved to be highly potent against the HER2-positive cell line, whilst having potency greater than two orders of magnitude lower for the HER2-negative MCF-7 (Fig. 3, top). PD-MMAE ADCs 3 and 4 were found to reduce HCC1954 cell viability with low nM to pM IC50 values. Whilst the effect of having a cleavable linker made a large difference for the PD-conjugates, equi-drug loaded non-cleavable linker NGM ADC 1 showed potency comparable to cleavable linker PD-MMAE ADC 4 – which tallies with our previous results.26
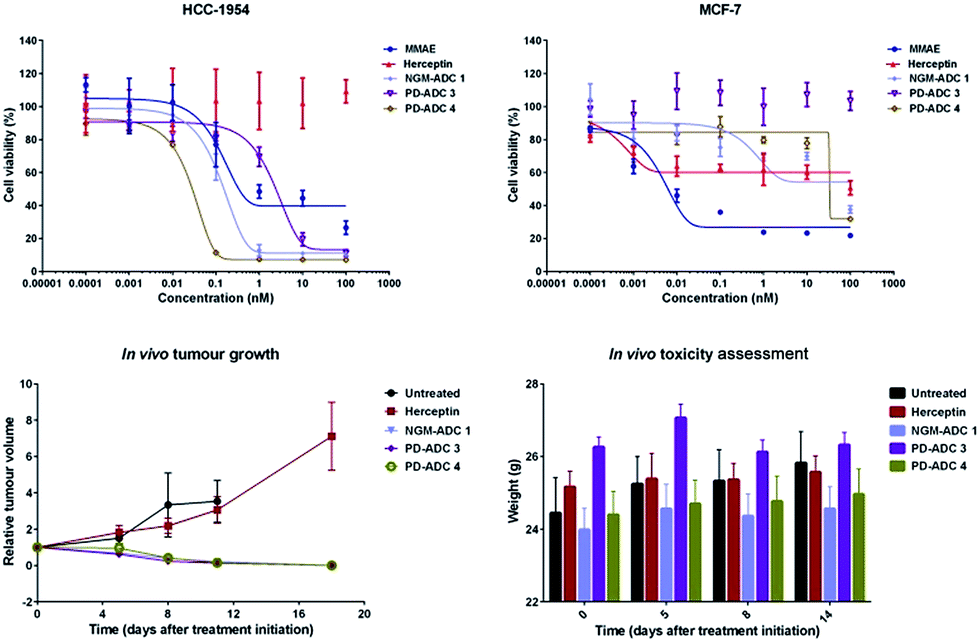 | ||
| Fig. 3 In vitro potency of MMAE, Herceptin, and ADCs 1, 3 & 4 on HCC1954 cells, and MCF-7 cells. Cell viability was plotted against antibody/MMAE concentration. In vivo efficacy of ADCs 1, 3 and 4 showing relative tumour volume during course of treatment, and mouse weight during study. | ||
We then evaluated the in vivo efficacy of our ADCs in a human breast cancer xenograft model using the HCC1954 cell line. The ADCs were dosed at 20 mg kg−1 on days 1, 8 and 15, which is in line with other interchain disulfide bridging ADCs.17,38 Gratifyingly, all ADCs achieved tumour inhibition soon after the first day of therapy (Fig. 3, bottom left). Tumour inhibition was sustained during and after dosing, leading to complete regression 18 days after start of therapy. No tumour recurrence was observed during the remainder of the study, up to day 45, when the study was terminated. Notably, trastuzumab alone was not efficacious and showed no improvement over the untreated vehicle. Furthermore, the ADCs were well tolerated with no significant change in body weight observed (Fig. 3, bottom right). As we dosed at a relatively high level, and the advantages of having such homogenous DAR 4 conjugates are so well characterised and understood, we were not aiming to see a significant difference in the in vivo potency of PD-ADCs 3–4 and NGM-ADC 1 but rather show that our platforms can be: (i) successfully employed in an in vivo setting with almost certain downstream advantages in terms of PK profile, tolerability, stability, clearance rate and therapeutic index, and (ii) that it is useful but not essential for the ADCs to have a cleavable linker in order to make efficacious MMAE-based ADCs.
To conclude, MMAE-bearing PD reagents were synthesised and conjugated to trastuzumab to prepare novel PD-MMAE ADCs 3 and 4 with exceptional control towards DAR 4 species with state-of-the-art characterisation. The PD-modified antibody conjugates were also shown to be stable in serum and retain binding affinity. The PD-MMAE ADCs 3 and 4, as well as NGM-MMAE ADC 1, were shown to be active against a HER2-positive human breast cancer cell line in an in vitro assay in the sub-nM range, and showed excellent efficacy in a mouse xenograph model with the cleavable linker portion of the linker shown to not be essential to the activity of MMAE.
Acknowledgements
The authors acknowledge UCLB, Wellcome Trust, HEFCE, BBSRC and EPSRC for funding this research. We also acknowledge the UCL Chemistry Mass Spectrometry (MS) Facility (Dr Kersti Karu), the EPSRC UK National MS Facility (Swansea), and MRCT for NMR assistance. We also acknowledge Dr Erik Årstad and Dr Kerstin Sanders for support with fluorescence SEC-HPLC.Notes and references
- A. Beck and J. M. Reichert, MAbs, 2014, 6, 15–17 CrossRef PubMed.
- V. Chudasama, A. Maruani and S. Caddick, Nat. Chem., 2016, 8, 114–119 CrossRef CAS PubMed.
- L. Ducry and B. Stump, Bioconjugate Chem., 2010, 21, 5–13 CrossRef CAS PubMed.
- J. A. Flygare, T. H. Pillow and P. Aristoff, Chem. Biol. Drug Des., 2013, 81, 113–121 CAS.
- C. F. McDonagh, E. Turcott and L. Westendorf, et al., Protein Eng., Des. Sel., 2006, 19, 299–307 CrossRef CAS PubMed.
- A. A. Wakankar, M. B. Feeney and J. Rivera, et al., Bioconjugate Chem., 2010, 21, 1588–1595 CrossRef CAS PubMed.
- J. R. Junutula, K. M. Flagella and R. A. Graham, et al., Clin. Cancer Res., 2010, 16, 4769–4778 CrossRef CAS PubMed.
- J. R. Junutula, H. Raab and S. Clark, et al., Nat. Biotechnol., 2008, 26, 925–932 CrossRef CAS PubMed.
- S. O. Doronina, B. E. Toki and M. Y. Torgov, et al., Nat. Biotechnol., 2003, 21, 778–784 CrossRef CAS PubMed.
- Y. T. Adem, K. A. Schwarz and E. Duenas, et al., Bioconjugate Chem., 2014, 25, 656–664 CrossRef CAS PubMed.
- K. J. Hamblett, P. D. Senter and D. F. Chace, et al., Clin. Cancer Res., 2004, 10, 7063–7070 CrossRef CAS PubMed.
- C. A. Boswell, E. E. Mundo and C. Zhang, et al., Bioconjugate Chem., 2011, 22, 1994–2004 CrossRef CAS PubMed.
- D. Jackson, J. Atkinson and C. I. Guevara, et al., PLoS One, 2014, 9, e83865 Search PubMed.
- D. S. Wilbur, M.-K. Chyan and H. Nakamae, et al., Bioconjugate Chem., 2012, 23, 409–420 CrossRef CAS PubMed.
- N. S. Beckley, K. P. Lazzareschi and H.-W. Chih, et al., Bioconjugate Chem., 2013, 24, 1674–1683 CrossRef CAS PubMed.
- C. R. Behrens, E. H. Ha and L. L. Chinn, et al., Mol. Pharm., 2015, 12, 3986–3998 CrossRef CAS PubMed.
- P. Bryant, M. Pabst and G. Badescu, et al., Mol. Pharm., 2015, 12, 1872–1879 CrossRef CAS PubMed.
- B. Q. Shen, K. Xu and L. Liu, et al., Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS PubMed.
- E. R. Boghaert, K. M. Khandke and L. Sridharan, et al., Cancer Chemoth. Pharm., 2008, 61, 1027–1035 CrossRef CAS PubMed.
- A. D. Baldwin and K. L. Kiick, Bioconjugate Chem., 2011, 22, 1946–1953 CrossRef CAS PubMed.
- S. C. Alley, D. R. Benjamin and S. C. Jeffrey, et al., Bioconjugate Chem., 2008, 19, 759–765 CrossRef CAS PubMed.
- S. C. Jeffrey, P. J. Burke and R. P. Lyon, et al., Bioconjugate Chem., 2013, 24, 1256–1263 CrossRef CAS PubMed.
- J. C. Kern, M. Cancilla and D. Dooney, et al., J. Am. Chem. Soc., 2016, 138, 1430–1445 CrossRef CAS PubMed.
- E. S. Zimmerman, T. H. Heibeck and A. Gill, et al., Bioconjugate Chem., 2014, 25, 351–361 CrossRef CAS PubMed.
- L. Castañeda, A. Maruani and F. F. Schumacher, et al., Chem. Commun., 2013, 49, 8187–8189 RSC.
- J. P. M. Nunes, M. Morais and V. Vassileva, et al., Chem. Commun., 2015, 51, 10624–10627 RSC.
- H. Pye, M. A. Butt and H. W. Reinert, et al., Photochem. Photobiol. Sci., 2016, 15, 1227–1238 CAS.
- C. P. Ryan, M. E. B. Smith and F. F. Schumacher, et al., Chem. Commun., 2011, 47, 5452–5454 RSC.
- F. F. Schumacher, M. Nobles and C. P. Ryan, et al., Bioconjugate Chem., 2011, 22, 132–136 CrossRef CAS PubMed.
- F. F. Schumacher, J. P. M. Nunes and A. Maruani, et al., Org. Biomol. Chem., 2014, 12, 7261–7269 CAS.
- F. F. Schumacher, V. A. Sanchania and B. Tolner, et al., Sci. Rep., 2013, 3, 1525 Search PubMed.
- M. E. B. Smith, F. F. Schumacher and C. P. Ryan, et al., J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS PubMed.
- V. Chudasama, M. E. B. Smith and F. F. Schumacher, et al., Chem. Commun., 2011, 47, 8781–8783 RSC.
- (a) M. T. W. Lee, A. Maruani and J. R. Baker, et al., Chem. Sci., 2016, 7, 799–802 RSC; (b) M. T. W. Lee, A. Maruani and D. A. Richards, et al., Chem. Sci., 2017 10.1039/C6SC03655D.
- (a) A. Maruani, S. Alom and P. Canavelli, et al., Chem. Commun., 2015, 51, 5279–5282 RSC; (b) M. T. W. Lee, A. Maruani and V. Chudasama, J. Chem. Res., 2016, 40, 1–9 CrossRef CAS.
- A. Maruani, H. Savoie and F. Bryden, et al., Chem. Commun., 2015, 51, 15304–15307 RSC.
- A. Maruani, M. E. B. Smith and E. Miranda, et al., Nat. Commun., 2015, 6, 6645 CrossRef CAS PubMed.
- G. Badescu, P. Bryant and M. Bird, et al., Bioconjugate Chem., 2014, 25, 1124–1136 CrossRef CAS PubMed.
- C. A. Hudis, N. Engl. J. Med., 2007, 357, 39–51 CrossRef CAS PubMed.
- S. Verma, D. Miles and L. Gianni, et al., N. Engl. J. Med., 2012, 367, 1783–1791 CrossRef CAS PubMed.
- A. Mullard, Nat. Rev. Drug Discovery, 2013, 12, 329–332 CrossRef CAS PubMed.
- V. Bhaskar, D. A. Law and E. Ibsen, et al., Cancer Res., 2003, 63, 6387–6394 CAS.
- R. J. Sanderson, M. A. Hering and S. F. James, et al., Clin. Cancer Res., 2005, 11, 843–852 CAS.
- S. O. Doronina, B. A. Mendelsohn and T. D. Bovee, et al., Bioconjugate Chem., 2006, 17, 114–124 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, 1H and 13C NMR spectra for synthesised compounds, additional SDS-PAGE gels, LCMS chromatograms and other supplementary results. See DOI: 10.1039/c7ra00788d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |